

Background: Excessive light exposure and genetic mutations that act as “equivalent light” cause photoreceptor cell death.
Results: Prolonged transducin signaling, but not channel closure, induces endoplasmic reticulum stress.
Conclusion: Induction of UPR is a distinct cell death pathway caused by transducin signaling.
Significance: Manipulation of UPR may prolong photoreceptor cell survival in transducin-induced retinal light damage.
Keywords: G-protein, Photoreceptor, Retinal Degeneration, Rhodopsin, Unfolded Protein Response (UPR), Light Damage
Abstract
Phototransduction is a G-protein signal transduction cascade that converts photon absorption to a change in current at the plasma membrane. Certain genetic mutations affecting the proteins in the phototransduction cascade cause blinding disorders in humans. Some of these mutations serve as a genetic source of “equivalent light” that activates the cascade, whereas other mutations lead to amplification of the light response. How constitutive phototransduction causes photoreceptor cell death is poorly understood. We showed that persistent G-protein signaling, which occurs in rod arrestin and rhodopsin kinase knock-out mice, caused a rapid and specific induction of the PERK pathway of the unfolded protein response. These changes were not observed in the cGMP-gated channel knock-out rods, an equivalent light condition that mimics light-stimulated channel closure. Thus transducin signaling, but not channel closure, triggers rapid cell death in light damage caused by constitutive phototransduction. Additionally, we show that in the albino light damage model cell death was not associated with increase in global protein ubiquitination or unfolded protein response induction. Taken together, these observations provide novel mechanistic insights into the cell death pathway caused by constitutive phototransduction and identify the unfolded protein response as a potential target for therapeutic intervention.
Introduction
Light is an environmental factor that modulates the rate of many retinal diseases (1, 2), including age-related macular degeneration (3, 4). Excessive light exposure in itself is damaging to the retina (5, 6), and different experimental paradigms of light damage have been used to investigate the mechanism of photoreceptor cell apoptosis. The use of rhodopsin knock-out and RPE65-deficient mice clearly showed the requirement of rhodopsin and a functioning visual cycle in light damage (7). Downstream of rhodopsin, two distinct pathways are known (8). The first involves bright light activating a large amount of rhodopsin, R*. Early changes in this pathway include a large increase in intracellular Ca2+ within the rod photoreceptors (9) and activation of c-Fos and c-Jun (10). Cell death in this pathway is independent of the visual G-protein transducin but is prevented in the c-Fos-deficient mice (11). The second pathway involves constitutive G-protein signaling. In retinal rods, photolyzed rhodopsin activates several transducin molecules which in turn activate the effector enzyme PDE6. PDE6 hydrolyzes cGMP, leading to closure of the cGMP-gated (CNG) channel. Photolyzed rhodopsin is deactivated by rhodopsin kinase (GRK1)-mediated phosphorylation of serine and threonine residues at the carboxyl terminus of rhodopsin (12) followed by arrestin (ARR1) binding (13, 14). Therefore G-protein signaling is prolonged in the ARR1 or GRK1 knock-out mice, and the retina degenerates even under low ambient light (15, 16). Light damage in this pathway is prevented in the transducin Gtα (encoded by the GNAT1 gene) knock-out background, indicating a dependence on phototransduction (8, 17, 18). How constitutive G-protein signaling leads to photoreceptor cell death is not well understood but is an important question to address inasmuch as defects in ARR1 and GRK1 genes occur in the human population (19, 20), and certain mutations in other phototransduction genes generate “equivalent light” by driving the phototransduction cascade (21).
Light damage to photoreceptors in the albino BALB/c mice is caused by pathologically high levels of light-activated rhodopsin, R*, produced even under relatively low ambient illumination. Two factors contribute to generate high levels of R* in BALB/c mice: first, the lack of pigment in the iris that normally restricts the amount of light reaching the neural retina; and second, a robust RPE65 enzyme that rapidly regenerates the visual pigment rhodopsin (22). In contrast, the pigmented iris of C57/B6 and ARR1−/− mice (which are in C57/B6 background) reduces the amount of light reaching the retina by 2 orders of magnitude relative to that of albino mice (23). Further, the Met-450 variant of RPE65 expressed in C57/B6 mice is less active and slows the kinetics of rhodopsin regeneration by 4-fold when compared with the Leu-450 variant expressed by BALB/c mice (22). Thus the difference in the levels of R* generated by the same light exposure to BALB/c and C57 mice can be ≥400-fold, making the albino BALB/c mice an ideal light damage model to isolate the toxic effect of excessive R*, which generates reactive oxygen species and cause oxidative stress (2, 24).
In wild type mice, rhodopsin catalytic lifetime has been estimated to be as short as 40 ms (25). However, in the ARR1−/− rods, the light response is abnormally prolonged. Rhodopsin phosphorylation proceeds normally in the ARR1−/− rods and decreases the catalytic activity of rhodopsin by ∼70%; full recovery follows the time course of meta II decay, which on average is ∼42 s in mouse rods (14). Assuming in wild type rods that rhodopsin phosphorylation occurs halfway through its catalytic lifetime and the rate of transducin activation is 1,300 Gt*/s (26), the number of transducin activated is estimated to be 34 ([1,300 Gt*/s × 0.02 s] + [1.300 Gt*/s × 0.02 s × 0.3] = 34 Gt*). In contrast, the long R* lifetime in the ARR1−/− would activate 16,400 Gt (1,300 Gt*/s × 42 s × 0.3), which is ∼480-fold more than wild type. Although high levels of R* generated in the albino mouse model will also activate Gt*, the amount is expected to be lower than ARR1−/− [34Gt*/s × 100 (for pupil dilation) = 3,400 Gt*/s]. Thus ARR1−/− mice is an ideal model system to isolate the deleterious effect of prolonged transducin activation.
In the present study, we compared the initial molecular events following light exposure in the albino BALB/c and the pigmented ARR1−/− mice using an experimental paradigm that produced a similar degree of photoreceptor cell death in both models. We showed that prolonged G-protein signaling caused by the lack of ARR1 led to a rapid induction of the PERK arm of the unfolded protein response (UPR)2 followed by a coordinated increase in global protein ubiquitination and degradation of PDE6 and guanylyl cyclase activating protein 2 (GCAP2). These changes were prevented when G-protein signaling was abolished in the GNAT1−/− background. Transducin activation leads to closure of cyclic nucleotide-gated (CNG) channels. However, light damage did not occur in the CNG channel knock-out rods, suggesting that G-protein signaling, but not channel closure, initiates the cell death pathway. In the albino light damage model, light-exposed BALB/c retinas showed equivalent degradation of phototransduction proteins without an increase in protein ubiquitination or induction of ER stress. Our findings implicate an involvement of the UPR in constitutive G-protein-induced cell death.
EXPERIMENTAL PROCEDURES
Mice
All experimental procedures were performed in accordance with regulations established by the National Institutes of Health. The animal protocol was approved by the University of Southern California Institutional Animal Care and Use Committee. The ARR1−/−, GRK1−/−, GNAT1−/−, and Cngb1−/− transgenic mice were derived from 129sv ES cells and successive bred with C57/B6 mice for more than five generations. C57/B6 and BALB/c mice were purchased from the Jackson Laboratories. ARR1−/−, GRK1−/−, and BALB/c mice were born and reared in darkness to avoid light-dependent retinal degeneration. C57Bl/6 mice were reared in 12-h light/dark cycles and moved to darkness 1 week before the light exposure.
Light Exposure
Experiments were performed on 4-week-old mice. The mice were exposed to diffuse cool white fluorescent light at luminescence level of 5000 lux without pupil dilation and then euthanized at the following time points: 0.5, 1, 2, 3, 4, 5, 12, or 36 h after light onset. For some experiments, the mice were exposed to light for 12 h followed by 36 h in darkness.
Retinal Morphology
After mice were euthanized, the superior pole of their cornea was marked by cauterization before enucleation. The cornea and lens were removed, and the remaining eye cup was embedded into epoxy resin. The epon-embedded eyes were sectioned along vertical meridian into 1-μm sections and stained with Richardson stain, and morphometric measurements of outer nuclear layer thickness were performed as described previously (27). Images were acquired on a Zeiss Axioplan2 microscope using a 63× objective.
