

Abstract
Investigation of the human microbiome has become an important field of research facilitated by advances in sequencing technologies. The lung, which is one of the latest body sites being explored for the characterization of human-associated microbial communities, has a microbiome that is suspected to play a substantial role in health and disease. In this review, we provide an overview of the basics of microbiome studies. Challenges in the study of the lung microbiome are highlighted, and further attention is called to the optimization and standardization of methodologies to explore the role of the lung microbiome in health and disease. We also provide examples of lung microbial communities associated with disease or infection status and discuss the role of fungal species in the lung. Finally, we review studies demonstrating that the environmental microbiome can influence lung health and disease, such as the finding that the diversity of microbial exposure correlates inversely with the development of childhood asthma.
Keywords: lung, microbiome, chronic obstructive pulmonary disease, asthma, HIV/AIDS
The microbiome refers to the community of microbes that share a particular environment, and its study has become an important field of research in recent years. With the development of second- and third-generation sequencing technologies (commonly referred to as next-generation sequencing), it is now feasible to examine the composition of complex microbial populations, including unculturable microbes. The Human Microbiome Project (HMP) was initiated with the sequencing of more than 11,000 primary specimens from 18 body sites in 300 healthy individuals (1). These microbial communities are suspected to affect human physiology and immunity in different ways (2). For example, the gut microbiome was shown to be a contributing factor in obesity, with the balance tipped toward the phylum Bacteroidetes in lean individuals and the Firmicutes in obese subjects (3, 4).
Investigation of the lung microbiome is an emerging field, and its findings are altering the way we think about respiratory disease. The lung—not selected as one of the initial HMP body sites—was originally believed to be sterile, based on bacterial culture studies that failed to show bacterial growth (5). However, new evidence suggests that the lung has a microbiome that varies in health and disease (6, 7). Furthermore, bacteria, fungi, and viruses and their interactions may all be important in lung health and in the development of respiratory disease. In addition to the localized lung microbiome, daily interactions with microbial populations found in the environment add another layer of complexity, and the environmental microbiome has now been shown to be associated with the development of asthma and other allergic respiratory disorders (8). We review the basic approaches used in lung microbiome studies and discuss their limitations, explore the relationships of bacteria and fungi to respiratory diseases, and examine the role of the environment in shaping the microbiome with its effect on lung disease.
Lung Microbiome Basics, Challenges in the Field, and the Microbiome in Health and Disease
Terminology
An understanding of basic definitions used in the microbiome field is important for interpretation of research results, but exact definitions of terms are often debated in the literature. In general, a microbe is any microscopic life form and can include bacteria, fungi, protozoa, or viruses; the microbiota refers to the microbes in a given population; and the microbiome is the microbiota found in a particular habitat at a specific time. Although “microbiome” and “microbiota” are often used interchangeably, they refer to different ecological principles. The “microbial community” within a body location consists of microbes that interact functionally and metabolically. Characterization of the microbiome can be done by sequencing regions of a conserved gene, such as the hypervariable regions of the 16S ribosomal RNA gene (Figure 1) (9). A common classification used for the sequences obtained from a microbial population is the operational taxonomic unit (OTU), which is often used as a synonym for taxon. Typically it comprises sequences that are clustered based on a percent similarity threshold (often set at 97% or above); this serves as a proxy for species-level taxonomic assignment (10). Using the OTUs, microbial communities can be described with various metrics, including richness, evenness, dominance, and the diversity index (Table 1) (11). The terms abundance and ubiquity are both used to describe the diversity within a microbiome, but the latter refers particularly to the prevalence level of an OTU in the microbial community characterized (12).
Figure 1.

16S ribosomal DNA hypervariable regions. Nine hypervariable regions (V1–V9) were labeled on the secondary structure of 16 rDNA. Among them, both V1–V3 and V3–V5 regions are commonly used to characterize the bacterial microbiome. Reprinted by permission from Reference 42.
Table 1.
Metrics used in describing the microbiome of a population
| Diversity Metric | Characteristic |
|---|---|
| Richness | Number of OTUs |
| Evenness | Relative abundance of different OTUs |
| Dominance | Emergence of a single OTU |
| Diversity index | Calculated index of complexity based on richness and evenness |
Definition of abbreviation: OTU = operational taxonomic unit.
