

Abstract
Conformational motions of proteins are highly dynamic and intrinsically complex. To capture the temporal and spatial complexity of conformational motions and further to understand their roles in protein functions, an attempt is made to probe multidimensional conformational dynamics of proteins besides the typical one-dimensional FRET coordinate or the projected conformational motions on the one-dimensional FRET coordinate. T4 lysozyme hinge-bending motions between two domains along α-helix have been probed by single-molecule FRET. Nevertheless, the domain motions of T4 lysozyme are rather complex involving multiple coupled nuclear coordinates and most likely contain motions besides hinge-bending. It is highly likely that the multiple dimensional protein conformational motions beyond the typical enzymatic hinged-bending motions have profound impact on overall enzymatic functions. In this report, we have developed a single-molecule multiparameter photon stamping spectroscopy integrating fluorescence anisotropy, FRET, and fluorescence lifetime. This spectroscopic approach enables simultaneous observations of both FRET-related site-to-site conformational dynamics and molecular rotational (or orientational) motions of individual Cy3-Cy5 labeled T4 lysozyme molecules. We have further observed wide-distributed rotational flexibility along orientation coordinates by recording fluorescence anisotropy and simultaneously identified multiple intermediate conformational states along FRET coordinate by monitoring time-dependent donor lifetime, presenting a whole picture of multidimensional conformational dynamics in the process of T4 lysozyme open-close hinge-bending enzymatic turnover motions under enzymatic reaction conditions. By analyzing the autocorrelation functions of both lifetime and anisotropy trajectories, we have also observed the dynamic and static inhomogeneity of T4 lysozyme multidimensional conformational fluctuation dynamics, providing a fundamental understanding of the enzymatic reaction turnover dynamics associated with overall enzyme as well as the specific active-site conformational fluctuations that are not identifiable and resolvable in the conventional ensemble-averaged experiment.
Inroduction
T4 lysozyme, as a member of the lysozyme family produced by bacteriophage, breaks down the bacterial cell wall by catalyzing the hydrolysis of poly saccharide chains during infection of the bacteria.1−4 The enzyme specifically cleaves the glycosidic bonds connecting the repeating subunits of cell walls between N-acetylglucosamine and N-acetylmuramic acid that are substituted with peptide side chains.2 The three-dimensional structure is clearly organized with two domains connected by a long α-helix (Figure 1). The active site cleft, where hydrolysis of the glycosidic linkage takes place, is located at the interface between the two domains. It has been widely accepted that T4 lysozyme exhibit hinge-bending conformational motions, referring to rotation of one domain relative to the other domain along an axis running through the interface of the two domains.3,4 Both ensemble-level and single-molecule measurements have revealed hinge-bending motions in which the opening of the active site cleft is within a nanometer.5,6 As we have reported previously,6,7 the T4 lysozyme enzymatic reaction involves complex conformational state changes in the enzymatic turnovers. A simplified Michaelis–Menten type of mechanism can be presented as
| 1 |
where E, S, ES, ES*, and EP represent enzyme, substrate, nonspecific enzyme–substrate complex, specific or active enzyme–substrate complex, and enzyme–product complex, respectively. The process of forming the active complex of ES* involves multiple conformational states. The process of E + S → ES → ES* essentially involves the enzyme active site opening up to take the substrate, forming a nonspecific ES complex, and binding down to form the active complex of ES* ready to react followed by turnover to EP. The process of reaction and product releasing ES* → EP → E + P may not involve significant enzymatic active site conformational changes.
Figure 1.

Multidimensional conformational motions of wild-type T4 lysozyme (PDB-code, 3LZM), including hinge-bending motions along α-helix and rotational motions of each domain. Cy3 and Cy5 are covalently labeled to two cysteines: Cys 54 on N-domain and Cys 97 on C-domain. Cy3-Cy5 labeled T4 lysozyme is tethered through an amine-to-sulfhydryl bifunctional cross-linker molecule to thiol-functionalized glass coverslip surface. The approximate ∼4–5 nm spacer allows free rotation of single T4 lysozyme without perturbation or confinement from the modified surface. Distance changes between the two labeling sites involved in hinge-bending conformational motions can be monitored by tracing the dynamic fluctuations of donor lifetime during the FRET process. Besides hinge-bending motions along α-helix line, the two domains of T4 lysozyme also exhibit other types of conformational motions, for example, rotational motions. The rotational motions can be probed by single-molecule fluorescence anisotropy.
While T4 lysozyme hinge-bending motions have been extensively probed by single-molecule FRET (fluorescence resonance energy transfer) spectroscopy, the domain motion of T4 lysozyme is rather complex and contains motions besides hinge-bending. It is reasonable to assume that the hinge-bending motion in nature involves multiple coupled nuclear coordinates that can be projected to a nuclear coordinate associated with the α-helix. In order to capture the complexity of T4 lysozyme conformational motions, herein we probe multidimensional conformational dynamics beyond the one-dimensional FRET coordinate.
Single-molecule spectroscopy is a powerful approach for mechanistic understanding of complex and fluctuating biological processes by resolving time-dependent dynamic process and allowing exploration of hidden heterogeneity beyond nonsynchronized ensemble-averaged measurements.8−23 Single-molecule FRET spectroscopy sensitive to single-molecule conformational fluctuation dynamics has offered possible direct observations of biological conformational dynamics by rendering spatial and temporal information between donor and acceptor fluorophores placed within a certain proximity on individual molecules of interest.8,24 This approach has made significant and extensive contributions to the understanding of complex biological dynamics through the perspectives of heterogeneous dynamics of protein molecules, nucleic acids, and their interactions with other molecules.8,16−18,25−28 For example, single-molecule FRET has been used to study RNA folding pathways,17 hairpin ribozymes intimidate states,29 DNA bubbles kinetics,30 epidermal growth factor receptor (EGFR) dimerization,31 and conformational dynamics of enzymes.6,7,32,33 Lu and co-workers have focused on conformational dynamics of T4 lysozymes and have observed conformational bunching effects in the process of T4 lysozyme hinge-bending.6,7
For complex biological systems, such as protein–protein interactions, ion channel receptor activations, protein folding and aggregations, and protein conformational fluctuations under enzymatic reactions, it is more than often that multiple conformational nuclear coordinates simultaneously play critical roles in regulating and gating biological functions. Under such complex multiple coordinate conformational dynamic rate processes, one-dimensional FRET may be insufficient to characterize the intrinsic complexity of molecular dynamics. Hence, it is desirable to have advanced FRET techniques capable of probing more than one-dimensional dynamic information. For example, some efforts have been made to develop three-color and four-color FRET in which more than one FRET pair are used to probe multiple site-to-site distance changes at a time.34−37 Nevertheless, multicolor FRET requires fluorophores with high photostability and clear spectral separation, which are much more difficult to achieve than one FRET pair, especially at single-molecule level. In addition, the fact that energy transfer between donor and acceptor obeys orientation dependence (κ2) as expected for a dipole–dipole interaction has seldom been took into account, instead, the assumption κ2 = 2/3 (the fluorephores undergo freely rotation in a time much shorter than fluorescence lifetimes)38 is widely used for approximate distance estimate. It has been experimentally proved that fluorescence energy transfer between Cy3 and Cy5 depends on the orientation.39 It has also been reported that the degree of position accuracy relies on the rotational mobility of single molecules.40 Therefore, the understanding of orientation effect could result in more accurate FRET distance approximation and molecular angular information for position determination. For this purpose, several groups have shed light on molecular orientations (or rotations) via imaging and fluorescence anisotropy (or polarization) spectroscopy. For example, out-of-focus and in-focus images have been combined to refer three-dimensional single molecule orientations.41 Dual-color and dual-polarization images of single molecules have been captured by CCD (charge-coupled device) camera.42 A multiparameter single-molecule fluorescence spectroscopy,43,44 photon distribution analysis,45 along with structural modeling46 have been developed to quantitatively describe single-molecule FRET in which several FRET-related parameters such as distance, molecular orientation, and dye quenching (or bleaching) are taken into account. Lu and co-workers have demonstrated the use of single-molecule nanosecond anisotropy to study spatially confined rotational diffusion dynamics of individual tethered proteins47 and nanosecond protein motion dynamics.48
The possibility of probing multidimensional conformational dynamics of complex biological systems calls for FRET-anisotropy correlated measurements on demand. Fluorescence anisotropy not only allows for estimating orientation effect on FRET or position determination for imaging mentioned above but also allows for acquiring information about the motion of the fluorophore, the rotational dynamics of subdomains, or the entire proteins. The methods of measuring rotational or tilting motions by fluorescence anisotropy in single molecules have been reviewed and discussed.49−51 Lu and co-workers have successfully probed nanosecond protein motions of Calmodulin and T4 lysozyme by single-molecule fluorescence anisotropy.47,48 Unfortunately, most of fluorescence anisotropy research have been either limited to the study of orientation effect for FRET, or limited to the rotational dynamics of molecules; although the potential ability of fluorescence anisotropy in the identification of multiple conformational states of biological molecules has been mentioned.52 Nevertheless, the potential abilities of correlated single-molecule FRET and fluorescence anisotropy for direct observing multidimensional conformational dynamics have not been demonstrated yet, to our best knowledge. In this article, we report our new technical approach integrating single-molecule FRET, photo stamping spectroscopy and fluorescence anisotropy to study multicoordinate conformational dynamics of T4 lysozyme under enzymatic reaction conditions. From conformational dynamics perspectives, this approach enables us to simultaneously probe multidimensional or multicoordinate conformational dynamics of proteins, including FRET coordinate motions probed by FRET pair and rotational motions monitored by fluorescence anisotropy.