Immunofluorescence
Eye cups were prepared as described above and placed in 4% formaldehyde, 0.5% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.2, for 1 h and rinsed four times for 15 min each time in 0.1 m cacodylate buffer. The eye cups were then cryoprotected in 30% sucrose overnight and then embedded in O.C.TTM (Tissue-Tech®) and sectioned at 10-μm thickness in a cryostat (CM 3050 S; Leica Microsystems). The sections were treated for 2 min room temperature with 0.2 mg/ml proteinase K in blocking buffer (2% bovine serum albumin, 2% goat serum, 0.3% Triton X-100 in PBS) and heated to 65 °C for 10 s followed by five rinses with PBS. Blocking buffer was applied for 1 h and then replaced with rabbit polyclonal antibody against GCAP2 (28) or GCAP1 (made in our laboratory, both diluted 1:100 in blocking buffer) or a mouse monoclonal antibody against rhodopsin (1D4, gift from R. Molday). The sections were rinsed and incubated with a fluorescein- or Texas Red-labeled secondary antibody (Vector Laboratories). The images were obtained using a LSM 5 confocal microscope (Zeiss Microscopy).
Western Blot Analysis
The retina was dissected and homogenized in 150 μl of buffer (150 mm NaCl, 50 mm Tris, pH 8.0, 0.1% Nonidet P-40, 0.5% deoxycholic acid) containing 0.1 mm PMSF, complete mini protease inhibitor (Roche Applied Sciences), and 50 μm NaF. DNase I (30 units; Roche Applied Sciences) was then added and incubated at room temperature for 30 min. The total protein amount of each sample was determined by the BCATM protein assay kit (Thermo Scientific). An equal amount of retinal homogenate from each sample was electrophoresed on 4–12% Bis-Tris SDS-PAGE gel (Invitrogen) followed by transfer to nitrocellulose membrane (WhatmanTM; GE Healthcare Life Sciences) and incubated overnight with the following primary antibodies: rabbit anti-PDE polyclonal antibody (1:1000, Cytosignal, PAB-06800), rabbit anti-ROS-GC1 polyclonal antibody (1:500, Santa Cruz, sc50512), mouse anti-Gtα antibody (1:5000, EMD4Biosciences, 371740), rabbit polyclonal anti-GCAP1 and GCAP2 antibodies (1:2000 and 1:1000, respectively, generated in our laboratory), mouse anti-ubiquitin monoclonal antibody (1:1000, Sigma, U0508), mouse anti-GRP78/Bip monoclonal antibody (1:1000, BD Biosciences, 610978), rabbit anti-phospho-eIF2α (Ser-51) polyclonal antibody (1:1000, Cell Signaling Technology, 9721), rabbit anti-ATF3 antibody (1:1000, Sigma-Aldrich, HPA001562), rabbit anti-CREB-2 (ATF4) antibody (1:500, Santa Cruz, sc200), rabbit anti-ATF6α antibody (1:500, Santa Cruz, sc22799), rabbit anti-IRE1α antibody (1:1000, Cell Signaling Technology, 3294), and mouse anti-actin antibody (1:5000, Millipore, MAB1501). For detection of phototransduction proteins (PDE, GC1, Gtα, GCAP1, and GCAP2), as well as ubiquitin and actin, membranes were then incubated with fluorescently labeled secondary antibodies (1:10,000, LI-COR Biosciences, P/N926-31081) at room temperature for 1 h and detected by Odyssey infrared imaging system. Densitometric scanning of each band was followed by quantitative analysis using ImageJ. GRP78, p-eIF2α, ATF3, ATF4, ATF6α, and IRE1α signals were detected using Amersham Biosciences ECL Western blotting detection reagent (GE Healthcare Life Sciences). The films were scanned, and the intensity of the bands was quantified using Photoshop. The proteasome-associated proteins were isolated by a proteasome isolation kit (Calbiochem, 539176) following the manufacturer's protocol and detected by the Western blot analysis as described above. Quantitation of Western data were obtained using at least three independent sets of experiments obtained from independent mice (n ≥ 3).
Biochemical Characterization of GCAPs Aggregates
Individual retinas from dark-adapted or light-exposed mice were isolated under infrared light and lysed in 200 μl of ice-cold buffer (PBS, pH 7.5, 5 mm EDTA, 1%Triton X-100) with protease inhibitor mixture (Roche Applied Science) for 1 h at 4 °C on a nutator. Insoluble material was recovered by centrifugation at 13,000 × g for 15 min and dissolved in 100 μl of SDS loading buffer at room temperature for 10 min and briefly homogenized to disperse the pellet. Equal fractions of the samples were subjected to SDS-PAGE followed by Western blot analysis.
RESULTS
Time Course of Retinal Degeneration in Two Different Mouse Models of Light Damage
ARR1−/−, BALB/c, and C57 mice were exposed to 5,000-lux white fluorescent light without pupil dilation for the indicated times (Fig. 1). In ARR1−/− retina, morphology was largely intact at 12 h except for a few pycnotic nuclei (Fig. 1A, arrows, top row). At 36 h, the nuclei appeared misaligned, and the outer segment structure was disrupted. When the mice exposed to 12 h of constant light were allowed to recover in darkness for 36 h, retinal structure recovered, but the thickness of the outer nuclear layer (ONL) was reduced by ∼20% because of cell death. Light exposure also led to substantial disruption of albino BALB/c retinas and a reduction of ONL thickness (Fig. 1A, middle row). Quantification of ONL thickness across the entire span of the retina is shown in Fig. 1B. The ONL thickness was not statistically different between BALB/c and ARR1−/− retinas except for the 12-h light + 36-h dark time point where the ARR1−/− retinas were slightly thinner.
FIGURE 1.

Light exposure causes rapid photoreceptor cell death in ARR1−/− and BALB/c mice. 1-month-old ARR1−/−, BALB/c, and C57/B6 mice were exposed to 5000-lux white light, pupils undilated, for 12 (12h Light) or 36 h (36h Light). Another group was dark adapted for 36 h after 12 h of light exposure (12h L+36h D). A, retinal morphology of the indicated mice before and after light exposure. Arrows point to pyknotic nuclei. No changes in retinal morphology were detected in C57/B6 mice. Retinal layers are depicted by the rod photoreceptor cell diagram: os, outer segment; is, inner segment; onl, outer nuclear layer. Scale bar, 20 μm. B, morphometric measurements of outer nuclear layer thickness were taken at 20 equal distance positions from superior (S) to inferior (I) poles of the retina. Each point represents mean ± S.D. (n ≥ 3 independent mice) per group. We performed ANOVA for ONL thickness in the central region, which showed significant p values for the 36-h light (p < 0.0009) and 12-h light up to the 36-h dark time points (p < 0.002). These were followed up by pairwise comparisons using t test. For the 36-h light time point, both ARR1−/− and BALB/c were significantly different from C57 (p < 0.006 and p < 0.004, respectively), whereas values for ONL thickness for ARR1−/− and BALB/c were not statistically different. For 12-h light to 36-h dark condition, the p values are as follows: C57 versus ARR1−/−, p < 0.004; C57 versus BALB/c, p < 0.04; and ARR1−/− versus BALB/c, p < 0.02.