Challenges in the Field
Multiple methodological issues are encountered when studying the lung microbiome; some are common to the microbiome field in general, whereas others are related specifically to lung studies. For example, primer selection is a considerable topic of discussion in all microbiome studies. Among the nine hypervariable regions (V1–V9) in the 16S rRNA gene, both the V1–V3 and V3–V5 regions are commonly used to characterize bacterial diversity, but these two regions occasionally give inconsistent and differing profiles (Figure 1) (9). Analogous to the amplification and sequencing of the 16S rRNA regions to characterize the bacterial microbiota, sequencing of fungal species relies on conserved target genes such as the 18S or the internal transcribed spacer of the rRNA gene (13). The choice of target locus is determined in part by ease of amplification and by the specificity required. For example, the 18S is more efficiently amplified and is better conserved across species, making it a good choice for phylogenetic analyses; the internal transcribed spacer, on the other hand, has better genus-level discrimination because it is highly diverse across species (13). Thus, findings can be influenced by the gene targeted, a fact that should be kept in mind when comparing results across studies.
In addition to the selection of the target gene region, the issue of reagent contamination in samples is common to all microbiome studies but is of particular importance in the lung, where the biomass of some lung sample types, such as bronchoalveolar lavage (BAL), tends to be low as compared with other body habitats, such as the gut or even the skin. Indeed, although samples should always be collected carefully using sterile reagents and tools, it is challenging to prepare 100% contaminant-free DNA, considering the high sensitivity of polymerase chain reaction (PCR) and next-generation sequencing techniques (14). Studies use different methods to attempt to account for contamination, but no one approach is widely accepted. Lung studies also face the unique issue of contamination from the oral cavity when passing the bronchoscope through the mouth. Potential methods to address oral contamination include use of two bronchoscopes and use of statistical methods such as the neutral model to select organisms enriched in the lung environment (15, 16). Possible contamination from reagents and equipment should also be included as systematic controls in the analysis.
Several other methods used in DNA isolation and amplification also vary across projects and may influence results. First, the specimen type sampled may influence findings. For example, use of bronchial washes, airway brushes, or lung tissue can give different results. In addition, the depth of sequencing can affect readouts about community composition. For example, if the number of reads per sample is too limited, it may lead to a skewed understanding of the lung microbiome because of undersampling. Finally, enrichment or purification of the target organism may also affect the yield of sequencing. In the case of the virome, for example, a failure to exclude host cell or bacterial nucleic acids may largely decrease the sequencing depth of viral genomes. A possible way to enrich for viral-like particles is to perform density gradient ultracentrifugation and nuclease digestion, but these become costly efforts that often do not efficiently exclude all human and bacterial DNA (17).
HIV-infected Populations
The Lung HIV Microbiome Project (LHMP) is currently the largest ongoing study on the lung microbiome (https://lhmp.bsc.gwu.edu/). The LHMP is funded by the National Heart, Lung, and Blood Institute to investigate the respiratory tract microbiome in HIV-infected and -uninfected individuals with and without underlying lung diseases, such as chronic obstructive pulmonary disease (COPD) and pulmonary arterial hypertension. Recent progress in this project includes the finding that, based on BAL samples, there is increased colonization of the lungs with Tropheryma whipplei in HIV-infected individuals as compared with the HIV-uninfected population and that antiretroviral therapy resulted in a significant decline in relative abundance of T. whipplei (18).
HIV-uninfected Populations
The composition of the “normal” bacterial microbiome in the lung in HIV-uninfected persons without underlying lung disease has been investigated in a small number of studies. Charlson and colleagues, using a two-bronchoscope technique in six subjects, found that the BAL microbiome primarily reflected the oral microbiome, potentially from contamination during bronchoscopy, although there could also be ongoing microaspiration that could result in intermittent detection of bacteria in the lung (16). Lung-specific OTUs were infrequent, and each identified OTU appeared to be unique to a single individual in this small study. The LHMP examined 64 participants and found that, although oral wash and BAL samples shared similar microbial communities as observed for the Charlson study, certain species were enriched in either the mouth or the lung (15). The lung microbiome was also described by Erb-Downward and colleagues in a study of 14 subjects (including 10 “healthy” smokers) sampling BAL and multiple tissue sites in explanted lung lobes. This study found genera such as Pseudomonas, Streptococcus, Prevotella, Fusobacterium, Haemophilus, Veillonella, and Porphyromonas as part of the core organisms in the normal lung (19). A high heterogeneity of different microhabitats in various areas of the lung was also revealed in this study. For example, although Haemophilus dominated in the left upper lobe of one subject, there was minimal detection in all other microanatomic areas of the lung lobe.
