

Abstract
Heterochromatin protein 1 (HP1) is a conserved nonhistone chromosomal protein with functions in euchromatin and heterochromatin. Here we investigated the diffusional behaviors of HP1 isoforms in mammalian cells. Using fluorescence correlation spectroscopy (FCS) and fluorescence recovery after photobleaching (FRAP) we found that in interphase cells most HP1 molecules (50–80%) are highly mobile (recovery halftime: t1/2 ≈ 0.9 s; diffusion coefficient: D ≈ 0.6–0.7 μm2 s-1). Twenty to 40% of HP1 molecules appear to be incorporated into stable, slow-moving oligomeric complexes (t1/2 ≈ 10 s), and constitutive heterochromatin of all mammalian cell types analyzed contain 5–7% of very slow HP1 molecules. The amount of very slow HP1 molecules correlated with the chromatin condensation state, mounting to more than 44% in condensed chromatin of transcriptionally silent cells. During mitosis 8–14% of GFP-HP1α, but not the other isoforms, are very slow within pericentromeric heterochromatin, indicating an isoform-specific function of HP1α in heterochromatin of mitotic chromosomes. These data suggest that mobile as well as very slow populations of HP1 may function in concert to maintain a stable conformation of constitutive heterochromatin throughout the cell cycle.
INTRODUCTION
The genomic DNA within the eukaryotic nucleus is organized into structurally distinct domains that regulate gene expression and chromosome behavior (Lamond and Earnshaw, 1998). Chromosomes are composed of two types of domains: heterochromatin and euchromatin (Cohen and Lee, 2002; Grewal and Moazed, 2003). Constitutive heterochromatic domains at centromeres and telomeres consist of repetitive DNA and are largely transcriptionally silent. Euchromatin defines the gene-rich and transcriptionally active region of the cell nucleus (Grewal and Elgin, 2002). Heterochromatin mediates many diverse functions in the cell nucleus, including centromere function, gene silencing, regulation of gene expression, and nuclear organization. At centromeres, heterochromatin is required for proper sister chromatid cohesion and mitotic segregation (Bernard et al., 2001; Peters et al., 2001; Nonaka et al., 2002; Hall et al., 2003). Smaller heterochromatin domains are involved in epigenetic regulation of gene expression during development and cellular differentiation (Cavalli, 2002; Grewal and Moazed, 2003). Heterochromatic inactivation of one of the two X chromosomes, giving rise to the Barr body, is essential in dosage compensation in somatic cells of female mammals (Avner and Heard, 2001). The link between heterochromatin and transcriptional silencing has been firmly established by detailed analysis of the phenomenon PEV (position effect variegation), in which a gene is silenced by positioning it abnormally close to heterochromatin (Wallrath and Elgin, 1995).
The establishment of heterochromatin requires the physical coupling of histone-modifying activities and structural proteins at specific genomic sites (Richards and Elgin, 2002). The “histone code” hypothesis predicts that these activities act in concert to regulate chromatin function (Strahl and Allis, 2000; Jenuwein and Allis, 2001). Deacetylation and subsequent methylation of the lysine residue at position 9 of histone H3 (K9-H3) is a prerequisite for the formation of constitutive heterochromatin (Nakayama et al., 2001; Hall et al., 2002; Shankaranarayana et al., 2003). Methylation of K9-H3 (meK9-H3) creates a specific binding site for heterochromatin protein HP1 (Bannister et al., 2001; Jacobs et al., 2001; Lachner et al., 2001; Nielsen et al., 2002). The formation and spreading of heterochromatin is believed to be driven by oligomerization of HP1 on meK9-H3 modified nucleosomes concomitant with further recruitment of methyltransferases (Rea et al., 2000; Hall et al., 2002). Sequential cycles of HP1 binding, H3 deacetylation, and methylation then result in the spreading of these “silencing” proteins along the chromatin fiber (Hoppe et al., 2002; Luo et al., 2002; Rusche et al., 2002). Methylation-independent binding of HP1 to histones H3 and H1 (Nielsen et al., 2001a), and interaction of HP1 with DNA and RNA (Sugimoto et al., 1996; Zhao et al., 2000; Maison et al., 2002; Muchardt et al., 2002; Meehan et al., 2003) suggest that additional types of HP1 interactions could also be involved in heterochromatin formation at the nucleosomal level. In addition, heterochromatic silencing and HP1 localization also depend on the RNAi machinery (Pal-Bhadra et al., 2004).
