

Abstract
Conserved signaling pathways that activate the mitogen-activated protein kinases (MAPKs) are involved in relaying extracellular stimulations to intracellular responses. The MAPKs coordinately regulate cell proliferation, differentiation, motility, and survival, which are functions also known to be mediated by members of a growing family of MAPK-activated protein kinases (MKs; formerly known as MAPKAP kinases). The MKs are related serine/threonine kinases that respond to mitogenic and stress stimuli through proline-directed phosphorylation and activation of the kinase domain by extracellular signal-regulated kinases 1 and 2 and p38 MAPKs. There are currently 11 vertebrate MKs in five subfamilies based on primary sequence homology: the ribosomal S6 kinases, the mitogen- and stress-activated kinases, the MAPK-interacting kinases, MAPK-activated protein kinases 2 and 3, and MK5. In the last 5 years, several MK substrates have been identified, which has helped tremendously to identify the biological role of the members of this family. Together with data from the study of MK-knockout mice, the identities of the MK substrates indicate that they play important roles in diverse biological processes, including mRNA translation, cell proliferation and survival, and the nuclear genomic response to mitogens and cellular stresses. In this article, we review the existing data on the MKs and discuss their physiological functions based on recent discoveries.
Cells recognize and respond to extracellular stimuli by engaging specific intracellular programs, such as the signaling cascade that leads to activation of the mitogen-activated protein kinases (MAPKs). All eukaryotic cells possess multiple MAPK pathways, which coordinately regulate diverse cellular activities running the gamut from gene expression, mitosis, and metabolism to motility, survival and apoptosis, and differentiation. To date, five distinct groups of MAPKs have been characterized in mammals: extracellular signal-regulated kinases (ERKs) 1 and 2 (ERK1/2), c-Jun amino-terminal kinases (JNKs) 1, 2, and 3, p38 isoforms α, β, γ, and δ, ERKs 3 and 4, and ERK5 (reviewed in references 25 and 103). Since Saccharomyces cerevisiae possesses six different MAPKs, the relative complexity of the human genome suggests that there are probably several additional vertebrate MAPK subfamilies (118). The most extensively studied groups of vertebrate MAPKs to date are the ERK1/2, JNKs, and p38 kinases.
MAPKs can be activated by a wide variety of different stimuli, but in general, ERK1 and ERK2 are preferentially activated in response to growth factors and phorbol esters, while the JNK and p38 kinases are more responsive to stress stimuli ranging from osmotic shock and ionizing radiation to cytokine stimulation (reviewed in reference 147) (Fig. 1). Although each MAPK has unique characteristics, a number of features are shared by the MAPK pathways studied to date. Each family of MAPKs is composed of a set of three evolutionarily conserved, sequentially acting kinases: a MAPK, a MAPK kinase (MAPKK), and a MAPKK kinase (MAPKKK). The MAPKKKs, which are serine/threonine kinases, are often activated through phosphorylation and/or as a result of their interaction with a small GTP-binding protein of the Ras/Rho family in response to extracellular stimuli (36, 98). MAPKKK activation leads to the phosphorylation and activation of a MAPKK, which then stimulates MAPK activity through dual phosphorylation on threonine and tyrosine residues located in the activation loop of kinase subdomain VIII. Once activated, MAPKs phosphorylate target substrates on serine or threonine residues followed by a proline; however, substrate selectivity is often conferred by specific interaction motifs located on physiological substrates. Furthermore, MAPK cascade specificity is also mediated through interaction with scaffolding proteins which organize pathways in specific modules through simultaneous binding of several components.
FIG. 1.

Signaling cascades leading to activation of the MKs. Mitogens and cellular stresses lead to activation of the ERK1/2 and p38 cascades, which in turn phosphorylate and activate the five subgroups of MKs.
The wide range of functions of the MAPKs are mediated through phosphorylation of several substrates, including phospholipases, transcription factors, and cytoskeletal proteins. MAPKs also catalyze the phosphorylation and activation of several protein kinases, termed MAPK-activated protein kinases (MKs), which represent an additional enzymatic and amplification step in the MAPK catalytic cascades. The MK family comprises the ≈90-kDa ribosomal S6 kinases (RSKs), the mitogen- and stress-activated kinases (MSKs), the MAPK-interacting kinases (MNKs), MAPK-activated protein kinases 2 and 3 (MK2 and -3, formally termed MAPKAP-K2 and -3), and MAPK-activated protein kinase 5 (MK5, formally termed MAPKAP-K5). The MKs are related kinases that mediate a wide range of biological functions in response to mitogens and stress stimuli. Because of the lack of specific inhibitors against these kinases, it has been a challenge to dissect their exact biological functions and to differentiate those from direct MAPK functions. This review will address our current understanding of the properties, structure, and regulation of the MKs and discuss their physiological functions based on studies with knockout mice and recently identified substrates.
MAPK FAMILIES
ERK1/2 Module
Properties.
The mammalian ERK1/2 module, also known as the classical mitogen kinase cascade, consists of the MAPKKKs A-Raf, B-Raf, and Raf-1, the MAPKKs MEK1 and MEK2, and the MAPKs ERK1 and ERK2 (Fig. 1). ERK1 and ERK2 have 83% amino acid identity and are expressed to various extents in all tissues (reviewed in reference 25). They are strongly activated by growth factors, serum, and phorbol esters and to a lesser extent by ligands of the heterotrimeric G protein-coupled receptors, cytokines, osmotic stress, and microtubule disorganization (112). MEKK1/2/3 and c-Mos kinases are also known to act as MAPKKKs in this pathway. While the proto-oncogene c-mos appears to play an important role during meiosis, gene disruption studies suggest that MEKK1/2/3 may have limited impact on or redundant contributions to activation of the ERK1/2 pathway (238, 242).
Activation mechanisms.
Typically, cell surface receptors such as tyrosine kinases (RTK) and G protein-coupled receptors transmit activating signals to the Raf/MEK/ERK cascade through different isoforms of the small GTP-binding protein Ras (reviewed in references 19 and 234). Activation of membrane-associated Ras is achieved through recruitment of SOS (son of sevenless), a Ras-activating guanine nucleotide exchange factor. SOS stimulates Ras to change GDP to GTP, allowing it to interact with a wide range of downstream effector proteins, including isoforms of the serine/threonine kinase Raf (63). The exact mechanism of Raf activation is still elusive but is known to require Ras binding as well as multiple phosphorylation events at the membrane (reviewed in reference 28). Regulation of both Ras and Raf is crucial for the proper maintenance of cell proliferation, as activating mutations in these genes lead to oncogenesis (28). Indeed, Ras has been shown to be mutated in 30% of all human cancers, while B-Raf is mutated in 60% of malignant melanomas (reviewed in references 42 and 125). Activated Raf binds to and phosphorylates the dual specificity kinases MEK1 and -2, which in turn phosphorylate ERK1/2 within a conserved Thr-Glu-Tyr (TEY) motif in their activation loop. Amplification through the signaling cascade is such that it is estimated that activation of solely 5% of Ras molecules is sufficient to induce full activation of ERK1/2 (71).
In S. cerevisiae, the MAPK modules are neatly organized by scaffolding proteins that ensure efficiency and specificity in the signaling cascades (226). The yeast scaffolding protein Ste5p selectively binds a MAPKKK (Ste11p), a MAPKK (Ste7p), and a MAPK (Fus3p) and couples them to their upstream activators. In mammals, no homologues of Ste5p have yet been identified, but the scaffolding protein JIP-1 appears to have similar functions in the JNK cascade of vertebrates (reviewed in reference 103). Several proteins have been shown to interact with members of the ERK cascade, including the scaffold proteins MP-1 and KSR and the modulators CNK and RKIP, resulting in stimulation or inhibition of the ERK1/2 cascade (reviewed in reference 148).
Substrates and functions.
ERK1/2 are distributed throughout quiescent cells, but upon stimulation, a significant population of ERK1/2 accumulates in the nucleus (24, 66, 111). While the mechanisms involved in nuclear accumulation of ERK1/2 remain elusive, nuclear retention, dimerization, phosphorylation, and release from cytoplasmic anchors have been shown to play a role (reviewed in reference 152). ERK1/2 signaling has been implicated as a key regulator of cell proliferation, and for this reason, inhibitors of the ERK pathway are entering clinical trials as potential anticancer agents (97). Two structurally unrelated compounds are commonly used to specifically inhibit the ERK1/2 pathway in cultured cells. Both U0126 and PD98059 are noncompetitive inhibitors of MEK1/2/5 and prevent stimulation-mediated activation of ERK1/2/5 (reviewed in reference 3) (Fig. 1).
Activated ERK1 and ERK2 phosphorylate numerous substrates in all cellular compartments, including various membrane proteins (CD120a, Syk, and calnexin), nuclear substrates (SRC-1, Pax6, NF-AT, Elk-1, MEF2, c-Fos, c-Myc, and STAT3), cytoskeletal proteins (neurofilaments and paxillin), and several MKs (reviewed in reference 25). RSKs, MSKs, and MNKs represent three kinase subfamilies of ERK1/2 substrates. While MSKs and MNKs have been shown to be activated by both the ERK1/2 and p38 pathways, RSK family members are exclusively activated by the ERKs (56). These ERK1/2-activated kinases will be discussed in greater detail below.
p38 MAPK Module
Properties.
p38 (also known as CSBP, mHOG1, RK, and SAPK2) is the archetypal member of the second MAPK-related pathway in mammalian cells (73, 108). The p38 module consists of several MAPKKKs, including MEKKs 1 to 4, (MEKK1-4), MLK2 and -3, DLK, ASK1, Tpl2 (also termed Cot), and Tak1, the MAPKKs MEK3 and MEK6 (also termed MKK3 and MKK6, respectively), and the four known p38 isoforms (α, β, γ, and δ) (Fig. 1) (reviewed in reference 103). p38α has 50% amino acid identity with ERK2 and bears significant homology to the product of the budding yeast hog1 gene, which is activated in response to hyperosmolarity (73, 108, 163). In mammalian cells, the p38 isoforms are strongly activated by environmental stresses and inflammatory cytokines but not appreciably by mitogenic stimuli. Most stimuli that activate p38 also activate JNK, but only p38 is inhibited by the anti-inflammatory drug SB203580, which has been extremely useful in delineating the function of p38 (108).
Activation mechanisms.
MEK3 and MEK6 are activated by a plethora of MAPKKKs which become activated in response to various physical and chemical stresses, such as oxidative stress, UV irradiation, hypoxia, ischemia, and various cytokines, including interleukin-1 (IL-1) and tumor necrosis factor alpha (reviewed in reference 25). MEK3 and MEK6 show a high degree of specificity for p38, as they do not activate ERK1/2 or JNK. MEK4 (also known as MKK4 and Sek1) is a known JNK kinase that possesses some MAPKK activity toward p38, suggesting that MEK4 represents a site of integration for the p38 and JNK pathways (14, 123). While MEK6 activates all p38 isoforms, MEK3 is somewhat selective, as it preferentially phosphorylates the p38α and p38β isoforms. The specificity in p38 activation is thought to result from the formation of functional complexes between MEK3/6 and different p38 isoforms and the selective recognition of the activation loop of p38 isoforms by MEK3/6 (47). Activation of the p38 isoforms results from the MEK3/6-catalyzed phosphorylation of a conserved Thr-Gly-Tyr (TGY) motif in their activation loop. The structures of inactive and active (phosphorylated) p38α have been solved by X-ray crystallography. The phosphorylated TGY motif and the length of the activation loop were found to differ in ERK2 and JNK, which likely contributes to the substrate specificity of p38 (219, 230).
Substrates and functions.
p38 was shown to be present in both the nucleus and cytoplasm of quiescent cells, but upon cell stimulation, the cellular localization of p38 is not well understood. Some evidence suggests that, following activation, p38 translocates from the cytoplasm to the nucleus (156), but other data indicate that activated p38 is also present in the cytoplasm of stimulated cells (6).
A large body of evidence indicates that p38 activity is critical for normal immune and inflammatory responses. p38 is activated in macrophages, neutrophils, and T cells by numerous extracellular mediators of inflammation, including chemoattractants, cytokines, chemokines, and bacterial lipopolysaccharide (143). p38 participates in macrophage and neutrophil functional responses, including respiratory burst activity, chemotaxis, granular exocytosis, adherence, and apoptosis, and also mediates T-cell differentiation and apoptosis by regulating gamma interferon production (143). p38 also regulates the immune response by stabilizing specific cellular mRNAs involved in this process. For instance, with SB203580 and constitutively active forms of p38 and MEK3/6, it has been shown that p38 regulates the expression of many cytokines, transcription factors, and cell surface receptors (143).
While the exact mechanisms involved in p38 immune functions are starting to emerge, activated p38 has been shown to phosphorylate several cellular targets, including cytosolic phospholipase A2, the microtubule-associated protein Tau, and the transcription factors ATF1 and -2, MEF2A, Sap-1, Elk-1, NF-κB, Ets-1, and p53 (103). p38 also activates several MKs, including MSK1 and -2, MNK1 and -2, and MK2 and -3, which will be discussed in greater detail below.
JNK Module
Properties.
The first member of the JNK family was originally isolated from rat livers injected with cycloheximide (104). JNK1, JNK2, and JNK3 (also known as SAPKγ, SAPKα, and SAPKβ, respectively) exist in 10 or more different spliced forms and are ubiquitously expressed, although JNK3 is present primarily in the brain. The JNKs are strongly activated in response to cytokines, UV irradiation, growth factor deprivation, DNA-damaging agents, and, to a lesser extent, some G protein-coupled receptors, serum, and growth factors (103).
Activation mechanisms.
Like ERK1/2 and p38, JNK activation requires dual phosphorylation on tyrosine and threonine residues within a conserved Thr-Pro-Tyr (TPY) motif. The MAPKKs that catalyze this reaction are known as MEK4 and MEK7, which are themselves phosphorylated and activated by several MAPKKKs, including MEKK1-4, MLK2 and -3, Tpl-2, DLK, TAO1 and -2, TAK1, and ASK1 and -2 (reviewed in reference 103) (Fig. 1).
Substrates and functions.
Like ERK1/2 and p38, the JNKs may relocalize from the cytoplasm to the nucleus following stimulation (127). A well-known substrate for JNKs is the transcription factor c-Jun. Phosphorylation of c-Jun on Ser63 and Ser73 by JNK leads to increased c-Jun-dependent transcription (reviewed in reference 225). Several other transcription factors have been shown to be phosphorylated by the JNKs, such as ATF-2, NF-ATc1, HSF-1, and STAT3 (25, 103). While some cytoplasmic targets of JNK are known, the fact that stimulated JNK does not exhibit exclusive nuclear localization suggests that many other cytoplasmic substrates remain to be identified. Intriguingly, there is currently no known JNK-activated MK.
DOCKING INTERACTIONS IN THE MAPK CASCADES
Description of Docking Sites
D domains.
It has become apparent that efficiency and specificity of signaling within the MAPK cascades are partly achieved through specialized docking motifs present in scaffold proteins, MAPKKs, MAPKs, and their substrates. The δ-domain in c-Jun was the first identified motif involved in MAPK docking (200), which occurs in addition to the transient enzyme-substrate interaction through the active site of the kinase. Subsequently, sequences related to the δ-domain were identified in other transcription factors, including the MAPK-regulated bZIP, ETS, and MAD (reviewed in reference 180). These domains were called docking domains (D domains or D-boxes) and shown to increase substrate phosphorylation by MAPKs. Although D domains can sometimes be recognized by more than one class of MAPKs, they are thought to increase signaling specificity. By examining the primary sequence of the regions involved in MAPK docking, it was found that D domains are characterized by a cluster of positively charged residues surrounded by hydrophobic residues (reviewed in reference 202). D domains can lie either upstream or downstream of the phosphoacceptor site and have been shown to be present on many MAPK substrates, including the MKs. Aside from MAPK substrates, D domains have also been identified in many MAPK regulatory proteins, which include upstream activating kinases (MAPKKs), phosphatases (PTP-SL, STEP, and MKPs), and scaffold proteins (KSR) across different species (reviewed in references 48).
DEF domains.
Analysis of the Caenorhabditis elegans ETS transcription factor LIN-1 and the scaffold protein KSR-1 revealed the presence of a second class of MAPK docking site that consists of the Phe-Xaa-Phe-Pro sequence, where Xaa is any amino acid and one of the Phe residues can also be a Tyr residue (50, 81, 129). This domain, termed DEF (docking site for ERK and FXFP), has been reported to be recognized only by ERK1/2 and typically lies C-terminal to the phosphoacceptor site. DEF domains are required for efficient substrate phosphorylation by ERK1/2 and have been found in many substrates of the mammalian, C. elegans, and Drosophila melanogaster ERK1/2 orthologs (50, 82, 129). DEF and D domains can sometimes be found on the same protein, such as for Elk-1 and KSR, where these domains may act synergistically to strengthen MAPK interaction.
CD and ED motifs.
While it is not clear what region of ERK1/2 interacts with DEF domains, two groups independently identified a conserved C-terminal common docking (CD) motif outside the catalytic domain of ERK, p38, and JNK involved in D domain interactions (166, 200). The CD motif contains acidic and hydrophobic residues that are necessary to establish hydrophobic and electrostatic interactions with the positively charged and hydrophobic residues of D domains, respectively (48, 200). The CD motif has been shown to mediate MAPK interactions with their upstream activators and downstream substrates, suggesting that utilization of the interaction motifs is heavily controlled during the MAPK activation cascade. Interestingly, the Drosophila ERK/Rolled sevenmaker allele is mutated in the CD domain and has been shown to be less sensitive to dephosphorylation, suggesting that the CD domain may also direct binding with presently uncharacterized ERK phosphatases (13).
Other regions of MAPKs have also been shown to play important roles in D domain interactions. Similar to the CD motif, the ERK docking (ED) motif is located opposite the MAPK active center and is thought to regulate binding specificity (202). Exchange of only two residues within the ED site of ERK2 altered its binding specificity to that of p38, rendering ERK2 capable of binding to MK3 (201). The complementary mutations in p38 did not revert the docking specificity of p38 for that of RSK2, suggesting that additional residues must participate in this interaction. Accordingly, another groupidentified a docking groove near the CD and ED motifs of p38 that is required for D domain interactions with MEF2A and MEK3b (21). Mutations of residues within the docking groove disrupted p38 binding to the D domain of MEF2A, suggesting that the homologous residues in ERK1/2 and JNK may also be involved in D domain interaction. Finally, several laboratories have reported a role for the N-terminal domain of MAPKs in docking specificity, but the exact requirement for this region remains to be determined (reviewed in reference 48).
Docking Interactions with MKs
Properties.
Almost all MKs possess a docking motif of the D domain class, containing the sequence Leu-Xaa-Xaa-Arg-Arg followed by one or several basic residues (Fig. 2). This docking sequence is necessary for ERK1/2 docking to RSKs (62, 165, 189) and MNK2B (170), ERK1/2 and p38 binding to MSK2 (207), as well as p38 docking to MK5 (179). It is likely that MAPK docking specificity arises from variations in the D domain sequence and from the potential involvement of other unidentified regions within the MKs. Indeed, a recent study demonstrated that replacement of the ERK1/2 docking site in RSK2 with the p38 docking site of MK2 converted RSK2 into a stress-activated kinase in vivo (188), indicating that specificity of activation results from the type of D domain. It has already been noted that the number of contiguous basic residues (Lys and Arg) correlates with MAPK specificity (189).
FIG. 2.

Amino acid alignments of D domains found in MKs. The ERK1/2 and p38 kinase binding regions, termed D domains, are found in most MKs and are required for efficient activation of the MKs by mitogens and cellular stresses. The D domains, shown in boldface, are characterized by a stretch of positively charged residues surrounded by hydrophobic residues. Some D domains mediate specific interaction with ERK1/2 or p38, while others are necessary for interaction with both upstream activators. In some MKs, the D domain region overlaps an NLS sequence (underlined).
