

Abstract
Neuronal transmission of information requires polarized distribution of membrane proteins within axonal compartments. Membrane proteins are synthesized and packaged in membrane-bounded organelles (MBOs) in neuronal cell bodies and later transported to axons by microtubule-dependent motor proteins. Molecular mechanisms underlying targeted delivery of MBOs to discrete axonal subdomains (i.e. nodes of Ranvier or presynaptic terminals) are poorly understood, but regulatory pathways for microtubule motors may be an essential step. In this work, pharmacological, biochemical and in vivo experiments define a novel regulatory pathway for kinesin-driven motility in axons. This pathway involves enzymatic activities of cyclin-dependent kinase 5 (CDK5), protein phosphatase 1 (PP1) and glycogen synthase kinase-3 (GSK3). Inhibition of CDK5 activity in axons leads to activation of GSK3 by PP1, phosphorylation of kinesin light chains by GSK3 and detachment of kinesin from transported cargoes. We propose that regulating the activity and localization of components in this pathway allows nerve cells to target organelle delivery to specific subcellular compartments. Implications of these findings for pathogenesis of neurodegenerative diseases such as Alzheimer's disease are discussed.
Keywords: axonal transport, CDK5, GSK-3, kinesin, protein phosphatase PP1
Introduction
Mature axons lack protein synthesis machinery, so proteins and membrane components necessary for maintaining neuronal functions need to be transported from the cell body into axons by axonal transport (Brady, 1993). Fast anterograde axonal transport (FAT) delivers many different membrane proteins packaged in membrane-bounded organelles (MBOs) to specific subdomains within axons. For example, sodium channels are delivered to nodes of Ranvier, whereas synaptic vesicles are provided to presynaptic terminals. Specialized delivery of MBOs at biochemically and structurally distinct domains within neurons implies the existence of regulated targeting mechanisms (Morfini et al, 2001).
The molecular motor kinesin is essential for fast anterograde transport of a variety of MBOs in neurons (Brady, 1995). Kinesin is a heterotetramer with two heavy and two light chains. Heavy chains (KHCs) are responsible for generation of force, comprising ATPase and microtubule (MT)-binding domains. Kinesin light-chain subunits (KLCs) are essential for kinesin binding to transported cargoes and are implicated in both cargo specificity (Cyr et al, 1991; Stenoien and Brady, 1997; Khodjakov et al, 1998; Tsai et al, 2000) and regulation of motility (Tsai et al, 2000; Morfini et al, 2001, 2002b). Molecular mechanisms for regulating kinesin-based motility have begun to emerge.
Kinesin is a phosphoprotein in vivo (Hollenbeck, 1993; Morfini et al, 2001), so regulation of kinesin activity may involve specific protein kinases and phosphatases. Different kinesin functions may be affected by phosphorylating specific sites within kinesin (Donelan et al, 2002; Morfini et al, 2002b). One property of kinesin regulated by kinases in vivo is association with cargoes, and specific phosphorylation of KLCs leads to release of kinesin from MBOs (Morfini et al, 2002b).
Glycogen synthase kinase 3 (GSK3) phosphorylates KLCs and promotes kinesin release from MBOs (Morfini et al, 2002b). GSK3 is a ubiquitous kinase with roles in the Wnt pathway, intracellular trafficking, adhesion and apoptosis (Frame and Cohen, 2001). Multiple mechanisms for inactivating GSK3 are known (Woodgett, 2001), but less is known about pathways for activation. GSK3 can interact with kinesin and inhibits fast anterograde (kinesin-based), but not retrograde (dynein-based), axonal transport in extruded squid axoplasm (Morfini et al, 2002b). This finding suggested that GSK3 must be inactive in much of the axon and that local pathways for modulating its activity must exist. Selective activation of GSK3 in specific neuronal compartments may allow delivery of specific cargo to discrete subcellular functional domains in neurons, but relatively little is known about localized regulation of GSK3 kinase activity.
As with GSK3 activation, inhibiting CDK5 kinase in axons specifically reduced transport rates for anterograde, but not retrograde, FAT (Ratner et al, 1998). This was shown by pharmacological and biochemical manipulations of CDK5 activity. The targets and molecular basis for CDK5-mediated regulation of kinesin-driven motility were not defined, because kinesin lacks CDK5 consensus sites. One possibility was a functional link between CDK5 and GSK3. The present work identifies a novel mode of GSK3 activation in neurons within axonal compartments. A pathway is defined that links activities of CDK5 and protein phosphatase 1 (PP1) to GSK3. Inhibition of CDK5 leads to activation of PP1 and GSK3, resulting in the release of kinesin from MBOs. We propose that regulating the distribution and activities of one or more elements in this pathway allows neurons to deliver kinesin cargoes selectively to different subcellular domains.
Results
Inhibiting CDK5 reduces kinesin-based motility
FAT was analyzed in extruded axoplasm from squid giant axons, an in vivo model for the study of FAT that was instrumental in the discovery of kinesins (Brady, 1985; Vale et al, 1985). Extruded axoplasms continue bidirectional MBO transport with properties essentially unchanged from intact axons (Brady et al, 1985). Video-enhanced microscopic techniques allow quantitative analysis of MBO movement for both directions of FAT. Typically, anterograde rates are 1.5–2.0 μm/s and retrograde rates are 1–1.2 μm/s in perfused axoplasms (see Figure 1A–C). These rates are maintained with little or no reduction for >1 h with control buffers (Brady et al, 1985).
Figure 1.
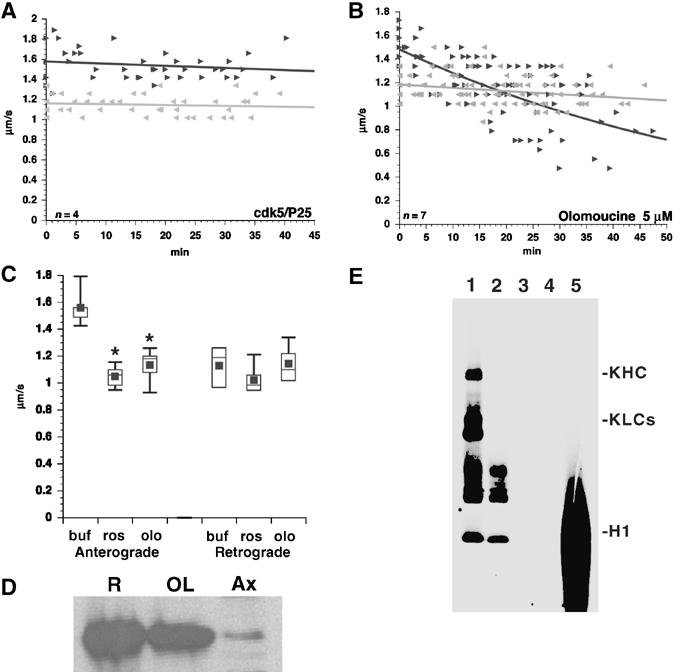
Sustained CDK5 activity is required for maintenance of kinesin-driven motility in axons. (A) Perfusion of active CDK5/p25 (1 μM) into axoplasm had no effect on either anterograde (dark) or retrograde (gray) FAT rates. (B) Perfusion of the CDK5 inhibitor Olo (5 μM) decreases anterograde, but not retrograde, FAT. (C) Average transport rates for anterograde and retrograde FAT in standard perfusion buffer (buf) or CDK5 inhibitors roscovitine (0.7 μM, ros), and Olo (5 μM, olo). Note inhibitory effect of CDK5 inhibitors on anterograde, but not retrograde, FAT. Differences (*) are significant at P⩽0.0001. (D) Anti-CDK5 detects endogenous CDK5 in squid optic lobe (OL) and isolated axoplasm (Ax). Rat brain (R) lysate is a positive control. (E) Kinesin is not directly phosphorylated by CDK5. Purified rat brain kinesin was incubated with recombinant casein kinase 2 (CK2 (1)) or CDK5/p25 (3). CDK5/p25 failed to phosphorylate kinesin despite displaying strong kinase activity toward H1 histone (5). CK2 phosphorylates both kinesin heavy (KHC) and light (KLC) chains (Donelan et al, 2002). Control reactions with CK2 (2) or GST-CDK5/P25 (4) alone are also shown. Longer exposures show P25 autophosphorylation.