In this work, we exploit the multidimensional aspect of T4 lysozyme conformational dynamics and demonstrate the potential of our new correlated single-molecule multidimensional photon stamping spectroscopy.
Materials and Methods
Materials
Wild-type T4 lysozyme plasmid was bought from Addgene, which was authorized by Prof. Brian Matthews from the University of Oregon. Two cysteines (residue 54 on N-terminal domain and residue 97 on C-terminal domain) of the wild-type T4 lysozyme are accessible to thiolation reactions. A Cy3-Cy5 FRET pair (GE Healthcare) was nonselectively labeled on these two cysteines. The crystal structure of wild-type Cy3-Cy5 labeled T4 lysozyme is shown in Figure 1. The individual donor–acceptor labeled T4 lysozyme can be easily distinguished by four-channel optical images as shown in Figure 2, because only donor–acceptor labeled molecules can simultaneously exhibit four emission spots (dual color and dual polarization for each color). The substrate for T4 lysozyme used in our experiments is peptidoglycan that is the major component of the bacterial cell wall that T4 lysozyme breaks. Peptidoglycan isolated from Micrococcus luteus was purchased from Sigma-Aldrich and was directly used without further purification. The substrate was suspended to a final concentration of 25 μg/mL in pH 7.3 PBS buffer during single-molecule experimental measurements.
Figure 2.

Single-molecule multiparameter photon stamping spectroscopy. (A) Experimental home-built four-channel single-molecule setup for measuring multidimensional conformational dynamics. Basically, it consists of an inverted confocal epi-illumination configured microscopy, a femtosecond pulse laser, four Si avalanche photodiode detectors, a time-correlated single photon counting module, and several optics. 532 nm green linearly polarized pulse laser is used to excite Cy3-Cy5 labeled T4 lysozyme. The emissions (yellow and red) from Cy3 and Cy5 are discriminated by dichroic mirrors. The polarization of the light emitted is further distinguished into parallel (∥) and vertical (⊥) components (relative to the polarization of the laser excitation) by two polarizing beam splitter cubes. (B) Single-molecule photon counting images of individual Cy3-Cy5 labeled T4 lysozymes. Dual-color (Cy3 and Cy5) and dual-polarization (∥ and ⊥) images are captured. DM: dichroic mirror; APD: avalanche photodiode; PBS: polarizing beam splitter cubes; LPF: long pass filter; WP: wave plate; L1/L2: lens; PC: personal computer.
Single-Molecule Sample Preparation and Measurements
In our single-molecule FRET experiments, T4 lysozyme was tethered through a bifunctional NHS-PEG6-Maleimide cross-linker to a modified glass coverslip surface. This cross-linker is functional between primary amines (NH2) and sulfhydryl (SH) groups in which the N-hydroxysuccinimide ester (NHS) group reacts specifically with primary amino groups of lysine to form stable amide bonds, and the maleimide group reacts with sulfhydryl to form stable thioether bonds. The glass coverslip was first sonicated with acetone for half an hour, followed by rinsing with alcohol solution and distilled water several times. The clean coverslip was treated overnight with a 10% (v/v) mixture of 3-mercaptopropyl-trimethoxysilane and isobutyl-trimethoxysilane (1/1000 ratio) in 15.0 mL of dimethyl sulfoxide (DMSO). After rinsing with ethanol and water, the coverslip was put in the pH 7.3 PBS buffer solution for 1 h to remove unreacted solvent. The coverslip was then incubated with 40.0 μL of 250 mM bifunctional cross-linker stock solution (NHS-PEG6-Malemiade, Thermo Scientific) in 12.0 mL of PBS buffer solution for 2 h at 4 °C. The amine-to-sulfhydryl cross-linkers with hydrophilic polyethylene glycol (PEG) spacer arms can be attached to the glass coverslip surface. After additional washing, the coverslip was incubated with 0.66 nM T4 lysozyme in the PBS buffer for 2 h at 4 °C. After the linkage between amine-reactive group of NHS-PEG6-Malemiade and primary amine group of T4 lysozyme’s lysine, the tethered enzyme sample was assembled on the glass coverslip surface. During our single-molecule measurements, the assembled T4 lysozyme on the coverslip was further incubated with 25.0 μg/mL substrate for half an hour at room temperature in PBS buffer solution (pH 7.3) to fulfill the engagement. The effective Trolox-oxygen scavenger solution, which contains 0.8% d-glucose, 1.0 mg/mL glucose oxidase, 0.04 mg/mL catalase, and 1.0 mM Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid),24,25,53,54 was added to the above sample chamber to prevent the possible photobleaching or quenching of the Cy3-Cy5-labeled T4 lysozyme molecules.
Single-Molecule FRET
Förster resonance energy transfer (FRET) refers to the nonradiative energy transfer from a donor molecule to an acceptor molecule, arising from a dipole–dipole interaction between the electronic states of the donor and the acceptor. Energy transfer occurs when the oscillations of an optically induced electronic coherence on the donor are resonant with the electronic energy gap of the acceptor.55,56,59 FRET efficiency is sensitive to the interdistance between the donor and acceptor in the range of 30–80 Å, although FRET is not necessarily accurate in measuring absolute distances, as often being specified as a “spectroscopic ruler”,6,7,17,24,29−32 single-molecule FRET is sensitive to probe the temporal fluctuation dynamics of the distance changes, such as the conformational changes of the biomolecules. The energy transfer efficiency (EFRET) is given by
| 2 |
where r is the separate distance between donor and acceptor and R0 (the Förster radius) is the critical distance at which energy transfer is equal to 50%. R0, as expressed in eq 3, is a function of the orientation factor κ2, the donor–acceptor spectral overlap J, the donor quantum yield ΦD, and the refractive index of the medium n.24,55 κ2 is the orientation factor that varies between 0 and 4 and is defined by the relative orientation of the donor emission and acceptor absorption dipoles. For molecules which rotate very fast, κ2 is taken its average value ⟨κ2⟩, which equals 2/3 for isotropic rotation. In general, κ2 is given by eq 4, in which θT is the angle between the donor emission dipole and the acceptor absorption dipole, θD is the angle between the donor–acceptor connection line and the donor emission dipole, and θA is the angle between the donor–acceptor connection line and the acceptor absorption dipole.
| 3 |
| 4 |
Any process that influences the distance of donor-to-acceptor affects the FRET rate or efficiency, which enables us to probe the conformational fluctuation dynamics of DNA, RNA, and proteins.6,7,17,24,29−32,57,58 For example, FRET can be used to sense the distance changes between donor and acceptor that have been labeled on a host molecule or two different molecules, and then the conformational changes of one host molecule or the relative motions of two molecules can be monitored by tracing the FRET efficiency. In single-molecule FRET measurements, EFRET from donor to acceptor reflects mutual distance changes, resulting in the capability of probing single molecules conformational dynamics in real time by tracking EFRET changes.24,56
The detection of EFRET, usually by ratio-metric methods, can be generally classified into intensity-based FRET and lifetime-based FRET.
| 5 |
where IA is the acceptor fluorescence intensity and ID is the donor fluorescence intensity in FRET process. τDA and τD are donor lifetime in the presence (τDA) and in absence (τD) of acceptor, respectively.59,60 FRET detection on the basis of donor lifetime is more effective and less sensitive to local environment fluctuations,26 especially in AFM tip-enhanced single-molecule spectroscopic and imaging measurements where amplified fluorescence signal by metal tip reflection exists.25,61 In the photon stamping spectroscopy, each detected photon is stamped with two parameters: a chronic arrival time and a delay time related to femtosecond laser pulse excitation. In this work, we treat photons distributions detected in each 10 ms (ms) bins as a Poisson distribution which gives the mean of delay times in each distribution as the lifetime τDA.