Generally, ERK1/2-specific MKs have two contiguous basic residues, ERK1/2- and p38-specific MKs have three or four contiguous basic residues, and p38-specific MKs have four or five contiguous basic residues (Fig. 2). Furthermore, the amount and location of hydrophobic residues within the D domain may also regulate the specificity of the MAPK. Indeed, the p38-activated MKs tend to have at least two hydrophobic residues before the stretch of basic residues (Fig. 2). Interestingly, the CD motif in p38 kinases also contains more contiguous acidic residues than ERK1/2, but mutational analyses of these motifs and flanking sequences are warranted to further assess the regions responsible for ERK1/2 and p38 specificity. Because the basic residues in D domains are clustered and overlap defined nuclear localization sequences (NLSs) in MSK1 and -2, MK2 and -3, and MK5 (Fig. 2), the functional results obtained with D domain mutants will have to be analyzed with caution.
Regulation of docking.
The regulation of MAPK docking to the MKs is not well understood. Several MKs have been shown to bind to ERK1/2 and/or p38 in quiescent cells and to dissociate following cell stimulation. For instance, RSK family members bind to ERK1/2 in quiescent cells, but following stimulation, the complex transiently dissociates for the duration of ERK1/2 activation (165). In fact, differences have even been noted between different RSK isoforms, with RSK1 having the most regulated interaction with ERK1/2. MSK1 and -2, MK2, and MK5 have been shown to reside in the nucleus of quiescent cells (6, 38, 44, 149, 179). Following stimulation, MK2 and MK5 translocate into the cytoplasm, but their relation to p38 at this point remains elusive (6, 179). Recent evidence indicates that p38 docking to MK5 masks its NLS and thereby promotes its nuclear export (179), suggesting that docking and localization are interrelated for MK5.
The mechanisms involved in complex dissociation are currently unknown, but mutagenesis analysis revealed that autophosphorylation of a residue located near the D domain of RSK1 was required for the regulated release of ERK1/2 (165). MSK1 and MSK2 also contain a homologous residue near their D domains, suggesting that these kinases may also regulate ERK1/2 and p38 docking through autophosphorylation of their C termini. Interestingly, MNK1 has been shown to interact with dephosphorylated ERK2 with greater affinity (221), suggesting that MNK1 activation may also regulate MAPK docking.
MAPK-ACTIVATED PROTEIN KINASES
The MK family comprises 11 members that are generally activated by mitogens through the ERK1/2 kinase cascade (RSK1, RSK2, RSK3, RSK4, and MNK2), stress stimuli through the p38 kinase cascade (MK2, MK3, and possibly MK5), or both (MSK1, MSK2, and MNK1) (Fig. 1). Of these, the RSKs and MSKs possess two distinct and functional kinase domains within the same polypeptide, termed the N-terminal kinase domain (NTKD) and C-terminal kinase domain (CTKD). Conversely, the MNKs, MK2 and -3, and MK5 have a single kinase domain that is mostly homologous to the CTKD of the RSKs and MSKs (Fig. 3). All of these kinases have similar activation loop sequences that are targeted for phosphor-ylation by either ERK1/2 or p38 (Fig. 4). Based on their sequence homologies, the MKs can be classified into five subgroups, the RSKs, MSKs, MNKs, MK2 and -3, and MK5 (Fig. 5), and each subgroup will be discussed in greater detail below.
FIG. 3.

Schematic representation of the overall structure of the MKs. While RSK1, -2, -3, and -4 and MSK1 and -2 are composed of two nonidentical kinase domains, the MNKs and MK2, -3, and -5 are single-kinase-domain proteins that display homology to the CTKD of RSKs and MSKs. The NTKDs of the RSKs and MSKs are members of the AGC family of kinases, which also require sequences known as the turn and hydrophobic motif for activity. Also necessary for activation of all MKs are the phosphorylation sites indicated by a black circle. The amino acid composition of each MK and its alternatively spliced variants refers to the human protein. The MK5A isoform has been omitted from this figure because it differs from MK5B only by the lack of two residues.
FIG. 4.

Alignment of the amino acid sequences of the single-kinase domain MKs (MNK1 and -2, MK2 and -3 and MK5) and the CTKDs of RSK1 and MSK1. The sequences comprising the kinase domain and its subregions are boxed and reveal the region of highest homology. The conserved activation loop threonine residue within the sequence Leu-Xaa-Thr-Pro is shown by an arrow, and the D domain region is identified by a line.
FIG. 5.

Phylogenetic tree of MK family members. The root of the tree is marked with a circle. All sequences used for the construction of this tree were human, and alternatively spliced isoforms of MNK2, MK2, and MK5 were also included in the analysis. The relative similarities between all MKs reflected by this tree suggest that the MKs comprise five groups, the RSK, MSK, MNK, MK2 and -3, and MK5 subfamilies. The Clustal X program was used to generate the multiple alignment on which the tree was based.
RSK Subfamily
Discovery.
RSK1 was the first identified MK and was discovered in Xenopus laevis extracts as a kinase that phosphorylated the 40S ribosomal subunit protein S6 (49). RSK homologues are present in most vertebrate species and have received several names over the years (Table 1) but are now generally called p90 ribosomal S6 kinases (RSKs). The RSK family contains four human isoforms (RSK1, RSK2, RSK3, and RSK4), which show 73 to 80% amino acid identity (Fig. 3). RSK-related molecules have also been identified in C. elegans (T01H8.1) and D. melanogaster (RPS6 protein kinase II), which show about 60% amino acid identity to RSK1 (223), but no RSK homologues have been found in S. cerevisiae to date. As mentioned above, the RSK homologues are particular among the serine/threonine kinases in that they contain two distinct and functional kinase domains (52, 91). This property is shared by the MSKs, which are a family of RSK cousins that will be discussed in the following section. The CTKD (also termed D2) of the RSKs belongs to the calcium- and calmodulin-dependent kinase (CaMK) group of kinases and is the domain most conserved in members of the MK family (Fig. 4). The NTKD (also termed D1) of the RSKs belongs to the AGC family of kinases, which includes protein kinase A, protein kinase C, protein kinase B (also called Akt), and the p70 ribosomal S6 kinases 1 and 2 (S6K1 and S6K2). The RSKs and MSKs are the only AGC members to be part of the MK family based on the homology of their NTKDs. Although S6K1 and S6K2 display substantial homology to the NTKD of the RSKs (about 40% amino acid identity), they are not members of the MK family because they lack the CTKD of the RSKs and MSKs.
TABLE 1.
Human diseases potentially associated with MK inactivation and phenotypes of knockout mice
| MK | Other name(s) | Human gene location | Associated diseasea | Phenotype of knockout miceb |
|---|---|---|---|---|
| RSK1 | HU-1, MAPKAP-K1A, S6K-alpha 1, p90RSK1 | 1p36-p35 | ND | |
| RSK2 | HU-3, MAPKAP-K1B, MRX19, S6K-alpha 3, ISPK-1, p90RSK2 | Xp22.13 | Coffin-Lowry syndrome (210) | RSK2 knockout mice weigh 10% less and have slightly impaired cognition and coordination skills (43) |
| RSK3 | HU-2, MAPKAP-K1C, S6K-alpha 2, p90RSK3 | 6q27 | ND | |
| RSK4 | S6K-alpha 6, p90RSK4 | Xq21 | Possible role in nonspecific X-linked mental retardation (241) | ND |
| MSK1 | MSPK1, RLPK | 14q31-q32.1 | No obvious phenotype (2) | |
| MSK2 | RSK-B | 11q11-q13 | Located in the locus of Bardet-Beidl syndrome (248) | MSK2 and MSK1/2 double knockouts have no obvious phenotypes (228) |
| MNK1 | 1p34.1 | ND | ||
| MNK2 | 19p13.3 | ND | ||
| MK2 | MAPKAP-K2 | 1q32.1 | MK2 knockout mice show increased resistance to endotoxic shock and increased susceptibility to infections (101, 109) | |
| MK3 | MAPKAP-K3, 3pK | 3p21.2 | Commonly deleted in small-cell lung cancer (186) | ND |
| MK5 | MAPKAP-K5, PRAK | 12q24.12 | No obvious phenotype (183); the MK2/MK5 double knockout has a phenotype similar to that of single MK2 knockout mice (183) |
Coffin-Lowry syndrome has been shown to be associated with RSK2 inactivation, but the role of RSK4 and MSK2 in mental retardation and Bardet-Beidl syndrome, respectively, remains to be determined.
ND, not determined or nonexistent.
Structure and expression.
All four human RSK isoforms have a similar structure that consists of two distinct kinase domains joined by a linker region of about 100 amino acids and N- and C-terminal ends (Fig. 3). Results from our group and others have elucidated the distinct roles of both kinase domains in RSK1 and RSK2. While the NTKD was found to be responsible for substrate phosphorylation, both kinase domains were shown to autophosphorylate on sites important for RSK1 and RSK2 activation (10, 52, 217). All RSK isoforms also contain a C-terminally located D domain that is responsible for docking and activation by ERK1/2 (Fig. 3) (62, 189). As mentioned above, the D domain of the RSKs consists of Leu-Ala-Gln-Arg-Arg (Fig. 2), where only the Leu and Arg residues are necessary for ERK1/2 docking (165). Two additional basic residues located C-terminal to the D domain also contribute to ERK1/2 binding, but their presence was not found to be essential for activation of RSK1 (165). The RSK isoforms and ERK1/2 have long been shown to interact in cells and to dissociate upon activation (79, 178, 246). We have recently shown that RSK1 dissociates from ERK1/2 through the autophosphorylation of a serine residue located near the D domain of RSK1, providing a mechanism for the regulated docking of ERK1/2 (165).
RSK1, RSK2, RSK3, and ERK1/2 are usually present in the cytoplasm of quiescent cells, but upon stimulation, a significant portion of these proteins translocate to the nucleus of activated HeLa, COS-7, and HEK293 cells (24, 111, 159, 213, 247). RSK3 is the only human isoform to possess a potential NLS (Lys-Lys-Xaa10-Leu-Arg-Arg-Lys-Ser-Arg) (Fig. 3), whereXaa10 is a stretch of any 10 amino acids, but it remains unknown whether this domain is functional or if other regions are required for nuclear translocation of the RSKs. The small death effector domain protein PEA-15 has been shown to inhibit RSK2 nuclear translocation, but the biological function of this interaction remains unknown (213). Activated RSK2 can also be found in the cytoplasm of stimulated cells, suggesting that RSK2 substrates may exist in both nuclear and cytoplasmic compartments (24). It appears likely that the RSKs possess protein substrates in all cellular compartments, including the cytosol, nucleus, and plasma membranes, and that their localization is regulated via multiple mechanisms.
Analysis of the expression of the RSK isoforms has revealed differential expression patterns. Northern analysis of rsk1 expression showed that this isoform is present in most tissues but is expressed at higher levels in the kidney, lung, and pancreas (243). Analysis of rsk2 mRNA revealed that the alternative use of two different polyadenylation sites gave rise to two transcripts of 3.5 and 8.5 kb (243). The rsk2 and rsk3 transcripts and proteins are expressed in most tissues, including heart, brain, placenta, liver, kidney, and pancreas, with predominant expression in skeletal muscle (43, 243, 247). The rsk4 transcript is also expressed ubiquitously but was shown to be present in high levels in fetal tissues and in adult brain and kidney (241). Northern analysis also revealed the presence of two secondary rsk4 transcripts (5 and 9 kb), but whether these transcripts result from alternative splicing or alternative polyadenylation remains unknown (241). Interestingly, the reported mouse and rat RSK4 sequences contain an additional N-terminal region that is not found in human RSK4 or in other RSK family members. Since human RSK4 was originally identified by positional cloning, this suggests that human RSK4 may also contain this additional domain at its N terminus. However, the function of this potentially novel domain remains to bedetermined.
Activation mechanisms.
All RSK isoforms, including the C. elegans and D. melanogaster homologues of RSK, contain six phosphorylation sites that have been shown in RSK1 and RSK2 to be responsive to mitogen stimulation (35) (Fig. 6). Mutational analysis revealed that four of these sites (Ser221, Ser363, Ser380, and Thr573 in human RSK1) are essential for RSK1 activation (35) (Fig. 7). Of these, Ser363 and Ser380 are located within sequences conserved among most AGC kinases, the turn motif and hydrophobic motif, respectively (138). Phosphorylation of the turn motif in RSK1 and RSK2 is essential for kinase activity and is thought to be mediated by ERK1/2 because it is located within a proline-directed phosphorylation site. However, results from our group indicate that this site is not efficiently phosphorylated by ERK1 in vitro (159), and alanine substitution of Pro364 only slightly reduces mitogen-stimulated RSK1 activation (P. Roux, unpublished results), suggesting that another kinase that does not require a proline at position +1 is able to phosphorylate this site in vivo. Interestingly, protein kinases A and C have been shown to autophosphorylate on the homologous site in vivo (4), and this site is modified by a heterologous kinase in Akt (5, 206). It is currently unclear which kinase(s) is responsible for phosphorylating the turn motif in RSK isoforms, and further studies are needed to understand the regulation of this site. Interestingly, the turn motif in protein kinase C has been shown to act as a phosphorylation-dependent docking site for heat shock protein 70 (HSP70) (61), suggesting that Ser363 may have a similar function during RSK1 activation.
FIG. 6.

Signaling cascades leading to activation of RSKs and MSKs. RSK1, -2, -3, and -4 are activated by two inputs originating from the ERK1/2 and PDK1 enzymes. Similarly, MSK1 and -2 are activated by ERK1/2 but also by stimuli that activate the p38 kinases. Despite their relatively high homology, the RSKs and MSKs appear to have different biological functions.
FIG. 7.

Schematic representation of RSK1 compared to the related MSK1. The known phosphorylation sites on RSK1 and MSK1 are indicated by solid (essential site) and open (nonessential site) circles. The amino acid alignment and phosphorylated residues of RSK1 and MSK1 refer to the human proteins. While the kinases responsible for the phosphorylation of most sites within RSK1 and MSK1 are known, the identity of the kinase(s) responsible for the phosphorylation of several other essential sites remains unknown.
Serine/threonine kinase 3′-phosphoinositide-dependent kinase 1 (PDK1) was identified by our group (160) and others (88) as the kinase responsible for phosphorylation of the activation loop site (Ser221 in human RSK1) located in the NTKD of the RSKs (Fig. 7). The hydrophobic motif in RSK1 (P. Roux, unpublished results) and RSK2 (57) was subsequently shown to mediate PDK1 docking when phosphorylated, allowing PDK1 to phosphorylate and activate the NTKD of RSK1 and RSK2 (10, 35). PDK1 is required for the activation of many AGC kinases. For instance, mitogen stimulation of PDK1-null embryonic stem cells does not lead to the activation of either RSK1, Akt, or S6K1 (229). Phosphorylation of the CTKD activation loop of the RSK isoforms is thought to be mediated by ERK1/2 (195), but mutation of this site (Thr573 in RSK1) to alanine only partially reduces RSK1 and RSK2 activation (10, 29, 165). Aside from phosphorylating the activation loop site, ERK1/2 may also promote RSK1 activation by mediating its translocation to the plasma membrane, where it can be activated by membrane-associated kinases (159). Indeed, recent data suggest that the simple myristoylation of an RSK1 mutant incapable of binding ERK1/2 increases its level of phosphorylation at Ser363 and Ser380, suggesting that membrane translocation of the RSKs plays an important part in their activation process (159). However, the exact domains within RSKs that are involved in their cellular localization before and after stimulation remain unknown.
The sequence of events during activation of the RSKs remains elusive, but the mechanisms of activation are starting to emerge and appear to require well-ordered phosphorylation events (Fig. 7). Upon mitogen stimulation, activated ERK1/2 is thought to phosphorylate RSK1 on Thr573, which is partly required for CTKD activation, and possibly also on Ser363 (turn motif) and Thr359 (site with no associated function) (35, 165). As mentioned above, the role of ERK1/2 in activation of the RSKs may also be to bring the RSK isoforms into proximity with membrane-associated kinases that phosphorylate Ser363 and/or Ser380 (159; P. Roux, unpublished results). The activated CTKD then phosphorylates the hydrophobic motif (Ser380) of the RSKs, which creates a docking site for PDK1, the enzyme that phosphorylates the activation loop site (Ser221) and thereby activates the NTKD. While the CTKD has definitely been shown to phosphorylate Ser380 (29, 217), the involvement of the NTKD or other kinases in the phosphorylation of this site remains unknown. Interestingly, the homologous site in Akt has been shown to be potentially regulated by autophosphorylation (206) and by a heterologous kinase localized to lipid rafts (78), suggesting that the NTKD of the RSKs or a membrane-associated kinase may also phosphorylate the RSK isoforms at this site. Consistent with this, we have found that a CTKD-inactive mutant of RSK1 is still partially responsive to mitogen and phosphorylated at Ser380, agreeing with the idea that the hydrophobic motif may be regulated by multiple inputs (P. Roux, unpublished results).
The importance of the hydrophobic motif in activation of the RSKs was recently underscored by the identification of a phosphate-binding pocket in the kinase domain of many AGC kinases, which is used for intramolecular interaction with their own phosphorylated hydrophobic motif (55). This interaction was shown to induce a synergistic stimulation of RSK2 catalytic activity, whereas mutation of the phosphate-binding pocket led to a reduction in the overall kinase activity of RSK2. PDK1 also contains such a binding motif in its kinase domain but does not have a hydrophobic motif. Using embryonic stem cells with a knock-in mutation in the phosphate-binding pocket of PDK1, Alessi and colleagues have recently shown that the binding pocket in PDK1 is essential for activation of RSK2, S6K1, and SGK, suggesting that PDK1 uses this motif to interact with the phosphorylated hydrophobic motif of these kinases (34).
Finally, RSK1 activity has been shown to be regulated through its interaction with 14-3-3β (20). 14-3-3 proteins interact with a wide variety of cellular proteins, including protein kinases, receptor proteins, enzymes, and small G proteins, generally through a phosphoserine-containing motif (240). 14-3-3β binds to phosphorylated Ser154 of RSK1 and thereby negatively regulates its catalytic activity (20). Mutation of Ser154 to alanine increased both basal and serum-stimulated RSK1 activity, indicating that 14-3-3β normally represses RSK1 activity. Because the RSKs have been shown to interact with their upstream activators (ERK1/2 and PDK1), various downstream substrates, and regulatory proteins such as 14-3-3β and PEA-15, they appear to function as scaffold proteins that allow multiple proteins to come together and form a signaling network.
Substrates and functions.
As mentioned above, RSK1 was first discovered as a kinase that phosphorylated the ribosomal subunit protein S6. However, several lines of evidence indicated that S6 is not a major physiological target of the RSKs (30; reviewed in reference 120). Indeed, several groups have shown that S6K1 and S6K2 were the physiological S6 kinases in somatic cells, because treatment of cells with the mTOR (mammalian target of rapamycin) inhibitor rapamycin was shown to fully inhibit mitogen-induced S6 phosphorylation without affecting the activity of the RSKs (30, 87). Thus, it is generally thought that the RSKs may only modulate S6 phosphorylation under certain specific cellular circumstances (120).
The important physiological roles played by the RSKs have been underscored by the discovery that defects in the rsk2 gene are the cause of Coffin-Lowry syndrome (Table 1). Coffin-Lowry syndrome is an X-linked dominant disorder characterized by psychomotor and growth retardation and facial, hand, and skeletal malformations (84, 210). Numerous mutations have now been identified in the rsk2 gene, most of which are predicted or have been shown to result in truncated or inactive RSK2 proteins (40, 83). Fibroblasts derived from Coffin-Lowry syndrome patients have been useful in determining the function of RSK2 with respect to this human disease; however, differences found between these cells and RSK2-deficient mouse fibroblasts suggest that Coffin-Lowry syndrome may be a multivariable disease and may not be the ideal system with which to study RSK2 function. Both the rsk2 and rsk4 genes are located on chromosome X, and recent data also implicate rsk4 in nonspecific X-linked mental retardation (Table 1), but definitive evidence remains to be provided for rsk4 (241).