Light Exposure Causes Distinct Profiles of Phototransduction Protein Degradation in the ARR1−/− and BALB/c Retinas
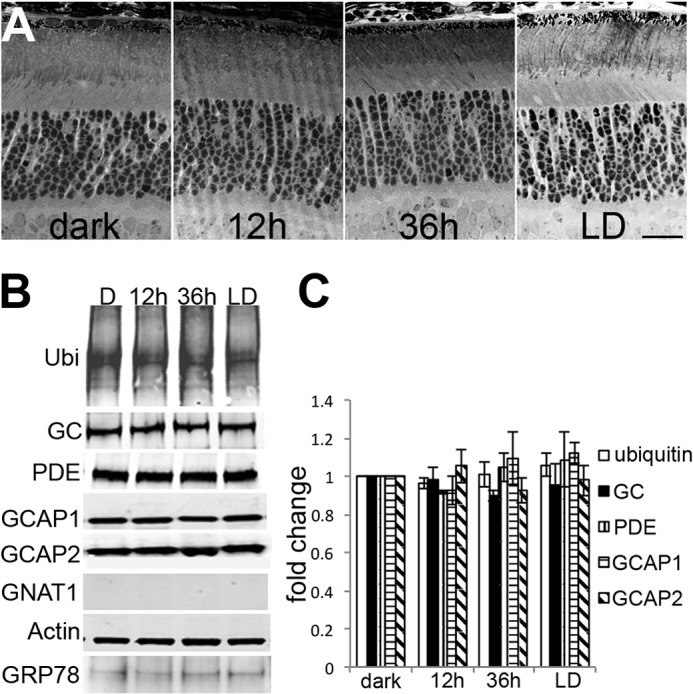
As a first step toward characterizing the initial molecular events downstream of rhodopsin activation, Western blots were performed to compare the levels of phototransduction proteins in the ARR1−/− and BALB/c retinas. In the control C57/B6 samples, light exposure had little effect on the level of guanylyl cyclase 1 (GC), rod PDE6 catalytic αβ subunits (PDEαβ), guanylate cyclase activating protein 1 (GCAP1), or rod transducin (GNAT1). However, the level of GCAP2 was significantly reduced after 12 and 36 h of light exposure (p < 0.03 and p < 0.003, respectively, by two-tailed Student's t test). The GCAPs are Ca2+-binding proteins that inhibit GC activity in the Ca2+-bound state and activate GC in the Ca2+-free state (28, 29). The CNG channels are gated open by high [cGMP] in the dark-adapted state. Ca2+ enters the outer segment through these open channels and is extruded by Na+/Ca2+-K+ exchanger 1 (NCKX1) (30). Closure of CNG channels following light exposure causes a drop in intracellular [Ca2+], which leads to GC stimulation by GCAPs to synthesize cGMP. Ca2+-free GCAP2 appears to be structurally unstable in vitro (31) and perhaps unstable in vivo as well (Fig. 2A; see also below). However, the selective degradation of GCAP2 alone had no apparent effect on retinal morphology in the control C57/B6 retina (Fig. 1). This decrease in GCAP2 was also observed in the ARR1−/− retina after 12 h of light exposure, together with PDE6αβ (Fig. 2B, p < 0.002 and p < 0.003, respectively). No significant changes were observed for GC1, GCAP1, or GNAT1. In the BALB/c retina, the levels of the majority of the proteins assayed were lowered after 12 h of light exposure (Fig. 2C, p < 0.05 for all protein assayed except for GC, which did not reach statistical significance). At the 36-h time points all of the phototransduction protein levels were further reduced in both ARR1−/− and BALB/c retinas, a result consistent with the ongoing cell death occurring at the later time points (Fig. 1).
FIGURE 2.

A–C, distinct degradation profiles of phototransduction proteins in light-exposed C57/B6 (A), ARR1−/− (B), and BALB/c (C) mice. Light exposure conditions were the same as in Fig. 1. The left panels show Western blots of GC1, PDE6αβ, GCAP1, GCAP2, and the α-subunit of rod transducin (GNAT1) in retinal extracts obtained from the indicated mice. Relative fold changes of these proteins normalized to the dark-adapted values, as well as levels of actin is shown in the bar graphs (mean ± S.E., n ≥ 3). D, Western blots of the GTPase-activating protein proteins PDE6γ, Gβ5, and RGS9 (left panel). Both the Gβ5L and the Gβ5S isoforms were recognized by the Gβ5 antibody. The signals were quantified and normalized to the dark-adapted values and actin levels as described above (mean ± S.E., n ≥ 3). E, global increase in ubiquitinated proteins was observed after 12 h of light exposure in ARR1−/− retinas but not in BALB/c retinas. A proteasome pull down of retinal extracts obtained after 12 h of light exposure showed PDE6αβ and GCAP2 to be associated with this protein degradation machinery in the ARR1−/− sample. The BALB/c extract also showed low levels of GCAP2 associated with the proteasome. The bar graph shows the relative fold change of protein ubiquitination levels compared with unexposed control levels in light-exposed ARR1−/− and Blab/C retinas (means ± S.E., n ≥ 3).
PDE activation by Gtα-GTP also sets in motion GTP hydrolysis by the GTPase-activating protein complex consisting of PDEγ, Gβ5L, and RGS9 (for regulator of G-protein signaling). These interactions are increased as a consequence of constitutive transducin activation by photolyzed rhodopsin. We compared the levels of GTPase-activating proteins between the dark basal level and after 12 h of light exposure to see whether increased interaction of these proteins may have caused their destabilization (Fig. 2D). Interestingly, the level of PDEγ was reduced by more than 70% in the light-exposed ARR1−/− retinas, but not Gβ5L or RGS9. In the BALB/c retina, all of the proteins in the GTPase-activating protein complex were decreased (Fig. 2D). Thus a specific down-regulation of all three subunits of PDE6 occurred in the light-exposed ARR1−/− retinas.
We next investigated whether the rapid protein degradation is through the ubiquitin-proteasome system (UPS). Phototransduction proteins are highly abundant proteins expressed by the rod cells. Notably, overload or impairment of the UPS has been implicated in many types of retinal degenerations (32–35). To see whether this is the case, the global ubiquitinated proteins and proteasome-associated proteins were examined by Western blots (Fig. 2E, upper and lower panel, respectively). A 2-fold increase in ubiquitinated proteins was observed in the ARR1−/− retina after 12 h of light exposure. These levels decreased at 36 h despite ongoing degeneration (Fig. 2E, quantification of ubiquitin levels is shown on the bar graph to the right). Thus excessive transducin signaling in the ARR1−/− retina caused many proteins other than GCAP2 and PDE6 to be marked for ubiquitin-mediated degradation. This was not observed in the BALB/c samples at any time points. A proteasome pulldown was carried out and probed with antibodies against the phototransduction proteins (Fig. 2E, lower panel). Of the list of proteins shown in Fig. 2 (A–C), only PDE6 and GCAP2 were associated with the proteasome at the 12-h time point (Fig. 2E, lower panel). These results suggest a specific degradation of PDE6 and GCAP2 through the ubiquitin-proteasome system. For the BALB/c retinas, only a weak signal was observed for GCAP2 (Fig. 2E), but not other proteins. Taken together, the large-scale degradation of the abundant phototransduction proteins PDE6 and GCAP2 in addition to other ubiquitinated proteins may have overloaded the UPS. This pathway was not utilized in the BALB/c retina, although global protein degradation did occur following light exposure.
Light Exposure Causes Aggregation of GCAP2 Protein
Structurally unstable proteins tend to form aggregates in the cell. This possibility was investigated using immunofluorescence microscopy on retinal sections prepared from C57/B6, ARR1−/−, and BALB/c mice to visualize GCAP2 localization (Fig. 3A). GCAP2 immunoreactivity appeared evenly distributed throughout all compartments of the photoreceptor cell layer in the dark-adapted retina from all three lines of mice. In the C57/B6 retina, this pattern was unchanged after 3 h of light exposure. However, after 48 h of light exposure, puncta can be seen to be concentrated at the level of the connecting cilia (arrow, cc). In the ARR1−/− retina, the puncta are visible by 3 h, and their density in the outer segment increased after 12 h of light exposure. The BALB/c retina also displayed GCAP2-positive puncta after light exposure. In both ARR1−/− and BALB/c retinas, the appearance of puncta is correlated with a loss of signal in the ONL (Fig. 3A).
FIGURE 3.