Conventional kinesin is the most abundant kinesin family member in mammalian brain and squid axoplasm (Wagner et al, 1989) and is essential for MBO movement in axoplasm (Brady et al, 1990; Stenoien and Brady, 1997). Studies with olomoucine (Olo) (Ratner et al, 1998) first provided evidence that kinases were important for FAT regulation. Olo and roscovitine are two well-characterized inhibitors of cdc2-like kinases, with IC50 values determined for >28 different kinases (Vesely et al, 1994; Bain et al, 2003). Cdc2 kinase family members (cdc2/cyclin A, CDK5/P35, etc.) are particularly sensitive to Olo (IC50 <10 μM) and roscovitine (IC50 ⩽0.25 μM). The major cdc2-like kinase in mature, postmitotic neurons is CDK5 (Hellmich et al, 1992; Lew and Wang, 1995). Immunoblots detect CDK5 in squid axoplasm (Figure 1D), comparable to distributions reported in mammalian axons (Tsai et al, 1993; Pigino et al, 1997). Axoplasmic CDK5 is active (Takahashi et al, 1995) and perfusion of 1 μM active GST-CDK5/p25 into axoplasm did not affect vesicle motility (Figure 1A). Perfusion of 5 μM Olo (IC50 CDK5=3 μM) (Figure 1B and C; see also Ratner et al, 1998) or 0.7 μM roscovitine (IC50 ros=0.25 μM) (Figure 1C) into axoplasm significantly reduced anterograde, but not retrograde, transport. These studies show that active CDK5 is required for kinesin-based motility in axons.
CDK5 inhibition increases KLC phosphorylation
Specific inhibition of anterograde, but not retrograde, FAT by CDK5 suggested that kinesin-based motility is modulated by CDK5. However, recombinant CDK5/p25 did not phosphorylate either KHC or KLC purified from rat brain (Figure 1E). CDK5/P25 also failed to phosphorylate recombinant KHC and KLC (not shown); so potential CDK5 sites were not phosphorylated in purified rat brain kinesin. This suggested that CDK5 does not directly phosphorylate kinesin.
Next we determined whether kinesin phosphorylation changed in cells when CDK5 was inhibited. Primary cortical neurons were radiolabeled and kinesin was immunoprecipitated from lysates. KHCs and KLCs were both phosphorylated (Figure 2A), but Olo treatment of cultured neurons increased KLC phosphorylation by 20% (Figure 2B). CDK5 activity in mammals requires activation by one of two activator subunits, p35 and p39 (Ko et al, 2001). To rule out nonspecific effects of Olo, we did metabolic labeling of cultured cortical neurons from mice lacking both p35 and p39 subunits (i.e. lacking CDK5 activity) As with Olo, KLC phosphorylation increased in double knockout (KO) neurons relative to wild type (Figure 2B). KLCs have multiple phosphorylation sites consistent with modification by different protein kinases. Some sites appear to be constitutively phosphorylated (Hollenbeck, 1993; Morfini et al, 2002b), but at least one has increased phosphorylation when CDK5 activity is reduced by Olo or absence of CDK5 activators. Kinesin is not a substrate for CDK5, so effects of reducing CDK5 activity on kinesin-driven motility and KLC phosphorylation must be indirect.
Figure 2.
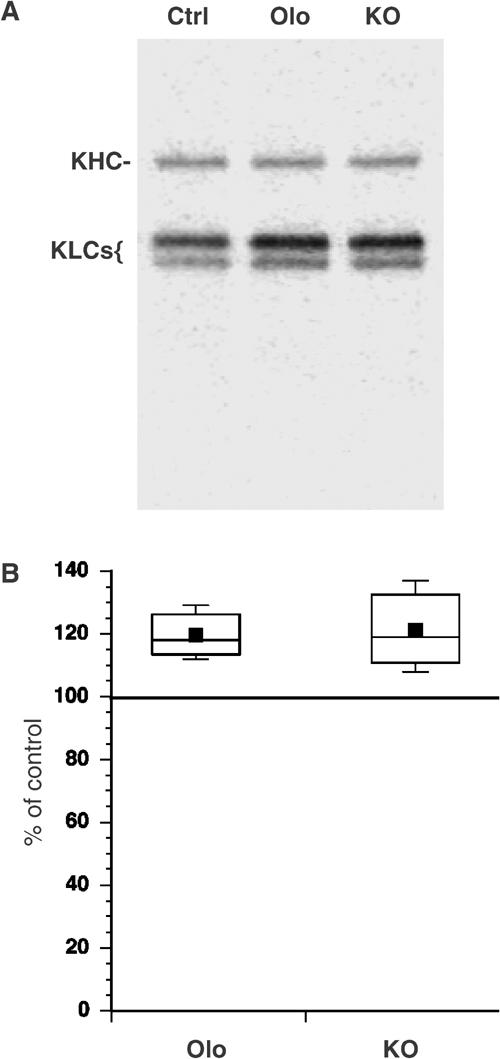
Inhibiting CDK5 increases KLC phosphorylation. (A) Representative autoradiogram showing immunoprecipitated–radiolabeled kinesin from wild-type (Ctrl), Olo-treated (Olo) or p35/p39 KO primary cultures of cortical neurons. (B) Phosphoimager quantitation showed increased KLC phosphorylation in neurons derived from p35−/− and p39−/− double KO and wild-type mouse embryos treated with Olo. Differences were significant at P⩽0.01.
CDK5 inhibition results in GSK3 activation
To determine whether CDK5 activates kinases in axoplasm, isolated axoplasm±5 μM Olo were incubated with [32P]ATP and histone H1, a substrate for many protein kinases in vitro. Phosphorylation of H1 and the major neurofilament subunits increased in Olo-treated axoplasm extracts (Figure 3A), so inhibiting CDK5 activates other protein kinase(s). To identify the kinases involved, peptide substrates for specific kinases (extracellular-activated kinase (ERK), casein kinase I and GSK3) were tested for an ability to block Olo-induced increases in H1 histone phosphorylation in axoplasm (Figure 3B). Only CREB phosphopeptide (CREBp), a specific GSK3 substrate (Wang et al, 1994b), significantly reduced phosphorylation of H1 histone (lane 5, Figure 3B). To evaluate GSK3 activity in Olo-treated axoplasm, kinase assays specific for GSK3 were performed. Samples treated with 5 μM Olo (Figure 3C) showed increased GSK3 activity relative to untreated axoplasms (P=0.001 in a pooled t-test). Neither Olo nor roscovitine affected GSK3 activity in kinase assays in vitro (Figure 3D), even at 50 μM Olo. Thus, inhibition of axonal CDK5 results in GSK3 activation.
Figure 3.
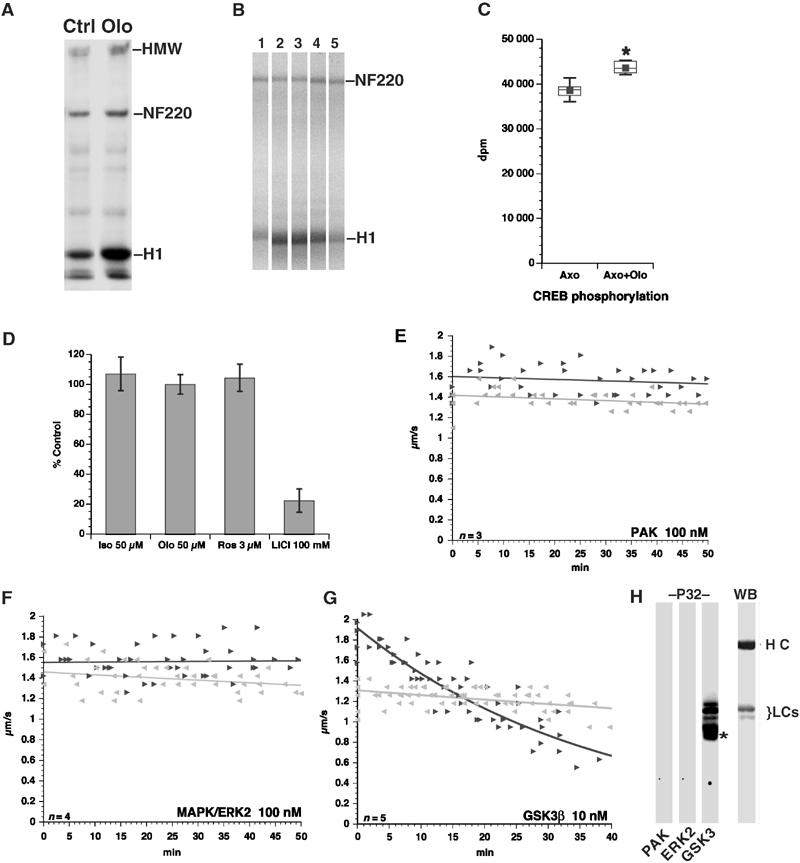
Inhibiting CDK5 activates GSK3. (A) Axoplasms were treated with DMSO (Ctrl) or 5 μM Olo and radiolabeled ATP using histone H1 (H1) as a phosphate acceptor. Autoradiogram shows that Olo increases H1 phosphorylation. Neurofilament heavy chain (NF220) and HMW neurofilament also exhibit increased phosphorylation. (B). Control (1) or Olo-treated (2–5) axoplasms prepared as in (A) were incubated with no peptide (1, 2), ERK peptide (3), CK1 peptide (4) or CREBp (5). Only CREBp prevented Olo-induced increases in histone H1 phosphorylation. (C) GSK3 kinase activity was measured in axoplasm extracts using CREBp as substrate. CREBp phosphorylation increased relative to control axoplasms (Axo) with Olo (Axo+Olo). Increase is significant (P=0.0017; pooled t-test (*)). (D) Effect of 50 μM Iso-Olo (Iso), 50 μM Olo (Olo), 3 μM roscovitine (Ros) and 100 mM LiCl on GSK3 phosphorylation of CREBp in vitro. Values are expressed as percent of GSK3 activity without inhibitors. Vesicle motility assays in isolated axoplasm show effects of PAK (E), ERK2 (F) and GSK3 (G) kinase activities on FAT. Note the specific inhibitory effect on anterograde FAT of GSK3, but not PAK or ERK2, similar to that of CDK5 inhibition. (H) Autoradiogram (P32) shows that GSK3, but not PAK or ERK2, phosphorylates KLCs. Immunoblot (WB) shows position of kinesin heavy (HCs) and light chains (LCs). Asterisk (*) indicates autophosphorylated GSK3.