HP1 proteins are highly conserved through evolution, as orthologues are found in yeasts, plants, and animals. In mammals, three isoforms of HP1 (α, β, and γ) have been identified (Eissenberg and Elgin, 2000; Jones et al., 2000). HP1 proteins are characterized by an amino-terminal chromo domain, a short variable region, and a chromo shadow domain. Several HP1-interacting proteins have been identified, including proteins involved in gene regulation, replication, and chromatin remodeling (Eissenberg and Elgin, 2000; Li et al., 2002). The HP1 chromo domain is a specific interaction motif for meK9-H3 (Bannister et al., 2001; Lachner et al., 2001), whereas the chromo shadow domain forms homo- and heteromeres (Brasher et al., 2000; Nielsen et al., 2001a). The ability of HP1 proteins to oligomerize and interact with several other chromatin components is consistent with the hypothesis that they induce transcriptional silencing through the formation of concatenated multimeric complexes that cross-link large chromatin areas (Locke et al., 1988; Cowieson et al., 2000; Wang et al., 2000). Consequently, HP1 proteins accumulate at blocks of constitutive heterochromatin, such as centromeres, in diverse eukaryotes (Wreggett et al., 1994; Ekwall et al., 1995; Kellum et al., 1995; Minc et al., 2000). HP1 has also been implicated in the regulation of euchromatic genes (Dillon and Festenstein, 2002). Recent studies demonstrated that K9-H3 methylation and HP1 are recruited to specific promoters for gene silencing (Nielsen et al., 2001b; Ayyanathan et al., 2003). In Drosophila, HP1 and histone modifiers were shown to regulate specific genes (Hwang et al., 2001), probably dependent on their chromosomal localization (Greil et al., 2003). HP1 localization studies have revealed a banded pattern across a small number of euchromatic sites on Drosophila chromosomes (James et al., 1989; Sun et al., 2000; Piacentini et al., 2003). These observations indicate that, although HP1 is primarily concentrated in constitutive heterochromatin, specific locations throughout the euchromatin are also under its control. All these findings contributed to the view that HP1 plays a central role in creating a stable and inaccessible heterochromatic compartment in which transcriptionally inactive regions of chromatin are densely packed and inaccessible to the transcription machinery (Grewal and Elgin, 2002).
The combination of high-resolution optical microscopy combined with green fluorescent protein technology has opened the possibility to observe the kinetics of cellular components in living cells (Lippincott-Schwartz et al., 2001). Employing kinetic microscopy, such as fluorescence recovery after photobleaching (FRAP) provided important new insights into cellular functions, particularly the dynamic nature of nuclear protein functions (Heun et al., 2001; Houtsmuller and Vermeulen, 2001; Misteli, 2001; Hager et al., 2002). Photobleaching experiments demonstrated that HP1 proteins are very mobile in the nucleus of living cells, suggesting dynamic HP1 binding as a mechanism of heterochromatin maintenance (Cheutin et al., 2003; Festenstein et al., 2003). In this report we have used fluorescence correlation spectroscopy (FCS), a technique to determine the diffusion coefficient by monitoring the Brownian movement of individual proteins (Elson, 2001; Schwille, 2001; Hess et al., 2002) and FRAP in combination in order to assess the full spectrum of HP1 kinetics in living cells. These analyses reveal at least three differently mobile HP1 populations including a fraction of very slow molecules in constitutive heterochromatin of mammalian cells.