GCAP2 is aggregation prone in the light-exposed retina. A, GCAP2 immunofluorescence (green) is uniformly distributed in the photoreceptor cell layer in retinal sections from dark-adapted C57/B6, ARR1−/−, and BALB/c mice. Following light exposure, brightly staining GCAP2 puncta appeared in the area at the level of the connecting cilium and outer segment layer in the C57/B6 retinal section (48 h, arrow). Puncta appeared earlier in the outer segment layer of ARR1 retinal sections and their number increased with time (3 and 12 h). GCAP2-positive aggregates also appeared in the BALB/c retina after light exposure, although the puncta appeared denser in the inner segment. The location of cell nuclei, visualized by DAPI staining, is shown on the right side of each panel. B, GCAP1 does not form puncta following light exposure. These sections were co-stained with GCAP1 (green), rhodopsin (red), and DAPI (blue). All three signals are shown on the right half of each photomicrograph, whereas the left half shows only GCAP1. In both the ARR1−/− and BALB/c retinas, light damage caused rhodopsin mislocalization to the outer nuclear layer. GCAP1 distribution is unchanged following light exposure in the ARR1−/− retina. Interestingly, GCAP1 (asterisk), as well as rhodopsin-positive vesicles (arrows), appeared in the RPE of BALB/c mice after light damage. os, outer segment; cc, connecting cilium; is, inner segment. Scale bar, 20 μm. C, retinal homogenates from the indicated mice were separated into detergent-soluble (S) and -insoluble fractions (P). The signals were quantified and the P/S ratio is shown in the bar graph (n ≥ 3; error bars, S.E.).
To see whether the light-induced aggregation of GCAP2 is specific to this Ca2+-binding protein, immunofluorescence of GCAP1, a functionally related Ca2+-binding protein, was also performed (Fig. 3B). In the dark-adapted retina of ARR1−/− and BALB/c mice, GCAP1 is evenly distributed in the different compartments of rods and cones, similar to GCAP2. This pattern remained unchanged by light exposure, a result consistent with the notion that formation of GCAP2-positive puncta reflects its tendency to misfold and aggregate under low intracellular Ca2+concentration as a consequence of constitutive phototransduction. Interestingly, GCAP1 immunoreactivity (green) appeared in the retinal pigmented epithelium (RPE) in the BALB/c, but not ARR1−/−, retina after light exposure (Fig. 3B, asterisk). This is more clearly seen in a higher magnification photomicrograph (Fig. 3B, BALB/c, right top panel) that showed diffuse GCAP1 immunoreactivity within the RPE together with rhodopsin-positive (red) vesicles (arrows). Thus different modes of protein degradation were observed: in BALB/c mice, the RPE appears to play an active role, whereas in the ARR1−/− model of light damage, protein degradation appears to occur within rod cells through protein ubiquitination.
Aggregation of GCAP2 was further evaluated by SDS-PAGE immunoblot analysis of detergent-soluble and detergent-insoluble retinal extracts from dark-adapted and light-exposed ARR1−/− and BALB/c mice (Fig. 3C). GCAP1 was carried as a control. Both GCAP2 and GCAP1 were in the detergent-soluble fraction (S) in the dark-adapted retinas from ARR1 and BALB/c mice. After 12 h of light exposure, GCAP2 shifted dramatically to the detergent-insoluble fraction (P) in the ARR1−/−, as well as the BALB/c retina, whereas GCAP1 remained predominantly in the soluble fraction. These results are consistent with the notion that light-induced fall in [Ca2+] destabilized GCAP2 structure, leading to its aggregation and degradation through the proteasomal pathway.
Constitutive Phototransduction Activates Endoplasmic Reticulum (ER) Stress
Overload of the UPS is known to cause ER stress and induce the UPR and vice versa (36). To examine this possibility, markers corresponding to three major arms of the UPR were analyzed by Western blots. These include RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK), inositol-requiring kinase 1 (IRE1α), and activating transcription factor 6 (ATF6α), and glucose regulatory protein 78 (GRP78), which binds and regulates their activity (37). In the ARR1−/− retinas, the levels of GRP78 and proteins downstream from the PERK pathway, such as phosphorylated eukaryotic translation initiation factor 2 α (p-eIF2α), ATF3, and ATF4 were markedly increased at the 12-h time point (Fig. 4, p < 0.05, two-tailed Student's t test) when morphologic changes were minimal (Fig. 1). In the BALB/c retinas, a slight elevation of ATF4 was observed but did not reach statistical significance; no changes were detected in the other proteins (Fig. 4). To further refine the time course of the UPR and to see whether the IRE1α or the ATF6α pathways were induced at an earlier time, ARR1−/− and BALB/c mice were exposed to light for 0.5, 1, 2, 3, 4, or 5 h. As can be seen in Fig. 5, increases in ATF4, p-eIF2α, protein ubiquitination, and ATF3 levels were detected at these early time points (p < 0.001, one-way ANOVA), beginning with ATF4 and p-eIF2α at 0.5 h, followed by protein ubiquitination and ATF3 at 1 and 2 h, respectively. The levels of these proteins continued to increase thereafter. The induction of p-eIF2α and ATF4, which are early responders of the PERK arm of ER stress, prior to protein ubiquitination suggests that ER stress occurred prior to proteasomal stress. No changes were observed for GRP78, ATF6, or IRE1α (p > 0.7, one-way ANOVA). The lack of change in GRP78 levels from 0 to 5 h in the ARR1−/− retinas suggests that the increase seen later at 12 h is likely due to positive feedback from UPR activation. No changes in ATF6 or IRE1α levels indicate a specific induction of the PERK arm of the UPR in the ARR1−/− light damage model. No changes in the three pathways of ER stress were observed in the light-exposed control C57 mice (data not shown) or BALB/c retinas except for a slight but significant elevation in p-eIF2α (p < 0.025, one-way ANOVA).
FIGURE 4.
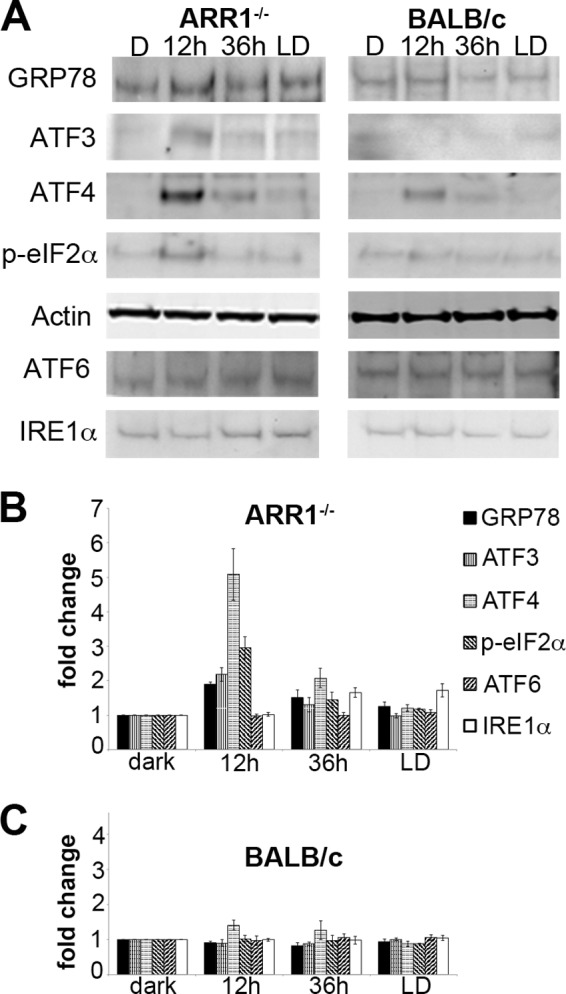
Constitutive transducin activation induces the UPR. A, Western blots of whole retinal homogenates prepared from the indicated mice probed with the indicated antibodies. The signal intensity was quantified. B and C, the relative fold changes of protein levels were normalized to actin and to the dark-adapted values and plotted for ARR1−/− (B) and BALB/c (C) retinas (means ± S.E., n ≥ 3).
FIGURE 5.