Fluorescence Anisotropy
Fluorescence anisotropy is capable of determining the rotational correlation time of the fluorescence probe, thus providing insights into the motions of probe, and orientation/rotation or mobility of subdomains or the entire molecule.49,56,62 Changes in probe’s orientation reflect the rotation or mobility of the target macromolecule where the probe is attached. The fluorescence anisotropy r(t) is defined as the difference between the vertically and horizontally polarized fluorescence emission divided by the total fluorescence emission, given by
| 6 |
where I∥ (t) and I⊥(t) are the fluorescence intensities of the parallelly (∥) and horizontally (⊥) polarized emission components under vertically polarized excitation. G is the correct coefficient compensation for the different instrumental detection efficiencies of the various polarized components of the emission, accounting for the ratio of the detection system sensitivities for vertically and horizontally polarized light. In our experiments, bright fluorescence microspheres (0.1 μm, 540/560 nm orange spheres, Invitrogen Molecular Probes) were used to measure the G factor by using horizontally polarized excitation. With the horizontally polarized excitation, the excited-state distribution of the molecules is rotated to lie along this observation axis, so that both the horizontally and vertically polarized components are orthogonal to the incident polarization and the intensities of collected signal are equivalent. G factors are averagely estimated to be ∼1.36 and ∼2.36 for donor and acceptor, respectively. The unbalanced G factor for donor and acceptor are most likely due to the detection discrepancy of different detectors and the bias of the optics response to different colors. Typical anisotropy values are in the range from −0.2 for probes with unrestricted motion to 0.4 for those that are immobile. Several factors can depolarize the measured anisotropy to values lower than 0.4 (the maximum theoretical values), such as the numerical aperture of the objective, the angle difference between the absorption and emission dipole, molecular rotation related rotational diffusion, and et al.63
In terms of rotational diffusion process, the expected anisotropy is given by the Perrin equation55,56
| 7 |
where r0 (assumed to be 0.40 here) is the fundamental anisotropy in the absence of rotational diffusion, rDA is donor’s anisotropy, and θ is the rotational correlation time for the diffusion process. We treat donor’s anisotropy in the similar way to donor lifetime described in eq 5: rDA is the mean of fluorescence anisotropy in each 10 ms bins measured based on eq 6. Fluorescence anisotropy provides information about detailed motions of Cy3 or Cy5 and conformational dynamics of the T4 lysozyme system being studied here.
Autocorrelation Function Analysis of Fluctuation Dynamics
We use autocorrelation function to analyze lifetime fluctuation and anisotropy fluctuation. The time-dependent correlation strength of lifetime as well as anisotropy trajectory is evaluated by autocorrelation function given by
| 8 |
where Xi is the donor lifetime (τDA) or anisotropy (rDA), the signal variable in time-dependent lifetime or anisotropy trajectory; i is the index number of data point; t is the time lag; X̅ is the mean value of lifetime or anisotropy in each calculation. By using autocorrelation analysis, the donor lifetime and anisotropy fluctuation decays (τ) or rates (1/τ) can be identified, providing insights into the T4 lysozyme conformational fluctuation dynamics.
New Approach of Four-Channel Single-Molecule Microscopy
Combining FRET, lifetime, polarization, and anisotropy at single-molecule sensitivity, we show our compact design for probing multidimensional conformational motions. Compared to two-color6,64 or two-polarization concepts,47 our new approach combines color discrimination and polarization using a four-channel single-molecule epi-illuminated microscopy, as shown in Figure 2A. In short, a femtosecond pulse laser (Ti: Sapphire Mira 900F/P, Coherent Inc.) is combined with an optical parametric oscillator (OPO BASIC, Coherent Inc.) as well as frequency doubling β-barium borate crystal (BBO) to generate linear-polarized pulse laser. The 532 nm linearly polarized pulse laser is aligned and focused into coverslip-solution interface by a confocal objective (Plan-APOCHROMAT, 1.40 NA, 63 × , Carl Zeiss) to excite molecules. A piezoelectric scanning stage (Nano-H100, MCL) with a positioning resolution of 0.2 nm is further used to scan the coverslip surface and locate individual molecules. The fluorescence signals from single molecule are separated to two pathways (two color) in wavelength ranges below and above 640 nm after passing through a filter (HQ545LP, Chroma) and two dichroic mirrors (545dcxru, 640dcxr, Chroma). Signals in each pathway are further divided into four-channel parallel and perpendicular components for each color by using two polarizing beam splitter cubes (PBS 201 420–680 nm, PBS 202 620–1000 nm, respectively). The photons from four-channel (parallel and perpendicular for both donor and acceptor) are detected by four APDs-Si avalanche photodiode (SPCM-AQR-14, PerkinElmer Optoelectronics). The time-stamped photon information is recorded through multichannel detector router (HRT-82, Becker & Hickl GmbH) to a time-correlated single photon counting module (SPC-830, Becker & Hickl GmbH) and a personal computer. Figure 2B shows the single-molecule photon counting images of individual Cy3-Cy5 labeled T4 lysozymes in which dual-color (donor and acceptor) and dual-polarization (∥ and ⊥) are captured by four APDs. Four images are taken simultaneously with an illumination of 532 nm and scanning area of 20 μm by 20 μm.
Results and Discussion
Figure 3 shows the single-molecule studies of multidimensional conformational motions of T4 lysozyme by recording real-time anisotropy trajectory and donor lifetime trajectory. In our single-molecule multiparameter spectroscopy, time-resolved FRET fluctuations detected by donor lifetime and the correlated fluorescence anisotropy are simultaneously recorded. As described before, each detected photon is stamped with two parameters: a chronic arrival time and a delay time related to femtosecond laser pulse excitation.26,47,48 Our data analyses are primarily based on those two temporal parameters recorded by single-molecule photon stamping spectroscopy.26 In this work, we have used donor lifetime (τDA) to probe FRET fluctuations26,65 and fluorescence anisotropy (rDA) to monitor domain rotational motions49,56,62 of Cy3-Cy5 labeled T4 lysozyme. Figure 3B shows a typical single-molecule lifetime trajectory, {τDA(t)}. The donor lifetime fluctuates from time to time, implying dynamic change of interdomain distance along FRET coordinate. The wide Gaussian-like distribution of lifetime (the right panel in Figure 3B) indicates the existence of multiple conformational intermediate states characterized with different interdomain distance along α-helix, from FRET perspective.
Figure 3.

Multidimensional conformational motions of T4 lysozyme probed by single-molecule multiparameter spectroscopy: dynamic anisotropy and lifetime-based FRET. All the data are collected from single Cy3-Cy5 labeled T4 lysozyme under enzymatic reactions with 25.0 μg/mL peptidoglycan and are simultaneously recorded by four-channel single photon stamping spectroscopy. (A) Single-molecule real-time anisotropy trajectory {rDA(t)} and distribution recorded within 100 s. Each dot represents the average anisotropy in every 10 ms binning of Cy3, calculated on the basis of eq 6. The distribution of anisotropy is shown in the right panel. (B) Single-molecule real-time donor lifetime trajectory {τDA(t)}. Each dot is the average of photon delay times in each 10 ms binning of Cy3 collected from single-molecule photon stamping. (C) 2D joint distribution between anisotropy in (A) and lifetime in (B). We note that the band widths of both lifetime and anisotropy distributions, 1.0 ns and 0.25, are significantly larger than the measurement error bars of ±0.30 ns and ±0.05, respectively. The broadness of the distributions of the lifetime and anisotropy represent intrinsic physical inhomogeneity beyond the measurement error bars.
Multiple conformational intermediate states have often been suggested as the general feature of enzymatic mechanisms and protein motions.6,7,66−77 In single-molecule studies, time-binned FRET or lifetime trajectory has been reliably used to identify the presence of multiple intermediate states corresponding to well-defined stable FRET values in the case of well-separated, low-noise, two-state or three-state systems.17,26,65,78,79 Nevertheless, for complex biological systems, due to the influence from local environmental fluctuations, thermal fluctuations, measurement short noise, photophysical effects, and conformational dynamic or static heterogeneity coupled with fluctuating catalytic activity, multiple intermediate states are often buried in a wide-distributed Gaussian-like distribution.7,80 In this work, the single-molecule lifetime trajectories are limited by 10 ms binning time and the other facts discussed above, resulting in that conformational intermediate states do not present clear separation in the lifetime distribution in Figure 3B. Furthermore, we have performed advanced quantitative analysis of lifetime, to be discussed later in this paper (Figures 4 and 5), to further resolve buried multiple intermediate states.
Figure 4.

Dynamic and static disorder of T4 lysozyme multidimensional conformational fluctuations along FRET coordinate and orientation coordinates via autocorrelation analysis of lifetime and anisotropy. (A) Histogram of fluctuation decays derived from the autocorrelation function of donor lifetime trajectories. (Inset) Typical nonexponential autocorrelation function calculated from a single molecule fluorescence lifetime trajectory {τDA(t)}. (B) Histogram of fluctuation decays derived from the autocorrelation function of donor anisotropy trajectories. (Inset) Typical nonexponential autocorrelation function originated from a single molecule fluorescence anisotropy trajectory {rDA(t)}. For autocorrelation functions of lifetime or anisotropy, τ represents the fluctuation decay. Correspondingly, 1/τ represents the fluctuation rate, that is, conformational fluctuation rate along FRET coordinate or multiple orientation coordinates under T4 lysozyme enzymatic reactions. ACF: autocorrelation function.
Figure 5.

Two-component donor lifetime decays associated with two major open and close conformational states. (A) The representative histograms of photon delay times of donor in a single-molecule measurement. Biexponential fitting (red curve) gives the best estimate to the experimental data (gray), and two-component donor lifetime decays (τ1 and τ2) are observed. (B) The distribution of two-component lifetime decays of donor. Narrowly distributed faster component (τ1) and widely distributed slower component (τ2) are revealed. (C) Statistical results of the two-component feature derived from (B). Mean and standard deviation of lifetime decays are illustrated. The results of two-component lifetime imply that T4 lysozyme exhibits open-close hinge-bending conformational motions characterized with two-component lifetime decays. The distinct distributions of each component even give a further implication that close state is rigid and spatially narrowly distributed while the open states involve flexible and broadly distributed conformational fluctuations.