The substrate specificity of RSK1 for target phosphorylation has been determined with synthetic peptide libraries and found to require the minimum sequences Arg/Lys-Xaa-Arg-Xaa-Xaa-pSer/Thr or Arg-Arg-Xaa-pSer/Thr, where pSer is phosphoserine/phosphothreonine(110). These analyses also revealed that RSK1 prefers to phosphorylate Ser rather than Thr residues by a factor of about 5, and consistent with this, the majority of RSK1 and RSK2 substrates found to date are phosphorylated on Ser residues. A number of RSK functions can be deduced from the nature of their substrates, and data from many groups point towards roles for the RSKs in transcriptional regulation, cell cycle regulation, and cell survival (Fig. 8). Although more substrates have been identified for RSK2, most studies have not determined whether the other RSK isoforms could also phosphorylate the same targets, indicating that many currently known substrates may be shared by different RSK family members.
FIG. 8.

Substrates of the MKs. Upon activation, the RSKs, MSKs, MNKs, and MK2, -3, and -5 phosphorylate several substrates and regulate many biological responses, including mRNA translation, cell proliferation and survival, and the nuclear response to stress and mitogen stimulation. The list of substrates indicated in this figure is not exhaustive but emphasizes the many important substrates identified to date.
(i) Transcriptional regulation by RSK.
Immediately after their activation and translocation, the RSKs and the ERKs are capable of phosphorylating multiple transcription factors that contribute to the induction of immediate-early (IE) genes (reviewed in reference 205). For example, the transcription factors serum response factor (SRF) (15, 161) and CREB (cAMP response element-binding protein) (12, 65, 239) and possibly the chromatin-associated histone H3 (168) are substrates of the RSKs that participate in the immediate-early response. Many immediate-early gene products, such as c-Fos (22), c-Jun (24), and Nur77 (52), are also phosphorylated by the RSKs, providing a dual mechanism of immediate-early gene control. The posttranslational modification of these immediate-early gene products depends upon sustained RSK activation. Indeed, extensive evidence has been presented supporting a model in which certain immediate-early gene products can act as molecular sensors for ERK and RSK nuclear localization, signal duration, and signal strength (128, 129). For example, after mitogen-stimulated synthesis of c-Fos, ERK1/2 and RSK proteins are known to phosphorylate two Ser residues in the extreme C terminus of c-Fos (22). Phosphorylation of c-Fos by ERK1/2 and RSKs enhances its stability, promotes further phosphorylation events, and increases its growth-promoting effects (23, 129, 141). This hyperphosphorylation of c-Fos occurs only when the activation of RSK and ERK proteins is sustained (128, 129).
Several kinases phosphorylate CREB at Ser133, including protein kinase A, Akt, and MSK1/2, but a controversial report taking advantage of Coffin-Lowry syndrome patient-derived fibroblasts indicated that RSK2 was the major CREB kinase activated by epidermal growth factor (39). In these cells, epidermal growth factor failed to increase transcription of the c-fos gene, which was suggested to result from altered CREB-mediated transcription (39). To gain better insight into the physiological role of RSK2, mice deficient in RSK2 that were generated by homologous recombination yielded slightly different results (15). While transcription of the c-fos gene was also altered in fibroblasts derived from RSK2-null mice, CREB phosphorylation was found to be normal following platelet-derived growth factor and IGF-1stimulation (15). Although stimulation of Elk-1 phosphorylation was unaltered in either human or mouse RSK2-deficient cells, the transcriptional activity of both Elk-1 and serum response factor was reduced in mouse cells, suggesting that altered c-fos transcription may in fact result from defects in the activation of Elk-1 and SRF (15).
Differences between the human and mouse models may be due to species-specific compensation for the loss of RSK2 by other RSK isoforms, and for this reason, mice deficient in RSK2 may not be the ideal model for Coffin-Lowry syndrome. Unlike Coffin-Lowry syndrome patients, RSK2-deficient mice do not display any major cerebral abnormalities, suggesting that Coffin-Lowry syndrome may result from several mutations affecting normal brain development, which include RSK2 inactivation (15). However, it was recently demonstrated that RSK2-deficient mice may have impaired learning abilities (43), but the exact role of RSK2 in mouse brain development and whether these mice can be used as a partial model for Coffin-Lowry syndrome remains to be determined.
RSK1 also interacts with the ETS transcription factor ER81 and enhances ER81-dependent transcription by phosphorylating Ser191 and Ser216 (236). ER81 performs many essential functions in homeostasis, signaling response, and development, implying that RSK1 is also involved in these processes. The transcription initiator factor TIF-1A also becomes phosphorylated by ERK1/2 and RSK2 following serum stimulation on two Ser residues that are important for TIF-1A function (245). TIF-1A is required for RNA polymerase I transcription and rRNA synthesis, suggesting that RSK2 and ERK1/2 regulate transcription initiation during growth-promoting conditions. Estrogen receptor α is an ERK1/2 substrate that becomes activated following stimulation with growth factors. RSK1 was also shown to associate with and to phosphorylate estrogen receptor α on Ser167, which increases estrogen receptor α-mediated transcription (90). Another transcription factor, microphthalmia (Mi), has been shown to be phosphorylated by the RSKs (237) and has been linked to malignant melanoma (145). As mentioned above, the MAPKKK B-Raf was shown to be activated in 60% of melanomas (125), suggesting that the RSKs may be potential therapeutic targets for the treatment of melanomas.
Finally, stimulation of the ERK pathway promotes the interaction between RSK1 and the transcriptional coactivator CREB-binding protein (CBP) (130). CBP and its paralog p300 are large molecules that facilitate complex formation between different components of the transcriptional machinery. RSK1 interaction with CBP was found to modulate its function (130, 220), but the exact outcome of this interaction remains to be determined. Ectopic expression of the RSK1 binding region of CBP in PC12 cells inhibits NGF-mediated transcription of c-fos and neurite outgrowth, suggesting that RSK1 binding to CBP is important for these processes (130). Interestingly, CBP and p300 are known to associate with several transcription factors also known to be RSK1 and RSK2 substrates, such as CREB, c-Fos, c-Jun, ER81, and NF-κB, suggesting that RSK1 binding to CBP may provide a second mechanism of transcriptional control.
(ii) RSK and cell cycle control.
In addition to contributing to the immediate-early gene response during the G0/G1 phase of the cell cycle, a recent report demonstrated that RSK1 and RSK2 may promote G1-phase progression through the phosphorylation of the cyclin-dependent kinase (CDK) inhibitor p27kip1. Phosphorylation of p27kip1 by RSK1 and RSK2 was found to promote its association with 14-3-3 and prevent its translocation to the nucleus (59). Under growth arrest conditions, nuclear p27kip1 negatively regulates G1 progression by sequestering and inactivating cyclin E- or cyclin A-CDK2 complexes. Through phosphorylation of Thr198, which is also recognized by Akt (58), RSK1- and RSK2-mediated inhibition of p27kip1 nuclear translocation may promote G1 progression within mammalian cells.
ERK1/2 and the RSKs may also regulate progression through the G2 phase of the cell cycle. The role of ERK1/2 in cell cycle regulation has been demonstrated by many groups with the preferred model of Xenopus oocyte maturation (reviewed in reference 131). Immature oocytes are usually arrested in the G2 phase of the first meiotic cell division. Addition of progesterone induces synthesis of the MAPKKK c-Mos, which in turn activates the MAPK cascade leading to M-phase entry and subsequent maturation to an unfertilized egg. M-phase entry is controlled in part by Cdc2, which is a CDK normally kept in check by dual phosphorylation on both Thr14 and Thr15 by the inhibitory kinase Myt1. RSK2 has been shown to be the prominent RSK isoform in Xenopus oocytes (8), and with this model system, RSK2 has been shown to contribute to the control of the meiotic cell cycle at several critical points (174).
One mechanism by which RSK2 participates in the progression of oocytes through the G2/M phase of meiosis I is through phosphorylation and inhibition of the Myt1 kinase (144). The importance of RSK2 is such that constitutively activated RSK2 can mediate meiosis I entry even in the absence of progesterone or MAPK activation (67). RSK2 regulates meiosis I entry by binding to and phosphorylating the C terminus of Myt1, reducing its ability to inhibit the kinase activity of Cdc2/cyclin B1 complexes (144). It remains unknown whether this mechanism is conserved in other species, but recent efforts demonstrated that Akt can also act as a Myt1 kinase in starfish oocytes (142). RSK2 may also be important for progression of mammalian somatic cells through the G2/M phase of mitosis because ERK1/2 activity was shown to be required in synchronized NIH 3T3 fibroblasts (235).
Another way by which RSK2 can modulate the meiotic cell cycle in Xenopus laevis is through mediating MAPK-mediated metaphase II arrest, an activity known as cytostatic factor. Two groups have found that Xenopus RSK1 and RSK2 are essential for cytostatic factor by showing that activated RSK2 causes cytostatic factor even when MAPK is inactive and that depletion of RSK2 from oocyte extracts removes cytostatic factor activity (9, 68). RSK1 was later shown to phosphorylate and activate the kinase Bub1 in vitro, a mediator of anaphase-promoting complex inhibition (177), suggesting that RSK1-mediated Bub1 activation contributes, at least in part, to metaphase II arrest (211).
Finally, RSK2 also modulates the meiotic cell cycle through phosphorylation of histone H3, a process that requires the activation of the MAPK-RSK pathway during oocyte maturation (173). MAPK-mediated histone H3 phosphorylation has been shown to contribute to chromatin remodeling during cell cycle progression and also results in increased transcriptional regulation of several genes (31). Although RSK1 and RSK2 can phosphorylate histone H3 in vitro (173), it remains unknown whether the RSKs or another H3 kinase, such as aurora B, can directly phosphorylate histone H3 in this system. In mammalian cells, RSK2 has been shown to mediate mitogen-stimulated phosphorylation of histone H3. Fibroblasts derived from Coffin-Lowry syndrome patients and RSK2-deficient mouse embryonic stem cells display attenuated histone H3 phosphorylation in response to epidermal growth factor (168). However, recent evidence coming from the use of MSK1 and MSK2 knockout cells is currently casting some doubt on the physiological role of RSK2 in histone H3 phosphorylation, and recent attempts to reproduce the experiments in fibroblasts from Coffin-Lowry syndrome patients have failed (190). Indeed, a completely normal histone H3 phosphorylation response was seen in fibroblasts from Coffin-Lowry syndrome patients, which compromises the major piece of evidence supporting the role of RSK2 in mitogen-stimulated histone H3 phosphorylation (190).
(iii) RSK promotes survival.
RSK1 and RSK2 have also been shown to regulate survival in proliferating as well as in differentiated cells. For example, RSK2 has been found to promote the survival of primary cortical neurons through both transcription-dependent and -independent mechanisms (12). Neurotrophic factor-stimulated RSK2 phosphorylates the proapoptotic protein Bad on Ser112, thereby repressing its death-promoting activity (12). Similar regulation of Bad phosphorylation on Ser112 was seen in a hematopoietic cell line, where RSK1-mediated survival required Bad phosphorylation and inactivation (184). RSK2-mediated phosphorylation of the transcription factor CREB on Thr133 was also found to promote the survival of primary cortical neurons through increased transcription of survival-promoting genes (12, 65, 239).
RSK1 promotes the survival of hepatic stellate cells by phosphorylating mouse C/EBPβ on Thr217 in response to the hepatotoxin CCI4 (17).Phosphorylation of Thr217 was suggested to create a functional XEVD caspase-inhibitory box (K-phospho-T217-VD) that binds and inhibits caspases 1 and 8, and consistent with this, the cell-permeating tetrapeptide KE217VD was also shown to inhibit CCI4-mediated apoptosis of stellate cells (17). Interestingly, phosphorylation of this site was also shown to promote cell proliferation in response to transforming growth factor alpha (18), suggesting that RSK1 may be involved in this process. Two other studies indicated that RSK1 may also promote survival through the activation of the transcription factor NF-κB (64, 175). NF-κB comprises a family of heterodimeric transcription factors that are key regulators of a broad range of genes involved in inflammatory responses, proliferation, and apoptosis. RSK1 was shown to phosphorylate the NF-κB inhibitor IκBα on Ser32 (64, 175), suggesting that RSK1 may modulate NF-κB-dependent survival and proliferation.
(iv) Other targets of RSK.
In addition to transcriptional regulators and cell cycle controllers, the RSKs have been shown to phosphorylate many other targets involved in diverse cellular processes. Upon metabotropic glutamate receptor activation, RSK1 phosphorylates several protein targets within polyribosomes, including the known RSK1 substrate glycogen synthase 3β (GSK3β) (1, 196). Phosphorylation of GSK3β on Ser9 by RSK1 inhibits its kinase activity and results in increased protein translation through the release of inhibition of the GSK3β-regulated translation initiation factor eIF2B (reviewed in reference 33). RSK2 also phosphorylates another downstream substrate, the Na+/H+ exchanger isoform 1 (NHE-1) (198). NHE-1 is a key member of a family of exchangers that regulate intracellular pH and cell volume (197). RSK2-mediated phosphorylation of NHE-1 on Ser703 was found to regulate mitogen-dependent Na+/H+ exchange and intracellular pH (198).
The tumor suppressor LKB1 (also known as STK11), which is mutated in Peutz-Jeghers syndrome, represents another phosphorylation target of RSK2 (167). LKB1 was recently shown to phosphorylate and activate at least 12 protein kinases of the AMP-activated protein kinase subfamily (115, 181). Phosphorylation of LKB1 on Ser431 by RSK2 did not affect its activity, membrane association, or prenylation but was found to be necessary for LKB1-mediated growth suppression through unknown mechanisms (167). As LKB1 is likely an important player regulating cell proliferation, it will be interesting to determine the mechanisms involved in RSK2-mediated modulation of its function.
RSK2 was also shown to phosphorylate the cell adhesion molecule L1 on Ser1152 (232). L1 is a phosphoprotein that becomes hyperphosphorylated during periods of high neuronal activity, suggesting the involvement of RSK2 in neurite outgrowth. Recently, RSK1 was suggested to play a role in membrane ruffling, as the cytoskeleton-associated protein filamin A was found to be phosphorylated on Ser2152 by RSK1 (233). Ser2152 was previously shown to be phosphorylated by PAK1 and to be necessary for membrane ruffling in response to PAK1 activation (212), suggesting that RSK1 may play a similar role in cytoskeletal reorganization.
Proper regulation of the Ras/ERK pathway through phosphorylation-mediated negative feedback has been demonstrated to occur at many levels, including Ras, Raf-1, and MEK1 and -2 (19, 28). Despite having lower ERK1/2 protein levels, fibroblasts from RSK2-deficient mice display higher and more sustained phosphorylation of ERK1/2 in response to exercise and insulin (43), suggesting that RSK2 inhibits the ERK1/2 signaling cascade. A possible mechanism by which RSK2 can do this is through phosphorylation of SOS (and possibly Raf-1) in response to epidermal growth factor treatment (41, 94). RSK2 would therefore prevent further activation of the ERK1/2 pathway through phosphorylation and inactivation of its upstream activators. Interestingly, epidermal growth factor-mediated stimulation of Akt is also higher in RSK2-deficient cells (43), suggesting that Akt may compensate for the loss of RSK2 or that RSK2 is also involved in feedback inhibition of the phosphatidylinositol 3-kinase/Akt pathway through an unknownmechanism.
MSK Subfamily
Discovery.
Mitogen- and stress-activated kinases 1 and 2 (MSK1 and MSK2) were both identified in 1998 through genomewide homology searches (38, 137) and as a result of a two-hybrid screen with p38α as the bait (149). Human MSK1 (RSK-like protein kinase, or RLPK) and MSK2 (RSK-B) are 75% identical and, like the RSK enzymes, have two distinct kinase domains (Fig. 3). Although the MSKs have relatively high levels of homology to the RSKs (40% identity), they are thought to form a subfamily of MKs activated by both mitogens and stress stimuli (Fig. 6). MSK homologues have also been identified in different species. While no homologues have been found in S. cerevisiae to date, the C. elegans hypothetical protein kinase C54G4 and the D. melanogaster kinase JIL-1 are homologous to human MSK1, with 50 and 45% amino acid identity, respectively (89). Although the role of C54G4 is unknown, JIL-1 mediates histone H3 phosphorylation in D. melanogaster (89), which appears to be an evolutionarily conserved function for MSK1 and -2 (190). Similar to the RSKs, the NTKD of MSK1 and -2 belongs to the AGC family of kinases and is most similar to S6K1 and S6K2 (40% amino acid identity). The CTKD of MSK1 and -2 has a CaMK-like sequence and is mostly homologous to the kinase domain of MK2 and -3 (about 40% amino acid identity) (Fig. 4).
Structure and expression.
MSK1 and MSK2 share the major structural characteristics of the RSKs, including two distinct catalytic domains, a linker region of about 100 amino acids, and relatively similar C- and N-terminal regions (Fig. 3). Analogous to the RSKs, the NTKD of MSK1 and -2 is considered to be mainly responsible for substrate phosphorylation, and the CTKD is involved in MSK1 and -2 activation through autophosphorylation of the kinase. Both MSK isoforms contain a D domain (Leu-Ala-Lys-Arg-Arg-Lys) in their C terminus that lies within a region found to be necessary for MSK2 docking to ERK1/2 and p38 (Fig. 2) (207).
Unlike the RSKs, MSK1 and -2 are usually present in the nuclei of quiescent cells (38, 149, 207). The C-terminal region of the MSKs contains a functional bipartite NLS (Lys-Arg-Xaa14-Lys-Arg-Arg-Lys-Gln-Lys in MSK2) (Fig. 2) which confers on MSK1 and MSK2 an almost exclusive nuclear localization (38, 149). Although it is unknown whether the MSKs translocate to different cellular locations following activation, expression of MSK2 was found to regulate the location of ectopically expressed p38α and ERK1 (149). For instance, in the absence of MSK2, both p38α and ERK1 localized to the nucleus and cytoplasm, but upon expression of MSK2 constructs containing an intact D domain, p38α and ERK1 shifted to a more nuclear localization (149). These results indicate that MSK1 and MSK2 may control the cellular localization of their upstream activators, ERK1/2 and p38, a finding that was also observed with the closely related kinase MK2 (6).
Analysis of MSK1 and MSK2 expression in different tissues revealed that they are ubiquitously expressed, with predominant expression of the msk1 and msk2 mRNAs in the brain, heart, placenta, and skeletal muscles (38). Interestingly, the msk2 gene maps to the BBS1 locus on chromosome 11 (248). Bardet-Beidl syndrome (BBS) is a genetically heterogeneous disorder characterized primarily by retinal dystrophy, obesity, polydactyly, renal malformations, and learning disabilities (92). Although MSK2 inactivation has not yet been shown to contribute to Bardet-Beidl syndrome, it is worth noting that the clinical symptoms are reminiscent of Coffin-Lowry syndrome, suggesting that inactivation of RSK2 and MSK2 may contribute to similar disorders.
Activation mechanisms.
MSK1 and MSK2 are potently activated in vivo by ERK1/2 and p38 but not JNK (38, 149), indicating that both mitogens and stress stimuli lead to the activation of MSK1 and -2. Indeed, specific inhibitors of p38α and p38β (SB203580) and MEK1 and -2 (U0126) block MSK1 and -2 activation, depending on the origin of the stimuli (Fig. 6). Although the site-specific and sequential phosphorylation events during MSK1 and -2 activation have not yet been fully characterized, the four key phosphorylation sites present in RSKs are conserved in MSK1 and -2 (Fig. 7), suggesting that they have similar activation mechanisms (56). Indeed, changing all four residues to Ala inactivates MSK2 in response to MEK1 and MEK6 activation (208). Two of the four essential sites in MSK1 and -2 (Ser360 within the turn motif and Thr581 in the CTKD activation loop of MSK1) are followed by a proline residue, suggesting that they are likely ERK1/2 and p38 phosphorylation sites (Fig. 7). The two other phosphorylation sites are homologous to most AGC kinases and lie within the hydrophobic motif (Ser376) and the NTKD activation loop (Ser212) of MSK1. Inactivation of either kinase domain through mutation of conserved residues completely blocks the N-terminal kinase activity of MSK1 and -2 (38, 149, 208). Similar mutations in the CTKD of RSK1 and RSK2 have been shown to only partly reduce the activity of RSK1 and RSK2 against exogenous substrates (10, 29, 165), indicating that, unlike the RSKs, MSK1 and -2 activation critically requires CTKD activation (29). Since RSK1 translocates to the plasma membrane for full activation (159), the different requirements of these kinases for CTKD activity could be explained by their different cellular localizations.