MATERIALS AND METHODS
Plasmids
Full-length HP1 cDNAs fragments were obtained by PCR from plasmids pIND-HP1α, pIND-HP1β, pGADGH-HP1γ (wild-type), and pCMV-HP1γ-HA (C59R point mutation) and subcloned into pCR®2.1-TOPO vectors (Invitrogen, Carlsbad, CA). Plasmids were kindly provided by H. Worman, Columbia University, NY, (HP1α and HP1β), and H. Ariga, Hokkaido University, Sapporo, Japan (HP1γ plasmids). Inserts in TOPO vectors were verified by sequencing and subcloned into pEGFP-C1 (CLONTECH, Palo Alto, CA) to generate the expression vectors pEGFP-C1-HP1α, -HP1β, -HP1γ wild-type, and HP1γ(C59R) mutant, respectively. For generation of glutathione-S-transferase (GST) fusion vectors, HP1γ wild-type was cloned as an EcoRI/NotI fragment into pGEX4T-3, and HP1γ(C59R) was cloned as an EcoRI/XhoI fragment into pGEX4T-1.
Cell Culture and Drug Treatment
HEp-2 cells and NIH3T3 cells obtained from the American Tissue Culture Collection (ATCC, Rockville, MD), and primary human fibroblasts obtained from R. Kinne (Experimental Rheumatology Unit, Friedrich-Schiller-University, Jena) were cultured in Dulbecco′s modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum in a 10% CO2 atmosphere at 37°C. HEp-2 cell lines stably expressing GFP-HP1 fusion proteins were generated by adding 1 mg/ml gentamicin (G418, Calbiochem, La Jolla, CA) to the growth medium 24 h after transfection over a period of 3 weeks with medium changes every 2–3 days. In some experiments, cells were treated with 500 ng/ml trichostatin A for 24 h or 20 μg/ml actinomycin D for 2 h.
Antibodies
The following primary antibodies were used for indirect immunofluorescence analyses and Western blotting: mouse monoclonal antibodies HP1α (2HP-2G9), HP1β (1MOD 1A9), HP1γ (2MOD 1G6; all Euromedex, Souffelweyersheim, France); human anti-HP1 autoimmune serum recognizing all three HP1 isoforms (a kind gift from A. v. Mikecz, Heinrich-Heine-University, Düsseldorf, Germany); rabbit antibody H238 (sc-5621, Santa Cruz Biotechnology, Santa Cruz, CA) to detect PML nuclear bodies; rabbit antibody against acetylated histone H3 (06-599; Upstate Biotechnology, Lake Placid, NY); rabbit antibody against di-methylated lysine at position 9 of histone H3 (07-212, Upstate Biotechnology); human CREST sera against centromeres (Kiesslich et al., 2002); mAb against GFP (sc-9996, Santa Cruz Biotechnology).
Immunocytochemistry
Cells grown on coverslips were fixed by incubation in 3.7% paraformaldehyde for 10 min at room temperature followed by 3–5 min permeabilization in 0.5% Triton X-100. Immunofluorescence was performed as previously described (Kiesslich et al., 2002). To detect endogenous HP1 proteins cells were permeabilized in 0.25% Triton X-100 on ice for 3 min before fixation. For dual immunofluorescence staining, primary antibodies from different sources (mouse, rabbit, or human) were used simultaneously and detected with species-specific secondary antibodies linked to fluorescein or rhodamin (Jackson ImmunoResearch, West Grove, PA). DNA was stained with ToPro-3 (Molecular Probes, Eugene, OR)
In Vivo Immunolabeling of Nascent RNA Transcripts
Fluoro-Uridine (Fl-U) incorporation assays to detect newly synthesized RNA were performed as previously described (Kiesslich et al., 2002). Briefly, cells were grown on coverslips until subconfluency. Fl-U was added to the culture medium (2 mM working concentration) and coverslips were removed for fixation after 5 min. Subsequently, cells were fixed in 3.7% formaldehyde for 10 min and permeabilized in 1% Triton-X-100 for 3 min at room temperature. The halogenated nucleotide was detected with a rat anti-bromo-deoxyuridine antibody (MAS250, Harlan Sera-Lab, Loughborough, England).