The rotational flexibility of the donor molecule Cy3 on T4 lysozyme fluctuates significantly in the course of the enzymatic reaction turnovers, revealed in our single-molecule anisotropy analysis. Figure 3A shows a typical single-molecule anisotropy trajectory, {rDA(t)} showing that the single-molecule anisotropy fluctuates in a wide range from negative to the maximum value of 0.4. This is most likely associated with the dye molecule fast spinning motions, the slow subdomain motions, and entire protein motions of the T4 lysozyme tethered with the dye molecule.55,56 Those three types of motions are all involved but contribute differently to the overall anisotropy measured from the experiments. If there are no rotational diffusion restrictions from the subdomains or the whole enzyme, the Cy3 probe on T4 lysozyme should exhibit fast wobbling spin rotation and the resulting Cy3 lifetime should be in the subnanosecond range consistant with a free Cy3 dye in solution.81,82 Our experimental results of lifetime trajectory and distribution (Figure 3A) exclude this scenario because most of the measured lifetimes of Cy3 are in the nanosecond range instead of the subnanosecond range. Similar behavior of the lack of rotational freedom and nanoseconds lifetime range of tethered dye molecules have also been reported for Cy3 covalently linked to DNA.81 The reported Cy3 lifetime is around 10 times larger than the fluorescence lifetime of the free dye in solution, which is similar to the result of Cy3 on T4 lysozyme in our measurements. The lifetime of Cy3 is dependent on physical and chemical properties of surroundings. The longer lifetime of Cy3 is directly related to local environment changes deviated from an aqueous environment, such as viscosity. For example, when Cy3 is attached to a strand of DNA or a protein, an additional increase in local viscosity may occur, leading to a lifetime increase.83,84 The interaction between Cy3 and DNA molecules has also been reported to play a role in longer Cy3 lifetime than that measured in aqueous solution.81 Therefore, it is most likely that the increase in local viscosity and the interaction between Cy3 and T4 lysozyme result in longer Cy3 lifetime which evidence the lack of Cy3 free spin and wobbling rotational freedom identified in our anisotropy measurements. The joint distribution of lifetime and anisotropy (Figure 3C) further implies that the measured overall anisotropy is not dominated by free dye rotation but rather by restricted rotation, indicating that the Cy3 dye probe is significantly regulated by interactions with the T4 lysozyme protein matrix. Presumably, there are two possibilities that may contribute to the restricted rotation: the enzyme–surface interaction and domain rotations of enzyme. Previous studies have reported that the tethered single T4 lysozyme to the hydrocarbon modified surface is mostly in solution phase, and the interaction between enzyme and surface is weak or insignificant in impacting enzyme rotations, giving rise to the absence of rotational rate fluctuations for most of the time during the measurements.47 Therefore, the restricted rotation of Cy3 characterized by the dynamic and fluctuating anisotropy is most likely regulated by the domain rotations of enzyme. Figure 3B,C gives a broad anisotropy distribution, implying different rotational flexibility of Cy3 regulated by T4 lysozyme domain motions. The different dye rotational flexibility has been found in the study of HIV-1 reverse transcriptase, and they have attributed broad rotational correlation time distribution probably to the clamping of the finger and thumb domains during polymerase activity.44 In our work, different Cy3 rotational flexibility, reflected in the broad anisotropy distribution in Figure 3B,C, is likely regulated by T4 lysozyme domain rotations during the interdomain hinge-bending conformational motions.
T4 lysozyme exhibits well-known hinge-bending conformational motions in which the distance changes between the opening and closing of the active site cleft are about 4–6 Å, revealed in crystal structure analyses, MD simulation, and our previous single-molecule spectroscopic analysis.2,4,6,7,47,85−87 Conceivably, T4 lysozyme conformational motions involve more than just hinge-bending along its α-helix. Furthermore, even the hinge-bending motions themselves are actually complex fluctuating conformational motions involving multiple conformations with distinct domain orientations or hinge-bending angles along multiple nuclear coordinates. For example, it has been suggested that T4 lysozyme populates multiple intermediates states with distinct hinge-bending angles trapped in the crystal structures of T4 lysozyme mutants.88
Our single-molecule T4 lysozyme anisotropy fluctuation result (Figure 3) suggests that there are a wide range of domain-rotational mobility, indicating different dominant orientations of the domains from time to time, consistent with distinct hinge-bending angles. Typically, single-molecule FRET spectroscopy only probes the conformational motions associated with the FRET donor–acceptor distance changes, and most likely, such FRET probed conformational motions are actually the projections of much more complex conformational motions of the examined enzyme molecules on the FRET sensitive coordinate. Nevertheless, besides hinge-bending motions along coordinate probed by single-molecule FRET spectroscopy,6,7 T4 lysozyme actually exhibits complex and fluctuating rotational motions along multiple orientation coordinates, leading to a comprehension of the multidimensional conformational motions associated with the T4 lysozyme enzymatic reaction turnovers.
T4 lysozyme multidimensional hinge-bending conformational motion dynamics presents dynamic and static inhomogeneity, revealed and identified by autocorrelation analysis of the single-molecule lifetime trajectories {τDA(t)} and anisotropy time trajectories {rDA(t)}. Autocorrelation function analysis has been extensively applied to identify inhomogeneous fluctuation rates of single-molecule electron transfer,89,90 energy transfer fluctuations,6,91,92 and protein conformational changes.6,9,10,93 We have analyzed the autocorrelation functions, C(t), of lifetime and anisotropy trajectories for 40 Cy3-Cy5 labeled T4 lysozyme molecules under the enzymatic reaction conditions. Figure 4 shows the autocorrelation analysis results of lifetime fluctuation decays (Figure 4A) and anisotropy fluctuation decays (τ) (Figure 4B). Most of the autocorrelation functions of FRET donor lifetime trajectories (Figure 4A, inset) show nonexponential fluctuation decays, implying the dynamic disorder of FRET energy transfer, i.e., the conformational fluctuation rate along FRET-probed hinge-bending coordinate changes from time to time under enzymatic condition in one single-molecule measurement. We have analyzed the autocorrelation functions by biexponential fitting and observed a wide range of fluctuation decays from milliseconds to seconds (Figure 4A), suggesting a static disorder of conformational fluctuation rate change from molecule to molecule. Autocorrelation functions of anisotropy trajectories also give similar results in terms of nonexponential and inhomogeneity of fluctuation correlation function decays (Figure 4B). The nonexponential conformational fluctuation dynamics and the wide-range rate constant distributions of the single-molecule anisotropy indicate dynamic disorder and static disorder of conformational rotational fluctuation along multiple orientation coordinates, respectively.
The flexibility of the hinge-bending conformational coordinates regulated by substrate binding to the enzymatic active site most likely contributes to the inhomogeneous and complex fluctuation dynamics of lifetime and anisotropy. The flexibility of conformations associated with the process of forming the nonspecific binding complex (E + S → ES), the process of enzyme closing down to form the active complex of ES → ES*, and the following enzymatic reaction of ES* → EP, involves complex local environment and molecular structures of substrate as well as the enzymatic active site. The conformational motions involve multiple coordinates in nature and are regulated by a fluctuating multiple coordinate energy landscape defined by the dynamically changing and statically inhomogeneous molecular interactions as well as local electric field in the process of open-close hinge-bending enzymatic turnovers.
Donor lifetime decays exhibit two major distributions, associated with T4 lysozyme open and close states during the hinge-bending motions of the enzymatic active site. Mean lifetime trajectory and distribution (Figure 3A) do not necessarily give a clear separation of multiple intermediate states but rather present a Gaussian-like distribution, due to the limitations from the local environment fluctuations and the time-resolution of our single-molecule spectroscopy. Most likely, the convolution of the multiple Poisson processes gives rise to the overall wide Gaussian-like distribution.7 To further resolve the intermediate conformational states, we have performed analysis of the temporal decays including all the photons in a single-molecule donor lifetime trajectories. Figure 5A shows the representative single-molecule lifetime decay curves of the FRET donor. Two-component lifetime decays, a faster one (τ1) and a slower one (τ2), are derived from biexponential fits. We note that the lifetime decays (τ1 and τ2) in Figure 5A reflects different properties of the enzymatic conformational dynamics from the fluctuation decays (τ) in Figure 4. As a lifetime-based FRET measurement expressed in eq 5, FRET donor lifetime decays are derived by fitting donor photon delay times histogram, essentially a time-correlated single photon counting distribution, to identify the FRET efficiency reflecting the major conformational states along FRET coordinates, whereas the fluctuation decays are calculated by the autocorrelation analysis of mean lifetimes τDA to characterize the conformational fluctuations and conformational flexibility. We attribute the two-component FRET donor lifetime decays corresponding to two different FRET efficiency values, to two major conformational states, that is, open and close states along FRET coordinates: in each enzymatic reaction cycle, the enzyme active site opens up to interact with the substrate forming a nonspecific enzyme–substrate complex (E + S → ES) and then closes down to form a specific enzyme–substrate complex (ES → ES*) ready to react and turnover the substrate to product.