CDK5 inhibition may also activate ERK (Wang et al, 1994b) and P21 kinase (PAK) (Nikolic et al, 1998). These observations prompted us to compare effects of ERK2, PAK and GSK3 on vesicle motility. Neither 100 nM of active ERK2 nor 100 nM of PAK catalytic fragment (Figure 3E and F) affected FAT in extruded axoplasm, suggesting that neither is a likely mediator for inhibition of kinesin motility. As with Olo, perfusion of active GSK3 at 10 nM profoundly inhibited anterograde FAT (Figure 3G). This effect correlates with phosphorylation of KLC and release of kinesin from transported cargo (Morfini et al, 2002b). Finally, in vitro kinase assays showed that GSK3, but not PAK or ERK2, directly phosphorylates KLCs (Figure 3H).
Reduced anterograde FAT by CDK5 inhibition requires GSK3 activation
Given that inhibiting CDK5 increases GSK3 activity and KLC phosphorylation, CDK5 and GSK3 could be part of a common pathway for regulating FAT. Previous studies showed that CREBp at 0.5 mM blocks the action of GSK3 on FAT (Morfini et al, 2002b). Co-perfusion of 5 μM Olo and 0.5 mM CREBp into axoplasm prevented inhibition of anterograde FAT by Olo (Figure 4A and B). Perfusion of 1 mM CREBp alone had no effect on either anterograde or retrograde transport (Figure 4B). Thus, GSK3 activation is required for reduced anterograde FAT due to CDK5 inhibition.
Figure 4.
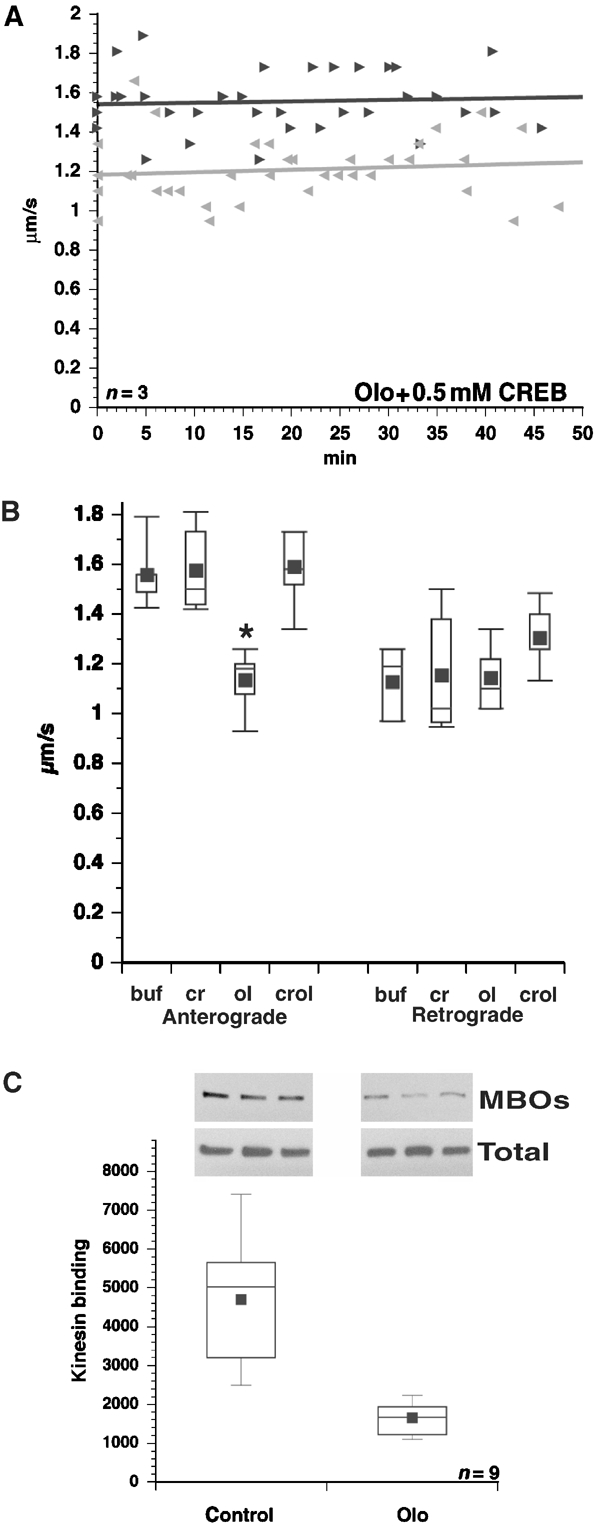
GSK3 mediates reduced kinesin-driven motility due to CDK5 inhibition. (A) Co-perfusion of 0.5 mM CREBp and 5 μM Olo abrogates the effects of Olo on FAT. (B) Average transport rates are shown for anterograde and retrograde FAT with control buffer (buf), 1 mM CREBp (cr), 5 μM Olo (ol) and 5 μM Olo plus CREBp (crol). Note that CREBp blocks the effect of Olo on anterograde FAT. CREBp alone did not affect either direction of transport. Asterisk denotes P<0.001. (C) Inhibiting CDK5 in cortical neurons reduces the amount of kinesin associated with microsomes (MBOs) but not total kinesin (Total). Representative immunoblots of KHC with H2 and fluorescent secondary antibodies are shown. The Y-axis shows pixel values from Typhoon scans.
KLC phosphorylation by GSK3 promotes kinesin detachment from MBOs (Morfini et al, 2002b). To evaluate effects of CDK5 inhibition on kinesin association with MBOs, primary cultured cortical neurons were treated with vehicle or Olo. Cells were harvested and MBOs were isolated by subcellular fractionation. MBO-associated kinesin levels were significantly decreased in Olo-treated cortical neurons, but overall kinesin levels remained unchanged (Figure 4C), consistent with GSK3 activation.
CDK5 inhibition leads to GSK3β Ser9 dephosphorylation
Phosphorylations at Ser9 of GSK3β (Ser21 of GSK3α) and Tyr216 regulate GSK3 kinase activity (Wang et al, 1994a). Phosphorylation at Ser9 inactivates GSK3 and dephosphorylation activates it. Autophosphorylation at Tyr216 (Cole et al, 2004) also enhances GSK3 activity, but only if Ser9 is dephosphorylated (Wang et al, 1994a). Thus, one can follow activation of GSK3 by measuring either dephosphorylation of Ser9 or phosphorylation at Tyr216. Effects of CDK5 inhibition on GSK3β phosphorylation at these regulatory residues were analyzed in PC12 cells and cortical neurons. Immunoblots were probed for GSK3 phosphorylation at Ser9 or Tyr216. Addition of Olo, but not vehicle or iso-Olo, to PC12 cells or cortical neurons resulted in time-dependent dephosphorylation of phosphoSer9 (Figure 5A). Corresponding increases were seen in phosphorylation at Tyr216 (not shown), consistent with increased GSK3 activity. Parallel immunoblot analysis of tau, an endogenous substrate for GSK3 in neurons (Lovestone et al, 1996), showed increased immunoreactivity with pS396 and PHF-1, but not Tau-5, in Olo-treated cells. PHF-1 and pS396 recognize tau at phosphoepitopes modified by GSK3 (Bhat et al, 2003), whereas Tau-5 is unaffected by tau phosphorylation (Figure 5B). Interestingly, phosphorylation of tau at these sites is generally associated with tau not being bound to MTs.
Figure 5.