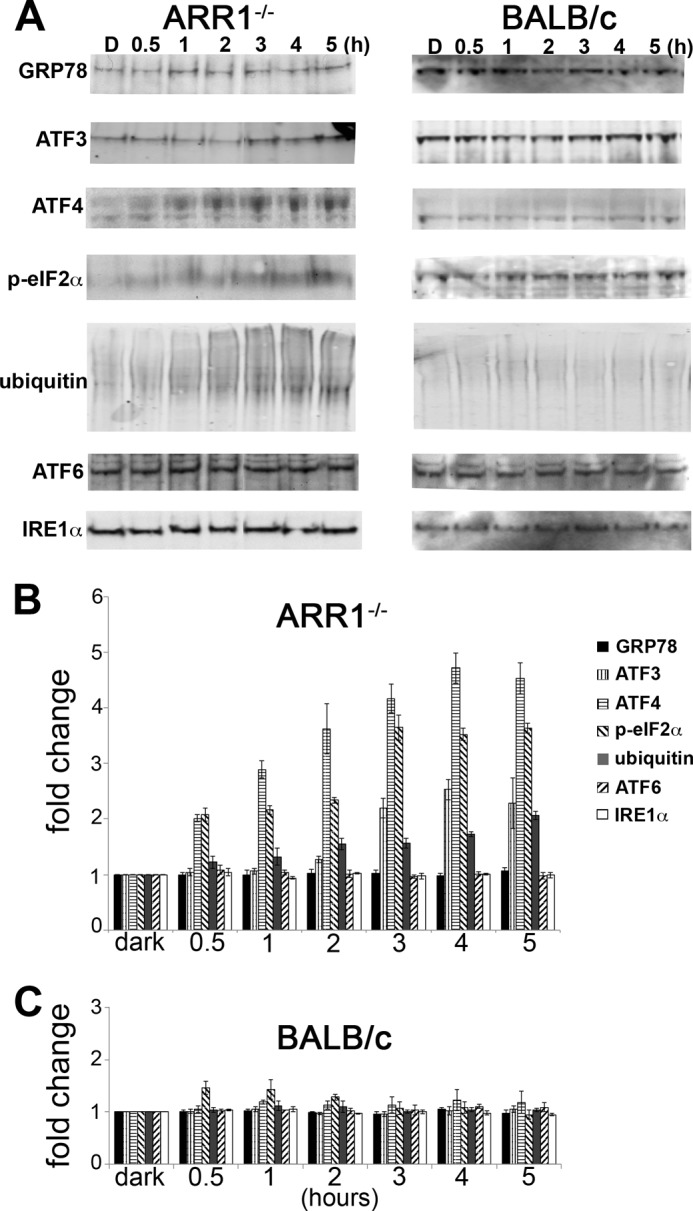
Early time course of UPR activation by constitutive transducin activation in the ARR1−/− retina. A, Western blots were performed on retinal extracts from ARR1−/− and BALB/c mice that were dark-adapted or exposed to light for 0.5, 1, 2, 3, 4, or 5 h. B and C, the signals were quantified and the relative fold changes of protein levels were normalized to actin and to the dark-adapted values and plotted for ARR1−/− (B) and BALB/c (C). The values represent the means ± S.E. (n ≥ 3). Induction of ATF4 and p-elf2α were the first to be detected at 0.5 h, followed by protein ubiquitination and ATF3. No changes were observed for IRE1α or ATF6. Little change in the UPR proteins was observed in the light-exposed BALB/c retinas.
To provide further evidence that induction of ER stress is caused by constitutive phototransduction, mice lacking rhodopsin kinase (GRK1) were exposed to light under the same conditions. The absence of rhodopsin phosphorylation leads to prolonged light responses (16), similar to ARR1−/−. As can be seen in Fig. 6, induction of ER stress by light exposure in the GRK1−/− retinas followed a similar time course as the ARR1−/− mice. At the 12-h time point, a coordinated up-regulation of GRP78, ATF3, ATF4, p-eIFα, and ubiquitinated proteins were observed. This is followed by a decline at longer time points at 36- and 12-h light exposure/36-h dark recovery (Fig. 6). At the 3-, 4-, and 5-h time points, up-regulation of GRP78 has not yet occurred, but robust induction of the PERK arm of the UPR was well underway (Fig. 6). Together, the results from ARR1−/− and GRK1−/− mice strongly implicate a rapid and specific induction of the PERK pathway as an initiating event in photoreceptor cell death caused by constitutive phototransduction.
FIGURE 6.

Constitutive transducin activation caused by the lack of GRK1 leads to induction of ER stress sensors and increase of protein ubiquitination similar to ARR1−/−. A, retinas from dark-adapted mice (D) or mice exposed to 12 or 36 h of constant light or from mice exposed to light for 12 h and dark-adapted for 36 h (LD) were prepared for Western blots and probed with antibodies against GRP78, ATF3, ATF4, p-elf2α, IRE1α, ATF6, and actin. The levels of these proteins after a shorter time course of light exposure were also evaluated (3, 4, and 5 h). B, quantification of the protein levels for the indicated time courses (means ± S.E., n ≥ 3).
To further establish transducin activation as the direct inducer of ER stress, we investigated the effects of Gtα (GNAT1) null background on light-triggered UPR activation in the ARR1−/− retina. No changes in retinal morphology were observed in the ARR1−/−, GNAT−/− double knock-out mice after 12 h or longer time points (Fig. 7A). Absence of G-protein signaling also circumvented up-regulation of protein ubiquitination and degradation of PDE and GCAP2 that occurred in the ARR1−/− retina. The UPR was not induced as evidenced by the lack of changes in the GRP78 levels (Fig. 7, B and C). In sum, these results show that constitutive activation of G-protein signaling rapidly initiates the PERK arm of the UPR and implicates this induction as an early event in the initiation of cell death.
FIGURE 7.
Retinal degeneration and protein degradation in the light-exposed ARR1−/− retina was prevented in the transducin (GNAT1) knock-out background. A, retinal morphology of ARR1−/−GNAT1−/− double knock-out mice was unchanged after light exposure. B, global protein ubiquitination (Ubi) levels, phototransduction, and GRP78 protein levels in ARR1−/−GNAT1−/− retinas did not change after light exposure. D, dark adapted; LD, 12 h light exposed followed by 36 h dark adaptation. C, the relative fold changes of phototransduction protein levels compared with dark-adapted control levels in ARR1−/−GNAT1−/− retinas (means ± S.E., n ≥ 3).
Once activated, GTα-GTP binds the inhibitory γ-subunit of PDE6, which in turn hydrolyzes cGMP, leading to closure of CNG channels. The CNG channel is composed of three α subunits and one β subunit (38). Two previous studies have shown that knock-out of Cngb1 dramatically reduced the level of the α subunit as well as the β subunit, which in turn diminished the current through the CNG channel (39, 40). This condition has also been referred to as equivalent light because it mimics the light-induced closure of the CNG channels (21). To see whether transducin activation or channel closure leads to cell death, we used the same light damage paradigm on a CNG channel knock-out mouse line (Fig. 8) that we generated through disruption of the Cngb1 gene locus that ablated expression of Cngb1 but not GARP, a product from alternative splicing of the same gene (Fig. 8B). In 1-month-old Cngb1 knock-out mice, the outer nuclear layer is largely similar in thickness to that of wild type C57 mice. However, the outer segment structure is shortened and less organized, suggesting a structural role as previously proposed (40). Light exposure had no effect on the retinal structure of Cngb1 knock-out mice (Fig. 8A) nor changed the level of GC, PDE, GCAP1, GCAP2, or GNAT (Fig. 8C). Also similar to C57 mice, expression pattern of UPR markers did not change (Fig. 8D). These results implicate constitutive transducin signaling, rather than channel closure, as the underlying cause of rapid light-induced rod cell death.
FIGURE 8.

Absence of light damage in the Cngb1−/− retina. A, retinal morphology of Cngb1−/− mice before and after the light damage protocol. Scale bar, 20 μm. B, Western blot of whole retinal homogenate prepared from Cngb1−/− and WT mice. Two different concentrations were loaded per sample. The blot was probed with monoclonal antibody 4B1 made against the GARP epitope. C, Western blots of whole retinal homogenates prepared from Cngb1−/− mice following light damage protocol probed for the indicated phototransduction proteins. The signals were quantified and plotted in the bar graph below (means ± S.E., n ≥ 3). No significant differences were detected by ANOVA. D, Western blots of the retinal homogenates probed with the indicated UPR markers. ARR1−/− retinal homogenates from 3-h light-exposed mice were included as positive control (+) for some lanes. The signals were quantified and plotted in the bar graph (means ± S.E., n ≥ 3). No significant differences were detected by ANOVA. D, dark adapted; LD, 12 h light exposed followed by 36 h dark adaptation.