Our result of the two-component donor lifetime decays (Figure 5B,C) corresponding to two distinct FRET efficiency distributions is consistent with hinge-bending motion that opens and closes the active site cleft along the α-helix.6,7 From the distributions of two-component lifetime decays of donor fluorescence, we have identified narrowly distributed faster component (τ1) and widely distributed slower component (τ2) (Figure 5B). Figure 5C shows the mean and standard deviation of the distributions. The results of two-component lifetime decays imply that T4 lysozyme exhibits open-close hinge-bending conformational motions associated with two-component FRET donor lifetime: the faster component (τ1), when the active-site is closed and the FRET efficiency is high, is relatively narrowly distributed, and the slower component (τ2), when the active-site is open and the FRET efficiency is low, is widely distributed. The distinct distribution of each component gives a further indication that close state is rigid and spatially narrowly distributed, and in contrast, the open states involve flexible and broadly distributed conformational fluctuations. On the basis of Michaelis–Menten mechanism in eq 1, in the process of forming the active complex ES* (E + S → ES → ES*), the enzyme involves active site opening up to intake the substrate to form the nonspecific enzyme–substrate complex ES, and binding down to form the active complex ES*. During this whole open-close hinged-bending rate process, the enzyme essentially involves multiple steps associated with multiple conformational intermediate states. In the process of catalytic reaction and product releasing (ES* → EP → E + P), corresponding to the rigid and narrowly distributed close states, the enzyme may not exhibit significant enzymatic active site conformational changes. In terms of T4 lysozyme hinge-bending open-close conformational motions, our result of two-component lifetime decays is consistent with both ensemble-level measurements3,4 and single-molecule fluorescence measurements.6,7,47,88,94,95 The different modes of open and close motions also agree with the recent study of conformational dynamics of T4 lysozyme through an electric circuit by means of attaching single molecules to single-walled carbon nanotube field-effective transistors,96,97 that is, T4 lysozyme closes up in a single step while the open process requires a minimum of two steps.96
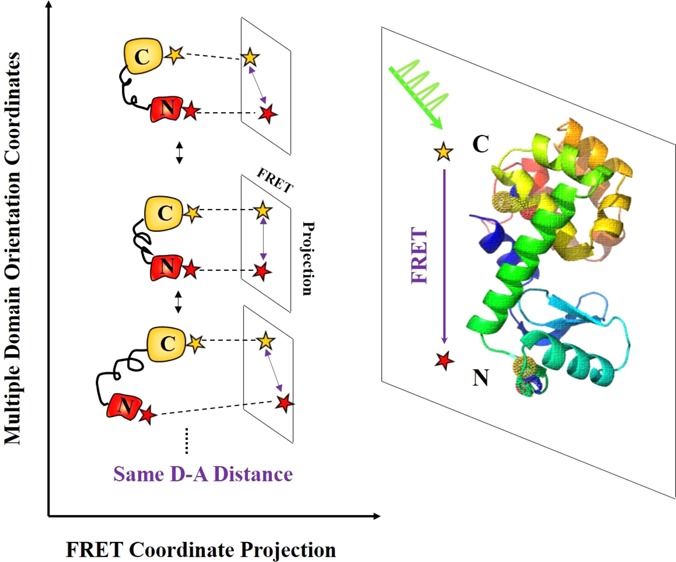
Our work provides a new insight into T4 lysozyme conformational dynamics from a multiple dimensional perspective. Along the domain orientation coordinates, significantly different T4 lysozyme conformations can have similar or same donor-to-acceptor (D-A) distance; therefore, the different T4 lysozyme conformations may not necessarily be identifiable from the FRET signal alone associated with this D–A sensitive coordinate (Figure 6). Typically, FRET measurement is sensitive to the projected FRET-coordinate changes from the multiple T4 lysozyme intermediates states associated with different domain orientation coordinates. The multiple intermediate states involved in the active-site open-close hinge-bending motions and the hinge-bending conformational motions are intrinsically multidimensional, allowing for repositioning and reorienting the subdomains in forming the active enzyme–substrate complex conformational state ready for a hydrolysis turnover reaction. While the hinge-bending motions along α-helix require spatial proximity of two domains to interact with the substrate within the active site, the domain rotational orientation motions are predicted to be important for the enzyme to function,98−100 allowing the substrate to enter and the products to leave the active site. Our results of T4 lysozyme conformational dynamics obtained from the single-molecule multiparameter photon-counting spectroscopy highlight the potential significance of probing the multidimensional conformational motions along multiple coordinates for characterizing the enzyme–substrate interactions and catalytic efficiency.
Figure 6.

Conceptual presentation of T4 lysozyme multidimensional conformational dynamics. T4 lysozyme exhibits multiple conformational intermediate states during open–close hinge-bending motions, involving multiple domain orientation coordinates and FRET coordinate. FRET-coordinate projections of multiple T4 lysozyme conformations associated with different domain orientation coordinates are presented. Along domain orientation coordinates, different enzyme conformations can have the same D–A distance, which can be undetectable and hidden in a conventional single-molecule FRET spectroscopic measurement.
In the past decades, there have been intensive efforts to investigate and understand how the enzymes work and are capable of changing the biological activity pathways and enhancing a biological reaction rate by as much as 1016 times. Over the years, it has been recognized that the conformational motions are essential for the catalytic functions of enzymes.98,99,101−105 For example, molecular dynamics (MD) simulation and statistical modeling have made significant contributions to characterize conformational motions and reaction fluctuations of enzymes in enzyme-catalyzed reactions.106−115 More often than not, subtle conformational changes even play a crucial role in enzyme functions, and these protein conformations are highly dynamic rather than being static, involving in multiple intermediate states and multiple conformational coordinates.7,33,70,99,101,116−120 The approach, experimental results, and discussion presented in this report are probably just a beginning step in expanding the studies and interpretations of new information about dissecting both the spatially and temporally complex enzymatic conformational dynamics under enzymatic reactions. Apparently, there is still a long way toward a detailed and quantitative analysis of function-related conformational motions. For example, FRET and anisotropy results suggest the existence of multidimensional conformational motions that are important to enzymatic activities, such as the hinged-bending motions under T4 lysozyme enzymatic reactions. Additional efforts are still on demand to give direct spatial and temporal characterizations of the exact angles and positions of multidimensional conformational coordinates. Temporal transitions of multiple intermediate states associated with FRET coordinate and orientation coordinates or the coupling between them are still unclear. The complementary MD simulations will likely be helpful and supportive to address those issues.
Conclusions
In this report, we have provided a new insight into T4 lysozyme conformational dynamics from a multiple dimensional perspective. The multidimensional conformational probing from our correlated single-molecule FRET and anisotropy measurements implies that T4 lysozyme exhibits much complex conformational motions along multiple orientations and nuclear coordinates beyond hinge-bending coordinate (α-helix). Significant information about the complex conformational motions are hidden by using only a conventional single-molecule one-dimensional FRET analysis that is primarily sensitive to the motions projected from the complex and real conformational motions to the FRET distance sensitive coordinate. The results of FRET donor lifetime decays and correlated anisotropy suggest that T4 lysozyme open states involve flexible and broadly distributed conformational fluctuations while the close state is more rigid. In addition, the dynamic and static inhomogeneity of multidimensional conformational fluctuations have been revealed by nonexponential features of autocorrelation functions of both lifetime and anisotropy. The developed single-molecule multiparameter photon stamping spectroscopy provides a possible access to probe multidimensional conformational motions of complex enzymatic systems, such as T4 lysozyme, by means of simultaneous acquisition of FRET, fluorescence anisotropy, and FRET donor fluorescence lifetime. There is still a high call for experimentally technical approaches which are capable of probing the complex enzymatic conformational fluctuations without ensemble-averaging as well as measurement synchronization, and multiple parameter measurements with the sensitivity of analyzing the enzyme rotational motion, translational diffusion, intramolecular domain motions, and intermolecular interactions. Our approach reported here holds the promise to characterize not only the enzymatic active site conformational fluctuations and enzyme–substrate interactions but also the overall enzyme matrix motions surrounding the active site. Evidently, such overall enzyme conformation fluctuations and multiple coordinate in nature, likely play important roles in establishing the catalytic reaction pathways and the overall enzymatic reaction energy landscape.