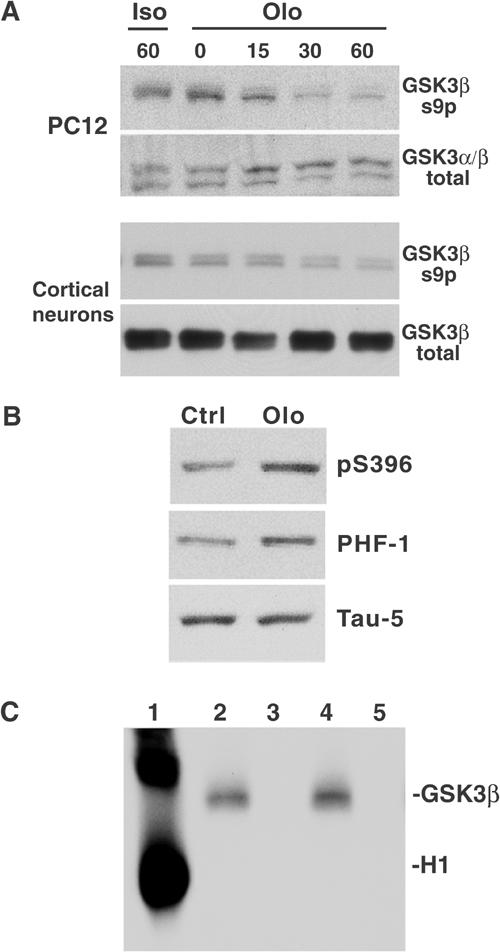
Inhibition of CDK5 leads to GSK3β Ser9 dephosphorylation/activation. (A) PC12 cells or primary cortical neurons were treated with iso-Olo (Iso) or Olo (Olo) for the indicated times (min). Samples were immunoblotted with total GSK3β or GSK3β Ser9p antibodies. (B) Immunoblots with PHF-1 and pS396 antibodies show increased tau phosphorylation at Ser396 in Olo-treated (Olo) cortical neurons relative to vehicle-treated (Ctrl) ones. These epitopes are GSK3 sites in vitro and in vivo. Similar levels of total tau (Tau-5) indicate equal protein loading. (C) CDK5 does not phosphorylate GSK3 Ser9. Histone H1 (lane 1), GSK3β wild type (lanes 2 and 4) or GSK3β kinase-dead (lanes 3 and 5) were incubated with (lanes 1–3) or without CDK5/25 (lanes 4–5). Autoradiogram shows that CDK5/p25 phosphorylated histone (H1) but not GSK3 (GSK3β). Note: Wild-type GSK3β autophosphorylation in lanes 2 and 4 does not increase in the presence of CDK5 (lane 2).
CDK5 might directly phosphorylate GSK3β at Ser9, but no 32P was incorporated into recombinant wild-type or kinase-dead GSK3β incubated with CDK5/p25 in vitro (lanes 2–5, Figure 5C). This same CDK5/P25 was strongly active against histone H1 (lane 1, Figure 5C). CDK5/P25 also failed to phosphorylate and activate recombinant PKB, a kinase that phosphorylates GSK3 in vivo. No changes in PKB phosphorylation were observed in Olo-treated cells (not shown). These experiments suggested that CDK5 keeps a pool of GSK3β in an inactive Ser9-phosphorylated form by an indirect mechanism.
CDK5 activity inhibits PP1 activity
CDK5 inhibition could activate a protein phosphatase that dephosphorylates Ser9 and activates GSK3β. To test this, Olo and okadaic acid were co-perfused into axoplasm (Figure 6A). Okadaic acid is a strong inhibitor of PP1 and PP2 serine–threonine phosphatases (Hardie et al, 1991). Okadaic acid (50 μM) had no effect on kinesin motility (Bloom et al, 1993), but co-perfusion of 20 nM okadaic acid and Olo prevented inhibition of anterograde FAT (Figure 6A). This suggested that Olo effects on kinesin-based motility involved activation of a Ser–Thr protein phosphatase. Consistent with this, incubating cortical neurons with okadaic acid (Figure 6C) or cantharidin (not shown) increased GSK3β Ser9 phosphorylation.
Figure 6.
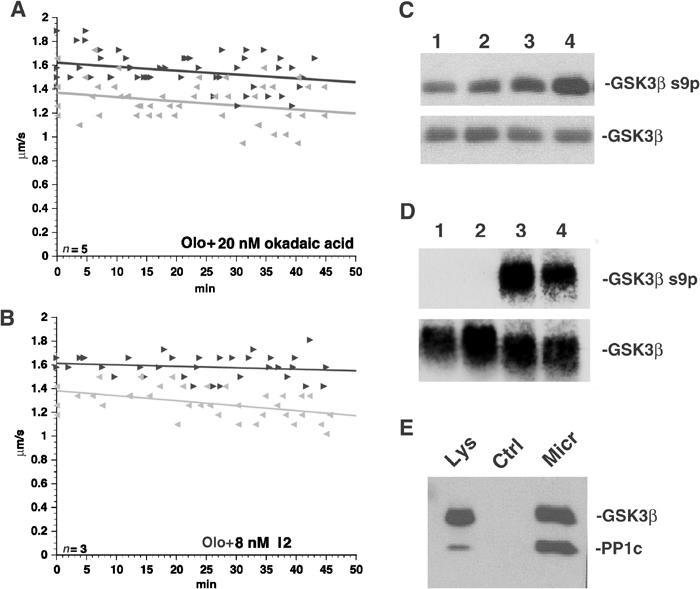
Reduced kinesin-driven motility due to inhibition of CDK5 depends on PP1 and PP1 activates GSK3. (A) Co-perfusion of Olo and 20 nM okadaic acid prevented inhibition of FAT by Olo. (B) Co-perfusion with 8 nM I2 also blocked Olo effects, suggesting that PP1 mediates Olo-induced inhibition of kinesin-driven motility. (C) Incubation of cortical neurons with 0, 5, 20 and 50 nM okadaic acid (lanes 1–4) shows dose-dependent increases in GSK3β Ser9 phosphorylation, suggesting a role for serine–threonine protein phosphatases in GSK3β regulation. (D) Recombinant PP1 can dephosphorylate Ser9 of GSK3β. Recombinant GSK3β was incubated for 30 min with 100 μM ATP to allow autophosphorylation. PP1 catalytic subunit (lanes 1 and 2) or vehicle (lanes 3 and 4) was added to autophosphorylated GSK3β and immunoblotted with total GSK3β or GSK3β pSer9 antibodies. (E) Immunoblots show that microcystin–Sepharose (Micr), but not control Sepharose (Ctrl), pulls down GSK3, suggesting association between GSK3 and phosphatases. Note an increase in the PP1/GSK3β ratio between rat brain lysate (Lys) and microcystin–Sepharose lanes.
IC50 values of okadaic acid for PP1 and PP2 are 10 and 0.1 nM respectively. So these studies did not identify which phosphatase type was activated by CDK5 inhibition. To determine this, inhibitor-2 (I2), a small polypeptide that inhibits PP1 with an IC50 of 2 nM but not PP2 (Cohen et al, 1988; Haystead et al, 1989), was co-perfused with Olo. I2 blocked inhibition of anterograde FAT by Olo (Figure 6B). PP1 also dephosphorylates GSK3β at Ser9 in vitro (Figure 6D), suggesting that PP1 mediates Olo effects on kinesin-based motility.
To see if CDK5, GSK3 and PP1 interact, protein phosphatases were affinity purified by microcystin–Sepharose (Moorhead et al, 1994). Microcystin-, but not control Sepharose, pulled down GSK3 with PP1 from mouse brain extracts (Figure 6E). PP1 to GSK3 ratios were nearly equal in microcystin pulldowns but quite different in lysates (GSK3≫PP1), suggesting a pool of GSK3 in a PP1 complex. These results indicate that CDK5 inhibition increases GSK3β activity by activating PP1 to dephosphorylate GSK3β at Ser9.
CDK5, PP1 and GSK3 colocalize at sites of active vesicle delivery
Distribution of GSK3, PP1 and CDK5 was evaluated in cell bodies, neurites and growth cones, a main site for membrane addition in growing neurites (Craig et al, 1995). All three were detected in cell bodies, neuritic shafts and growth cones of cultured neurons (Figure 7A). At higher magnification (Figure 7C), CDK5 and GSK3 showed extensive overlap with PP1 in central areas of growth cones. Central region localization, a site where vesicles accumulate, is distinct from both MTs and microfilaments (Dailey and Bridgman, 1991; Morfini et al, 2002b). Immunoblot analysis of isolated growth cone particles (Pfenninger et al, 1983) confirmed the presence of CDK5, PP1 and active GSK3 in growth cones (Figure 7B). In summary, CDK5, PP1 and GSK3 exhibit a similar distribution in neurites and were found in neuronal compartments where delivery of membrane proteins actively takes place.
Figure 7.