DISCUSSION
Our findings on ARR1−/− and GRK1−/− mice provide a link between constitutive G-protein signaling and photoreceptor cell death. Analysis of the markers of the three arms of UPR indicated a specific and rapid up-regulation of the PERK pathway; by 0.5-h post-light exposure, an increase in the signaling components ATF4 and p-eIF2α was already apparent, followed by protein ubiquitination by 1 h and an increase of ATF3 by 2 h (Fig. 5). That G-protein signaling caused these changes is demonstrated by their complete absence when ARR1−/− was placed into the GNAT−/− background (Fig. 7). Little changes in the UPR stress markers were detected in the BALB/c light damage model, indicating a different mechanism of cell death. These results are consistent with our previous studies on light-induced transcriptional changes using expression arrays, which showed that ATF4 and ATF3 mRNA levels were increased several folds in the ARR1−/−, but not BALB/c retinas, after 3 h of light exposure (42).
Extensive work in different tissues has shown that upon PERK activation, eIF2α is phosphorylated. Phosphorylated eIF2α inhibits general protein translation, thereby reducing the amount of protein synthesis in the ER, and selectively up-regulates ATF4 translation (37). ATF4 is a transcription factor that activates ATF3 expression, as well as other UPR-target genes that act in concert in an attempt to reestablish cellular homeostasis (43). The ARR1−/− light damage results are in complete agreement with this signaling cascade. Similar results obtained from GRK1−/− mice provide further validation to the involvement of PERK activation in this cell death pathway.
Phototransduction terminates at closure of CNG channels. The retinas from Cngb1 knock-out mice mimic this equivalent light condition and were used in the same light damage paradigm to determine whether the cell death signal originates from transducin activation or channel closure. Light exposure had little effect on retinal structure and did not induce UPR in the Cngb1−/− retina. The Cngb1−/− retina exhibits a time-dependent, slow rate of retinal degeneration similar to previously published mouse lines (39, 40). These results suggest that excessive transducin activation, but not channel closure, triggers rapid rod cell death through UPR induction.
The instability of the PDEα and β subunits that we observed in the light-exposed ARR1−/− and GRK1−/− retinas is likely caused by persistent sequestration of the γ subunit by the excessive amount of activated transducin, leading to structural instability of PDEαβ followed by proteasome degradation. Two pieces of evidence support the notion that PDE6γ stabilizes the PDE6αβ subunits. First, genetic ablation of PDE6γ in mice led to a 4-fold reduction in the levels of PDE6αβ subunits and abolition of enzyme activity of the remaining molecules, resulting in cGMP accumulation and a rapid rate of retinal degeneration (44). Second, down-regulation of PDEαβ catalytic subunits was also observed in two different transgenic mouse lines that express either a GTP hydrolysis-deficient human cone transducin (45) or mouse rod transducin (46), suggesting that the mutant transducin, locked in the active conformation, sequestered mouse rod PDE6γ, leading to structural instability and degradation of PDEαβ. The ability of cone transducin to bind rod PDE6γ is supported by our recent study demonstrating that mouse cone transducin activated rod PDE6 with similar efficiency (47). Normal retinal morphology in the GTP hydrolysis-deficient transducin mutant mice indicates that PDE instability alone does not trigger cell death.
Light exposure also lowered GCAP2 levels in C57/B6, ARR1−/−, and BALB/c retinas. Phototransduction lowers intracellular Ca2+, and Ca2+-free GCAP2 appeared to be aggregation-prone in vitro (31). The appearance of GCAP2-positive puncta following light exposure (Fig. 3A) and its partitioning into detergent-insoluble fraction is consistent with the notion that Ca2+-free GCAP2 is structurally unstable in vivo and formed aggregates. Such misfolded proteins become ubiquitinated and is targeted to the proteasome for degradation (Fig. 2E). Because the light-exposed C57/B6 retinas also lowered GCAP2 levels and formed GCAP2-positive puncta in retinal sections, degradation of GCAP2 alone appeared to have a negligible effect on photoreceptor cell survival. However, inasmuch as GCAP stimulation of GC decreases dark-adapted sensitivity (28), this light-induced decrease in GCAP2 concentration may have functional consequences for dark adaption following bright light exposure. In the light-exposed ARR1−/− retina, GCAP2 degradation occurs in conjunction with that of PDE, as well as many other proteins conjugated, to ubiquitin. When combined, they may overload the capacity of the proteasome and contribute to the demise of photoreceptors.
Although light exposure did cause a global decrease in phototransduction proteins in the BALB/c retinas, their degradation was not through the UPS pathway inasmuch as no increase in protein ubiquitination was observed, and UPR was not induced during the early initiation phase of the light damage. Recent studies show that RPE is adversely affected by light exposure (48). We observed the involvement of the RPE in the phagocytosis of GCAP1 and rhodopsin-positive membranes (Fig. 3). Acute induction of phagocytosis of outer segment proteins may contribute to RPE stress. Additionally, protein degradation in BALB/c retinas may occur through calcium-stimulated calpain activity, which in turn activates downstream caspases that are capable of cleaving intracellular proteins (49).
As mentioned previously, phototransduction lowers intracellular Ca2+ in rods. We recently showed that Ca2+-free GCAP2 is retained in the inner segment compartment (50) and may be the basis for the accumulation of GCAP2 in the inner segment of BALB/c retinas, whereas the paucity of GCAP2 aggregates in the ARR1−/− retina at this location may reflect its clearance by UPS. At first look, the low amount of GCAP2 aggregates at the inner segment may seem at odds with UPS activation in the ARR1−/− light damage model. It is noteworthy that, although protein aggregation is a sign of impairment of UPS, this impairment is unlikely to be a consequence of direct choking of proteasomes by protein aggregates. Rather, UPS impairment occurs prior to the coalescence of aggregated proteins, and it has been suggested that sequestration of aggregates may be a protective response (51).
The retinas from the transgenic mice that expressed the GTP hydrolysis-deficient transducin in rods appeared morphologically intact (45, 46), whereas activation of excessive numbers of transducin in retinas from ARR1−/− mice caused retinal degeneration. What could be the basis behind the difference between the two mouse models? It may be of relevance that transducin undergoes rapid GTP-GDP turnover in the ARR1−/− rods, an energy consuming process, whereas the transducin mutant is slow to hydrolyze bound GTP. Also, as mentioned previously, the lack of ARR1 leads to a >400-fold increase in the number of transducin activated per R* (16,400 Gt*/R* versus 34 Gt*/R*). This energy consumption is further amplified by the cycles of cGMP hydrolysis and cGMP synthesis from GTP by the powerful catalytic activities of PDE6 and GC, respectively. Inasmuch as photoreceptors have very a stringent energy requirement (52), we speculate that the increased energy consumption may have caused metabolic stress leading to UPR induction. This hypothesis may appear to contradict the predicted energy consumption in darkness and light in rods, where it was calculated that light exposure reduces ATP consumption by 75% (53). However, that value was derived for light intensities within the operating range of rods. In contrast, the light regime used in our study, compounded with excessive transducin activation in the ARR1−/− rods, are well beyond the operating range of the rod.
In conclusion, our data suggest the following model in the initiation of ER stress by constitutive phototransduction (Fig. 9): greatly amplified cycles of transducin activation and subsequent increased PDE6/GC activity triggered metabolic stress through excessive GTP utilization. This is followed by an increase in global protein ubiquitination that included PDE6 as well as GCAP2. The degradation of these abundant phototransduction proteins together with other ubiquitinated proteins marked for degradation caused proteasome overload, which in turn exacerbated ER stress in a vicious cycle. When the UPR failed to restore homeostasis, apoptosis ensued. In addition to constitutive G-protein signaling, there are naturally occurring rhodopsin mutations in human patients afflicted with autosomal dominant retinitis pigmentosa that have a tendency to misfold, and it has been demonstrated that these mutants also induce the UPR (32, 36, 54). Therefore, strategies to control ER stress, either by overexpression of GRP78 (55) or application of small molecules that modulate UPR (41, 56, 57), may be effective in the treatment of these types of retinal degenerative diseases.
FIGURE 9.