Acknowledgments
This work is supported by NIH/NIGMS and Ohio Eminent Scholar endowment. We thank Yufan He for his stimulating discussions on sample preparation and data analysis.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Matthews B. W. Studies on Protein Stability with T4 Lysozyme. Adv. Protein Chem. 1995, 46, 249–278. [DOI] [PubMed] [Google Scholar]
- Meroueh S. O.; Bencze K. Z.; Hesek D.; Lee M.; Fisher J. F.; Stemmler T. L.; Mobashery S. Three-Dimensional Structure of the Bacterial Cell Wall Peptidoglycan. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 4404–4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber H. R.; Matthews B. W. A Mutant T4 Lysozyme Displays Five Different Crystal Conformations. Nature 1990, 348, 263–266. [DOI] [PubMed] [Google Scholar]
- Mchaourab H. S.; Oh K. J.; Fang C. J.; Hubbell W. L. Conformation of T4 Lysozyme in Solution. Hinge-Bending Motion and the Substrate-Induced Conformational Transition Studied by Site-Directed Spin Labeling. Biochemistry 1997, 36, 307–316. [DOI] [PubMed] [Google Scholar]
- Zhang X. J.; Wozniak J. A.; Matthews B. W. Protein Flexibility and Adaptability Seen in 25 Crystal Forms of T4 Lysozyme. J. Mol. Biol. 1995, 250, 527–552. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Hu D. H.; Vorpagel E. R.; Lu H. P. Probing Single-Molecule T4 Lysozyme Conformational Dynamics by Intramolecular Fluorescence Energy Transfer. J. Phys. Chem. B 2003, 107, 7947–7956. [Google Scholar]
- Wang Y.; Lu H. P. Bunching Effect in Single-Molecule T4 Lysozyme Nonequilibrium Conformational Dynamics under Enzymatic Reactions. J. Phys. Chem. B 2010, 114, 6669–6674. [DOI] [PubMed] [Google Scholar]
- Ha T.; Enderle T.; Ogletree D. F.; Chemla D. S.; Selvin P. R.; Weiss S. Probing the Interaction between Two Single Molecules: Fluorescence Resonance Energy Transfer between a Single Donor and a Single Acceptor. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 6264–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H. P.; Xun L. Y.; Xie X. S. Single-Molecule Enzymatic Dynamics. Science 1998, 282, 1877–1882. [DOI] [PubMed] [Google Scholar]
- Xie X. S.; Lu H. P. Single-Molecule Enzymology. J. Biol. Chem. 1999, 274, 15967–15970. [DOI] [PubMed] [Google Scholar]
- Moerner W. E.; Orrit M. Illuminating Single Molecules in Condensed Matter. Science 1999, 283, 1670–1676. [DOI] [PubMed] [Google Scholar]
- Moerner W. E. New Directions in Single-Molecule Imaging and Analysis. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 12596–12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X. S. Single-Molecule Spectroscopy and Dynamics at Room Temperature. Acc. Chem. Res. 1996, 29, 598–606. [Google Scholar]
- Ishii Y.; Yanagida T. Single Molecule Detection in Life Sciences. Single Mol. 2000, 1, 5–16. [Google Scholar]
- Moerner W. E. A Dozen Years of Single-Molecule Spectroscopy in Physics, Chemistry, and Biophysics. J. Phys. Chem. B 2002, 106, 910–927. [Google Scholar]
- Kim H. D.; Nienhaus G. U.; Ha T.; Orr J. W.; Williamson J. R.; Chu S. Mg2+-Dependent Conformational Change of RNA Studied by Fluorescence Correlation and FRET on Immobilized Single Molecules. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 4284–4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X. W.; Bartley L. E.; Babcock H. P.; Russell R.; Ha T. J.; Herschlag D.; Chu S. A Single-Molecule Study of RNA Catalysis and Folding. Science 2000, 288, 2048–2051. [DOI] [PubMed] [Google Scholar]
- Ha T.; Zhuang X. W.; Kim H. D.; Orr J. W.; Williamson J. R.; Chu S. Ligand-Induced Conformational Changes Observed in Single RNA Molecules. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 9077–9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. L.; Cao J. S. Direct Measurements of Memory Effects in Single-Molecule Kinetics. J. Chem. Phys. 2002, 117, 10996–11009. [Google Scholar]
- Andoy N. M.; Zhou X. C.; Choudhary E.; Shen H.; Liu G. K.; Chen P. Single-Molecule Catalysis Mapping Quantifies Site-Specific Activity and Uncovers Radial Activity Gradient on Single 2D Nanocrystals. J. Am. Chem. Soc. 2013, 135, 1845–1852. [DOI] [PubMed] [Google Scholar]
- Zhou X. C.; Xu W. L.; Liu G. K.; Panda D.; Chen P. Size-Dependent Catalytic Activity and Dynamics of Gold Nanoparticles at the Single-Molecule Level. J. Am. Chem. Soc. 2010, 132, 138–146. [DOI] [PubMed] [Google Scholar]
- English B. P.; Min W.; van Oijen A. M.; Lee K. T.; Luo G. B.; Sun H. Y.; Cherayil B. J.; Kou S. C.; Xie S. X. Ever-Fluctuating Single Enzyme Molecules: Michaelis-Menten Equation Revisited. Nat. Chem. Biol. 2006, 2, 168–168. [DOI] [PubMed] [Google Scholar]
- Lee C. L.; Lin C. T.; Stell G.; Wang J. Diffusion Dynamics, Moments, and Distribution of First-Passage Time on the Protein-Folding Energy Landscape, with Applications to Single Molecules. Phys. Rev. E 2003, 67, 041905. [DOI] [PubMed] [Google Scholar]
- Roy R.; Hohng S.; Ha T. A Practical Guide to Single-Molecule FRET. Nat. Methods 2008, 5, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y. F.; Lu M. L.; Cao J.; Lu H. P. Manipulating Protein Conformations by Single-Molecule AFM-FRET Nanoscopy. ACS Nano 2012, 6, 1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y. F.; Lu M. L.; Lu H. P. Single-Molecule Photon Stamping FRET Spectroscopy Study of Enzymatic Conformational Dynamics. Phys. Chem. Chem. Phys. 2013, 15, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T. Single-Molecule Fluorescence Methods for the Study of Nucleic Acids. Curr. Opin. Struct. Biol. 2001, 11, 287–292. [DOI] [PubMed] [Google Scholar]
- Kulinski T.; Wennerberg A. B. A.; Rigler R.; Provencher S. W.; Pooga M.; Langel U.; Bartfai T. Conformational Analysis of Galanin using End to End Distance Distribution Observed by Forster Resonance Energy Transfer. Eur. Biophys. J. Biophys. 1997, 26, 145–154. [DOI] [PubMed] [Google Scholar]
- Tan E.; Wilson T. J.; Nahas M. K.; Clegg R. M.; Lilley D. M. J.; Ha T. A Four-Way Junction Accelerates Hairpin Ribozyme Folding via a Discrete Intermediate. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 9308–9313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabanayagam C. R.; Eid J. S.; Meller A. Long Time Scale Blinking Kinetics of Cyanine Fluorophores Conjugated to DNA and its Effect on Forster Resonance Energy Transfer. J. Chem. Phys. 2005, 224708. [DOI] [PubMed] [Google Scholar]
- Sako Y.; Minoguchi S.; Yanagida T. Single-Molecule Imaging of EGFR Signalling on the Surface of Living Cells. Nat. Cell Biol. 2000, 2, 168–172. [DOI] [PubMed] [Google Scholar]
- Brasselet S.; Peterman E. J. G.; Miyawaki A.; Moerner W. E. Single-Molecule Fluorescence Resonant Energy Transfer in Calcium Concentration Dependent Cameleon. J. Phys. Chem. B 2000, 104, 3676–3682. [Google Scholar]
- Lerch H. P.; Rigler R.; Mikhailov A. S. Functional Conformational Motions in the Turnover Cycle of Cholesterol Oxidase. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 10807–10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohng S.; Joo C.; Ha T. Single-Molecule Three-Color FRET. Biophys. J. 2004, 87, 1328–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Lee J.; Hohng S. Single-Molecule Three-Color FRET with both Negligible Spectral Overlap and Long Observation Time. PLoS One 2010, 5, e12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein I. H.; Steinhauer C.; Tinnefeld P. Single-Molecule Four-Color FRET Visualizes Energy-Transfer Paths on DNA Origami. J. Am. Chem. Soc. 2011, 133, 4193–4195. [DOI] [PubMed] [Google Scholar]
- Lee J.; Lee S.; Ragunathan K.; Joo C.; Ha T.; Hohng S. Single-Molecule Four-Color FRET. Angew. Chem., Int. Ed. 2010, 49, 9922–9925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale R. E.; Eisinger J.; Blumberg W. E. The Orientational Freedom of Molecular Probes. The Orientation Factor in Intramolecular Energy Transfer. Biophys. J. 1979, 26, 161–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal A.; Arslan S.; Okumus B.; Wilson T. J.; Giraud G.; Norman D. G.; Ha T.; Lilley D. M. Orientation Dependence in Fluorescent Energy Transfer between Cy3 and Cy5 Terminally Attached to Double-Stranded Nucleic Acids. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 11176–11181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew M. D.; Backlund M. P.; Moerner W. E. Rotational Mobility of Single Molecules Affects Localization Accuracy in Super-Resolution Fluorescence Microscopy. Nano Lett. 2013, 13, 3967–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartko A. P.; Dickson R. M. Imaging Three-Dimensional Single Molecule Orientations. J. Phys. Chem. B 1999, 103, 11237–11241. [Google Scholar]