During activation of the RSKs, phosphorylation of the hydrophobic motif creates a docking site for PDK1, allowing it to phosphorylate the activation loop of the NTKD. These phosphorylation sites are also conserved within MSK1 and MSK2 (Fig. 7), implying similar activation mechanisms. However, recent data indicate that MSK1 becomes fully activated in PDK1-null embryonic stem cells (229), suggesting that a kinase other than PDK1 phosphorylates the activation loop of MSK1 and -2. The high homology between the activation loop sequences of the RSKs and MSKs suggests that the kinase responsible for the phosphorylation of this site may be similar or related to PDK1. Although it is possible that the MSKs regulate their own activation loop through autophosphorylation, it seems more likely that an unknown PDK1-like kinase phosphorylates this site in vivo. Analogous to the RSKs, the MSKs have a phosphate-binding pocket within the NTKD that mediates intramolecular interaction with the phosphorylated hydrophobic motif (34, 55). This pocket was shown to be essential for MSK1 kinase activation, which underscores the importance of hydrophobic motif phosphorylation in MSK1 and -2 activation. It is clear that further experiments will be necessary to characterize the regulation of the hydrophobic motif in MSK1 and -2 and to identify the kinase responsible for the phosphorylation of this site.
According to their homology to the RSKs, the MSKs may also contain two additional phosphorylation sites. Based on work from our group (165), the role of the site located near the C terminus of MSK2 (Ser737) may be to regulate its association with ERK1/2 and p38. Although the D domain of MSK1 and -2 is located near the C terminus, MSK1 and MSK2 have an additional 20 and 50 amino acids C-terminal to the docking site, respectively, suggesting that these regions may contain additional elements necessary for ERK1/2 and p38 association (Fig. 7).
Substrates and functions.
The substrate specificity of the MSKs resembles that of the RSKs, with the minimal consensus substrate sequence Arg-Xaa-Xaa-pSer/Thr (38), indicating that the MSKs and RSKs may have similar targets. MSK1 and MSK2 are localized in the nucleus of quiescent and activated cells (38, 149), which suggests that they may preferentially phosphorylate nuclear substrates. Consistent with this, studies from newly engineered MSK1-, MSK2-, and MSK1/MSK2-deficient mice indicated that MSK1 and -2 play active roles in transcriptional regulation and in the nuclear response to stress and mitogens (2, 176, 228) (Fig. 8).
(i) Transcriptional regulation by MSK.
Upon activation, MSK1 and -2 are capable of phosphorylating multiple transcription factors and nuclear proteins, thereby increasing their stability or activity. As mentioned for the RSKs, the transactivation potential of the related transcription factors CREB and ATF1 depends on phosphorylation of a Ser residue in the kinase-inducible domain (Ser133 and Ser63 for CREB and ATF1, respectively). Both sites are phosphorylated in response to growth factors and cellular stresses, via the ERK1/2 and p38 pathways, respectively. Upon their discovery, MSK1 and MSK2 were shown to phosphorylate CREB on Ser133 in vitro with a Km much lower than that of the protein kinases RSK1 and MK2 (38, 149), raising the possibility that MSK1 rather than RSK1 mediates the mitogen-stimulated phosphorylation of CREB. Since then, work by several groups has supported the involvement of MSK1 and -2 in CREB phosphorylation with different pathway inhibitors (72, 107).
Further supporting the role of MSK1 and -2 as the mitogen- and stress-induced kinases responsible for CREB phosphorylation, knockout mice for MSK1, MSK2, and both kinases have recently been engineered (2, 176, 228). Stress-induced phosphorylation of CREB and ATF1 was found to be completely impaired in primary embryonic fibroblasts derived from MSK1/MSK2 knockout animals (228). Importantly, mitogen-induced phosphorylation of CREB and ATF1 was partially inhibited in these cells, suggesting that the RSKs may also contribute to the mitogenic response leading to CREB and ATF1 phosphorylation. As mentioned above, CREB activation participates in the transcriptional activation of several immediate-early genes, such as c-fos, junB, and egr1 (116). The knockout of both MSK1 and MSK2 resulted in a 50% reduction in c-fos and junB transcription in response to stress but only a minimal reduction in response to mitogenic stimulation (228), suggesting again that MSK1 and -2 regulate the immediate-early genes in response to stress stimuli.
Similar to RSK1, MSK1 has been shown to mediate NF-κB-dependent transcription through phosphorylation of the NF-κB isoform p65 on Ser276 (216). Loss of MSK1 and -2 in mouse fibroblasts resulted in reduced tumor necrosis factor-mediated activation of NF-κB, suggesting that stress-stimulated activation of MSK1 and -2 is important for the activation of NF-κB-dependent transcription (216). MSK1 has also been shown to phosphorylate the transcription factors ER81 (86) and STAT3 (227, 244); however, the use of MSK1/MSK2-null mouse fibroblasts will be necessary to elucidate the role of MSK1 and -2 in stress- and mitogen-mediated activation of ER81 and STAT3 because other MKs have also been shown to phosphorylate these factors.
(ii) MSK mediates the nucleosomal response.
The nucleosomal response refers to the rapid phosphorylation of histone H3 on Ser10 and HMG-14 on Ser6 that occurs concomitantly with immediate-early gene induction in response to a wide variety of stimuli (204). The fact that the ERK1/2- and p38-mediated signaling pathways culminate in a common nucleosomal response suggests that there may be a common nuclear effector. Although RSK1 and RSK2 have been shown to phosphorylate histone H3 in vitro and in vivo (168, 173), the MSKs were recently shown to be the prominent kinases involved in the nucleosomal response of somatic cells (190). Indeed, stress- and mitogen-induced phosphorylation of histone H3 and HMG-14 was found to be completely inhibited in primary embryonic fibroblasts from MSK1/MSK2 knockout animals (190), ruling out the possible involvement of other kinases such as the RSKs (37).
(iii) Other targets of MSK.
The MSKs have been shown to phosphorylate many more targets involved in diverse cellular processes. MSK1 and/or MSK2 has recently been shown to phosphorylate Bad (182), Akt (140), and 4E-BP1 (114) in response to UV irradiation, but the biological role of MSK1 and -2 in these processes remains to be determined.
MNK Subfamily
Discovery.
MAPK-interacting kinases 1 and 2 (MNK1 and -2) form a subfamily of MKs that were discovered in 1997 as a result of a two-hybrid screen designed to identify ERK2 binding proteins (221) and in a novel phosphorylation screen looking for ERK1 substrates (60). Human MNK1 has approximately 70% amino acid identity with MNK2, which differs from MNK1 mostly within its C-terminal region (Fig. 3 and 4). Homologues of the MNKs have been identified in other species. MNK1 displays relatively high homology (51% amino acid identity) to a large protein kinase in D. melanogaster termed LK6, which was found to localize to centrosomes and to regulate microtubule organization (93). MNK1 also has homology (46% amino acid identity) to the hypothetical protein kinase R166.5 found in C. elegans, but as with many MKs, no homologues of MNK1 and MNK2 have been identified in S. cerevisiae. The MNK1 and -2 catalytic domain belongs to the CaMK family of kinases and is most similar to the CTKD of the RSKs (33% amino acid identity) and MK2 and -3 (221) (Fig. 4).
Structure and expression.
While the mnk1 gene encodes only one isoform of 424 amino acids, transcription of the mnk2 gene generates two spliced isoforms, referred to as MNK2A and MNK2B, which are 465 and 414 amino acids in length, respectively (Fig. 3) (187). MNK2B lacks the C-terminal 52 amino acid containing the D domain (Fig. 4) and has been shown to be a poor substrate for ERK1/2 and p38 in vitro (170). The D domain of MNK1 consists of Leu-Ala-Arg-Arg-Arg (Fig. 2) and mediates the interaction of MNK1 with both ERK1/2 and p38 (60, 221). The D domain present in MNK2A contains only two contiguous basic residues (Leu-Ala-Gln-Arg-Arg), which is subtly different from the D domain in MNK1 (Fig. 2). This domain is highly reminiscent of the D domain found in RSKs, and consistent with this, MNK2A has been shown to interact only with ERK1/2 (221). The regulation of ERK1/2 and p38 interaction with the MNKs is currently unknown, but MNK1 has been shown to selectively bind unphosphorylated inactive ERK2 (221), suggesting that the complex between the two kinases may be regulated in a phosphorylation-dependent manner, similar to regulation by the RSKs (165).
While MNK1 and MNK2A localize primarily to the cytoplasm, MNK2B is enriched in the nucleus of quiescent cells. All three proteins contain a polybasic sequence in their N terminus that functions as a potent NLS (170). With a two-hybrid strategy, the NLS of MNK1 has been shown to mediate binding to importin-α, which is an intracellular receptor protein that mediates nuclear import (146, 222). The cytoplasmic localization of MNK1 can be explained by the presence of a CRM1-type nuclear export signal (NES) in its C terminus (Leu-Ala-Asp-Gly-Leu-Cys-Ser-Met-Lys-Leu) (146). The NES of MNK1 is functional, because leptomycin B (which blocks CRM1-dependent nuclear export) was shown to trap MNK1 in the nucleus, revealing that MNK1 is actively shuttling between the cytoplasm and the nucleus (121, 146). The C terminus of MNK2B does not contain such a motif, which correlates with its mostly nuclear localization. Because MNK2A lacks critical residues important for NES function, its cytoplasmic localization has been more of a mystery, but recent data suggest that its nuclear export is regulated by the intramolecular interaction between its N and C termini, which masks and unmasks critical regions necessary for nuclear export (170).
MNK1 and MNK2 are expressed in all adult tissues with the exception of the brain, where levels are greatly reduced (221). The expression of both proteins is especially abundant in skeletal muscles.
Activation mechanisms.
Although ERK1/2 and p38 are both capable of phosphorylating MNK1, MNK2A, and, to a lesser extent, MNK2B in vitro, MNK1 is the only isoform that can be activated by the p38 pathway in vivo (221) (Fig. 9). MNK1 has low basal activity in cells and is responsive to both stress- and mitogen-stimulated pathways (60, 221). MNK2A and, to a lesser extent, MNK2B appear to have high basal activity compared to MNK1 in quiescent cells (170), but the role of MNK2A and MNK2B in resting cells is unknown. Despite the fact that MNK2A possesses a potential ERK docking domain, both MNK2 isoforms are only marginally regulated by mitogen stimulation (170), indicating that further investigation will be necessary to uncover the specific roles of the MNKs in quiescent and stimulated cells.
FIG. 9.

Signaling cascades leading to activation of the MNKs. MNK1 is activated by two inputs originating from the ERK1/2 and p38 kinases, but MNK2 is only stimulated by ERK1/2. The limited number of substrates identified for the MNKs suggests that they play roles in mRNA translation.
Phosphopeptide analysis of MNK1 and MNK2 revealed the presence of several MAPK-stimulated phosphorylation sites (169, 222). Two of these proline-directed sites (Thr255 and Thr385 in the activation loop in MNK1) are homologous to residues in MK2 and -3 that have been shown to be phosphorylated by p38 and necessary for MK2 activity (7), suggesting similar activation mechanisms (Fig. 3). Substitution of both sites with Ala residues in MNK1 and -2 results in inactive kinases (170, 221, 222), and phosphomimetic mutation of Thr385 was found to activate MNK1 (60), indicating the importance of these sites in MNK1 and -2 activation. Deletion of the C-terminal 91 amino acids containing the D domain was also shown to render MNK1 inactive (60), suggesting that ERK1/2 and/or p38 docking and phosphorylation of two important regulatory sites are required for efficient MNK1 and MNK2 activation.
Substrates and functions.
Regulation of protein synthesis by the eukaryotic initiation factor 4F (eIF4F) complex plays an important role in controlling cell growth and proliferation (reviewed in reference 191). In higher eukaryotes, the eIF4F complex consists of three subunits, eIF4A, eIF4E, and eIF4G. eIF4E recruits mRNAs to the complex through binding of their 5′ cap structure, the N7-methylguanosine cap. eIF4E also interacts with the scaffold protein eIF4G, which recruits the RNA helicase eIF4A to the complex, unwinds the secondary structure in the mRNA, and facilitates translation initiation by the 40S ribosomal complex. The activity of eIF4E is considered to be partly regulated by phosphorylation at Ser209, and mitogens and stresses have been shown to induce eIF4E phosphorylation through ERK1/2 and p38 (53).
MNK1 was found to be recruited to the eIF4F complex through its association with the C terminus of eIF4G (155), making it a likely candidate kinase that mediates eIF4E phosphorylation (Fig. 8). Consistent with this idea, MNK1 and MNK2 were later found to phosphorylate eIF4E at Ser209 in response to stress and mitogen stimulation (95, 169, 221). MNK1 can also phosphorylate eIF4G and bind to phosphoprotein p97 in vitro, but whether these events also occur in vivo remains to be determined (154). The biological significance of eIF4E phosphorylation is not completely understood. Through an unknown mechanism, phosphorylated eIF4E was reported to interact more efficiently with eIF4G (16), and phosphorylation of eIF4E has been shown to both increase (126) and diminish (172) its affinity for the cap structure of the mRNA. Recent findings show that MNK1- and MNK2-mediated phosphorylation of eIF4E on Ser209 correlates with reduced cap-dependent translation (95). Thus, it seems clear that future work aimed at analyzing the role of the MNKs in translation control, for example, through the generation of MNK1 and -2 knockout mice and fibroblasts, will likely help to clarify the function of eIF4E phosphorylation (171).
MNK2B but not MNK1 or MNK2A binds to estrogen receptor β (187), which is highly reminiscent of the phosphorylation of estrogen receptor α by RSK1 (90). Addition of the steroid estradiol increased the interaction between MNK2B and estrogen receptor β, but whether MNK2B phosphorylates estrogen receptor β to affect its function remains unknown.
MK2 and -3 Subfamily
Discovery.
MK2 was discovered in 1992 by Philip Cohen and colleagues as a protein kinase activated by ERK1/2 from rabbit skeletal muscle (192). MK3 was later discovered by a two-hybrid screen looking for interactors of p38 (122) and as the product of a gene commonly deleted in small-cell lung cancer cells (Table 1) (186). MK2 homologues have been identified in D. melanogaster and C. elegans, which show about 60% amino acid identity with human MK2 (106). Interestingly, the yeast protein kinases Rck1 and Rck2 show some homology with MK2 (about 35% amino acid identity) and represent the sole MK-related kinases identified to date in the S. cerevisiae genome (157). Rck1 and Rck2 have been shown to play roles in the inhibition of meiosis in S. cerevisiae, but no such functions have yet been attributed to MK2. MK2 is highly homologous to MK3 (75% amino acid identity), which suggests that they form a subfamily of related kinases (Fig. 5). The kinase domains of MK2 and -3 are most similar (35 to 40% identity) to CaMK, phosphorylase b kinase, and the CTKD of the RSK isoforms (193, 249) (Fig. 4).
Structure and expression.
While the mk2 gene encodes two alternatively spliced transcripts of 370 amino acids (MK2A) and 400 amino acids (MK2B) (27, 192), the mk3 gene encodes only one transcript of 382 amino acids (186). These proteins are highly homologous, yet MK2A possesses a shorter C-terminal region. The C terminus of MK2B contains a functional bipartite NLS (Lys-Lys-Xaa10-Lys-Arg-Arg-Lys-Lys) that is not present in the shorter MK2A isoform (Fig. 3), indicating that alternative splicing determines the cellular localization of the MK2 isoforms. MK3 possesses a similar NLS, but the functionality of this domain has not yet been tested by mutational analysis. The NLS found in both MK2B and MK3 also encompasses a D domain (Leu-Leu-Lys-Arg-Arg-Lys-Lys in MK2B) (Fig. 2) that was shown to mediate the specific interaction of MK2B and MK3 with p38α and p38β (188). MK2B and MK3 also possess a functional NES located N-terminal to the NLS and D domain (Fig. 3). The NES in MK2B is sufficient to trigger nuclear export following stimulation, which can be inhibited by leptomycin B (6, 44). The sequence N-terminal to the catalytic domain in MK2 and MK3 is proline rich and contains one (MK3) or two (MK2) putative Src homology 3 (SH3) domain-binding sites, which have been shown, for MK2, to mediate binding to the SH3 domain of c-Abl in vitro (45, 151). Recent data suggest that this domain is involved in MK2-mediated cell migration (102).
While MK2A is mostly present in the cytoplasm, MK2B and MK3 are predominantly located in the nucleus of quiescent cells (44, 134). Upon stress stimulation, both MK2B and MK3 are rapidly exported to the cytoplasm via a CRM1-dependent mechanism (6, 44, 179). Nuclear export of MK2B appears to be mediated by kinase activation, as phosphomimetic mutation of Thr334 within the activation loop of the kinase enhances the cytoplasmic localization of MK2B (44). For this reason, it is thought that MK2B and MK3 contain a constitutively active NLS and a phosphorylation-regulated NES.
MK2 and MK3 appear to be ubiquitously expressed, with predominant expression in the heart, skeletal muscle, and kidney tissues (186, 192).
Activation mechanisms.
ERK1/2 and p38 were first reported to activate MK2 and MK3 in vitro (7, 46, 83, 117), but it was later found that ERK1 and -2 are not physiological MK2 and -3 kinases. Thus, MK2 and MK3 activity is potently stimulated by various activators of p38α and p38β (54, 70, 122, 163) (Fig. 10).
FIG. 10.
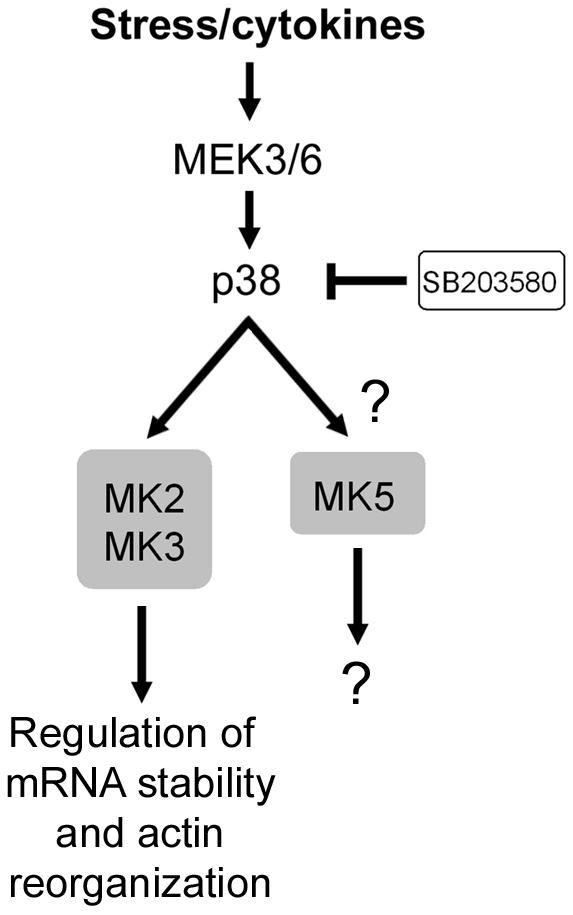
Signaling cascades leading to activation of MK2 and -3 and MK5. MK2 and -3 have been shown to be activated by p38. Conversely, MK5 was initially shown to be regulated by p38, but recent data have not been able to confirm these results. While no bona fide substrates of MK5 have been identified to date, MK2 and -3 have been found to regulate mRNA stability and actin reorganization.
p38 was found to mediate the in vitro and in vivo phosphorylation of MK2 on four proline-directed sites, Thr25, Thr222, Ser272, and Thr334 (7), of which only Thr25 is not conserved in MK3. The function of phosphorylated Thr25 is unknown, but its location between the two SH3 domain-binding sites suggests that it may regulate protein-protein interactions. Thr222 in MK2 (Thr201 in MK3) is located in the activation loop of the kinase domain and has been shown to be essential for MK2 and MK3 kinase activity (7, 46, 117). Thr334 in MK2 (Thr313 in MK3) is located C-terminal to the catalytic domain and is essential for kinase activity (117, 192). The crystal structure of MK2 has been resolved and suggests that Thr334 phosphorylation may serve as a switch for MK2 nuclear import and export (124). It is also thought that phosphorylation of Thr334 would weaken or interrupt binding of the C terminus of MK2 to the catalytic domain, exposing the NES and promoting nuclear export. Such conformational changes in MK2 are also supported by a FRET analysis of a green fluorescence protein (GFP)-MK2 fusion protein, which revealed that activation of MK2 is accompanied by an open conformation of the protein which only becomes detectable in the cytoplasm of activated cells (133).