The lung is particularly susceptible to outside influences such as smoking, but the effect of smoking on the lung microbiome is not entirely clear. Morris and LHMP colleagues found that smoking altered the oral microbiome but did not significantly affect the lung microbiome (BAL samples) of “healthy” smokers, as compared with nonsmokers (15). The Erb-Downward study also suggests that there are no significant differences in the lung microbiome (BAL samples) based on smoking status (19).
Persons with Underlying Lung Disease
Pulmonary diseases, such as cystic fibrosis (CF) and COPD, may be associated with perturbations of the lung microbiome. In a study of serial sputum samples from six patients with CF, the diversity of the lung microbiota was found to decrease significantly after episodic antibiotic treatment for exacerbation. The microbiota then regained its original level of diversity during the convalescence stage when the patients were recovering from CF and, in some cases, were treated with maintenance antibiotics (20). In a study of lungs removed from patients with CF during transplantation, important differences were detected between the upper-airway specimens (throat swabs and sputum) and lung specimens (airway lavage), suggesting that samples from the upper respiratory tract are not representative of the lung biota (21). In addition to CF, COPD also appears to be associated with lung microbiome changes, demonstrated in a study on lung tissue samples showing that the genus Lactobacillus increases significantly in patients with severe COPD (22).
The Fungal Microbiome (Mycobiome) and Lung Disease
The majority of the research effort on the microbiome has focused on bacterial species, but, as mentioned earlier, other organisms are also likely to play an important role (13). Fungal communities in an environment—termed the mycobiome—are increasingly of interest, but relatively few studies have been published to date. Certain fungal species have been linked to human disease. Invasive fungal infections are increasing, particularly in the growing immunocompromised population and with widespread antibiotic usage (23). In addition to their direct effects on the host by shaping the immune response both locally and remotely, which could impact disease (Figure 2), fungi may be important because of their interactions with bacterial or viral biomes (23). Although fungal species are numerous (an estimated 1.5 million), only 5% of them have been formally classified, because the large majority cannot be cultured, making application of next-generation sequencing particularly useful (24).
Figure 2.
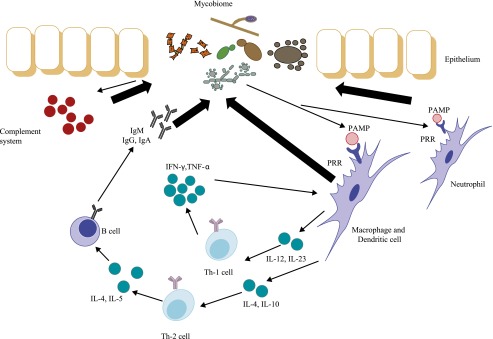
Interaction between the mycobiome and the immune system. The mycobiome shapes the immune response by the recognition between pathogen-associated molecular patterns (PAMPs) and pattern recognition receptors (PRRs) and the stimulation of the complement system and immune cell maturation. IFN = interferon; Ig = immunoglobulin; IL = interleukin; Th = T helper; TNF = tumor necrosis factor. Reprinted by permission from Reference 13.
Few studies of the respiratory mycobiome have been conducted to date. Ghannoum and colleagues characterized the core oral mycobiome from oral rinse samples in 20 healthy individuals (25). They detected 74 culturable and 11 nonculturable fungal genera, with Candida as the dominant genus (25). In a study of 21 lung transplant recipients and 6 healthy control subjects, transplant recipients showed a significantly decreased fungal richness in both oral wash and BAL (most of the recipients were receiving antibacterial and/or antifungal treatment medications at the time of sampling) (26). BAL in healthy control subjects had few sequence reads corresponding to fungal species, but Candida, Aspergillus, and Cryptococcus were found in transplant BALs (Figure 3). Another study of eight sputum samples from four patients with CF found a lower diversity and richness of bacterial and fungal microbiomes associated with worse clinical status, with a cooccurrence of Candida and Pseudomonas (27).
Figure 3.

Relative abundance of fungal taxa in the respiratory mycobiome after lung transplant. Oropharyngeal washes and bronchoalveolar lavages were collected from each patient. Pyrosequencing data of the internal transcribed spacer (ITS) were clustered into operational taxonomic units, which were then assigned to taxonomic classes. The colors represent the relative abundance of the fungal taxon within each sample. The top row is the concentration of ITS DNA post–polymerase chain reaction for each sample. Reprinted by permission from Reference 26.