Chromatin Fractionation, Immunoprecipitation, and Western Blots
Chromatin fractionation was carried out according to previously described protocols (Remboutsika et al., 1999). Briefly, HEp-2 cells were lysed in buffer N (0.3% NP40, 15 mM Tris-HCl, pH 7.5, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 1 mM dithiothreitol, 2 mM Na-vanadate, 250 mM sucrose, Complete protease inhibitor, Roche, Mannheim, Germany). Nuclear DNA was digested by incubation with increasing amounts of micrococcal nuclease (MNase). After centrifugation, supernatant fractions were removed and the pellets were resuspended in ice-cold 2 mM EDTA. For immunoprecipitation (IP), the same procedure was carried out as for nuclear fractionation using 125 U MNase in the sample. The supernatant was used for IP using protein A-Sepharose according to protocols described previously (von Mikecz et al., 2000). A rabbit anti-mouse bridging antibody (Jackson ImmunoResearch) was added in precipitations using mAB against GFP. Immunoprecipitates were resuspended in SDS loading buffer and analyzed by Western blotting. Protein samples or immunoprecipitates were electrophoresed on SDS-PAGE and transferred to a nitrocellulose membrane (Protran, Schleicher & Schuell, Dassel, Germany). The membrane was blocked in 5% milk powder in PBS-T (1× PBS with 0.1% Tween 20, pH 7.4) for 1 h, incubated with primary antibody (in PBS-T) for the same time, and washed three times in PBS-T. The membrane was then incubated with a peroxidase-conjugated secondary antibody (Jackson ImmunoResearch) at a dilution of 1:4000 in PBS-T for 45 min and afterward washed in PBS-T. Signal was detected using chemiluminescence reagent (ECL, Amersham, Freiburg, Germany) on imaging film (Biomax, Eastman-Kodak, Rochester, NY).
Recombinant Proteins
Full-length HP1γ and HP1γC59R were expressed as GST-tagged fusion proteins in Escherichia coli BL21(DE3) and purified as described previously (Smith and Johnson, 1988). Protein concentrations were determined using the microBSA protein assay kit (Pierce, Rockford, IL).
Electromobility Shift Assays
Protein-DNA binding reactions (20 μl) contained protein(s) at the concentrations indicated, 0.5 nM of a 180-base pair long double-stranded CEN-DNA (described in Hemmerich et al., 2000), 25 mM HEPES buffer at pH 7.6, 50% glycerine, and 10 mg/ml BSA (binding buffer). Reaction mixtures were kept on ice for 20 min and then run on 4% native polyacrylamide gels in 0.5× TBE (1× TBE contains 100 mM Tris, 83 mM borat, 0.1 mM EDTA, pH 8.0) at 20 mA at room temperature and stained in SYBR Gold nucleic acid stain solution (Molecular Probes). The fluorescence of the gel bands was visualized by UV light. Digital pictures were taken with a high-resolution CCD camera (Cybertec, Berlin, Germany). Band shift assays detecting protein-RNA interactions were performed exactly as described above with RNA transcribed from the CEN-DNA with RNA polymerase T7 (Promega, Madison, WI).
Microscopy
A LSM 510 laser scanning confocal microscope (Carl Zeiss, Jena, Germany) with 20 mW argon ion laser, a helium neon laser with a 543 nm line, and a helium neon laser with a 633 nm line was used. Samples were scanned using a 63× Plan-Apochromat oil immersion objective. Fluorescein, rhodamine, and Topro-3 dyes (to visualize DNA) were excited by laser light at 488, 543, and 633 nm wavelength, respectively. To avoid bleed-through effects in double- or triple-staining experiments, each dye was scanned independently in a multi-tracking mode.