While p38 is capable of activating MK2 and MK3 in the nucleus, the cellular location of p38 itself has been shown to be controlled by MK2 and possibly MK3. Following phosphorylation and activation of MK2, nuclear p38 has been shown to be exported to the cytoplasm in a complex with MK2 (6). The interaction between p38 and MK2 also appears to be important for p38 stabilization, because a recent report indicated that p38 levels are low in MK2-deficient cells and expression of a catalytically inactive MK2 protein restores p38 levels (102). These results suggest that activation and nuclear export of MK2 and -3 are coupled by a phosphorylation-dependent conformational switch that also dictates p38 stabilization and localization.
Substrates and functions.
MK2 shares many substrates with MK3 (Fig. 8); the two enzymes have comparable substrate preferences and phosphorylate peptide substrates with similar kinetic constants (32). The minimum sequence required for efficient phosphorylation by MK2 was found to be Hyd-Xaa-Arg-Xaa-Xaa-pSer/Thr, where Hyd is a bulky hydrophobic residue (193).
Experimental evidence supports a role for p38 in the regulation of cytokine biosynthesis (73, 108) and cell migration (76, 150). The targeted deletion of the mk2 gene in mice provided the unexpected result that, although p38 mediates the activation of many similar kinases, MK2 seems to be the key kinase responsible for these p38-dependent biological processes (101, 102). Loss of MK2 leads to a defect in lipopolysaccharide-induced synthesis of cytokines such as tumor necrosis factor alpha, interleukin-6, and gamma interferon (101) and also to changes in the migration of mouse embryonic fibroblasts, smooth muscle cells, and neutrophils (75, 102, 132). Consistent with a role for MK2 in inflammatory responses, MK2-deficient mice show increased susceptibility to Listeria monocytogenes infection (109) and reduced inflammation-mediated neuronal death following focal ischemia (218). Since the levels of p38 protein are also significantly reduced in MK2-deficient cells, it was necessary to distinguish whether these phenotypes were due solely to the loss of MK2 (100). To achieve this, MK2 mutants were expressed in MK2-deficient cells, and the results indicated that the catalytic activity of MK2 was not necessary to restore p38 levels but was required to regulate cytokine biosynthesis (102).
(i) Regulation of mRNA translation.
MK2 was shown to increase tumor necrosis factor alpha production by increasing the rate of translation of its mRNA (101, 132). Indeed, no significant reductions in the transcription, processing, and shedding of tumor necrosis factor alpha could be detected in MK2-deficient mice (101, 132). The p38 pathway is known to play an important role in regulating mRNA stability (99, 215), and MK2 represents a likely target of p38 mediating of this function (231). Recent data obtained from MK2-deficient mice indicated that the catalytic activity of MK2 is necessary for its effects on cytokine production and migration (102), suggesting that MK2 phosphorylates targets involved in mRNA stability. Consistent with this, MK2 has been shown to bind and/or phosphorylate the heterogeneous nuclear ribonucleoprotein (hnRNP) A0 (164), tristetraprolin (119), the poly(A)-binding protein PABP1 (11), and HuR (74, 209). These substrates are known to bind or copurify with mRNAs that contain AU-rich elements in the 3′ untranslated region (69), suggesting that MK2 may regulate the stability of AU-rich mRNAs such as tumor necrosis factor alpha. It is currently unknown whether MK3 plays similar functions, but lipopolysaccharide treatment of MK2-deficient fibroblasts completely abolished hnRNP A0 phosphorylation, suggesting that MK3 is not able to compensate for the loss of MK2 (164).
MK3 participates with MK2 in phosphorylation of the eukaryotic elongation factor 2 (eEF2) kinase, the enzyme that phosphorylates and inactivates eEF2 (96). eEF2 activity is critical for the elongation of mRNA during translation, and phosphorylation of eEF2 on Thr56 results in the termination of mRNA translation. MK2 and -3 phosphorylation of eEF2 kinase on Ser377 suggests that these enzymes can modulate eEF2 kinase activity and thereby regulate mRNA translation elongation (96).
(ii) Transcriptional regulation by MK2 and -3.
Similar to many MKs, nuclear MK2 contributes to the phosphorylation of CREB (199), serum response factor (77), and ER81 (85). Comparison of wild-type and MK2-deficient cells revealed that MK2 is the major SRF kinase induced by stress (77), suggesting a role for MK2 in the stress-mediated immediate-early response. Both MK2 and MK3 interact with basic helix-loop-helix transcription factor E47 in vivo and phosphorylate E47 in vitro (134). MK2-mediated phosphorylation of E47 was found to repress the transcriptional activity of E47 and thereby inhibit E47-dependent gene expression, suggesting that MK2 and -3 may regulate tissue-specific gene expression and cell differentiation (134).
(iii) Other targets of MK2 and -3.
Several other MK2 and -3 substrates have also been identified, suggesting that MK2 and -3 play diverse functions in several biological processes. With a functional proteomic approach consisting of mass spectrometric phosphoprotein identification following in vitro phosphorylation of neutrophil lysate by active recombinant MK2, the scaffolding protein 14-3-3ζ was identified as a physiological MK2 substrate (153). 14-3-3ζ was reported to interact with a number of components of cell signaling pathways, including protein kinases, phosphatases, and transcription factors (reviewed in reference 214). MK2-mediated phosphorylation of 14-3-3ζ on Ser58 was shown to compromise its binding activity (153), suggesting that MK2 may affect the regulation of several signaling molecules normally regulated by 14-3-3ζ.
With the same approach, MK2 was found to also interact with and to phosphorylate the p16 subunit of the seven-member Arp2 and -3 complex, p16-Arc, on Ser77 (185). p16-Arc plays roles in regulating the actin cytoskeleton, suggesting that MK2 may be involved in this process. Consistent with such a role for MK2, the small heat shock protein HSP27 (54, 163, 194), lymphocyte-specific protein LSP-1 (80), and vimentin (26) have been shown to be phosphorylated by MK2. HSP27 is of particular interest because it forms large oligomers which can act as molecular chaperones and protect cells from heat shock and oxidative stress (105). Upon phosphorylation, HSP27 loses its ability to form large oligomers and is unable to block actin polymerization (105, 162), suggesting that MK2-mediated phosphorylation of HSP27 serves a homeostatic function aimed at regulating actin dynamics that would otherwise be destabilized during stress (51, 70). Interestingly, MK3 was also shown to phosphorylate HSP27 in vitro and in vivo (117, 122), but its role during stressful conditions has not yet been addressed.
MK2 and MK3 can also phosphorylate 5-lipoxygenase, an enzyme that catalyzes the initial steps in the formation of the inflammatory mediators leukotrienes (224). Tyrosine hydroxylase (195), glycogen synthase (192), and Akt (158) were also shown to be phosphorylated by MK2. Finally, MK2 phosphorylates the tumor suppressor protein tuberin on Ser1210, creating a docking site for 14-3-3 (113). Tuberin and hamartin normally form a functional complex that negatively regulates cell growth by antagonizing mTOR-dependent signaling (203), suggesting that p38-mediated activation of MK2 may regulate cell growth by increasing 14-3-3 binding to tuberin.
MK5
Discovery.
MK5 (p38-regulated/activated protein kinase) was discovered through homology searches of expressed sequence tag database with the MK2 sequence as the query (136, 139). MK5 has 40% amino acid identity with MK2 and MK3, which suggests that it is a more distant homologue of these proteins (Fig. 5). Indeed, MK5 may have originated earlier during evolution from a common ancestral protein, because MK2 and -3 and MK5 have roughly the same amino acid relatedness to the MK2 homologues found in D. melanogaster and C. elegans. Like MK2 and MK3, the kinase domain of MK5 is most similar to that of CaMK-related kinases (Fig. 3). Because MK5 is a relatively new MK family member, less is known about its activation mechanism and function.
Structure and expression.
The mk5 gene encodes two alternatively spliced transcripts of 471 amino acids (MK5A) and 473 amino acids (MK5B), but the relevance of having two nearly identical isoforms is unknown (Fig. 5). MK5A and MK5B are similar in structure to MK2 and -3, possessing a kinase domain bordered with a short N-terminal region lacking the SH3 domain binding motif and a C-terminal region that contains a functional NLS, NES, and D domain (Fig. 3). The NLS (Arg-Lys-Arg-Lys) of MK5 was shown to be functional because alanine substitutions were found to disrupt the nuclear localization of MK5 (179). The NLS also overlaps a D domain (Ile-Leu-Arg-Lys-Arg-Lys-Leu-Leu) that mediates interaction with p38α and p38β (179). Interaction of p38 with MK5 was found to be weaker than with MK2, and consistent with this, MK5 does not appear to stabilize p38 in vivo (183). The NES of MK5 (Leu-Arg-Val-Ser-Leu-Arg-Pro-Leu-His-Ser) is functional and is sufficient to trigger CRM1-dependent nuclear export, as shown by leptomycin B experiments (179).
When overexpressed, MK5 localizes to the nucleus of quiescent cells, but upon cellular stress, MK5 translocates to the cytoplasm (135, 179). Interestingly, expression of exogenous p38α with MK5 has been shown to relocate MK5 to the cytoplasm, suggesting that p38 docking interferes with the function of its NLS (179). MK5 expression appears to be ubiquitous, with predominant expression in the heart and skeletal muscle (136, 139).
Activation mechanisms.
Upon its discovery, MK5 was found to be activated by p38α and p38β through phosphorylation of Thr182, located in the activation loop of the kinase domain (135, 136, 179) (Fig. 4). However, recent data have challenged these results, because MK5 activity does not appear to be significantly increased by stimulation of the p38 cascade (183) (Fig. 10). While MK2 was able to interact with p38 and become activated by extracellular stresses, endogenous MK5 did not display the same properties (183), indicating that more experimentation will be necessary to fully characterize MK5's activation mechanisms and functions. In addition, the generation of MK5-specific reagents will be necessary to distinguish MK5- from MK2 and -3-dependent functions.
Substrates and functions.
MK5 has originally been shown to share many substrates with MK2, such as HSP27 and glycogen synthase (136), but the recent description of mice rendered null for MK5 by homologous recombination has suggested the need for a reevaluation of these results (183) (Fig. 8). Despite the relatively high similarity between MK2 and MK5 (Fig. 4), MK5-deficient mice did not display any of the phenotypic changes seen in MK2-deficient animals (101). Although no MK5 protein or kinase activity was detected in the MK5 knockout mice, the animals were viable and fertile and did not display abnormalities in tissue morphology or behavior (183). The parallel analysis of MK5- and MK2-deficient fibroblasts revealed that MK2 but not MK5 is able to phosphorylate HSP27 at the physiological residue in vivo (183). Indeed, the loss of MK5 expression did not affect HSP27 phosphorylation induced by stress stimulation, indicating that MK2 is the sole regulator of stress-stimulated HSP27 phosphorylation in vivo (183). The authors also found that the MK5 antibody originally used to demonstrate the ability of MK5 to phosphorylate HSP27 actually cross-reacts with MK2 and MK3 (136), explaining the discrepancies between different reports (183). Because of the high homology between MK2 and MK5, future substrates of MK5 should be tested in MK5-deficient cells to determine whether they are also shared with MK2.
CONCLUSIONS AND PERSPECTIVES
Recent evidence indicates that, although they have relatively high homologies and are activated by similar mechanisms, each MK family member will exert specific functions within a cell. Currently available data suggest that ERK- and p38-responsive MKs generally function during mitogenic and stressful conditions, respectively. For example, one might predict that active p38 will stimulate MK2 and -3 activity, which in turn will regulate actin reorganization and mRNA stability in response to various physical and chemical stresses. MK2 is also responsible for increasing the mRNA stability of several cytokines that are required in response to viral or bacterial insults that activate the p38 signaling cascade. Another example involves ERK1/2-mediated activation of the RSKs, which are usually thought of as having roles in cell survival and proliferation in response to mitogenic signaling.
Activation of the RSKs during mitogenic conditions seems logical, but it is more difficult to understand the need for a cell to have kinases that respond to both mitogenic and stress stimuli. While the RSKs and MK2 and -3 are activated solely by ERK1/2 and p38, respectively, the MSKs and MNKs are functionally positioned to integrate these two signaling pathways, indicating that cellular stresses and mitogens induce specific and overlapping responses. One can imagine that the MSK1- and -2-mediated regulation of histone H3 and HMG-14 is necessary to allow either ERK1/2- or p38-mediated effectors to stimulate transcription. In the same vein, activation of the MNKs would result in increased mRNA translation, which may be necessary for both the ERK1/2 and p38 signaling cascades to exert their functions. Does this mean that MSK1 and -2 and MNK1 and -2 have more diverse and important functions because these kinases are required in response to both mitogens and stress stimuli? At present, this question cannot be answered. Data from MK2, RSK2, and MSK1 and -2 knockout mice indicate that MK2 knockout mice have a stronger phenotype than RSK2 knockout mice, but interestingly, MSK1 and -2 knockout animals have a relatively mild phenotype. Clearly, the generation of additional reagents that inhibit specific members of the MK family will be necessary to answer this question.
The diverse biological functions of the many ERK- and p38-regulated MKs are generated by their unique structural properties and temporal and spatial regulation. For example, the differential cellular distribution of the RSKs and MSKs may dictate the identity of their targeted substrates. Similarly, alternative splicing of MK2A and MK2B appears to determine their localization by the presence or absence of a functional NLS, and this will also likely affect their regulation and the identity of the substrates targeted by these enzymes. With regard to the structural aspects of signaling, the RSKs have been shown to interact with several proteins. This property is highly reminiscent of a scaffold protein and suggests that the RSKs may be involved in creating specific signaling modules by bringing together upstream activating kinases, adaptor proteins, and downstream substrates.
The continual characterization of MK signaling complexes and the identification of novel substrates should reveal overlapping and unique biological functions for the various MKs. Along these lines, a rewarding task for the future will be to generate reagents to specifically inhibit the different MKs, allowing the contribution of each kinase to various cellular responses to be deciphered. Moreover, the generation of knockout mice for RSK1, RSK3 and RSK4, MNK1 and MNK2, and MK3, in addition to those already generated, should lead to new information about their specific roles in MAPK signaling. It is likely that we are just beginning to understand the cellular processes regulated by the MKs, and future studies should be most enlightening.
Acknowledgments
We thank members of the Blenis laboratory for helpful discussions and Rana Anjum, Andrew Tee, and Leon Murphy for critical reading of the manuscript.
P.P.R. is supported by a fellowship from the International Human Frontier Science Program Organization.