The fungus Pneumocystis jirovecii has been studied in association with lung disease, particularly COPD (28). Pneumocystis is an extracellular organism associated with Type I pneumocytes and cannot be cultured in the lab (29). Before the availability of next-generation sequencing, studies of Pneumocystis and lung disease were generally done using targeted nested PCR. Colonization with P. jirovecii (defined as detection by nested PCR without signs of Pneumocystis pneumonia) is increased in HIV-infected individuals and associated with airway obstruction independent of smoking history (30). In a nonhuman primate model of HIV, airway obstruction and airspace enlargement increased during coinfection with Pneumocystis (31). Similarly, greatest airspace enlargement occurred in mice with both Pneumocystis colonization and smoke exposure, suggesting an interaction of these factors in causing COPD-like changes (32).
Environmental Microbiome in Lung Health
In addition to the lung microbiome, the environmental microbiome also influences lung health and disease. The hygiene hypothesis, proposed years ago, hypothesized that individuals with a higher number of older siblings and thus living under “less hygienic” conditions have a lower risk of developing respiratory allergies (33). A more comprehensive look at this topic from a microbiome perspective was performed in recent years, exploring the effect of environmental microbes in causing current asthma epidemics (34).
Several epidemiological studies were launched to find the correlation between microbial exposure and asthma prevention. Comparing Finnish Karelians to Russian Karelians, who have a much lower prevalence of asthma, the house dust in the Russian homes showed higher amounts of bacteria and dominance of gram-positive bacteria (35). In examining several microbial markers, including muramic acid, endotoxin, and ergosterol, an inverse dose-dependent correlation was found between the atopy risk and the index of microbial components in dust in Finnish Karelian households (34). In an add-on study to the GABRIEL survey (a study including genetics, epidemiology, and immunology to identify key factors in the development of asthma in the European community [36]), a significantly lower prevalence of asthma, hay fever, and atopy was found in Amish and Swiss children living on farms compared with children living in more urban areas (37).
Maternal farm exposure during pregnancy is believed to shape the immune system of the offspring, either by increasing regulatory T-cell numbers and enhancing their function in infants (38) or by increasing the levels of cord blood proinflammatory cytokines, such as IFN-γ and tumor necrosis factor-α (39). The diversity of microbial exposure is inversely related to asthma development (8). Several nonpathogenic bacteria have been reported as candidates. In a mouse model of asthma, an intranasal application of Lactococcus lactis and Acinetobacter lwoffii successfully reversed airway hyperresponsiveness (40), and maternal exposure to Acinetobacter lwoffii also protected offspring from asthma (41).
Conclusions
The bacterial, fungal, and viral lung microbiomes as well as environmental microbiomes may all be important in lung health and disease. Investigations are starting to define the role of the microbiome in lung health and disease, but much remains to be discovered. Future challenges for the field include standardization of collection and sequencing methods, accounting for contamination, and developing standards for consistent presentation of data and analyses. The field would particularly benefit by expanding studies into the analysis of large and well-characterized cohorts, geographic and environmental diversity in sample collection, comparison of upper and lower airways, examination of longitudinal samples, and association of the lung microbiome with clinical outcomes.
Footnotes
Supported by National Heart, Lung, and Blood Institute/National Institutes of Health grants U01HL098962 (A.M., E.G.), K24HL087713 and U01HL098964 (L.H.), U01HL98961 (J.M.B.), and U01HL098960 (H.L.T.); the project HERA (Host–environment interactions in the protection from asthma and allergies), grant agreement number: 250268, European Research Council (E.v.M.); and the Chinese Scholarship Council (L.C.).
The views expressed in this article do not communicate an official position of the University of Pittsburgh or of the funding agency (National Heart, Lung, and Blood Institute/National Institutes of Health).