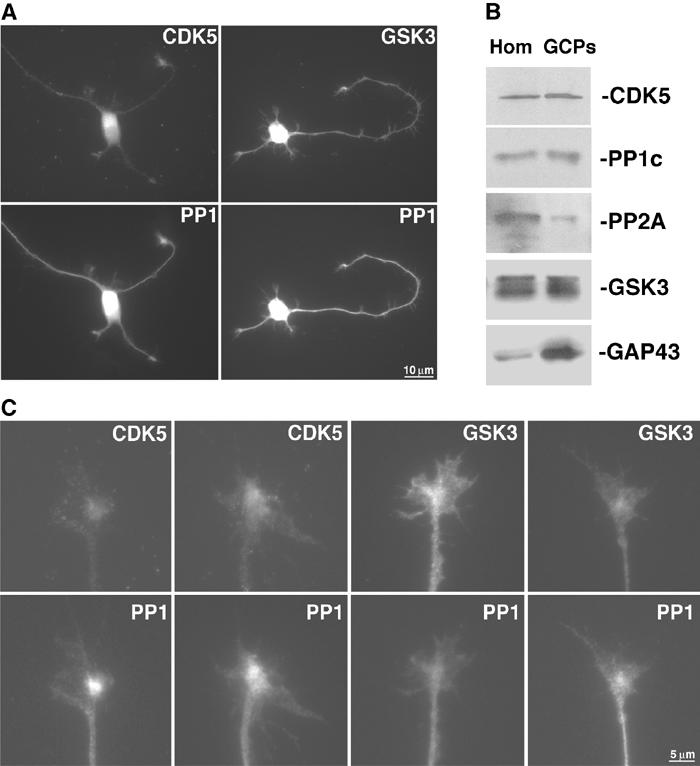
CDK5, GSK3 and PP1 colocalize in the centers of axonal growth cones. (A) Double immunostainings for CDK5 and PP1 (left) and total GSK3 and PP1 (right) show co-enrichment of CDK5, PP1 and GSK3 in growth cones of cultured neurons. All three enzymes are abundant in cell bodies and found all along the neurites. (B) Purified growth cone particles (GCPs) and rat brain homogenates (Hom) were analyzed by immunoblot. High levels of GAP43 show enrichment in GCP components. GSK3, CDK5 and PP1 are all present at significant levels. In contrast, PP2A is not enriched relative to homogenates. (C) Higher magnification of CDK5/PP1 and GSK3/PP1 shows colocalization in the centers of actively extending growth cones.
Discussion
Nerve cell survival and proper function require efficient delivery of proteins from cell body to axonal and dendritic processes. Neurons require accurate membrane protein compartmentalization to function, but little is known about how axonal subdomains are generated and maintained. Mechanisms must exist to allow MT-dependent motors to deliver different types of MBOs as needed to specific subcellular compartments. Phosphorylation and dephosphorylation appear to regulate kinesin-based motility (Hollenbeck, 1990; Morfini et al, 2001; Donelan et al, 2002), and kinase/phosphatase activities relevant to kinesin function have been identified.
GSK3 is implicated in the regulation of MBO delivery in neurons (Morfini et al, 2002b). Phosphorylation of KLC by GSK3 promotes removal of kinesin from its cargo. As a result, pathways leading to GSK3 activation are likely to inhibit kinesin-based motility. A major mode of GSK3 inactivation is phosphorylation of Ser9 in GSK3β or Ser21 in GSK3α. Ser9 phosphorylation can occur either through autophosphorylation (Wang et al, 1994a) or via the action of kinases, such as PKB/Akt (Woodgett, 1994). In addition, proteins without intrinsic enzymatic activity (i.e. AKAP220 and axin) act as scaffolds to bind GSK3 and limit its activity to a particular compartment or substrate (Ali et al, 2001). Multiple pathways for inactivating GSK3 and localizing its activity exist, but less is known about pathways for its activation. In adipocytes or epithelial cells, GSK3 may be constitutively active (Ali et al, 2001), but increased GSK3 activity in neurons leads to neurite retraction (Munoz-Montano et al, 1999), apoptosis (Lucas et al, 2001), increased embryonic lethality (Hoeflich et al, 2000) and behavioral abnormalities (Hernandez et al, 2002). This suggests that GSK3 activation in neuronal cells must be a transient and localized event.
CDK5 activity is essential for proper neuronal function (Smith et al, 2001). Lack of CDK5 is embryonic lethal (Ohshima et al, 1996), and deletion of both normal CDK5 activators (p35 and p39) has a phenotype comparable to CDK5 deletion (Ko et al, 2001). Animals survive to adulthood when only one activator is eliminated (Chae et al, 1997), but have defects in neuronal migration and axon pathfinding. Sustained CDK5 activity is required for normal kinesin-driven motility in neurons (Ratner et al, 1998), vesicle transport from Golgi to neurites (Paglini et al, 2001) and neurite outgrowth (Nikolic et al, 1998).
Both tau phosphorylation and GSK3 activity are increased in p35 KO mice (Hallows et al, 2003) and the tau sites with increased phosphorylation were GSK3 sites (Bhat et al, 2003). This is consistent with a mechanism in which CDK5 inhibits GSK3 activity. Further, P35/P39 play a role in localizing CDK5 activity, which appears critical for normal function of CDK5 in neurons (Humbert et al, 2000). Overexpressing p35/CDK5 and tau does not increase tau phosphorylation (Van den Haute et al, 2001), but overexpressing CDK5 with p25, a proteolytic fragment of p35 with an altered subcellular distribution, increases phosphorylation of some but not all CDK5 substrates (Cruz et al, 2003). Substrates with increased phosphorylation include tau epitopes modified by GSK3. This is consistent with reports of increased GSK3 activity in association with tangles found in mice overexpressing p25/CDK5 and tau (Noble et al, 2003). These observations led to suggestions that p25 targets CDK5 preferentially to pathogenic targets (Cruz et al, 2003; Noble et al, 2003) while reducing phosphorylation of normal substrates.
Evidence presented here suggests a novel pathway for activating GSK3 in axons that includes CDK5 and PP1. First, GSK3 activity is increased in axoplasm and cultured nerve cells treated with specific Cdk5 inhibitors. Second, both pharmacological and genetic elimination of CDK5 activity increases phosphorylation of KLC and tau, the latter at epitopes phosphorylated by GSK3. Third, kinesin associated with MBOs was significantly reduced by inhibition of CDK5, consistent with consequences of KLC phosphorylation by GSK3. Finally, CREBp, a peptide substrate for GSK3, prevented inhibition of kinesin-driven motility by Olo in axoplasm.
Although CDK5-mediated effects on transport require GSK3 activity, CDK5 does not act directly on GSK3. CDK5 failed to phosphorylate GSK3 or affect GSK3 activity in vitro, but CDK5 inhibition resulted in Ser9 dephosphorylation and activation of GSK3 in vivo. Inhibition of CDK5 in cortical neurons reduced GSK3 Ser9 phosphorylation. Conversely, inhibition of S/T phosphatases by cantharidin or okadaic acid significantly increased GSK3 phosphorylation at Ser9, implicating phosphatases in the regulation of GSK3 activity by Ser9 modification. PP1 dephosphorylated GSK3β Ser9 in vitro. More importantly, both okadaic acid and the selective PP1 inhibitor I2 prevent inhibition of anterograde FAT by Olo. PP1 is a major serine/threonine phosphatase in brain that regulates many cellular functions (Cohen, 2002). Thus, effects of CDK5 on vesicle motility in axoplasm depend on both PP1 and GSK3 activity.
Several lines of evidence indicate that GSK3, PP1 and CDK5 can physically interact in vivo. The three components of this pathway have overlapping distributions in specific neuronal domains, that is, neuritic growth cones. Microcystin binds PP1 but not GSK3 or CDK5, and microcystin resins pull down PP1, GSK3 and CDK5 (not shown) from brain lysates. Interestingly, PP1/GSK3 ratios are greater in microcystin pulldowns than in brain lysates. Similarly, PP1, GSK3 and CDK5 are found in high-molecular-weight complexes from brain (Lee et al, 1996; Agarwal-Mawal and Paudel, 2001; Tanji et al, 2002).
Our results indicate that normal CDK5 activity keeps a pool of PP1 inactive. CDK5 may suppress PP1 activity in several ways. Cdc2, a closely related kinase with in vitro substrate specificity nearly identical to CDK5 (Smith and Tsai, 2002), phosphorylates PP1 at T320 and inactivates PP1 catalytic subunit during mitosis (Dohadwala et al, 1994). However, CDK5 failed to phosphorylate PP1 in vitro (not shown). PP1 regulation in neurons involves a diverse set of regulatory partners. Over 50 regulatory subunits for PP1 have been described that target PP1 to specific subcellular locations, that include inhibitor-1, DARP-32 and spinophilin (Cohen, 2002). Many are potent regulators of PP1 activity and regulation often depends on phosphorylation. For example, phosphorylating DARP-32 or inhibitor-1 by PKA converts them into potent inhibitors of PP1 (Bibb et al, 1999). These precedents suggest that CDK5 phosphorylation of one or more neuronal proteins may confer PP1-specific inhibitory properties. Identification of specific PP1 partners regulated by CDK5 phosphorylation should complete mapping of this pathway.