REFERENCES
- 1.Angenstein, F., W. T. Greenough, and I. J. Weiler. 1998. Metabotropic glutamate receptor-initiated translocation of protein kinase p90rsk to polyribosomes: a possible factor regulating synaptic protein synthesis. Proc. Natl. Acad. Sci. USA 95:15078-15083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arthur, J. S., and P. Cohen. 2000. MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett. 482:44-48. [DOI] [PubMed] [Google Scholar]
- 3.Ballif, B. A., and J. Blenis. 2001. Molecular mechanisms mediating mammalian mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth Differ. 12:397-408. [PubMed] [Google Scholar]
- 4.Behn-Krappa, A., and A. C. Newton. 1999. The hydrophobic phosphorylation motif of conventional protein kinase C is regulated by autophosphorylation. Curr. Biol. 9:728-737. [DOI] [PubMed] [Google Scholar]
- 5.Bellacosa, A., T. O. Chan, N. N. Ahmed, K. Datta, S. Malstrom, D. Stokoe, F. McCormick, J. Feng, and P. Tsichlis. 1998. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene 17:313-325. [DOI] [PubMed] [Google Scholar]
- 6.Ben-Levy, R., S. Hooper, R. Wilson, H. F. Paterson, and C. J. Marshall. 1998. Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr. Biol. 8:1049-1057. [DOI] [PubMed] [Google Scholar]
- 7.Ben-Levy, R., I. A. Leighton, Y. N. Doza, P. Attwood, N. Morrice, C. J. Marshall, and P. Cohen. 1995. Identification of novel phosphorylation sites required for activation of MAPKAP kinase-2. EMBO J. 14:5920-5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhatt, R. R., and J. E. Ferrell, Jr. 2000. Cloning and characterization of Xenopus Rsk2, the predominant p90 Rsk isozyme in oocytes and eggs. J. Biol. Chem. 275:32983-32990. [DOI] [PubMed] [Google Scholar]
- 9.Bhatt, R. R., and J. E. Ferrell, Jr. 1999. The protein kinase p90 rsk as an essential mediator of cytostatic factor activity. Science 286:1362-1365. [DOI] [PubMed] [Google Scholar]
- 10.Bjorbaek, C., Y. Zhao, and D. E. Moller. 1995. Divergent functional roles for p90rsk kinase domains. J. Biol. Chem. 270:18848-18852. [DOI] [PubMed] [Google Scholar]
- 11.Bollig, F., R. Winzen, M. Gaestel, S. Kostka, K. Resch, and H. Holtmann. 2003. Affinity purification of ARE-binding proteins identifies polyA-binding protein 1 as a potential substrate in MK2-induced mRNA stabilization. Biochem. Biophys. Res. Commun. 301:665-670. [DOI] [PubMed] [Google Scholar]
- 12.Bonni, A., A. Brunet, A. E. West, S. R. Datta, M. A. Takasu, and M. E. Greenberg. 1999. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286:1358-1362. [DOI] [PubMed] [Google Scholar]
- 13.Bott, C. M., S. G. Thorneycroft, and C. J. Marshall. 1994. The sevenmaker gain-of-function mutation in p42 MAP kinase leads to enhanced signalling and reduced sensitivity to dual specificity phosphatase action. FEBS Lett. 352:201-205. [DOI] [PubMed] [Google Scholar]
- 14.Brancho, D., N. Tanaka, A. Jaeschke, J. J. Ventura, N. Kelkar, Y. Tanaka, M. Kyuuma, T. Takeshita, R. A. Flavell, and R. J. Davis. 2003. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 17:1969-1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruning, J. C., J. A. Gillette, Y. Zhao, C. Bjorbaeck, J. Kotzka, B. Knebel, H. Avci, B. Hanstein, P. Lingohr, D. E. Moller, W. Krone, C. R. Kahn, and D. Muller-Wieland. 2000. Ribosomal subunit kinase-2 is required for growth factor-stimulated transcription of the c-Fos gene. Proc. Natl. Acad. Sci. USA 97:2462-2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bu, X., D. W. Haas, and C. H. Hagedorn. 1993. Novel phosphorylation sites of eukaryotic initiation factor-4F and evidence that phosphorylation stabilizes interactions of the p25 and p220 subunits. J. Biol. Chem. 268:4975-4978. [PubMed] [Google Scholar]
- 17.Buck, M., V. Poli, T. Hunter, and M. Chojkier. 2001. C/EBPbeta phosphorylation by RSK creates a functional XEXD caspase inhibitory box critical for cell survival. Mol. Cell 8:807-816. [DOI] [PubMed] [Google Scholar]
- 18.Buck, M., V. Poli, P. van der Geer, M. Chojkier, and T. Hunter. 1999. Phosphorylation of rat serine 105 or mouse threonine 217 in C/EBP beta is required for hepatocyte proliferation induced by TGF alpha. Mol. Cell 4:1087-1092. [DOI] [PubMed] [Google Scholar]
- 19.Campbell, S. L., R. Khosravi-Far, K. L. Rossman, G. J. Clark, and C. J. Der. 1998. Increasing complexity of Ras signaling. Oncogene 17:1395-1413. [DOI] [PubMed] [Google Scholar]
- 20.Cavet, M. E., S. Lehoux, and B. C. Berk. 2003. 14-3-3beta is a p90 ribosomal S6 kinase (RSK) isoform 1-binding protein that negatively regulates RSK kinase activity. J. Biol. Chem. 278:18376-18383. [DOI] [PubMed] [Google Scholar]
- 21.Chang, C. I., B. E. Xu, R. Akella, M. H. Cobb, and E. J. Goldsmith. 2002. Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol. Cell 9:1241-1249. [DOI] [PubMed] [Google Scholar]
- 22.Chen, R.-H., C. Abate, and J. Blenis. 1993. Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 90:10952-10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen, R. H., P. C. Juo, T. Curran, and J. Blenis. 1996. Phosphorylation of c-Fos at the C-terminus enhances its transforming activity. Oncogene 12:1493-1502. [PubMed] [Google Scholar]
- 24.Chen, R.-H., C. Sarnecki, and J. Blenis. 1992. Nuclear localization and regulation of the erk- and rsk-encoded protein kinases. Mol. Cell. Biol. 12:915-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen, Z., T. B. Gibson, F. Robinson, L. Silvestro, G. Pearson, B. Xu, A. Wright, C. Vanderbilt, and M. H. Cobb. 2001. MAP kinases. Chem. Rev. 101:2449-2476. [DOI] [PubMed] [Google Scholar]
- 26.Cheng, T. J., and Y. K. Lai. 1998. Identification of mitogen-activated protein kinase-activated protein kinase-2 as a vimentin kinase activated by okadaic acid in 9L rat brain tumor cells. J. Cell Biochem. 71:169-181. [PubMed] [Google Scholar]
- 27.Chevalier, D., and B. G. Allen. 2000. Two distinct forms of MAPKAP kinase-2 in adult cardiac ventricular myocytes. Biochemistry 39:6145-6156. [DOI] [PubMed] [Google Scholar]
- 28.Chong, H., H. G. Vikis, and K. L. Guan. 2003. Mechanisms of regulating the Raf kinase family. Cell Signal. 15:463-469. [DOI] [PubMed] [Google Scholar]
- 29.Chrestensen, C. A., and T. W. Sturgill. 2002. Characterization of the p90 ribosomal S6 kinase 2 carboxyl-terminal domain as a protein kinase. J. Biol. Chem. 277:27733-27741. [DOI] [PubMed] [Google Scholar]
- 30.Chung, J., C. J. Kuo, G. R. Crabtree, and J. Blenis. 1992. Rapamycin-FKBP specifically blocks growth-dependent activation of and signalling by the 70kD S6 protein kinases. Cell 69:1227-1236. [DOI] [PubMed] [Google Scholar]
- 31.Clayton, A. L., and L. C. Mahadevan. 2003. MAP kinase-mediated phosphoacetylation of histone H3 and inducible gene regulation. FEBS Lett. 546:51-58. [DOI] [PubMed] [Google Scholar]
- 32.Clifton, A. D., P. R. Young, and P. Cohen. 1996. A comparison of the substrate specificity of MAPKAP kinase-2 and MAPKAP kinase-3 and their activation by cytokines and cellular stress. FEBS Lett. 392:209-214. [DOI] [PubMed] [Google Scholar]
- 33.Cohen, P., and S. Frame. 2001. The renaissance of GSK3. Nat. Rev. Mol. Cell. Biol. 2:769-776. [DOI] [PubMed] [Google Scholar]
- 34.Collins, B. J., M. Deak, J. S. Arthur, L. J. Armit, and D. R. Alessi. 2003. In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J. 22:4202-4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dalby, K. N., N. Morrice, F. B. Caudwell, J. Avruch, and P. Cohen. 1998. Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J. Biol. Chem. 273:1496-1505. [DOI] [PubMed] [Google Scholar]
- 36.Dan, I., N. M. Watanabe, and A. Kusumi. 2001. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 11:220-230. [DOI] [PubMed] [Google Scholar]
- 37.Davie, J. R. 2003. MSK1 and MSK2 mediate mitogen- and stress-induced phosphorylation of histone H3: a controversy resolved. Science STKE 2003:PE33. [DOI] [PubMed]
- 38.Deak, M., A. D. Clifton, L. M. Lucocq, and D. R. Alessi. 1998. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 17:4426-4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Cesare, D., S. Jacquot, A. Hanauer, and P. Sassone-Corsi. 1998. Rsk-2 activity is necessary for epidermal growth factor-induced phosphorylation of CREB protein and transcription of c-fos gene. Proc. Natl. Acad. Sci. USA 95:12202-12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delaunoy, J., F. Abidi, M. Zeniou, S. Jacquot, K. Merienne, S. Pannetier, M. Schmitt, C. Schwartz, and A. Hanauer. 2001. Mutations in the X-linked RSK2 gene (RPS6KA3) in patients with Coffin-Lowry syndrome. Hum. Mutat. 17:103-116. [DOI] [PubMed] [Google Scholar]
- 41.Douville, E., and J. Downward. 1997. EGF induced SOS phosphorylation in PC12 cells involves P90 RSK-2. Oncogene 15:373-383. [DOI] [PubMed] [Google Scholar]
- 42.Downward, J. 2003. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 3:11-22. [DOI] [PubMed] [Google Scholar]
- 43.Dufresne, S. D., C. Bjorbaek, K. El-Haschimi, Y. Zhao, W. G. Aschenbach, D. E. Moller, and L. J. Goodyear. 2001. Altered extracellular signal-regulated kinase signaling and glycogen metabolism in skeletal muscle from p90 ribosomal S6 kinase 2 knockout mice. Mol. Cell. Biol. 21:81-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engel, K., A. Kotlyarov, and M. Gaestel. 1998. Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J. 17:3363-3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Engel, K., K. Plath, and M. Gaestel. 1993. The MAP kinase-activated protein kinase 2 contains a proline-rich SH3-binding domain. FEBS Lett. 336:143-147. [DOI] [PubMed] [Google Scholar]
- 46.Engel, K., H. Schultz, F. Martin, A. Kotlyarov, K. Plath, M. Hahn, U. Heinemann, and M. Gaestel. 1995. Constitutive activation of mitogen-activated protein kinase-activated protein kinase 2 by mutation of phosphorylation sites and an A-helix motif. J. Biol. Chem. 270:27213-27221. [DOI] [PubMed] [Google Scholar]
- 47.Enslen, H., D. M. Brancho, and R. J. Davis. 2000. Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J. 19:1301-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Enslen, H., and R. J. Davis. 2001. Regulation of MAP kinases by docking domains. Biol. Cell 93:5-14. [DOI] [PubMed] [Google Scholar]
- 49.Erikson, E., and J. L. Maller. 1985. A protein kinase from Xenopus eggs specific for ribosomal protein S6. Proc. Natl. Acad. Sci. USA 82:742-746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fantz, D. A., D. Jacobs, D. Glossip, and K. Kornfeld. 2001. Docking sites on substrate proteins direct extracellular signal-regulated kinase to phosphorylate specific residues. J. Biol. Chem. 276:27256-27265. [DOI] [PubMed] [Google Scholar]
- 51.Fernando, P., L. A. Megeney, and J. J. Heikkila. 2003. Phosphorylation-dependent structural alterations in the small hsp30 chaperone are associated with cellular recovery. Exp. Cell Res. 286:175-185. [DOI] [PubMed] [Google Scholar]
- 52.Fisher, T. L., and J. Blenis. 1996. Evidence for two catalytically active kinase domains in pp90rsk. Mol. Cell. Biol. 16:1212-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flynn, A., R. G. Vries, and C. G. Proud. 1997. Signalling pathways which regulate eIF4E. Biochem. Soc. Trans. 25:192S. [DOI] [PubMed]
- 54.Freshney, N. W., L. Rawlinson, F. Guesdon, E. Jones, S. Cowley, J. Hsuan, and J. Saklatvala. 1994. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell 78:1039-1049. [DOI] [PubMed] [Google Scholar]
- 55.Frodin, M., T. L. Antal, B. A. Dummler, C. J. Jensen, M. Deak, S. Gammeltoft, and R. M. Biondi. 2002. A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J. 21:5396-5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frodin, M., and S. Gammeltoft. 1999. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol. Cell. Endocrinol. 151:65-77. [DOI] [PubMed] [Google Scholar]
- 57.Frodin, M., C. J. Jensen, K. Merienne, and S. Gammeltoft. 2000. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J. 19:2924-2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fujita, N., S. Sato, K. Katayama, and T. Tsuruo. 2002. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J. Biol. Chem. 277:28706-28713. [DOI] [PubMed] [Google Scholar]
- 59.Fujita, N., S. Sato, and T. Tsuruo. 2003. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J. Biol. Chem. 278:49254-49260. [DOI] [PubMed] [Google Scholar]
- 60.Fukunaga, R., and T. Hunter. 1997. MNK1, a new MAP kinase-activate protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 16:1921-1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao, T., and A. C. Newton. 2002. The turn motif is a phosphorylation switch that regulates the binding of Hsp70 to protein kinase C. J. Biol. Chem. 277:31585-31592. [DOI] [PubMed] [Google Scholar]
- 62.Gavin, A. C., and A. R. Nebreda. 1999. A MAP kinase docking site is required for phosphorylation and activation of p90(rsk)/MAPKAP kinase-1. Curr. Biol. 9:281-284. [DOI] [PubMed] [Google Scholar]
- 63.Geyer, M., and A. Wittinghofer. 1997. GEFs, GAPs, GDIs and effectors: taking a closer (3D) look at the regulation of Ras-related GTP-binding proteins. Curr. Opin. Struct. Biol. 7:786-792. [DOI] [PubMed] [Google Scholar]
- 64.Ghoda, L., X. Lin, and W. C. Greene. 1997. The 90-kDa ribosomal S6 kinase (pp90rsk) phosphorylates the N-terminal regulatory domain of IkappaBalpha and stimulates its degradation in vitro. J. Biol. Chem. 272:21281-21288. [DOI] [PubMed] [Google Scholar]
- 65.Ginty, D. D., A. Bonni, and M. E. Greenberg. 1994. Nerve growth factor activates a Ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell 77:713-725. [DOI] [PubMed] [Google Scholar]
- 66.Gonzalez, F. A., A. Seth, D. L. Raden, D. S. Bowman, F. S. Fay, and R. J. Davis. 1993. Serum-induced translocation of mitogen-activated protein kinase to the cell surface ruffling membrane and the nucleus. J. Cell Biol. 122:1089-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gross, S. D., A. L. Lewellyn, and J. L. Maller. 2001. A constitutively active form of the protein kinase p90Rsk1 is sufficient to trigger the G2/M transition in Xenopus oocytes. J. Biol. Chem. 276:46099-46103. [DOI] [PubMed] [Google Scholar]
- 68.Gross, S. D., M. S. Schwab, A. L. Lewellyn, and J. L. Maller. 1999. Induction of metaphase arrest in cleaving Xenopus embryos by the protein kinase p90Rsk. Science 286:1365-1367. [DOI] [PubMed] [Google Scholar]
- 69.Grzybowska, E. A., A. Wilczynska, and J. A. Siedlecki. 2001. Regulatory functions of 3′UTRs. Biochem. Biophys. Res. Commun. 288:291-295. [DOI] [PubMed] [Google Scholar]
- 70.Guay, J., H. Lambert, G. Gingras-Breton, J. N. Lavoie, J. Huot, and J. Landry. 1997. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J. Cell Sci. 110:357-368. [DOI] [PubMed] [Google Scholar]
- 71.Hallberg, B., S. I. Rayter, and J. Downward. 1994. Interaction of Ras and Raf in intact mammalian cells upon extracellular stimulation. J. Biol. Chem. 269:3913-3916. [PubMed] [Google Scholar]
- 72.Hamm, J., D. R. Alessi, and R. M. Biondi. 2002. Bi-functional, substrate mimicking RNA inhibits MSK1-mediated cAMP-response element-binding protein phosphorylation and reveals magnesium ion-dependent conformational changes of the kinase. J. Biol. Chem. 277:45793-45802. [DOI] [PubMed] [Google Scholar]
- 73.Han, J., J. D. Lee, L. Bibbs, and R. J. Ulevitch. 1994. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265:808-811. [DOI] [PubMed] [Google Scholar]
- 74.Han, Q., J. Leng, D. Bian, C. Mahanivong, K. A. Carpenter, Z. K. Pan, J. Han, and S. Huang. 2002. Rac1-MKK3-p38-MAPKAPK2 pathway promotes urokinase plasminogen activator mRNA stability in invasive breast cancer cells. J. Biol. Chem. 277:48379-48385. [DOI] [PubMed] [Google Scholar]
- 75.Hannigan, M. O., L. Zhan, Y. Ai, A. Kotlyarov, M. Gaestel, and C. K. Huang. 2001. Abnormal migration phenotype of mitogen-activated protein kinase-activated protein kinase 2-/- neutrophils in Zigmond chambers containing formyl-methionyl-leucyl-phenylalanine gradients. J. Immunol. 167:3953-3961. [DOI] [PubMed] [Google Scholar]
- 76.Hedges, J. C., M. A. Dechert, I. A. Yamboliev, J. L. Martin, E. Hickey, L. A. Weber, and W. T. Gerthoffer. 1999. A role for p38(MAPK)/HSP27 pathway in smooth muscle cell migration. J. Biol. Chem. 274:24211-24219. [DOI] [PubMed] [Google Scholar]
- 77.Heidenreich, O., A. Neininger, G. Schratt, R. Zinck, M. A. Cahill, K. Engel, A. Kotlyarov, R. Kraft, S. Kostka, M. Gaestel, and A. Nordheim. 1999. MAPKAP kinase 2 phosphorylates serum response factor in vitro and in vivo. J. Biol. Chem. 274:14434-14443. [DOI] [PubMed] [Google Scholar]
- 78.Hill, M. M., J. Feng, and B. A. Hemmings. 2002. Identification of a plasma membrane Raft-associated PKB Ser473 kinase activity that is distinct from ILK and PDK1. Curr. Biol. 12:1251-1255. [DOI] [PubMed] [Google Scholar]
- 79.Hsiao, K.-M., S.-Y. Chou, S.-J. Shih, and J. E. Ferrell, Jr. 1994. Evidence that inactive p42 mitogen-activated protein kinase and inactive Rsk exist as a heterodimer in vivo. Proc. Natl. Acad. Sci. USA 91:5480-5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang, C. K., L. Zhan, Y. Ai, and J. Jongstra. 1997. LSP1 is the major substrate for mitogen-activated protein kinase-activated protein kinase 2 in human neutrophils. J. Biol. Chem. 272:17-19. [DOI] [PubMed] [Google Scholar]
- 81.Jacobs, D., G. J. Beitel, S. G. Clark, H. R. Horvitz, and K. Kornfeld. 1998. Gain-of-function mutations in the Caenorhabditis elegans lin-1 ETS gene identify a C-terminal regulatory domain phosphorylated by ERK MAP kinase. Genetics 149:1809-1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jacobs, D., D. Glossip, H. Xing, A. J. Muslin, and K. Kornfeld. 1999. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 13:163-175. [PMC free article] [PubMed] [Google Scholar]
- 83.Jacquot, S., K. Merienne, D. De Cesare, S. Pannetier, J. L. Mandel, P. Sassone-Corsi, and A. Hanauer. 1998. Mutation analysis of the RSK2 gene in Coffin-Lowry patients: extensive allelic heterogeneity and a high rate of de novo mutations. Am. J. Hum. Genet. 63:1631-1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jacquot, S., K. Merienne, E. Trivier, M. Zeniou, S. Pannetier, and A. Hanauer. 1999. Coffin-Lowry syndrome: current status. Am. J. Med. Genet. 85:214-215. [PubMed] [Google Scholar]
- 85.Janknecht, R. 2001. Cell type-specific inhibition of the ETS transcription factor ER81 by mitogen-activated protein kinase-activated protein kinase 2. J. Biol. Chem. 276:41856-41861. [DOI] [PubMed] [Google Scholar]
- 86.Janknecht, R. 2003. Regulation of the ER81 transcription factor and its coactivators by mitogen- and stress-activated protein kinase 1 (MSK1). Oncogene 22:746-755. [DOI] [PubMed] [Google Scholar]
- 87.Jefferies, H. B., S. Fumagalli, P. B. Dennis, C. Reinhard, R. B. Pearson, and G. Thomas. 1997. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. EMBO J. 16:3693-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jensen, C. J., M. B. Buch, T. O. Krag, B. A. Hemmings, S. Gammeltoft, and M. Frodin. 1999. 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3-phosphoinositide-dependent protein kinase-1. J. Biol. Chem. 274:27168-27176. [DOI] [PubMed] [Google Scholar]