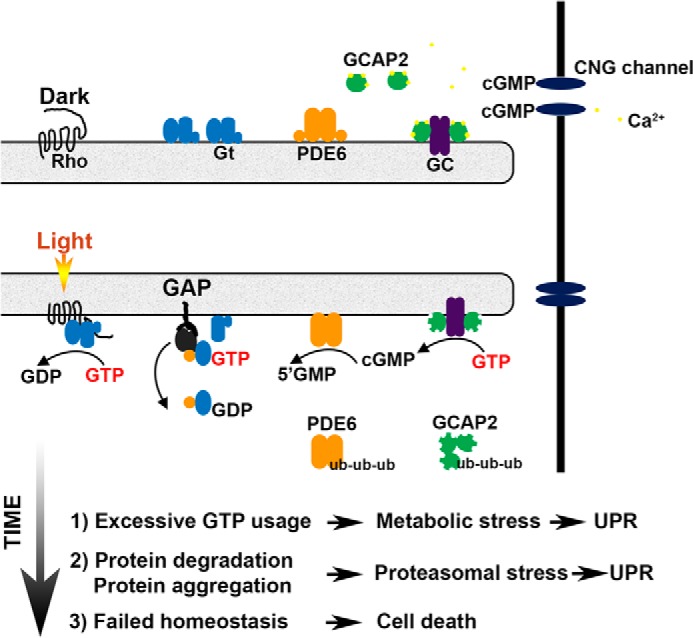
Model of UPR induction by amplified rounds of transducin activation. Defective rhodopsin shutoff in the ARR1−/− and GRK1−/− leads to excessive GTP utilization by transducin and GC/PDE. This in turn leads to metabolic stress, followed by proteasomal stress. When UPR fails to re-establish homeostasis, cell death ensues.
Acknowledgments
We thank Xiao (Helen) He for assistance with maintaining the mouse colony and for technical assistance with the preparation of histological specimen. We thank Dr. Robert Molday for providing the antibody 4B1 that recognizes the GARP domain on Cngb1.
This work was supported, in whole or in part, by National Institutes of Health Grants EY12155 (to J. C.) and EY03040 (to Doheny Eye Institute). This work was also supported by funds from the Beckman Initiative for Macular Research (to J. C.).
- UPR
- unfolded protein response
- ONL
- outer nuclear layer
- GC
- guanylyl cyclase
- UPS
- ubiquitin-proteasome system
- RPE
- retinal pigmented epithelium
- ER
- endoplasmic reticulum
- ANOVA
- analysis of variance
- CNG
- cyclic nucleotide-gated.
REFERENCES
- 1. LaVail M. M., Gorrin G. M., Yasumura D., Matthes M. T. (1999) Increased susceptibility to constant light in nr and pcd mice with inherited retinal degenerations. Invest. Ophthalmol. Vis. Sci. 40, 1020–1024 [PubMed] [Google Scholar]
- 2. Athanasiou D., Aguilà M., Bevilacqua D., Novoselov S. S., Parfitt D. A., Cheetham M. E. (2013) The cell stress machinery and retinal degeneration. FEBS Lett. 587, 2008–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mitchell P., Smith W., Wang J. J. (1998) Iris color, skin sun sensitivity, and age-related maculopathy: the Blue Mountains Eye Study. Ophthalmology 105, 1359–1363 [DOI] [PubMed] [Google Scholar]
- 4. Sui G. Y., Liu G. C., Liu G. Y., Gao Y. Y., Deng Y., Wang W. Y., Tong S. H., Wang L. (2013) Is sunlight exposure a risk factor for age-related macular degeneration? A systematic review and meta-analysis. Br. J. Ophthalmol. 97, 389–394 [DOI] [PubMed] [Google Scholar]
- 5. Noell W. K., Walker V. S., Kang B. S., Berman S. (1966) Retinal damage by light in rats. Invest. Ophthalmol. 5, 450–473 [PubMed] [Google Scholar]
- 6. Youssef P. N., Sheibani N., Albert D. M. (2011) Retinal light toxicity. Eye 25, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grimm C., Wenzel A., Hafezi F., Yu S., Redmond T. M., Remé C. E. (2000) Protection of Rpe65-deficient mice identifies rhodopsin as a mediator of light-induced retinal degeneration. Nat. Genet. 25, 63–66 [DOI] [PubMed] [Google Scholar]
- 8. Hao W., Wenzel A., Obin M. S., Chen C. K., Brill E., Krasnoperova N. V., Eversole-Cire P., Kleyner Y., Taylor A., Simon M. I., Grimm C., Remé C. E., Lem J. (2002) Evidence for two apoptotic pathways in light-induced retinal degeneration. Nat. Genet. 32, 254–260 [DOI] [PubMed] [Google Scholar]
- 9. Donovan M., Carmody R. J., Cotter T. G. (2001) Light-induced photoreceptor apoptosis in vivo requires neuronal nitric-oxide synthase and guanylate cyclase activity and is caspase-3-independent. J. Biol. Chem. 276, 23000–23008 [DOI] [PubMed] [Google Scholar]
- 10. Grimm C., Wenzel A., Hafezi F., Remé C. E. (2000) Gene expression in the mouse retina: the effect of damaging light. Mol. Vis. 6, 252–260 [PubMed] [Google Scholar]
- 11. Hafezi F., Steinbach J. P., Marti A., Munz K., Wang Z. Q., Wagner E. F., Aguzzi A., Remé C. E. (1997) The absence of c-fos prevents light-induced apoptotic cell death of photoreceptors in retinal degeneration in vivo. Nat. Med. 3, 346–349 [DOI] [PubMed] [Google Scholar]
- 12. Wilden U., Kühn H. (1982) Light-dependent phosphorylation of rhodopsin: number of phosphorylation sites. Biochemistry 21, 3014–3022 [DOI] [PubMed] [Google Scholar]
- 13. Wilden U., Hall S. W., Kühn H. (1986) Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc. Natl. Acad. Sci. U.S.A. 83, 1174–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu J., Dodd R. L., Makino C. L., Simon M. I., Baylor D. A., Chen J. (1997) Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature 389, 505–509 [DOI] [PubMed] [Google Scholar]
- 15. Chen J., Simon M. I., Matthes M. T., Yasumura D., LaVail M. M. (1999) Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness). Invest. Ophthalmol. Vis. Sci. 40, 2978–2982 [PubMed] [Google Scholar]
- 16. Chen C. K., Burns M. E., Spencer M., Niemi G. A., Chen J., Hurley J. B., Baylor D. A., Simon M. I. (1999) Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc. Natl. Acad. Sci. U.S.A. 96, 3718–3722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen J., Shi G., Concepcion F. A., Xie G., Oprian D., Chen J. (2006) Stable rhodopsin/arrestin complex leads to retinal degeneration in a transgenic mouse model of autosomal dominant retinitis pigmentosa. J. Neurosci. 26, 11929–11937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moaven H., Koike Y., Jao C. C., Gurevich V. V., Langen R., Chen J. (2013) Visual arrestin interaction with clathrin adaptor AP-2 regulates photoreceptor survival in the vertebrate retina. Proc. Natl. Acad. Sci. U.S.A. 110, 9463–9468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fuchs S., Nakazawa M., Maw M., Tamai M., Oguchi Y., Gal A. (1995) A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat. Genet. 10, 360–362 [DOI] [PubMed] [Google Scholar]
- 20. Yamamoto S., Sippel K. C., Berson E. L., Dryja T. P. (1997) Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat. Genet. 15, 175–178 [DOI] [PubMed] [Google Scholar]
- 21. Lisman J., Fain G. (1995) Support for the equivalent light hypothesis for RP. Nat. Med. 1, 1254–1255 [DOI] [PubMed] [Google Scholar]
- 22. Wenzel A., Reme C. E., Williams T. P., Hafezi F., Grimm C. (2001) The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J. Neurosci. 21, 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rapp L. M., Williams T. P. (1980) The role of ocular pigmentation in protecting against retinal light damage. Vision Res. 20, 1127–1131 [DOI] [PubMed] [Google Scholar]
- 24. Hunter J. J., Morgan J. I., Merigan W. H., Sliney D. H., Sparrow J. R., Williams D. R. (2012) The susceptibility of the retina to photochemical damage from visible light. Prog. Retin. Eye Res. 31, 28–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burns M. E., Pugh E. N., Jr. (2010) Lessons from photoreceptors: turning off G-protein signaling in living cells. Physiology 25, 72–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heck M., Hofmann K. P. (2001) Maximal rate and nucleotide dependence of rhodopsin-catalyzed transducin activation: initial rate analysis based on a double displacement mechanism. J. Biol. Chem. 276, 10000–10009 [DOI] [PubMed] [Google Scholar]
- 27. Concepcion F., Mendez A., Chen J. (2002) The carboxyl-terminal domain is essential for rhodopsin transport in rod photoreceptors. Vision Res. 42, 417–426 [DOI] [PubMed] [Google Scholar]