In a model encompassing these data (Figure 8), anterograde FAT depends on a regulated balance between kinase and phosphatase activities in specific cellular domains. Localized changes in the balance between kinase and phosphatase activities exist. For example, changes in NF phosphorylation occur at nodes of Ranvier, a compartment where Na channels are delivered to the plasma membrane (de Waegh et al, 1992). Local inhibition of CDK5 and local activation of GSK3 could promote delivery of newly synthesized material to a node. Possible mechanisms for local inhibition of CDK5 include the tumor suppressor APC (Ratner et al, 1998), P35 binding proteins like C42 (Ching et al, 2002) or CDK5 modification by other kinases (Matsuura and Wang, 1996). Localized inhibition of CDK5 by any of these might locally increase PP1 activity and activate GSK3. Such mechanisms would provide neurons with diverse modes for assembly and maintenance of subcellular compartments with unique biochemical identities. Additionally, this model could account for observed increases in GSK3 activity and cytoskeletal protein phosphorylation in p35 KO mice (Hallows et al 2003) as well as the opposing effects of CDK5 and GSK3 in APP processing (Ryder et al, 2003).
Figure 8.
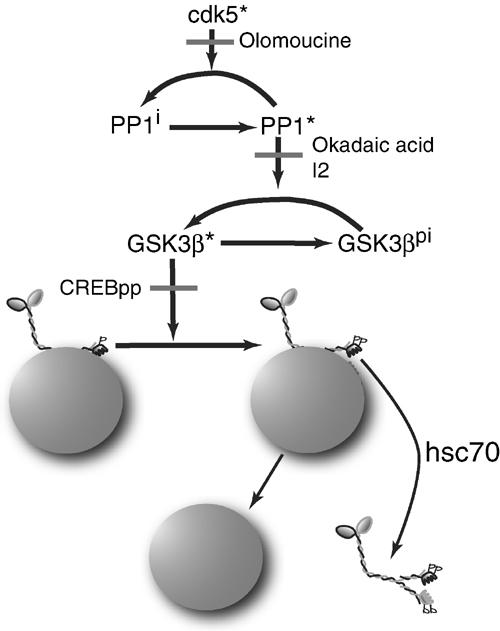
Schematic of a pathway for regulation of GSK3 activity and kinesin function in axons by CDK5 and PP1. KLCs on MBOs have a priming phosphorylation making them substrates for GSK3, but GSK3 is inactive in axons with active CDK5 and suppressed PP1 activity. CDK5 is normally active in axons, but local inhibition of CDK5 activity (Olo) (Ratner et al, 1998) allows local activation of PP1 that dephosphorylates and activates GSK3. PP1 inhibitors (okadaic acid, I2) blocked activation of GSK3. Further phosphorylation of KLCs by GSK3 makes kinesin subject to removal from MBOs by hsc70 and effects delivery of that cargo to an axonal domain. Kinesin phosphorylation is blocked by the addition of an inhibitor of GSK3 (CREBpp). Released kinesin is apparently degraded (Li et al, 1999). Consistent with this model, local changes in kinase/phosphatase activity were implicated in targeting Na channels to nodes of Ranvier (de Waegh et al, 1992).
Defects in protein transport may play critical roles in Alzheimer's disease (Price et al, 1996; Morfini et al, 2002a) and other neurodegenerative conditions (Borchelt et al, 1998; Williamson and Cleveland, 1999). In Alzheimer's disease, activities and phosphorylation patterns for CDK5 and GSK3 are altered (Mandelkow and Mandelkow, 1998; Morfini et al, 2002a). The role of GSK3 in regulating kinesin-based motility suggested that axonal transport might be a vulnerable step in Alzheimer's pathogenesis. Significantly, presenilin mutants causing familial Alzheimer's disease increase GSK3 activity, leading to defective transport of kinesin cargoes (Pigino et al, 2003). As CDK5 and GSK3 activity can be regulated via multiple pathways, different pathogenic mechanisms might converge to alter kinesin-based transport. Inappropriate activation of GSK3 could cause premature release of kinesin from its cargo and account for many changes seen in sick neurons. Due to their extreme polarization and complexity, neurons are especially vulnerable to decrements in FAT, and misregulation of kinesin-driven motility may be sufficient to promote the characteristic features of these neuropathies.
Materials and methods
Antibodies and reagents
For immunoblots, the following were used: H2 anti-KHC (1:2000) (Pfister et al, 1989), 63-90, anti-KLC (Stenoien and Brady, 1997), anti-GSK3β (Transduction Laboratories), anti-GSK3β phosphoSer9 (Cell Signaling) (against inactive GSK3β), anti-PP2A (C-20), anti-PP1 (E-9), anti-CDK5 (C-8) (Santa Cruz), anti-GAP-43 (91E12; Boehringer Mannheim), anti-tau (Tau-5, PHF-1 and pS396; Biosource) and anti-CDK5 (DC17; Upstate). Olo, okadaic acid (Calbiochem), CREBp, I2, CKI substrate, histone H1 (New England Biolabs) and ERK peptide (Upstate).
Recombinant kinases/phosphatases
GST-CDK5/GST-p25 expression plasmids (A Caceres) and purified GST-PAK catalytic fragment (X Bing and M Cobb) were gifts. GST fusion proteins were expressed and purified by glutathione-Sepharose (Sigma) affinity chromatography. Active ERK, PP1 (NE Biolabs) and His-tagged rat GSK3β (Sigma) were obtained commercially. His-tagged kinase-dead GSK3 construct was purified using TALON (Clontech). Recombinant kinases and phosphatases were tested in vitro with appropriate substrates before perfusion into axoplasm.
Kinase/phosphatase assays
CDK5 kinase assays were performed as described (Tsai et al, 1994). In Figure 2, 3–4 axoplasms were triturated in KB–ATP. Histone H1 (10 μM), Olo (10 μM), and CREBp, CK1 or ERK peptide substrates (250 μM) were added and reactions started by adding ATP (100 μM). Reactions (50 μl, 20 min at RT) were stopped by adding 2 × Laemmli buffer (50 μl), and were analyzed by SDS–PAGE and autoradiography. GSK3 activity in axoplasm was assayed by adding CREBp (200 μM) to axoplasm±Olo. Reactions were stopped by 50% TCA (20 μl) and spun at 14 000 rpm for 10 min to precipitate axoplasmic protein. Aliquots (10 μl) of the supernatant were spotted onto P81 paper. Filters were washed three times with 100 mM phosphoric acid and once with acetone. Radioactivity in peptides was measured by scintillation counting.
Cortical neurons cell culture and pharmacological inhibition
Primary cultured neurons were prepared as described previously (Beffert et al, 2002). Olo was added at 100 μM and okadaic acid at indicated doses. Drug treatments were typically 1–2 h. Iso-Olo or DMSO vehicle was added to control cultures.
Lysate preparation/immunoblot analysis
Cell cultures and tissues were homogenized in ROLB buffer (10 mM Hepes (pH 7.4), 0.5% Triton X-100, 80 mM β-glycerophosphate, 50 mM NaF, 2 mM Na-orthovanadate, 100 nM staurosporine, 100 nM K252a, 50 nM okadaic acid, 50 nM microcystin, 100 mM K-phosphate and mammalian protease inhibitor cocktail (Sigma). Lysates were clarified by centrifugation and protein level was determined by BCA (Pierce). Proteins were separated by SDS–PAGE and transferred to nitrocellulose as previously described (Beffert et al, 2002).
In vivo metabolic labeling and analysis
Neuronal cultures were prepared from rat or wild-type or p35−/−/p39−/− mouse embryos at day 16 of gestational age. A total of 6 × 106 cells were grown in 100 mm Petri dishes for 6–7 days in vitro. Media were changed and Olo or DMSO vehicle was added to cells. At 4 h, 1 mCi/dish [32P]phosphate (ICN) was added and incubated for 1 h. Media were discarded, cells homogenized in 1 ml of ROLB buffer and processed as above. Lysate aliquots (10 μl) were precipitated with 15% TCA and radioactivity in protein was assayed by scintillation counter. Equal counts were used for kinesin immunoprecipitation with H2 antibody (10 μg) (Donelan et al, 2002). Immunoprecipitates were separated by 7.5–16% SDS–PAGE. Gels were processed for phosphorimaging on a Typhoon (Amersham).
Motility in isolated axoplasm
Axoplasm was extruded from giant axons of the squid Loligo pealii (Marine Biological Laboratory) and transport measured as described previously (Brady et al, 1985; Morfini et al, 2002b).