- 89.Jin, Y., Y. Wang, D. L. Walker, H. Dong, C. Conley, J. Johansen, and K. M. Johansen. 1999. JIL-1: a novel chromosomal tandem kinase implicated in transcriptional regulation in Drosophila. Mol. Cell 4:129-135. [DOI] [PubMed] [Google Scholar]
- 90.Joel, P. B., J. Smith, T. W. Sturgill, T. L. Fisher, J. Blenis, and D. A. Lannigan. 1998. pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol. Cell. Biol. 18:1978-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jones, S. W., E. Erikson, J. Blenis, J. L. Maller, and R. L. Erikson. 1988. A Xenopus ribosomal protein S6 kinase has two apparent kinase domains that are each similar to distinct protein kinases. Proc. Natl. Acad. Sci. USA 85:3377-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Katsanis, N., J. R. Lupski, and P. L. Beales. 2001. Exploring the molecular basis of Bardet-Biedl syndrome. Hum. Mol. Genet. 10:2293-2299. [DOI] [PubMed] [Google Scholar]
- 93.Kidd, D., and J. W. Raff. 1997. LK6, a short lived protein kinase in Drosophila that can associate with microtubules and centrosomes. J. Cell Sci. 110:209-219. [DOI] [PubMed] [Google Scholar]
- 94.Kinuya, M., K. Takishima, and G. Mamiya. 2000. Detection of kinases that phosphorylate 14-3-3 binding sites of Raf-1 using in situ gel kinase assay. Biol. Pharm. Bull. 23:1158-1162. [DOI] [PubMed] [Google Scholar]
- 95.Knauf, U., C. Tschopp, and H. Gram. 2001. Negative regulation of protein translation by mitogen-activated protein kinase-interacting kinases 1 and 2. Mol. Cell. Biol. 21:5500-5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Knebel, A., C. E. Haydon, N. Morrice, and P. Cohen. 2002. Stress-induced regulation of eukaryotic elongation factor 2 kinase by SB 203580-sensitive and -insensitive pathways. Biochem. J. 367:525-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kohno, M., and J. Pouyssegur. 2003. Pharmacological inhibitors of the ERK signaling pathway: application as anticancer drugs. Prog. Cell Cycle Res. 5:219-224. [PubMed] [Google Scholar]
- 98.Kolch, W. 2000. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 351:289-305. [PMC free article] [PubMed] [Google Scholar]
- 99.Kontoyiannis, D., A. Kotlyarov, E. Carballo, L. Alexopoulou, P. J. Blackshear, M. Gaestel, R. Davis, R. Flavell, and G. Kollias. 2001. Interleukin-10 targets p38 MAPK to modulate ARE-dependent TNF mRNA translation and limit intestinal pathology. EMBO J. 20:3760-3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kotlyarov, A., and M. Gaestel. 2002. Is MK2 (mitogen-activated protein kinase-activated protein kinase 2) the key for understanding post-transcriptional regulation of gene expression? Biochem. Soc. Trans. 30:959-963. [DOI] [PubMed] [Google Scholar]
- 101.Kotlyarov, A., A. Neininger, C. Schubert, R. Eckert, C. Birchmeier, H. D. Volk, and M. Gaestel. 1999. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat. Cell Biol. 1:94-97. [DOI] [PubMed] [Google Scholar]
- 102.Kotlyarov, A., Y. Yannoni, S. Fritz, K. Laass, J. B. Telliez, D. Pitman, L. L. Lin, and M. Gaestel. 2002. Distinct cellular functions of MK2. Mol. Cell. Biol. 22:4827-4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kyriakis, J. M., and J. Avruch. 2001. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81:807-869. [DOI] [PubMed] [Google Scholar]
- 104.Kyriakis, J. M., and J. Avruch. 1990. pp54 microtubule-associated protein 2 kinase. A novel serine/threonine protein kinase regulated by phosphorylation and stimulated by poly-L-lysine. J. Biol. Chem. 265:17355-17363. [PubMed] [Google Scholar]
- 105.Lambert, H., S. J. Charette, A. F. Bernier, A. Guimond, and J. Landry. 1999. HSP27 multimerization mediated by phosphorylation-sensitive intermolecular interactions at the amino terminus. J. Biol. Chem. 274:9378-9385. [DOI] [PubMed] [Google Scholar]
- 106.Larochelle, S., and B. Suter. 1995. The Drosophila melanogaster homolog of the mammalian MAPK-activated protein kinase-2 (MAPKAPK-2) lacks a proline-rich N-terminus. Gene 163:209-214. [DOI] [PubMed] [Google Scholar]
- 107.Lee, C. W., J. S. Nam, Y. K. Park, H. K. Choi, J. H. Lee, N. H. Kim, J. Cho, D. K. Song, H. W. Suh, J. Lee, Y. H. Kim, and S. O. Huh. 2003. Lysophosphatidic acid stimulates CREB through mitogen- and stress-activated protein kinase-1. Biochem. Biophys. Res. Commun. 305:455-461. [DOI] [PubMed] [Google Scholar]
- 108.Lee, J. C., J. T. Laydon, P. C. McDonnell, T. F. Gallagher, S. Kumar, D. Green, D. McNulty, M. J. Blumenthal, J. R. Heys, S. W. Landvatter, and et al. 1994. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372:739-746. [DOI] [PubMed] [Google Scholar]
- 109.Lehner, M. D., F. Schwoebel, A. Kotlyarov, M. Leist, M. Gaestel, and T. Hartung. 2002. Mitogen-activated protein kinase-activated protein kinase 2-deficient mice show increased susceptibility to Listeria monocytogenes infection. J. Immunol. 168:4667-4673. [DOI] [PubMed] [Google Scholar]
- 110.Leighton, I. A., K. N. Dalby, F. B. Caudwell, P. T. Cohen, and P. Cohen. 1995. Comparison of the specificities of p70 S6 kinase and MAPKAP kinase-1 identifies a relatively specific substrate for p70 S6 kinase: the N-terminal kinase domain of MAPKAP kinase-1 is essential for peptide phosphorylation. FEBS Lett. 375:289-293. [DOI] [PubMed] [Google Scholar]
- 111.Lenormand, P., C. Sardet, G. Pages, G. L'Allemain, A. Brunet, and J. Pouyssegur. 1993. Growth factors induce nuclear translocation of MAP kinases (p42mapk and p44mapk) but not of their activator MAP kinase kinase (p45mapkk) in fibroblasts. J. Cell Biol. 122:1079-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lewis, T. S., P. S. Shapiro, and N. G. Ahn. 1998. Signal transduction through MAP kinase cascades. Adv. Cancer Res. 74:49-139. [DOI] [PubMed] [Google Scholar]
- 113.Li, Y., K. Inoki, P. Vacratsis, and K. L. Guan. 2003. The p38 and MK2 kinase cascade phosphorylates tuberin, the tuberous sclerosis 2 gene product, and enhances its interaction with 14-3-3. J. Biol. Chem. 278:13663-13671. [DOI] [PubMed] [Google Scholar]
- 114.Liu, G., Y. Zhang, A. M. Bode, W. Y. Ma, and Z. Dong. 2002. Phosphorylation of 4E-BP1 is mediated by the p38/MSK1 pathway in response to UVB irradiation. J. Biol. Chem. 277:8810-8816. [DOI] [PubMed] [Google Scholar]
- 115.Lizcano, J. M., O. Goransson, R. Toth, M. Deak, N. A. Morrice, J. Boudeau, S. A. Hawley, L. Udd, T. P. Makela, D. G. Hardie, and D. R. Alessi. 2004. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 23:833-843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lonze, B. E., and D. D. Ginty. 2002. Function and regulation of CREB family transcription factors in the nervous system. Neuron 35:605-623. [DOI] [PubMed] [Google Scholar]
- 117.Ludwig, S., K. Engel, A. Hoffmeyer, G. Sithanandam, B. Neufeld, D. Palm, M. Gaestel, and U. R. Rapp. 1996. 3pK, a novel mitogen-activated protein (MAP) kinase-activated protein kinase, is targeted by three MAP kinase pathways. Mol. Cell. Biol. 16:6687-6697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Madhani, H. D., and G. R. Fink. 1998. The riddle of MAP kinase signaling specificity. Trends Genet. 14:151-155. [DOI] [PubMed] [Google Scholar]
- 119.Mahtani, K. R., M. Brook, J. L. Dean, G. Sully, J. Saklatvala, and A. R. Clark. 2001. Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol. Cell. Biol. 21:6461-6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Martin, K. A., and J. Blenis. 2002. Coordinate regulation of translation by the PI 3-kinase and mTOR pathways. Adv. Cancer Res. 86:1-39. [DOI] [PubMed] [Google Scholar]
- 121.McKendrick, L., E. Thompson, J. Ferreira, S. J. Morley, and J. D. Lewis. 2001. Interaction of eukaryotic translation initiation factor 4G with the nuclear cap-binding complex provides a link between nuclear and cytoplasmic functions of the m7 guanosine cap. Mol. Cell. Biol. 21:3632-3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.McLaughlin, M. M., S. Kumar, P. C. McDonnell, S. Van Horn, J. C. Lee, G. P. Livi, and P. R. Young. 1996. Identification of mitogen-activated protein (MAP) kinase-activated protein kinase-3, a novel substrate of CSBP p38 MAP kinase. J. Biol. Chem. 271:8488-8492. [DOI] [PubMed] [Google Scholar]
- 123.Meier, R., J. Rouse, A. Cuenda, A. R. Nebreda, and P. Cohen. 1996. Cellular stresses and cytokines activate multiple mitogen-activated-protein kinase kinase homologues in PC12 and KB cells. Eur. J. Biochem. 236:796-805. [DOI] [PubMed] [Google Scholar]
- 124.Meng, W., L. L. Swenson, M. J. Fitzgibbon, K. Hayakawa, E. Ter Haar, A. E. Behrens, J. R. Fulghum, and J. A. Lippke. 2002. Structure of mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 suggests a bifunctional switch that couples kinase activation with nuclear export. J. Biol. Chem. 277:37401-37405. [DOI] [PubMed] [Google Scholar]
- 125.Mercer, K. E., and C. A. Pritchard. 2003. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim. Biophys. Acta 1653:25-40. [DOI] [PubMed] [Google Scholar]
- 126.Minich, W. B., M. L. Balasta, D. J. Goss, and R. E. Rhoads. 1994. Chromatographic resolution of in vivo phosphorylated and nonphosphorylated eukaryotic translation initiation factor eIF-4E: increased cap affinity of the phosphorylated form. Proc. Natl. Acad. Sci. USA 91:7668-7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mizukami, Y., K. Yoshioka, S. Morimoto, and K. Yoshida. 1997. A novel mechanism of JNK1 activation. Nuclear translocation and activation of JNK1 during ischemia and reperfusion. J. Biol. Chem. 272:16657-16662. [DOI] [PubMed] [Google Scholar]
- 128.Murphy, L. O., J. P. MacKeigan, and J. Blenis. 2004. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol. Cell. Biol. 24:144-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Murphy, L. O., S. Smith, R. H. Chen, D. C. Fingar, and J. Blenis. 2002. Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 4:556-564. [DOI] [PubMed] [Google Scholar]
- 130.Nakajima, T., A. Fukamizu, J. Takahashi, F. H. Gage, T. Fisher, J. Blenis, and M. R. Montminy. 1996. The signal-dependent coactivator CBP is a nuclear target for pp90RSK. Cell 86:465-474. [DOI] [PubMed] [Google Scholar]
- 131.Nebreda, A. R., and I. Ferby. 2000. Regulation of the meiotic cell cycle in oocytes. Curr. Opin. Cell Biol. 12:666-675. [DOI] [PubMed] [Google Scholar]
- 132.Neininger, A., D. Kontoyiannis, A. Kotlyarov, R. Winzen, R. Eckert, H. D. Volk, H. Holtmann, G. Kollias, and M. Gaestel. 2002. MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J. Biol. Chem. 277:3065-3068. [DOI] [PubMed] [Google Scholar]
- 133.Neininger, A., H. Thielemann, and M. Gaestel. 2001. FRET-based detection of different conformations of MK2. EMBO Rep. 2:703-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Neufeld, B., A. Grosse-Wilde, A. Hoffmeyer, B. W. Jordan, P. Chen, D. Dinev, S. Ludwig, and U. R. Rapp. 2000. Serine/threonine kinases 3pK and MAPK-activated protein kinase 2 interact with the basic helix-loop-helix transcription factor E47 and repress its transcriptional activity. J. Biol. Chem. 275:20239-20242. [DOI] [PubMed] [Google Scholar]
- 135.New, L., Y. Jiang, and J. Han. 2003. Regulation of PRAK subcellular location by p38 MAP kinases. Mol. Biol. Cell 14:2603-2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.New, L., Y. Jiang, M. Zhao, K. Liu, W. Zhu, L. J. Flood, Y. Kato, G. C. Parry, and J. Han. 1998. PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 17:3372-3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.New, L., M. Zhao, Y. Li, W. W. Bassett, Y. Feng, S. Ludwig, F. D. Padova, H. Gram, and J. Han. 1999. Cloning and characterization of RLPK, a novel RSK-related protein kinase. J. Biol. Chem. 274:1026-1032. [DOI] [PubMed] [Google Scholar]
- 138.Newton, A. C. 2003. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem. J. 370:361-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ni, H., X. S. Wang, K. Diener, and Z. Yao. 1998. MAPKAPK5, a novel mitogen-activated protein kinase (MAPK)-activated protein kinase, is a substrate of the extracellular-regulated kinase (ERK) and p38 kinase. Biochem. Biophys. Res. Commun. 243:492-496. [DOI] [PubMed] [Google Scholar]
- 140.Nomura, M., A. Kaji, W. Y. Ma, S. Zhong, G. Liu, G. T. Bowden, K. I. Miyamoto, and Z. Dong. 2001. Mitogen- and stress-activated protein kinase 1 mediates activation of Akt by ultraviolet B irradiation. J. Biol. Chem. 276:25558-25567. [DOI] [PubMed] [Google Scholar]
- 141.Okazaki, K., and N. Sagata. 1995. The Mos/MAP kinase pathway stabilizes c-Fos by phosphorylation and augments its transforming activity in NIH 3T3 cells. EMBO J. 14:5048-5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Okumura, E., T. Fukuhara, H. Yoshida, S. Hanada Si, R. Kozutsumi, M. Mori, K. Tachibana, and T. Kishimoto. 2002. Akt inhibits Myt1 in the signalling pathway that leads to meiotic G2/M-phase transition. Nat. Cell Biol. 4:111-116. [DOI] [PubMed] [Google Scholar]
- 143.Ono, K., and J. Han. 2000. The p38 signal transduction pathway: activation and function. Cell Signal. 12:1-13. [DOI] [PubMed] [Google Scholar]
- 144.Palmer, A., A. C. Gavin, and A. R. Nebreda. 1998. A link between MAP kinase and p34(cdc2)/cyclin B during oocyte maturation: p90(rsk) phosphorylates and inactivates the p34(cdc2) inhibitory kinase Myt1. EMBO J. 17:5037-5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Park, H. Y., and B. A. Gilchrest. 2002. More on MITF. J. Investig. Dermatol. 119:1218-1219. [DOI] [PubMed] [Google Scholar]
- 146.Parra-Palau, J. L., G. C. Scheper, M. L. Wilson, and C. G. Proud. 2003. Features in the N- and C-termini of the MAP kinase-interacting kinase Mnk1 mediate its nucleocytoplasmic shuttling. J. Biol. Chem. 278:44197-44204. [DOI] [PubMed]
- 147.Pearson, G., F. Robinson, T. Beers Gibson, B. E. Xu, M. Karandikar, K. Berman, and M. H. Cobb. 2001. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocrinol. Rev. 22:153-183. [DOI] [PubMed] [Google Scholar]
- 148.Peyssonnaux, C., and A. Eychene. 2001. The Raf/MEK/ERK pathway: new concepts of activation. Biol. Cell 93:53-62. [DOI] [PubMed] [Google Scholar]
- 149.Pierrat, B., J. S. Correia, J. L. Mary, M. Tomas-Zuber, and W. Lesslauer. 1998. RSK-B, a novel ribosomal S6 kinase family member, is a CREB kinase under dominant control of p38alpha mitogen-activated protein kinase (p38alphaMAPK). J. Biol. Chem. 273:29661-29671. [DOI] [PubMed] [Google Scholar]
- 150.Piotrowicz, R. S., E. Hickey, and E. G. Levin. 1998. Heat shock protein 27 kDa expression and phosphorylation regulates endothelial cell migration. FASEB J. 12:1481-1490. [DOI] [PubMed] [Google Scholar]
- 151.Plath, K., K. Engel, G. Schwedersky, and M. Gaestel. 1994. Characterization of the proline-rich region of mouse MAPKAP kinase 2: influence on catalytic properties and binding to the c-abl SH3 domain in vitro. Biochem. Biophys. Res. Commun. 203:1188-1194. [DOI] [PubMed] [Google Scholar]
- 152.Pouyssegur, J., V. Volmat, and P. Lenormand. 2002. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem. Pharmacol. 64:755-763. [DOI] [PubMed] [Google Scholar]
- 153.Powell, D. W., M. J. Rane, B. A. Joughin, R. Kalmukova, J. H. Hong, B. Tidor, W. L. Dean, W. M. Pierce, J. B. Klein, M. B. Yaffe, and K. R. McLeish. 2003. Proteomic identification of 14-3-3ζ as a mitogen-activated protein kinase-activated protein kinase 2 substrate: role in dimer formation and ligand binding. Mol. Cell. Biol. 23:5376-5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pyronnet, S., H. Imataka, A. C. Gingras, R. Fukunaga, T. Hunter, and N. Sonenberg. 1999. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J. 18:270-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Quan, L. T., A. Caputo, R. C. Bleackley, D. J. Pickup, and G. S. Salvesen. 1995. Granzyme B is inhibited by the cowpox virus serpin cytokine response modifier A. J. Biol. Chem. 270:10377-10379. [DOI] [PubMed] [Google Scholar]
- 156.Raingeaud, J., S. Gupta, J. S. Rogers, M. Dickens, J. Han, R. J. Ulevitch, and R. J. Davis. 1995. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 270:7420-7426. [DOI] [PubMed] [Google Scholar]
- 157.Ramne, A., E. Bilsland-Marchesan, S. Erickson, and P. Sunnerhagen. 2000. The protein kinases Rck1 and Rck2 inhibit meiosis in budding yeast. Mol. Gen. Genet. 263:253-261. [DOI] [PubMed] [Google Scholar]
- 158.Rane, M. J., P. Y. Coxon, D. W. Powell, R. Webster, J. B. Klein, W. Pierce, P. Ping, and K. R. McLeish. 2001. p38 Kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils. J. Biol. Chem. 276:3517-3523. [DOI] [PubMed] [Google Scholar]
- 159.Richards, S. A., V. C. Dreisbach, L. O. Murphy, and J. Blenis. 2001. Characterization of regulatory events associated with membrane targeting of p90 ribosomal S6 kinase 1. Mol. Cell. Biol. 21:7470-7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Richards, S. A., J. Fu, A. Romanelli, A. Shimamura, and J. Blenis. 1999. Ribosomal S6 kinase 1 (RSK1) activation requires signals dependent on and independent of the MAP kinase ERK. Curr. Biol. 9:810-820. [DOI] [PubMed] [Google Scholar]
- 161.Rivera, V. M., C. K. Miranti, R. P. Misra, D. D. Ginty, R. H. Chen, J. Blenis, and M. E. Greenberg. 1993. A growth factor-induced kinase phosphorylates the serum response factor at a site that regulates its DNA-binding activity. Mol. Cell. Biol. 13:6260-6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rogalla, T., M. Ehrnsperger, X. Preville, A. Kotlyarov, G. Lutsch, C. Ducasse, C. Paul, M. Wieske, A. P. Arrigo, J. Buchner, and M. Gaestel. 1999. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J. Biol. Chem. 274:18947-18956. [DOI] [PubMed] [Google Scholar]
- 163.Rouse, J., P. Cohen, S. Trigon, M. Morange, A. Alonso-Llamazares, D. Zamanillo, T. Hunt, and A. R. Nebreda. 1994. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 78:1027-1037. [DOI] [PubMed] [Google Scholar]
- 164.Rousseau, S., N. Morrice, M. Peggie, D. G. Campbell, M. Gaestel, and P. Cohen. 2002. Inhibition of SAPK2a/p38 prevents hnRNP A0 phosphorylation by MAPKAP-K2 and its interaction with cytokine mRNAs. EMBO J. 21:6505-6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Roux, P. P., S. A. Richards, and J. Blenis. 2003. Phosphorylation of p90 ribosomal S6 kinase (RSK) regulates extracellular signal-regulated kinase docking and RSK activity. Mol. Cell. Biol. 23:4796-4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rubinfeld, H., T. Hanoch, and R. Seger. 1999. Identification of a cytoplasmic-retention sequence in ERK2. J. Biol. Chem. 274:30349-30352. [DOI] [PubMed] [Google Scholar]
- 167.Sapkota, G. P., A. Kieloch, J. M. Lizcano, S. Lain, J. S. Arthur, M. R. Williams, N. Morrice, M. Deak, and D. R. Alessi. 2001. Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90(RSK) and cAMP-dependent protein kinase, but not its farnesylation at Cys(433), is essential for LKB1 to suppress cell growth. J. Biol. Chem. 276:19469-19482. [DOI] [PubMed] [Google Scholar]
- 168.Sassone-Corsi, P., C. A. Mizzen, P. Cheung, C. Crosio, L. Monaco, S. Jacquot, A. Hanauer, and C. D. Allis. 1999. Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science 285:886-891. [DOI] [PubMed] [Google Scholar]