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Proctor LM. The human microbiome project in 2011 and beyond. Cell Host Microbe. 2011;10:287–291. doi: 10.1016/j.chom.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Mitreva M. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 4.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 5.Beck JM, Young VB, Huffnagle GB. The microbiome of the lung. Transl Res. 2012;160:258–266. doi: 10.1016/j.trsl.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang YJ, Charlson ES, Collman RG, Colombini-Hatch S, Martinez FD, Senior RM. The role of the lung microbiome in health and disease. A National Heart, Lung, and Blood Institute workshop report. Am J Respir Crit Care Med. 2013;187:1382–1387. doi: 10.1164/rccm.201303-0488WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiley JP. Advancing respiratory research. Chest. 2011;140:497–501. doi: 10.1378/chest.11-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ege MJ, Mayer M, Normand A-C, Genuneit J, Cookson WO, Braun-Fahrländer C, Heederik D, Piarroux R, von Mutius E. Exposure to environmental microorganisms and childhood asthma. N Engl J Med. 2011;364:701–709. doi: 10.1056/NEJMoa1007302. [DOI] [PubMed] [Google Scholar]
- 9.Huse SM, Ye Y, Zhou Y, Fodor AA. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS ONE. 2012;7:e34242. doi: 10.1371/journal.pone.0034242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wooley JC, Godzik A, Friedberg I. A primer on metagenomics. PLOS Comput Biol. 2010;6:e1000667. doi: 10.1371/journal.pcbi.1000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang YJ, Lynch SV. The emerging relationship between the airway microbiota and chronic respiratory disease: clinical implications. Expert Rev Respir Med. 2011;5:809–821. doi: 10.1586/ers.11.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li K, Bihan M, Methé BA. Analyses of the stability and core taxonomic memberships of the human microbiome. PLoS ONE. 2013;8:e63139. doi: 10.1371/journal.pone.0063139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui L, Morris A, Ghedin E. The human mycobiome in health and disease. Genome Med. 2013;5:63. doi: 10.1186/gm467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, Bushman FD, Knight R, Kelley ST. Bayesian community-wide culture-independent microbial source tracking. Nat Methods. 2011;8:761–763. doi: 10.1038/nmeth.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, Flores SC, Fontenot AP, Ghedin E, Huang L. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187:1067–1075. doi: 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delwart E. A roadmap to the human virome. PLoS Pathog. 2013;9:e1003146. doi: 10.1371/journal.ppat.1003146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lozupone C, Cota-Gomez A, Palmer BE, Linderman DJ, Charlson ES, Sodergren E, Mitreva M, Abubucker S, Martin J, Yao G. Widespread colonization of the lung by Tropheryma whipplei in HIV infection. Am J Respir Crit Care Med. 2013;187:1110–1117. doi: 10.1164/rccm.201211-2145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, Young VB, Toews GB, Curtis JL, Sundaram B. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS ONE. 2011;6:e16384. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao J, Schloss PD, Kalikin LM, Carmody LA, Foster BK, Petrosino JF, Cavalcoli JD, VanDevanter DR, Murray S, Li JZ. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci USA. 2012;109:5809–5814. doi: 10.1073/pnas.1120577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goddard AF, Staudinger BJ, Dowd SE, Joshi-Datar A, Wolcott RD, Aitken ML, Fligner CL, Singh PK. Direct sampling of cystic fibrosis lungs indicates that DNA-based analyses of upper-airway specimens can misrepresent lung microbiota. Proc Natl Acad Sci USA. 2012;109:13769–13774. doi: 10.1073/pnas.1107435109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sze MA, Dimitriu PA, Hayashi S, Elliott WM, McDonough JE, Gosselink JV, Cooper J, Sin DD, Mohn WW, Hogg JC. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185:1073–1080. doi: 10.1164/rccm.201111-2075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iliev ID, Underhill DM. Striking a balance: fungal commensalism versus pathogenesis. Curr Opin Microbiol. 2013;16:366–373. doi: 10.1016/j.mib.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawksworth DL. The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol Res. 2001;105:1422–1432. [Google Scholar]