Immunocytochemistry
Immunocytochemical staining was performed as described previously (Morfini et al, 2002b).
Microcystin pulldown assays
Microcystin pulldown assays were performed as described previously (Schillace and Scott, 1999) on mouse brain lysates prepared in ROLB as above (3 mg/ml total protein).
Subcellular fractionation
Growth cone-enriched fractions were prepared from embryonic (E18) rat brain as described (Pfenninger et al, 1983; Morfini et al, 2002b). Microsomes were prepared largely as described previously for axoplasm (Morfini et al, 2002b), using cultured cells grown for 7 days in vitro (7 × 106/dish/experiment) treated with inhibitors and homogenized in HB buffer (10 mM Hepes (pH 7.4), 0.32 M sucrose, 5 mM EDTA, 50 nM okadaic acid, 100 nM staurosporine, 100 nM K252a and protease inhibitors). Equal amounts of protein were loaded and analyzed by immunoblot with Cy5 secondary antibodies in fluorescence mode on a Typhoon.
Statistical analysis
Experiments were repeated at least three times. Unless otherwise stated, data were analyzed by ANOVA and post hoc Student–Newman–Keul test to make all possible comparisons. Data were expressed as mean±s.e.m. and significance was assessed at P<0.05 (Supplementary data).
Supplementary Material
Supplementary data
Acknowledgments
This paper is dedicated to L Efremova (GM). The authors thank H Reyna and S Nguyen for excellent technical assistance, A Caceres (INIMEC, Argentina) for CDK5 constructs, H Eldar-Finkelman for GSK3 wild-type and kinase-dead constructs, X Bing and M Cobb (UT Southwestern) for PAK fragment, and J Herz for p39−/−, p35−/−mice. Research supported by grants from NINDS (NS23868, NS23320, NS41170 and NS43408) to STB.
References
- Agarwal-Mawal A, Paudel HK (2001) Neuronal Cdc2-like protein kinase (Cdk5/p25) is associated with protein phosphatase 1 and phosphorylates inhibitor-2. J Biol Chem 276: 23712–23718 [DOI] [PubMed] [Google Scholar]
- Ali A, Hoeflich KP, Woodgett JR (2001) Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev 101: 2527–2540 [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P (2003) The specificities of protein kinase inhibitors: an update. Biochem J 371: 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beffert U, Morfini G, Bock HH, Reyna H, Brady ST, Herz J (2002) Reelin-mediated signaling locally regulates PKB/Akt and GS. J Biol Chem 277: 49958–49968 [DOI] [PubMed] [Google Scholar]
- Bhat R, Xue Y, Berg S, Hellberg S, Ormo M, Nilsson Y, Radesater AC, Jerning E, Markgren PO, Borgegard T, Nylof M, Gimenez-Cassina A, Hernandez F, Lucas JJ, Diaz-Nido J, Avila J (2003) Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem 278: 45937–45945 [DOI] [PubMed] [Google Scholar]
- Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, Tsai LH, Kwon YT, Girault JA, Czernik AJ, Huganir RL, Hemmings HC Jr, Nairn AC, Greengard P (1999) Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature 402: 669–671 [DOI] [PubMed] [Google Scholar]
- Bloom GS, Richards BW, Leopold PL, Ritchey DM, Brady ST (1993) GTPγS inhibits organelle transport along axonal microtubules. J Cell Biol 120: 467–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Wong PC, Becher MW, Pardo CA, Lee MK, Xu ZS, Thinakaran G, Jenkins NA, Copeland NG, Sisodia SS, Cleveland DW, Price DL, Hoffman PN (1998) Axonal transport of mutant superoxide dismutase 1 and focal axonal abnormalities in the proximal axons of transgenic mice. Neurobiol Dis 5: 27–35 [DOI] [PubMed] [Google Scholar]
- Brady ST (1985) A novel brain ATPase with properties expected for the fast axonal transport motor. Nature 317: 73–75 [DOI] [PubMed] [Google Scholar]
- Brady ST (1993) Axonal dynamics and regeneration. In Neuroregeneration, Gorio A (ed) pp 7–36. New York, NY: Raven Press [Google Scholar]
- Brady ST (1995) A kinesin medley: biochemical and functional heterogeneity. Trends Cell Biol 5: 159–164 [DOI] [PubMed] [Google Scholar]
- Brady ST, Lasek RJ, Allen RD (1985) Video microscopy of fast axonal transport in isolated axoplasm: a new model for study of molecular mechanisms. Cell Motil 5: 81–101 [DOI] [PubMed] [Google Scholar]
- Brady ST, Pfister KK, Bloom GS (1990) A monoclonal antibody against kinesin inhibits both anterograde and retrograde fast axonal transport in squid axoplasm. Proc Natl Acad Sci USA 87: 1061–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH (1997) Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron 18: 29–42 [DOI] [PubMed] [Google Scholar]
- Ching YP, Pang AS, Lam WH, Qi RZ, Wang JH (2002) Identification of a neuronal Cdk5 activator-binding protein as Cdk5 inhibitor. J Biol Chem 277: 15237–15240 [DOI] [PubMed] [Google Scholar]
- Cohen P, Foulkes JG, Holmes CF, Nimmo GA, Tonks NK (1988) Protein phosphatase inhibitor-1 and inhibitor-2 from rabbit skeletal muscle. Methods Enzymol 159: 427–437 [DOI] [PubMed] [Google Scholar]
- Cohen PT (2002) Protein phosphatase 1—targeted in many directions. J Cell Sci 115: 241–256 [DOI] [PubMed] [Google Scholar]
- Cole A, Frame S, Cohen P (2004) Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem J 377: 249–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM, Wyborski RJ, Banker G (1995) Preferential addition of newly synthesized membrane protein at axonal growth cones. Nature 375: 592–594 [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH (2003) Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40: 471–483 [DOI] [PubMed] [Google Scholar]
- Cyr JL, Pfister KK, Bloom GS, Slaughter CA, Brady ST (1991) Molecular genetics of kinesin light chains: generation of isoforms by alternative splicing. Proc Natl Acad Sci USA 88: 10114–10118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey ME, Bridgman PC (1991) Structure and organization of membrane organelles along distal microtubule segments in growth cones. J Neurosci Res 30: 242–258 [DOI] [PubMed] [Google Scholar]
- de Waegh SM, Lee VM-Y, Brady ST (1992) Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell 68: 451–463 [DOI] [PubMed] [Google Scholar]
- Dohadwala M, da Cruz e Silva EF, Hall FL, Williams RT, Carbonaro-Hall DA, Nairn AC, Greengard P, Berndt N (1994) Phosphorylation and inactivation of protein phosphatase 1 by cyclin dependent kinases. Proc Natl Acad Sci USA 91: 6408–6412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donelan MJ, Morfini G, Julyan R, Sommers S, Hays L, Kajio H, Briaud I, Easom RA, Molkentin JD, Brady ST, Rhodes CJ (2002) Ca2+-dependent dephosphorylation of kinesin heavy chain on beta-granules in pancreatic beta-cells. Implications for regulated beta-granule transport and insulin exocytosis. J Biol Chem 277: 24232–24242 [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P (2001) GSK3 takes centre stage more than 20 years after its discovery. Biochem J 359: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallows JL, Chen K, DePinho RA, Vincent I (2003) Decreased cyclin-dependent kinase 5 (cdk5) activity is accompanied by redistribution of cdk5 and cytoskeletal proteins and increased cytoskeletal protein phosphorylation in p35 null mice. J Neurosci 23: 10633–10644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Haystead TA, Sim AT (1991) Use of okadaic acid to inhibit protein phosphatases in intact cells. Methods Enzymol 201: 469–476 [DOI] [PubMed] [Google Scholar]
- Haystead TA, Sim AT, Carling D, Honnor RC, Tsukitani Y, Cohen P, Hardie DG (1989) Effects of the tumour promoter okadaic acid on intracellular protein phosphorylation and metabolism. Nature 337: 78–81 [DOI] [PubMed] [Google Scholar]
- Hellmich MR, Pant HC, Wada E, Battey JF (1992) Neuronal cdc2-like kinase: a cdc2-related protein kinase with predominantly neuronal expression. Proc Natl Acad Sci USA 89: 10867–10871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez F, Borrell J, Guaza C, Avila J, Lucas JJ (2002) Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J Neurochem 83: 1529–1533 [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR (2000) Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406: 86–90 [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ (1990) Kinesin heavy and light chains are phosphorylated in vivo in neurons. J Cell Biol 115: 390a (abstract) [Google Scholar]
- Hollenbeck PJ (1993) Phosphorylation of neuronal kinesin heavy and light chains in vivo. J Neurochem 60: 2265–2275 [DOI] [PubMed] [Google Scholar]