- 28. Mendez A., Burns M. E., Sokal I., Dizhoor A. M., Baehr W., Palczewski K., Baylor D. A., Chen J. (2001) Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc. Natl. Acad. Sci. U.S.A. 98, 9948–9953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Palczewski K., Sokal I., Baehr W. (2004) Guanylate cyclase-activating proteins: structure, function, and diversity. Biochem. Biophys. Res. Commun. 322, 1123–1130 [DOI] [PubMed] [Google Scholar]
- 30. Schnetkamp P. P. (1986) Sodium-calcium exchange in the outer segments of bovine rod photoreceptors. J. Physiol. 373, 25–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ames J. B., Dizhoor A. M., Ikura M., Palczewski K., Stryer L. (1999) Three-dimensional structure of guanylyl cyclase activating protein-2, a calcium-sensitive modulator of photoreceptor guanylyl cyclases. J. Biol. Chem. 274, 19329–19337 [DOI] [PubMed] [Google Scholar]
- 32. Lin J. H., Walter P., Yen T. S. (2008) Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 3, 399–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shinde V. M., Sizova O. S., Lin J. H., LaVail M. M., Gorbatyuk M. S. (2012) ER stress in retinal degeneration in S334ter Rho rats. PLoS One 7, e33266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Salminen A., Kauppinen A., Hyttinen J. M., Toropainen E., Kaarniranta K. (2010) Endoplasmic reticulum stress in age-related macular degeneration: trigger for neovascularization. Mol. Med. 16, 535–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Choudhury S., Bhootada Y., Gorbatyuk O., Gorbatyuk M. (2013) Caspase-7 ablation modulates UPR, reprograms TRAF2-JNK apoptosis and protects T17M rhodopsin mice from severe retinal degeneration. Cell Death Dis. 4, e528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Griciuc A., Aron L., Ueffing M. (2011) ER stress in retinal degeneration: a target for rational therapy? Trends Mol. Med. 17, 442–451 [DOI] [PubMed] [Google Scholar]
- 37. Pfaffenbach K. T., Lee A. S. (2011) The critical role of GRP78 in physiologic and pathologic stress. Curr. Opin. Cell Biol. 23, 150–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhong H., Molday L. L., Molday R. S., Yau K. W. (2002) The heteromeric cyclic nucleotide-gated channel adopts a 3A:1B stoichiometry. Nature 420, 193–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hüttl S., Michalakis S., Seeliger M., Luo D. G., Acar N., Geiger H., Hudl K., Mader R., Haverkamp S., Moser M., Pfeifer A., Gerstner A., Yau K. W., Biel M. (2005) Impaired channel targeting and retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit CNGB1. J. Neurosci. 25, 130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Y., Molday L. L., Molday R. S., Sarfare S. S., Woodruff M. L., Fain G. L., Kraft T. W., Pittler S. J. (2009) Knockout of GARPs and the β-subunit of the rod cGMP-gated channel disrupts disk morphogenesis and rod outer segment structural integrity. J. Cell Sci. 122, 1192–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mockel A., Obringer C., Hakvoort T. B., Seeliger M., Lamers W. H., Stoetzel C., Dollfus H., Marion V. (2012) Pharmacological modulation of the retinal unfolded protein response in Bardet-Biedl syndrome reduces apoptosis and preserves light detection ability. J. Biol. Chem. 287, 37483–37494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roca A., Shin K. J., Liu X., Simon M. I., Chen J. (2004) Comparative analysis of transcriptional profiles between two apoptotic pathways of light-induced retinal degeneration. Neuroscience 129, 779–790 [DOI] [PubMed] [Google Scholar]
- 43. Rzymski T., Milani M., Singleton D. C., Harris A. L. (2009) Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 8, 3838–3847 [DOI] [PubMed] [Google Scholar]
- 44. Tsang S. H., Gouras P., Yamashita C. K., Kjeldbye H., Fisher J., Farber D. B., Goff S. P. (1996) Retinal degeneration in mice lacking the gamma subunit of the rod cGMP phosphodiesterase. Science 272, 1026–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Raport C. J., Lem J., Makino C., Chen C. K., Fitch C. L., Hobson A., Baylor D., Simon M. I., Hurley J. B. (1994) Downregulation of cGMP phosphodiesterase induced by expression of GTPase-deficient cone transducin in mouse rod photoreceptors. Invest. Ophthalmol. Vis. Sci. 35, 2932–2947 [PubMed] [Google Scholar]
- 46. Kerov V., Chen D., Moussaif M., Chen Y. J., Chen C. K., Artemyev N. O. (2005) Transducin activation state controls its light-dependent translocation in rod photoreceptors. J. Biol. Chem. 280, 41069–41076 [DOI] [PubMed] [Google Scholar]
- 47. Mao W., Miyagishima K. J., Yao Y., Soreghan B., Sampath A. P., Chen J. (2013) Functional comparison of rod and cone Gαt on the regulation of light sensitivity. J. Biol. Chem. 288, 5257–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cachafeiro M., Bemelmans A. P., Samardzija M., Afanasieva T., Pournaras J. A., Grimm C., Kostic C., Philippe S., Wenzel A., Arsenijevic Y. (2013) Hyperactivation of retina by light in mice leads to photoreceptor cell death mediated by VEGF and retinal pigment epithelium permeability. Cell Death Dis. 4, e781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wenzel A., Grimm C., Samardzija M., Remé C. E. (2005) Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog. Retin. Eye Res. 24, 275–306 [DOI] [PubMed] [Google Scholar]
- 50. Hoyo N. L., López-Begines S., Rosa J. L., Chen J., Méndez A. (2014) Functional EF-hands in neuronal calcium sensor GCAP2 determine its phosphorylation state and subcellular distribution in vivo, and are essential for photoreceptor cell integrity. PLoS Genet. 10, e1004480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bennett E. J., Bence N. F., Jayakumar R., Kopito R. R. (2005) Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol. Cell 17, 351–365 [DOI] [PubMed] [Google Scholar]
- 52. Chertov A. O., Holzhausen L., Kuok I. T., Couron D., Parker E., Linton J. D., Sadilek M., Sweet I. R., Hurley J. B. (2011) Roles of glucose in photoreceptor survival. J. Biol. Chem. 286, 34700–34711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Okawa H., Sampath A. P., Laughlin S. B., Fain G. L. (2008) ATP consumption by mammalian rod photoreceptors in darkness and in light. Curr. Biol. 18, 1917–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kunte M. M., Choudhury S., Manheim J. F., Shinde V. M., Miura M., Chiodo V. A., Hauswirth W. W., Gorbatyuk O. S., Gorbatyuk M. S. (2012) ER stress is involved in T17M rhodopsin-induced retinal degeneration. Invest. Ophthalmol. Vis. Sci. 53, 3792–3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gorbatyuk M. S., Knox T., LaVail M. M., Gorbatyuk O. S., Noorwez S. M., Hauswirth W. W., Lin J. H., Muzyczka N., Lewin A. S. (2010) Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. U.S.A. 107, 5961–5966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kudo T., Kanemoto S., Hara H., Morimoto N., Morihara T., Kimura R., Tabira T., Imaizumi K., Takeda M. (2008) A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 15, 364–375 [DOI] [PubMed] [Google Scholar]
- 57. Tsaytler P., Harding H. P., Ron D., Bertolotti A. (2011) Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332, 91–94 [DOI] [PubMed] [Google Scholar]