- 25.Ghannoum MA, Jurevic RJ, Mukherjee PK, Cui F, Sikaroodi M, Naqvi A, Gillevet PM. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 2010;6:e1000713. doi: 10.1371/journal.ppat.1000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charlson ES, Diamond JM, Bittinger K, Fitzgerald AS, Yadav A, Haas AR, Bushman FD, Collman RG. Lung-enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. Am J Respir Crit Care Med. 2012;186:536–545. doi: 10.1164/rccm.201204-0693OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delhaes L, Monchy S, Fréalle E, Hubans C, Salleron J, Leroy S, Prevotat A, Wallet F, Wallaert B, Dei-Cas E. The airway microbiota in cystic fibrosis: a complex fungal and bacterial community—implications for therapeutic management. PLoS ONE. 2012;7:e36313. doi: 10.1371/journal.pone.0036313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359:2355–2365. doi: 10.1056/NEJMra0800353. [DOI] [PubMed] [Google Scholar]
- 29.Limper AH, Thomas CF, Anders RA, Leof EB. Interactions of parasite and host epithelial cell cycle regulation during Pneumocystis carinii pneumonia. J Lab Clin Med. 1997;130:132–138. doi: 10.1016/s0022-2143(97)90089-5. [DOI] [PubMed] [Google Scholar]
- 30.Morris A, Alexander T, Radhi S, Lucht L, Sciurba FC, Kolls JK, Srivastava R, Steele C, Norris KA. Airway obstruction is increased in Pneumocystis-colonized human immunodeficiency virus-infected outpatients. J Clin Microbiol. 2009;47:3773–3776. doi: 10.1128/JCM.01712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shipley TW, Kling HM, Morris A, Patil S, Kristoff J, Guyach SE, Murphy JE, Shao X, Sciurba FC, Rogers RM. Persistent Pneumocystis colonization leads to the development of chronic obstructive pulmonary disease in a nonhuman primate model of AIDS. J Infect Dis. 2010;202:302–312. doi: 10.1086/653485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Christensen PJ, Preston AM, Ling T, Du M, Fields WB, Curtis JL, Beck JM. Pneumocystis murina infection and cigarette smoke exposure interact to cause increased organism burden, development of airspace enlargement, and pulmonary inflammation in mice. Infect Immun. 2008;76:3481–3490. doi: 10.1128/IAI.00165-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.von Hertzen L, Hyvärinen A, Laatikainen T, Mäkelä MJ, Nevalainen A, Vartiainen E, Haahtela T. Risk of atopy associated with microbial components in house dust. Ann Allergy Asthma Immunol. 2010;104:269–270. doi: 10.1016/j.anai.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 35.Pakarinen J, Hyvärinen A, Salkinoja‐Salonen M, Laitinen S, Nevalainen A, Mäkelä MJ, Haahtela T, Von Hertzen L. Predominance of Gram‐positive bacteria in house dust in the low‐allergy risk Russian Karelia. Environ Microbiol. 2008;10:3317–3325. doi: 10.1111/j.1462-2920.2008.01723.x. [DOI] [PubMed] [Google Scholar]
- 36.Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, Von Mutius E, Farrall M, Lathrop M, Cookson WO. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holbreich M, Genuneit J, Weber J, Braun-Fahrländer C, Waser M, von Mutius E. Amish children living in northern Indiana have a very low prevalence of allergic sensitization. J Allergy Clin Immunol. 2012;129:1671–1673. doi: 10.1016/j.jaci.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 38.Schaub B, Liu J, Höppler S, Schleich I, Huehn J, Olek S, Wieczorek G, Illi S, von Mutius E. Maternal farm exposure modulates neonatal immune mechanisms through regulatory T cells. J Allergy Clin Immunol. 2009;123:774–782. doi: 10.1016/j.jaci.2009.01.056. [DOI] [PubMed] [Google Scholar]
- 39.Pfefferle PI, Büchele G, Blümer N, Roponen M, Ege MJ, Krauss-Etschmann S, Genuneit J, Hyvärinen A, Hirvonen M-R, Lauener R. Cord blood cytokines are modulated by maternal farming activities and consumption of farm dairy products during pregnancy: the pasture study. J Allergy Clin Immunol. 2010;125:108–115. doi: 10.1016/j.jaci.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 40.Debarry J, Garn H, Hanuszkiewicz A, Dickgreber N, Blümer N, von Mutius E, Bufe A, Gatermann S, Renz H, Holst O. Acinetobacter lwoffii and Lactococcus lactis strains isolated from farm cowsheds possess strong allergy-protective properties. J Allergy Clin Immunol. 2007;119:1514–1521. doi: 10.1016/j.jaci.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 41.Conrad ML, Ferstl R, Teich R, Brand S, Blümer N, Yildirim AÖ, Patrascan CC, Hanuszkiewicz A, Akira S, Wagner H. Maternal TLR signaling is required for prenatal asthma protection by the nonpathogenic microbe Acinetobacter lwoffii f78. J Exp Med. 2009;206:2869–2877. doi: 10.1084/jem.20090845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beck JM. ABCs of the lung microbiome. Ann Am Thorac Soc. 2014;11:S1–S3. doi: 10.1513/AnnalsATS.201306-188MG. [DOI] [PMC free article] [PubMed] [Google Scholar]