- 169.Scheper, G. C., N. A. Morrice, M. Kleijn, and C. G. Proud. 2001. The mitogen-activated protein kinase signal-integrating kinase Mnk2 is a eukaryotic initiation factor 4E kinase with high levels of basal activity in mammalian cells. Mol. Cell. Biol. 21:743-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Scheper, G. C., J. L. Parra, M. Wilson, B. Van Kollenburg, A. C. Vertegaal, Z. G. Han, and C. G. Proud. 2003. The N and C termini of the splice variants of the human mitogen-activated protein kinase-interacting kinase Mnk2 determine activity and localization. Mol. Cell. Biol. 23:5692-5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Scheper, G. C., and C. G. Proud. 2002. Does phosphorylation of the cap-binding protein eIF4E play a role in translation initiation? Eur. J. Biochem. 269:5350-5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Scheper, G. C., B. van Kollenburg, J. Hu, Y. Luo, D. J. Goss, and C. G. Proud. 2002. Phosphorylation of eukaryotic initiation factor 4E markedly reduces its affinity for capped mRNA. J. Biol. Chem. 277:3303-3309. [DOI] [PubMed] [Google Scholar]
- 173.Schmitt, A., G. J. Gutierrez, P. Lenart, J. Ellenberg, and A. R. Nebreda. 2002. Histone H3 phosphorylation during Xenopus oocyte maturation: regulation by the MAP kinase/p90Rsk pathway and uncoupling from DNA condensation. FEBS Lett. 518:23-28. [DOI] [PubMed] [Google Scholar]
- 174.Schmitt, A., and A. R. Nebreda. 2002. Signalling pathways in oocyte meiotic maturation. J. Cell Sci. 115:2457-2459. [DOI] [PubMed] [Google Scholar]
- 175.Schouten, G. J., A. C. O. Vertegaal, S. T. Whiteside, A. Israel, M. Toebes, J. C. Dorsman, A. J. van der Eb, and A. Zantema. 1997. IκBα is a target for the mitogen-activated 90 kDa ribosomal S6 kinase. EMBO J. 16:3133-3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Schuck, S., A. Soloaga, G. Schratt, J. S. Arthur, and A. Nordheim. 2003. The kinase MSK1 is required for induction of c-fos by lysophosphatidic acid in mouse embryonic stem cells. BMC Mol. Biol. 4:6. [DOI] [PMC free article] [PubMed]
- 177.Schwab, M. S., B. T. Roberts, S. D. Gross, B. J. Tunquist, F. E. Taieb, A. L. Lewellyn, and J. L. Maller. 2001. Bub1 is activated by the protein kinase p90(Rsk) during Xenopus oocyte maturation. Curr. Biol. 11:141-150. [DOI] [PubMed] [Google Scholar]
- 178.Scimeca, J. C., T. T. Nguyen, C. Filloux, and E. Van Obbergen. 1992. Nerve growth factor-induced phosphorylation cascade in PC12 pheochromocytoma cells. Association of S6 kinase II with the mictrotubule-associated protein kinase, ERK1. J. Biol. Chem. 267:17369-17374. [PubMed] [Google Scholar]
- 179.Seternes, O. M., B. Johansen, B. Hegge, M. Johannessen, S. M. Keyse, and U. Moens. 2002. Both binding and activation of p38 mitogen-activated protein kinase (MAPK) play essential roles in regulation of the nucleocytoplasmic distribution of MAPK-activated protein kinase 5 by cellular stress. Mol. Cell. Biol. 22:6931-6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sharrocks, A. D., S. H. Yang, and A. Galanis. 2000. Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem. Sci. 25:448-453. [DOI] [PubMed] [Google Scholar]
- 181.Shaw, R. J., M. Kosmatka, N. Bardeesy, R. L. Hurley, L. A. Witters, R. A. DePinho, and L. C. Cantley. 2004. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 101:3329-3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.She, Q. B., W. Y. Ma, S. Zhong, and Z. Dong. 2002. Activation of JNK1, RSK2, and MSK1 is involved in serine 112 phosphorylation of Bad by ultraviolet B radiation. J. Biol. Chem. 277:24039-24048. [DOI] [PubMed] [Google Scholar]
- 183.Shi, Y., A. Kotlyarov, K. Laabeta, A. D. Gruber, E. Butt, K. Marcus, H. E. Meyer, A. Friedrich, H. D. Volk, and M. Gaestel. 2003. Elimination of protein kinase MK5/PRAK activity by targeted homologous recombination. Mol. Cell. Biol. 23:7732-7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Shimamura, A., B. A. Ballif, S. A. Richards, and J. Blenis. 2000. Rsk1 mediates a MEK-MAP kinase cell survival signal. Curr. Biol. 10:127-135. [DOI] [PubMed] [Google Scholar]
- 185.Singh, S., D. W. Powell, M. J. Rane, T. H. Millard, J. O. Trent, W. M. Pierce, J. B. Klein, L. M. Machesky, and K. R. McLeish. 2003. Identification of the p16-Arc subunit of the Arp 2/3 complex as a substrate of MAPK-activated protein kinase 2 by proteomic analysis. J. Biol. Chem. 278:36410-36417. [DOI] [PubMed] [Google Scholar]
- 186.Sithanandam, G., F. Latif, F. M. Duh, R. Bernal, U. Smola, H. Li, I. Kuzmin, V. Wixler, L. Geil, and S. Shrestha. 1996. 3pK, a new mitogen-activated protein kinase-activated protein kinase located in the small cell lung cancer tumor suppressor gene region. Mol. Cell. Biol. 16:868-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Slentz-Kesler, K., J. T. Moore, M. Lombard, J. Zhang, R. Hollingsworth, and M. P. Weiner. 2000. Identification of the human Mnk2 gene (MKNK2) through protein interaction with estrogen receptor beta. Genomics 69:63-71. [DOI] [PubMed] [Google Scholar]
- 188.Smith, J. A., C. E. Poteet-Smith, D. A. Lannigan, T. A. Freed, A. J. Zoltoski, and T. W. Sturgill. 2000. Creation of a stress-activated p90 ribosomal S6 kinase. The carboxyl-terminal tail of the MAPK-activated protein kinases dictates the signal transduction pathway in which they function. J. Biol. Chem. 275:31588-31593. [DOI] [PubMed] [Google Scholar]
- 189.Smith, J. A., C. E. Poteet-Smith, K. Malarkey, and T. W. Sturgill. 1999. Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J. Biol. Chem. 274:2893-2898. [DOI] [PubMed] [Google Scholar]
- 190.Soloaga, A., S. Thomson, G. R. Wiggin, N. Rampersaud, M. H. Dyson, C. A. Hazzalin, L. C. Mahadevan, and J. S. Arthur. 2003. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 22:2788-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sonenberg, N., and T. E. Dever. 2003. Eukaryotic translation initiation factors and regulators. Curr. Opin. Struct. Biol. 13:56-63. [DOI] [PubMed] [Google Scholar]
- 192.Stokoe, D., D. G. Campbell, S. Nakielny, H. Hidaka, S. J. Leevers, C. Marshall, and P. Cohen. 1992. MAPKAP kinase-2; a novel protein kinase activated by mitogen-activated protein kinase. EMBO J. 11:3985-3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Stokoe, D., B. Caudwell, P. T. Cohen, and P. Cohen. 1993. The substrate specificity and structure of mitogen-activated protein (MAP) kinase-activated protein kinase-2. Biochem. J. 296:843-849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Stokoe, D., K. Engel, D. G. Campbell, P. Cohen, and M. Gaestel. 1992. Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett. 313:307-313. [DOI] [PubMed] [Google Scholar]
- 195.Sutherland, C., J. Alterio, D. G. Campbell, B. Le Bourdelles, J. Mallet, J. Haavik, and P. Cohen. 1993. Phosphorylation and activation of human tyrosine hydroxylase in vitro by mitogen-activated protein (MAP) kinase and MAP-kinase-activated kinases 1 and 2. Eur. J. Biochem. 217:715-722. [DOI] [PubMed] [Google Scholar]
- 196.Sutherland, C., I. A. Leighton, and P. Cohen. 1993. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem. J. 296:15-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Takahashi, E., J. Abe, and B. C. Berk. 1997. Angiotensin II stimulates p90rsk in vascular smooth muscle cells. A potential Na+-H+ exchanger kinase. Circ. Res. 81:268-273. [DOI] [PubMed] [Google Scholar]
- 198.Takahashi, E., J. Abe, B. Gallis, R. Aebersold, D. J. Spring, E. G. Krebs, and B. C. Berk. 1999. p90(RSK) is a serum-stimulated Na+/H+ exchanger isoform-1 kinase. Regulatory phosphorylation of serine 703 of Na+/H+ exchanger isoform-1. J. Biol. Chem. 274:20206-20214. [DOI] [PubMed] [Google Scholar]
- 199.Tan, Y., J. Rouse, A. Zhang, S. Cariati, P. Cohen, and M. J. Comb. 1996. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 15:4629-4642. [PMC free article] [PubMed] [Google Scholar]
- 200.Tanoue, T., M. Adachi, T. Moriguchi, and E. Nishida. 2000. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol. 2:110-116. [DOI] [PubMed] [Google Scholar]
- 201.Tanoue, T., R. Maeda, M. Adachi, and E. Nishida. 2001. Identification of a docking groove on ERK and p38 MAP kinases that regulates the specificity of docking interactions. EMBO J. 20:466-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Tanoue, T., and E. Nishida. 2003. Molecular recognitions in the MAP kinase cascades. Cell Signal. 15:455-462. [DOI] [PubMed] [Google Scholar]
- 203.Tee, A. R., D. C. Fingar, B. D. Manning, D. J. Kwiatkowski, L. C. Cantley, and J. Blenis. 2002. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 99:13571-13576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Thomson, S., A. L. Clayton, C. A. Hazzalin, S. Rose, M. J. Barratt, and L. C. Mahadevan. 1999. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. 18:4779-4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Thomson, S., L. C. Mahadevan, and A. L. Clayton. 1999. MAP kinase-mediated signalling to nucleosomes and immediate-early gene induction. Semin. Cell Dev. Biol. 10:205-214. [DOI] [PubMed] [Google Scholar]
- 206.Toker, A., and A. C. Newton. 2000. Akt/protein kinase B is regulated by autophosphorylation at the hypothetical PDK-2 site. J. Biol. Chem. 275:8271-8274. [DOI] [PubMed] [Google Scholar]
- 207.Tomas-Zuber, M., J. L. Mary, F. Lamour, D. Bur, and W. Lesslauer. 2001. C-terminal elements control location, activation threshold, and p38 docking of ribosomal S6 kinase B (RSKB). J. Biol. Chem. 276:5892-5899. [DOI] [PubMed] [Google Scholar]
- 208.Tomas-Zuber, M., J. L. Mary, and W. Lesslauer. 2000. Control sites of ribosomal S6 kinase B and persistent activation through tumor necrosis factor. J. Biol. Chem. 275:23549-23558. [DOI] [PubMed] [Google Scholar]
- 209.Tran, H., F. Maurer, and Y. Nagamine. 2003. Stabilization of urokinase and urokinase receptor mRNAs by HuR is linked to its cytoplasmic accumulation induced by activated mitogen-activated protein kinase-activated protein kinase 2. Mol. Cell. Biol. 23:7177-7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Trivier, E., D. De Cesare, S. Jacquot, S. Pannetier, E. Zackai, I. Young, J. L. Mandel, P. Sassone-Corsi, and A. Hanauer. 1996. Mutations in the kinase Rsk-2 associated with Coffin-Lowry syndrome. Nature 384:567-570. [DOI] [PubMed] [Google Scholar]
- 211.Tunquist, B. J., M. S. Schwab, L. G. Chen, and J. L. Maller. 2002. The spindle checkpoint kinase bub1 and cyclin e/cdk2 both contribute to the establishment of meiotic metaphase arrest by cytostatic factor. Curr. Biol. 12:1027-1033. [DOI] [PubMed] [Google Scholar]
- 212.Vadlamudi, R. K., F. Li, L. Adam, D. Nguyen, Y. Ohta, T. P. Stossel, and R. Kumar. 2002. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat. Cell Biol. 4:681-690. [DOI] [PubMed] [Google Scholar]
- 213.Vaidyanathan, H., and J. W. Ramos. 2003. RSK2 activity is regulated by its interaction with PEA-15. J. Biol. Chem. 278:32367-32372. [DOI] [PubMed] [Google Scholar]
- 214.van Hemert, M. J., H. Y. Steensma, and G. P. van Heusden. 2001. 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. Bioessays 23:936-946. [DOI] [PubMed] [Google Scholar]
- 215.Vasudevan, S., and S. W. Peltz. 2001. Regulated ARE-mediated mRNA decay in Saccharomyces cerevisiae. Mol. Cell 7:1191-1200. [DOI] [PubMed] [Google Scholar]
- 216.Vermeulen, L., G. De Wilde, P. Van Damme, W. Vanden Berghe, and G. Haegeman. 2003. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 22:1313-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Vik, T. A., and J. W. Ryder. 1997. Identification of serine 380 as the major site of autophosphorylation of Xenopus pp90rsk. Biochem. Biophys. Res. Commun. 235:398-402. [DOI] [PubMed] [Google Scholar]
- 218.Wang, X., L. Xu, H. Wang, P. R. Young, M. Gaestel, and G. Z. Feuerstein. 2002. Mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 deficiency protects brain from ischemic injury in mice. J. Biol. Chem. 277:43968-43972. [DOI] [PubMed] [Google Scholar]
- 219.Wang, Z., P. C. Harkins, R. J. Ulevitch, J. Han, M. H. Cobb, and E. J. Goldsmith. 1997. The structure of mitogen-activated protein kinase p38 at 2.1-A resolution. Proc. Natl. Acad. Sci. USA 94:2327-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wang, Z., B. Zhang, M. Wang, and B. I. Carr. 2003. Persistent ERK phosphorylation negatively regulates cAMP response element-binding protein (CREB) activity via recruitment of CREB-binding protein to pp90RSK. J. Biol. Chem. 278:11138-11144. [DOI] [PubMed] [Google Scholar]
- 221.Waskiewicz, A. J., A. Flynn, C. G. Proud, and J. A. Cooper. 1997. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 16:1909-1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Waskiewicz, A. J., J. C. Johnson, B. Penn, M. Mahalingam, S. R. Kimball, and J. A. Cooper. 1999. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol. Cell. Biol. 19:1871-1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Wassarman, D. A., N. M. Solomon, and G. M. Rubin. 1994. The Drosophila melanogaster ribosomal S6 kinase II-encoding sequence. Gene 144:309-310. [DOI] [PubMed] [Google Scholar]
- 224.Werz, O., J. Klemm, B. Samuelsson, and O. Radmark. 2000. 5-lipoxygenase is phosphorylated by p38 kinase-dependent MAPKAP kinases. Proc. Natl. Acad. Sci. USA 97:5261-5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Weston, C. R., and R. J. Davis. 2002. The JNK signal transduction pathway. Curr. Opin. Genet. Dev. 12:14-21. [DOI] [PubMed] [Google Scholar]
- 226.Whitmarsh, A. J., and R. J. Davis. 1998. Structural organization of MAP-kinase signaling modules by scaffold proteins in yeast and mammals. Trends Biochem. Sci. 23:481-485. [DOI] [PubMed] [Google Scholar]
- 227.Wierenga, A. T., I. Vogelzang, B. J. Eggen, and E. Vellenga. 2003. Erythropoietin-induced serine 727 phosphorylation of STAT3 in erythroid cells is mediated by a MEK-, ERK-, and MSK1-dependent pathway. Exp. Hematol. 31:398-405. [DOI] [PubMed] [Google Scholar]
- 228.Wiggin, G. R., A. Soloaga, J. M. Foster, V. Murray-Tait, P. Cohen, and J. S. Arthur. 2002. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol. Cell. Biol. 22:2871-2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Williams, M. R., J. S. Arthur, A. Balendran, J. van der Kaay, V. Poli, P. Cohen, and D. R. Alessi. 2000. The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr. Biol. 10:439-448. [DOI] [PubMed] [Google Scholar]
- 230.Wilson, K. P., M. J. Fitzgibbon, P. R. Caron, J. P. Griffith, W. Chen, P. G. McCaffrey, S. P. Chambers, and M. S. Su. 1996. Crystal structure of p38 mitogen-activated protein kinase. J. Biol. Chem. 271:27696-27700. [DOI] [PubMed] [Google Scholar]
- 231.Winzen, R., M. Kracht, B. Ritter, A. Wilhelm, C. Y. Chen, A. B. Shyu, M. Muller, M. Gaestel, K. Resch, and H. Holtmann. 1999. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 18:4969-4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Wong, E. V., A. W. Schaefer, G. Landreth, and V. Lemmon. 1996. Involvement of p90rsk in neurite outgrowth mediated by the cell adhesion molecule L1. J. Biol. Chem. 271:18217-18223. [DOI] [PubMed] [Google Scholar]
- 233.Woo, M. S., Y. Ohta, I. Rabinovitz, T. P. Stossel, and J. Blenis. 2004. Ribosomal S6 kinase regulates phosphorylation of filamin A on an important regulatory site. Mol. Cell. Biol. 24:3025-3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wood, K., C. Sarnecki, T. M. Roberts, and J. Blenis. 1992. c-Ras mediates nerve growth factor receptor modulation of three signal-transducing protein kinases: MAP kinase, Raf-1 and RSK. Cell 68:1041-1050. [DOI] [PubMed] [Google Scholar]
- 235.Wright, J. H., E. Munar, D. R. Jameson, P. R. Andreassen, R. L. Margolis, R. Seger, and E. G. Krebs. 1999. Mitogen-activated protein kinase kinase activity is required for the G2/M transition of the cell cycle in mammalian fibroblasts. Proc. Natl. Acad. Sci. USA 96:11335-11340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wu, J., and R. Janknecht. 2002. Regulation of the ETS transcription factor ER81 by the 90-kDa ribosomal S6 kinase 1 and protein kinase A. J. Biol. Chem. 277:42669-42679. [DOI] [PubMed] [Google Scholar]
- 237.Wu, M., T. J. Hemesath, C. M. Takemoto, M. A. Horstmann, A. G. Wells, E. R. Price, D. Z. Fisher, and D. E. Fisher. 2000. c-Kit triggers dual phosphorylations, which couple activation and degradation of the essential melanocyte factor Mi. Genes Dev. 14:301-312. [PMC free article] [PubMed] [Google Scholar]
- 238.Xia, Y., C. Makris, B. Su, E. Li, J. Yang, G. R. Nemerow, and M. Karin. 2000. MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc. Natl. Acad. Sci. USA 97:5243-5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Xing, J., D. D. Ginty, and M. E. Greenberg. 1996. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 273:959-963. [DOI] [PubMed] [Google Scholar]
- 240.Yaffe, M. B. 2002. How do 14-3-3 proteins work?—Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 513:53-57. [DOI] [PubMed] [Google Scholar]
- 241.Yntema, H. G., B. van den Helm, J. Kissing, G. van Duijnhoven, F. Poppelaars, J. Chelly, C. Moraine, J. P. Fryns, B. C. Hamel, H. Heilbronner, H. J. Pander, H. G. Brunner, H. H. Ropers, F. P. Cremers, and H. van Bokhoven. 1999. A novel ribosomal S6-kinase (RSK4; RPS6KA6) is commonly deleted in patients with complex X-linked mental retardation. Genomics 62:332-343. [DOI] [PubMed] [Google Scholar]
- 242.Yujiri, T., S. Sather, G. R. Fanger, and G. L. Johnson. 1998. Role of MEKK1 in cell survival and activation of JNK and ERK pathways defined by targeted gene disruption. Science 282:1911-1914. [DOI] [PubMed] [Google Scholar]
- 243.Zeniou, M., T. Ding, E. Trivier, and A. Hanauer. 2002. Expression analysis of RSK gene family members: the RSK2 gene, mutated in Coffin-Lowry syndrome, is prominently expressed in brain structures essential for cognitive function and learning. Hum. Mol. Genet. 11:2929-2940. [DOI] [PubMed] [Google Scholar]
- 244.Zhang, Y., G. Liu, and Z. Dong. 2001. MSK1 and JNKs mediate phosphorylation of STAT3 in UVA-irradiated mouse epidermal JB6 cells. J. Biol. Chem. 276:42534-42542. [DOI] [PubMed] [Google Scholar]
- 245.Zhao, J., X. Yuan, M. Frodin, and I. Grummt. 2003. ERK-dependent phosphorylation of the transcription initiation factor TIF-IA is required for RNA polymerase I transcription and cell growth. Mol. Cell 11:405-413. [DOI] [PubMed] [Google Scholar]
- 246.Zhao, Y., C. Bjorbaek, and D. E. Moller. 1996. Regulation and interaction of pp90rsk isoforms with mitogen-activated protein kinases. J. Biol. Chem. 271:29773-29779. [DOI] [PubMed] [Google Scholar]
- 247.Zhao, Y., C. Bjorbaek, S. Weremowicz, C. C. Morton, and D. E. Moller. 1995. RSK3 encodes a novel pp90rsk isoform with a unique N-terminal sequence: growth factor-stimulated kinase function and nuclear translocation. Mol. Cell. Biol. 15:4353-4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Zhu, S., and D. S. Gerhard. 1998. A transcript map of an 800-kb region on human chromosome 11q13, part of the candidate region for SCA5 and BBS1. Hum. Genet. 103:674-680. [DOI] [PubMed] [Google Scholar]
- 249.Zu, Y. L., F. Wu, A. Gilchrist, Y. Ai, M. E. Labadia, and C. K. Huang. 1994. The primary structure of a human MAP kinase activated protein kinase 2. Biochem. Biophys. Res. Commun. 200:1118-1124. [DOI] [PubMed] [Google Scholar]