- Humbert S, Dhavan R, Tsai L (2000) p39 activates cdk5 in neurons, and is associated with the actin cytoskeleton. J Cell Sci 113 (Part 6): 975–983 [DOI] [PubMed] [Google Scholar]
- Khodjakov A, Lizunova EM, Minin AA, Koonce MP, Gyoeva FK (1998) A specific light chain of kinesin associates with mitochondria in cultured cells. Mol Biol Cell 9: 333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Humbert S, Bronson RT, Takahashi S, Kulkarni AB, Li E, Tsai LH (2001) p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J Neurosci 21: 6758–6771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Rosales JL, Tang D, Wang JH (1996) Interaction of cyclin-dependent kinase 5 (Cdk5) and neuronal Cdk5 activator in bovine brain. J Biol Chem 271: 1538–1543 [DOI] [PubMed] [Google Scholar]
- Lew J, Wang JH (1995) Neuronal cdc2-like kinase. Trends Biochem Sci 20: 33–37 [DOI] [PubMed] [Google Scholar]
- Li J-Y, Pfister KK, Brady ST, Dahlström A (1999) Axonal transport and distribution of immunologically distinct kinesin heavy chains in rat neurons. J Neurosci Res 58: 226–241 [PubMed] [Google Scholar]
- Lovestone S, Hartley CL, Pearce J, Anderton BH (1996) Phosphorylation of tau by glycogen synthase kinase-3 beta in intact mammalian cells: the effects on the organization and stability of microtubules. Neuroscience 73: 1145–1157 [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J (2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J 20: 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E (1998) Tau in Alzheimer's disease. Trends Cell Biol 8: 425–427 [DOI] [PubMed] [Google Scholar]
- Matsuura I, Wang JH (1996) Demonstration of cyclin-dependent kinase inhibitory serine/threonine kinase in bovine thymus. J Biol Chem 271: 5443–5450 [DOI] [PubMed] [Google Scholar]
- Moorhead G, MacKintosh RW, Morrice N, Gallagher T, MacKintosh C (1994) Purification of type 1 protein (serine/threonine) phosphatases by microcystin–Sepharose affinity chromatography. FEBS Lett 356: 46–50 [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Beffert U, Busciglio J, Brady ST (2002a) Fast axonal transport misregulation and Alzheimer's disease. Neuromol Med 2: 89–99 [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST (2002b) Glycogen Synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J 23: 281–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Richards B, Brady ST (2001) Regulation of kinesin: implications for neuronal development. Dev Neurosci 23: 364–376 [DOI] [PubMed] [Google Scholar]
- Munoz-Montano JR, Lim F, Moreno FJ, Avila J, Diaz-Nido J (1999) Glycogen synthase kinase-3 modulates neurite outgrowth in cultured neurons: possible implications for neurite pathology in Alzheimer's disease. J Alzheimers Dis 1: 361–378 [DOI] [PubMed] [Google Scholar]
- Nikolic M, Chou MM, Lu W, Mayer BJ, Tsai LH (1998) The p35/Cdk5 kinase is a neuron-specific Rac effector that inhibits Pak1 activity. Nature 395: 194–198 [DOI] [PubMed] [Google Scholar]
- Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K (2003) Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38: 555–565 [DOI] [PubMed] [Google Scholar]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna Pant HC, Brady RO, Martin LJ, Kulkarni AB (1996) Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA 93: 11173–11178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paglini G, Peris L, Diez-Guerra J, Quiroga S, Caceres A (2001) The Cdk5-p35 kinase associates with the Golgi apparatus and regulates membrane traffic. EMBO Rep 2: 1139–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfenninger K, Ellis L, Johnson M, Friedman L, Somlo S (1983) Nerve growth cones isolated from fetal rat brain. I. Subcellular fractionation and characterization. Cell 33: 573–584 [DOI] [PubMed] [Google Scholar]
- Pfister KK, Wagner MC, Stenoien D, Bloom GS, Brady ST (1989) Monoclonal antibodies to kinesin heavy and light chains stain vesicle-like structures, but not microtubules, in cultured cells. J Cell Biol 108: 1453–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Mattson MP, Brady ST, Busciglio J (2003) Alzheimer's presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci 23: 4499–4508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Paglini G, Ulloa L, Avila J, Caceres A (1997) Analysis of the expression, distribution and function of cyclin dependent kinase 5 (cdk5) in developing cerebellar macroneurons. J Cell Sci 110: 257–270 [DOI] [PubMed] [Google Scholar]
- Price DL, Borchelt DR, Wong PC, Pardo CA, Thinakaran G, Lee MK, Cleveland DW, Sisodia SS (1996) Neurodegenerative diseases and model systems. Cold Spring Harb Symp Quant Biol 61: 725–738 [PubMed] [Google Scholar]
- Ratner N, Bloom GS, Brady ST (1998) A role for Cdk5 kinase in fast anterograde axonal transport: novel effects of olomoucine and the APC tumor suppressor protein. J Neurosci 18: 7717–7726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder J, Su Y, Liu F, Li B, Zhou Y, Ni B (2003) Divergent roles of GSK3 and CDK5 in APP processing. Biochem Biophys Res Commun 312: 922–929 [DOI] [PubMed] [Google Scholar]
- Schillace RV, Scott JD (1999) Association of the type 1 protein phosphatase PP1 with the A-kinase anchoring protein AKAP220. Curr Biol 9: 321–324 [DOI] [PubMed] [Google Scholar]
- Smith DS, Greer PL, Tsai LH (2001) Cdk5 on the brain. Cell Growth Differ 12: 277–283 [PubMed] [Google Scholar]
- Smith DS, Tsai LH (2002) Cdk5 behind the wheel: a role in trafficking and transport? Trends Cell Biol 12: 28–36 [DOI] [PubMed] [Google Scholar]
- Stenoien DS, Brady ST (1997) Immunochemical analysis of kinesin light chain function. Mol Biol Cell 8: 675–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Amin N, Grant P, Pant HC (1995) P13suc1 associates with a cdc2-like kinase in a multimeric cytoskeletal complex in squid axoplasm. J Neurosci 15: 6222–6229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanji C, Yamamoto H, Yorioka N, Kohno N, Kikuchi K, Kikuchi A (2002) A-kinase anchoring protein AKAP220 binds to glycogen synthase kinase-3beta (GSK-3beta) and mediates protein kinase A-dependent inhibition of GSK-3beta. J Biol Chem 277: 36955–36961 [DOI] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness VSJ, Chae T, Harlow E (1994) p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 371: 419–423 [DOI] [PubMed] [Google Scholar]
- Tsai LH, Takahashi T, Caviness VS Jr, Harlow E (1993) Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development 119: 1029–1040 [DOI] [PubMed] [Google Scholar]
- Tsai M-Y, Morfini G, Szebenyi G, Brady ST (2000) Modulation of kinesin–vesicle interactions by Hsc70: implications for regulation of fast axonal transport. Mol Biol Cell 11: 2161–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Reese TS, Sheetz MP (1985) Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42: 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Haute C, Spittaels K, Van Dorpe J, Lasrado R, Vandezande K, Laenen I, Geerts H, Van Leuven F (2001) Coexpression of human cdk5 and its activator p35 with human protein tau in neurons in brain of triple transgenic mice. Neurobiol Dis 8: 32–44 [DOI] [PubMed] [Google Scholar]
- Vesely J, Havlicek L, Strnad M, Blow JJ, Donella-Deana A, Pinna L, Letham DS, Kato J-Y, Detivaud L, Leclerc S, Meijer L (1994) Inhibition of cyclin-dependent kinases by purine analogues. Eur J Biochem 224: 771–786 [DOI] [PubMed] [Google Scholar]
- Wagner MC, Pfister KK, Bloom GS, Brady ST (1989) Copurification of kinesin polypeptides with microtubule-stimulated Mg-ATPase activity and kinetic analysis of enzymatic processes. Cell Motil Cytoskeleton 12: 195–215 [DOI] [PubMed] [Google Scholar]
- Wang QM, Fiol CJ, DePaoli-Roach AA, Roach PJ (1994a) Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J Biol Chem 269: 14566–14574 [PubMed] [Google Scholar]
- Wang QM, Roach PJ, Fiol CJ (1994b) Use of a synthetic peptide as a selective substrate for glycogen synthase kinase 3. Anal Biochem 220: 397–402 [DOI] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW (1999) Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci 2: 50–56 [DOI] [PubMed] [Google Scholar]
- Woodgett JR (1994) Regulation and functions of the glycogen synthase kinase-3 subfamily. Semin Cancer Biol 5: 269–275 [PubMed] [Google Scholar]
- Woodgett JR (2001) Judging a protein by more than its name: GSK-3. Science's STKE, http://stke.sciencemag.org/cgi /content/full/sigtrans 2001/100/re12 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data