- Cognet L.; Harms G. S.; Blab G. A.; Lommerse P. H.; Schmidt T. Simultaneous Dual-Color and Dual-Polarization Imaging of Single Molecules. Appl. Phys. Lett. 2000, 77, 4052–4054. [Google Scholar]
- Eggeling C.; Berger S.; Brand L.; Fries J.; Schaffer J.; Volkmer A.; Seidel C. Data Registration and Selective Single-Molecule Analysis using Multi-Parameter Fluorescence Detection. J. Biotechnol. 2001, 86, 163–180. [DOI] [PubMed] [Google Scholar]
- Rothwell P.; Berger S.; Kensch O.; Felekyan S.; Antonik M.; Wöhrl B.; Restle T.; Goody R.; Seidel C. Multiparameter Single-Molecule Fluorescence Spectroscopy Reveals Heterogeneity of HIV-1 Reverse Transcriptase: Primer/Template Complexes. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 1655–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonik M.; Felekyan S.; Gaiduk A.; Seidel C. A. Separating Structural Heterogeneities from Stochastic Variations in Fluorescence Resonance Energy Transfer Distributions via Photon Distribution Analysis. J. Phys. Chem. B 2006, 110, 6970–6978. [DOI] [PubMed] [Google Scholar]
- Kalinin S.; Peulen T.; Sindbert S.; Rothwell P. J.; Berger S.; Restle T.; Goody R. S.; Gohlke H.; Seidel C. A. A Toolkit and Benchmark Study for FRET-Restrained High-Precision Structural Modeling. Nat. Methods 2012, 9, 1218–1225. [DOI] [PubMed] [Google Scholar]
- Hu D. H.; Lu H. P. Single-Molecule Nanosecond Anisotropy Dynamics of Tethered Protein Motions. J. Phys. Chem. B 2003, 107, 618–626. [Google Scholar]
- Tan X.; Hu D. H.; Squier T. C.; Lu H. P. Probing Nanosecond Protein Motions of Calmodulin by Single-Molecule Fluorescence Anisotropy. Appl. Phys. Lett. 2004, 85, 2420–2422. [Google Scholar]
- Rosenberg S. A.; Quinlan M. E.; Forkey J. N.; Goldman Y. E. Rotational Motions of Macromolecules by Single-Molecule Fluorescence Microscopy. Acc. Chem. Res. 2005, 38, 583–593. [DOI] [PubMed] [Google Scholar]
- Forkey J. N.; Quinlan M. E.; Goldman Y. E. Protein Structural Dynamics by Single-Molecule Fluorescence Polarization. Prog. Biophys. Mol. Biol. 2000, 74, 1–35. [DOI] [PubMed] [Google Scholar]
- Peterman E. J. G.; Sosa H.; Moerner W. E. Single-Molecule Fluorescence Spectroscopy and Microscopy of Biomolecular Motors. Annu. Rev. Phys. Chem. 2004, 55, 79–96. [DOI] [PubMed] [Google Scholar]
- Ha T.; Laurence T. A.; Chemla D. S.; Weiss S. Polarization Spectroscopy of Single Fluorescent Molecules. J. Phys. Chem. B 1999, 103, 6839–6850. [Google Scholar]
- Selvin P. R.Single-Molecule Techniques: a Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, 2008. [Google Scholar]
- He Y. F.; Li Y.; Mukherjee S.; Wu Y.; Yan H. G.; Lu H. P. Probing Single-Molecule Enzyme Active-Site Conformational State Intermittent Coherence. J. Am. Chem. Soc. 2011, 133, 14389–14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berberan-Santos M.Pioneering Contributions of Jean and Francis Perrin to Molecular Luminescence. New Trends in Fluorescence Spectroscopy; Springer: Berlin, 2001; Vol. 1, pp 7–33. [Google Scholar]
- Lakowicz J. R.Principles of Fluorescence Spectroscopy; Springer: Berlin, 2009. [Google Scholar]
- Schuler B.; Lipman E. A.; Eaton W. A. Probing the Free-Energy Surface for Protein Folding with Single-Molecule Fluorescence Spectroscopy. Nature 2002, 419, 743–747. [DOI] [PubMed] [Google Scholar]
- Talaga D. S.; Lau W. L.; Roder H.; Tang J. Y.; Jia Y. W.; DeGrado W. F.; Hochstrasser R. M. Dynamics and Folding of Single Two-Stranded Coiled-Coil Peptides Studied by Fluorescent Energy Transfer Confocal Microscopy. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 13021–13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J.; Snapp E.; Kenworthy A. Studying Protein Dynamics in Living Cells. Nat. Rev. Mol. Cell Biol. 2001, 2, 444–456. [DOI] [PubMed] [Google Scholar]
- Grecco H. E.; Verveer P. J. FRET in Cell Biology: Still Shining in the Age of Super-Resolution?. ChemPhysChem 2011, 12, 484–490. [DOI] [PubMed] [Google Scholar]
- Li H.; Yen C. F.; Sivasankar S. Fluorescence Axial Localization with Nanometer Accuracy and Precision. Nano Lett. 2012, 12, 3731–3735. [DOI] [PubMed] [Google Scholar]
- Gradinaru C. C.; Marushchak D. O.; Samim M.; Krull U. J. Fluorescence Anisotropy: from Single Molecules to Live Cells. Analyst 2010, 135, 452–459. [DOI] [PubMed] [Google Scholar]
- Vogel S. S.; Thaler C.; Blank P. S.; Koushik S. V.. Time Resolved Fluorescence Anisotropy. FLIM Microscopy in Biology and Medicine; Chapman and Hall/CRC: Boca Raton, FL, 2009; Vol. 1, pp 245–288. [Google Scholar]
- Liu R.; Hu D.; Tan X.; Lu H. P. Revealing Two-State Protein-Protein Interactions of Calmodulin by Single-Molecule Spectroscopy. J. Am. Chem. Soc. 2006, 128, 10034–10042. [DOI] [PubMed] [Google Scholar]
- Gopich I. V.; Szabo A. Theory of the Energy Transfer Efficiency and Fluorescence Lifetime Distribution in Single-Molecule FRET. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 7747–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes G. G. Multiple Conformational Changes in Enzyme Catalysis. Biochemistry 2002, 41, 8221–8228. [DOI] [PubMed] [Google Scholar]
- Zhuang X.; Rief M. Single-Molecule Folding. Curr. Opin. Struct. Biol. 2003, 13, 88–97. [DOI] [PubMed] [Google Scholar]
- Hammes G. G.; Benkovic S. J.; Hammes-Schiffer S. Flexibility, Diversity, and Cooperativity: Pillars of Enzyme Catalysis. Biochemistry 2011, 50, 10422–10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm V. L. Enzymatic Transition States, Transition-State Analogs, Dynamics, Thermodynamics, and Lifetimes. Annu. Rev. Biochem. 2011, 80, 703–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q.; Wang J. Single Molecule Conformational Dynamics of Adenylate Kinase: Energy Landscape, Structural Correlations, and Transition State Ensembles. J. Am. Chem. Soc. 2008, 130, 4772–4783. [DOI] [PubMed] [Google Scholar]
- Frauenfelder H.; Sligar S. G.; Wolynes P. G. The Energy Landscapes and Motions of Proteins. Science 1991, 254, 1598–1603. [DOI] [PubMed] [Google Scholar]
- Whitford P. C.; Sanbonmatsu K. Y.; Onuchic J. N. Biomolecular Dynamics: Order–Disorder Transitions and Energy Landscapes. Rep. Prog. Phys. 2012, 75, 076601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafiq S.; Rajbongshi B. K.; Nair N. N.; Sen P.; Ramanathan G. Excited State Relaxation Dynamics of Model Green Fluorescent Protein Chromophore Analogs: Evidence for Cis-Trans Isomerism. J. Phys. Chem. A 2011, 115, 13733–13742. [DOI] [PubMed] [Google Scholar]
- Sahu K.; Mondal S. K.; Ghosh S.; Roy D.; Sen P.; Bhattacharyya K. Femtosecond Study of Partially Folded States of Cytochrome C by Solvation Dynamics. J. Phys. Chem. B 2006, 110, 1056–1062. [DOI] [PubMed] [Google Scholar]
- Anand U.; Jash C.; Mukherjee S. Protein Unfolding and Subsequent Refolding: a Spectroscopic Investigation. Phys. Chem. Chem. Phys. 2011, 13, 20418–20426. [DOI] [PubMed] [Google Scholar]
- Anand U.; Jash C.; Boddepalli R. K.; Shrivastava A.; Mukherjee S. Exploring the Mechanism of Fluorescence Quenching in Proteins Induced by Tetracycline. J. Phys. Chem. B 2011, 115, 6312–6320. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Tang C.; Wang E.; Wang J. Exploration of Multi-State Conformational Dynamics and Underlying Global Functional Landscape of Maltose Binding Protein. PLoS Comput. Biol. 2012, 8, e1002471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokinsky G.; Rueda D.; Misra V. K.; Rhodes M. M.; Gordus A.; Babcock H. P.; Walter N. G.; Zhuang X. W. Single-Molecule Transition-State Analysis of RNA Folding. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 9302–9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopich I. V.; Szabo A. FRET Efficiency Distributions of Multistate Single Molecules. J. Phys. Chem. B 2010, 114, 15221–15226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoso Y.; Joyce C. M.; Potapova O.; Le Reste L.; Hohlbein J.; Torella J. P.; Grindley N. D. F.; Kapanidis A. N. Conformational Transitions in DNA Polymerase I Revealed by Single-Molecule FRET. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn M. E.; Connolly B. K.; Gurunathan K.; Levitus M. Fluorescence Properties and Photophysics of the Sulfoindocyanine Cy3 Linked Covalently to DNA. J. Phys. Chem. B 2007, 111, 11064–11074. [DOI] [PubMed] [Google Scholar]
- Levitus M.; Ranjit S. Cyanine Dyes in Biophysical Research: the Photophysics of Polymethine Fluorescent Dyes in Biomolecular Environments. Q. Rev. Biophys. 2011, 44, 123–151. [DOI] [PubMed] [Google Scholar]
- Ha T.; Tinnefeld P. Photophysics of Fluorescent Probes for Single-Molecule Biophysics and Super-Resolution Imaging. Annu. Rev. Phys. Chem. 2012, 63, 595–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramendia P. F.; Negri R. M.; Sanroman E. Temperature-Dependence of Fluorescence and Photoisomerization in Symmetrical Carbocyanines - Influence of Medium Viscosity and Molecular-Structure. J. Phys. Chem. 1994, 98, 3165–3173. [Google Scholar]
- de Groot B. L.; Hayward S.; van Aalten D. M. F.; Amadei A.; Berendsen H. J. C. Domain Motions in Bacteriophage T4 Lysozyme: A Comparison between Molecular Dynamics and Crystallographic Data. Proteins 1998, 31, 116–127. [DOI] [PubMed] [Google Scholar]
- Wang S. C.; Lee C. T. Enhanced Enzymatic Activity through Photoreversible Conformational Changes. Biochemistry 2007, 46, 14557–14566. [DOI] [PubMed] [Google Scholar]
- Hamill A. C.; Wang S. C.; Lee C. T. Probing Lysozyme Conformation with Light Reveals a New Folding Intermediate. Biochemistry 2005, 44, 15139–15149. [DOI] [PubMed] [Google Scholar]
- Yirdaw R. B.; McHaourab H. S. Direct Observation of T4 Lysozyme Hinge-Bending Motion by Fluorescence Correlation Spectroscopy. Biophys. J. 2012, 103, 1525–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. M.; Wang X. F.; Ghosh S. K.; Lu H. P. Probing Single-Molecule Interfacial Electron Transfer Dynamics of Porphyrin on TiO2 Nanoparticles. J. Am. Chem. Soc. 2009, 131, 1479–1487. [DOI] [PubMed] [Google Scholar]
- Yang H.; Luo G. B.; Karnchanaphanurach P.; Louie T. M.; Rech I.; Cova S.; Xun L. Y.; Xie X. S. Protein Conformational Dynamics Probed by Single-Molecule Electron Transfer. Science 2003, 302, 262–266. [DOI] [PubMed] [Google Scholar]
- Bonnet G.; Krichevsky O.; Libchaber A. Kinetics of Conformational Fluctuations in DNA Hairpin-Loops. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 8602–8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalet X.; Weiss S.; Jager M. Single-Molecule Fluorescence Studies of Protein Folding and Conformational Dynamics. Chem. Rev. 2006, 106, 1785–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenter G. K.; Lu H. P.; Xie X. S. Statistical Analyses and Theoretical Models of Single-Molecule Enzymatic Dynamics. J. Phys. Chem. A 1999, 103, 10477–10488. [Google Scholar]
- Lu H. P. Revealing Time Bunching Effect in Single-Molecule Enzyme Conformational Dynamics. Phys. Chem. Chem. Phys. 2011, 13, 6734–6749. [DOI] [PubMed] [Google Scholar]
- Hu D.; Lu H. P. Placing Single-Molecule T4 Lysozyme Enzymes on a Bacterial Cell Surface: toward Probing Single-Molecule Enzymatic Reaction in Living Cells. Biophys. J. 2004, 87, 656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y.; Moody I. S.; Sims P. C.; Hunt S. R.; Corso B. L.; Perez I.; Weiss G. A.; Collins P. G. Single-Molecule Lysozyme Dynamics Monitored by an Electronic Circuit. Science 2012, 335, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y.; Weiss G. A.; Collins P. G. Single Molecule Recordings of Lysozyme Activity. Phys. Chem. Chem. Phys. 2013, 15, 14879–14895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. X.; McCammon J. A. The Gates of Ion Channels and Enzymes. Trends Biochem. Sci. 2010, 35, 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes G. G. How do Enzymes Really Work?. J. Biol. Chem. 2008, 283, 22337–22346. [DOI] [PubMed] [Google Scholar]
- Rashin A. A.; Rashin A. H. L.; Jernigan R. L. Diversity of Function-Related Conformational Changes in Proteins: Coordinate Uncertainty, Fragment Rigidity, and Stability. Biochemistry 2010, 49, 5683–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus M.; McCammon J. A. Molecular Dynamics Simulations of Biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [DOI] [PubMed] [Google Scholar]
- Doshi U.; McGowan L. C.; Ladani S. T.; Hamelberg D. Resolving the Complex Role of Enzyme Conformational Dynamics in Catalytic Function. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 5699–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J. L. Catalysis by Enzyme Conformational Change as Illustrated by Orotidine 5 ’-monophosphate Decarboxylase. Curr. Opin. Struct. Biol. 2003, 13, 184–192. [DOI] [PubMed] [Google Scholar]
- Karplus M.; Mccammon J. A. Dynamics of Proteins - Elements and Function. Annu. Rev. Biochem. 1983, 52, 263–300. [DOI] [PubMed] [Google Scholar]
- McCammon J. A.; Gelin B. R.; Karplus M.; Wolynes P. G. The Hinge-Bending Mode in Lysozyme. Nature 1976, 262, 325–326. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Gan L.; Wang E.; Wang J. Exploring the Dynamic Functional Landscape of Adenylate Kinase Modulated by Substrates. J. Chem. Theory Comput. 2012, 9, 84–95. [DOI] [PubMed] [Google Scholar]
- Warshel A. Dynamics of Enzymatic-Reactions. Proc. Natl. Acad. Sci. U.S.A. 1984, 81, 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M. H. M.; Parson W. W.; Warshel A. Dynamical Contributions to Enzyme Catalysis: Critical Tests of a Popular Hypothesis. Chem. Rev. 2006, 106, 1737–1756. [DOI] [PubMed] [Google Scholar]
- Bruice T. C. A View at the Millennium: The Efficiency of Enzymatic Catalysis. Acc. Chem. Res. 2002, 35, 139–148. [DOI] [PubMed] [Google Scholar]
- Bruice T. C.; Benkovic S. J. Chemical Basis for Enzyme Catalysis. Biochemistry 2000, 39, 6267–6274. [DOI] [PubMed] [Google Scholar]
- Zhou R. H.; Huang X. H.; Margulis C. J.; Berne B. J. Hydrophobic Collapse in Multidomain Protein Folding. Science 2004, 305, 1605–1609. [DOI] [PubMed] [Google Scholar]
- Perez-Jimenez R.; Li J. Y.; Kosuri P.; Sanchez-Romero I.; Wiita A. P.; Rodriguez-Larrea D.; Chueca A.; Holmgren A.; Miranda-Vizuete A.; Becker K.; Cho S. H.; Beckwith J.; Gelhaye E.; Jacquot J. P.; Gaucher E. A.; Sanchez-Ruiz J. M.; Berne B. J.; Fernandez J. M. Diversity of Chemical Mechanisms in Thioredoxin Catalysis Revealed by Single-Molecule Force Spectroscopy. Nat. Struct. Mol. Biol. 2009, 16, 890–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J. L.; Cao J. S. Generalized Michaelis-Menten Equation for Conformation-Modulated Monomeric Enzymes. Adv. Chem. Phys. 2012, 146, 329–365. [Google Scholar]
- Whitford P. C.; Onuchic J. N.; Wolynes P. G. Energy Landscape along an Enzymatic Reaction Trajectory: Hinges or Cracks?. HFSP J. 2008, 2, 61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolynes P. Passage through Fluctuating Geometrical Bottlenecks. The General Gaussian Fluctuating Case. Chem. Phys. Lett. 1993, 212, 427–433. [Google Scholar]
- Grant B. J.; Gorfe A. A.; McCammon J. A. Large Conformational Changes in Proteins: Signaling and other Functions. Curr. Opin. Struct. Biol. 2010, 20, 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCammon J. A.; Harvey S. C.. Dynamics of Proteins and Nucleic Acids; Cambridge University Press: Cambridge, 1988. [Google Scholar]
- Lu Q.; Wang J. Kinetics and Statistical Distributions of Single-Molecule Conformational Dynamics. J. Phys. Chem. B 2009, 113, 1517–1521. [DOI] [PubMed] [Google Scholar]
- Hyeon C.; Jennings P. A.; Adams J. A.; Onuchic J. N. Ligand-Induced Global Transitions in the Catalytic Domain of Protein Kinase A. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 3023–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Xu L.; Xue K.; Wang E. Exploring the Origin of Power Law Distribution in Single-Molecule Conformation Dynamics: Energy Landscape Perspectives. Chem. Phys. Lett. 2008, 463, 405–409. [Google Scholar]