Fluorescence Correlation Spectroscopy Measurements
Fluorescence correlation spectroscopy (FCS) measurements were performed at 37°C on a LSM 510/ConfoCor 2 combi system (Carl Zeiss) using a C-Apochromat infinity-corrected 1.2 NA 40× water objective. Cells were seeded on 42-mm glass dishes (Saur Laborbedarf, Reutlingen, Germany) the day before the experiment and immediately before the measurement transferred to a live cell chamber (Pecon, Erbach, Germany). Excitation of GFP was performed with the 488 nm line of a 20 mW argon laser with 4.3 Ampere tube current attenuated by an acousto-optcal tunable filter (AOTF) to 0.1%. Under this condition, no influence of bleaching on the diffusion time was notable. The detection pinhole had a diameter of 70 μm and emission was recorded through a 505 nm long path filter. For intracellular measurements, the desired recording position was chosen in the LSM image using the automated stage positioning feature of the ConfoCor 2 system. Autocorrelation curves were derived from fluorescence fluctuation analysis using the ConfoCor 2 software. The principles of FCS in confocal systems have been previously described (Elson and Magde, 1974, Thompson, 1991; Rigler et al., 1993; Elson, 2001; Schwille, 2001; Bacia and Schwille, 2003; Haustein and Schwille, 2003). Briefly, the normalized form of the autocorrelation function is described as G(τ) = 〈δF(t)·δF(t+τ)〉/〈F(t)〉2, where 〈 〉 denotes the time average and δF(t) = F(t) - 〈F(t)〉 the fluctuations around the mean intensity. Autocorrelation curves were either fit to one or two component models of free diffusion in three dimensions with triplet function (Rigler et al., 1992) or to an anomalous diffusion model in three dimensions with triplet function (Schwille et al., 1999; Saxton, 2001) using Origin Software (Microcal Software Inc., Northhampton, MA). The analytical functions for the three models take the following forms: the one-component free diffusion model (Equation 1):
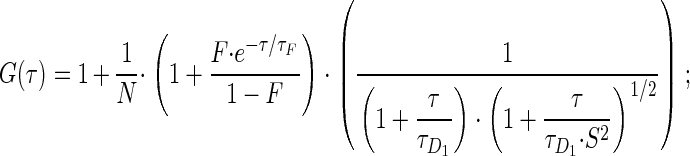 |
1 |
the two-component free diffusion model (Equation 2):
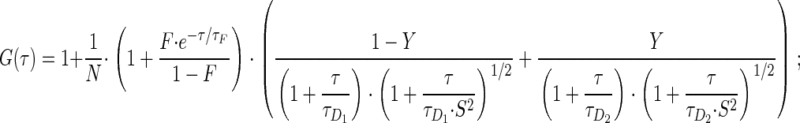 |
2 |
and the anomalous diffusion model (Equation 3):
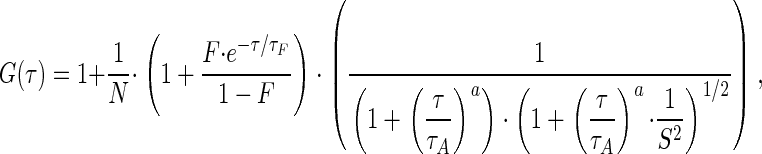 |
3 |
where N and F represent the total number of particles and the triplet fraction, respectively; τD, τA and τF the free diffusion time (the subscripts indicate the different molecule species), anomalous diffusion time and triplet time, respectively; Y and 1 - Y the fractions of species 2 and 1, respectively; α the temporal coefficient; τ the correlation time, and S = ωz/ωxy the structural parameter with ωz and ωxy, representing the half height and radius of the confocal volume that is approximated by a cylinder, respectively. In the ConfoCor 2 ωz = 0.75 μm and ωxy = 0.15 μm according to the supplier's information. The diffusion coefficient can be calculated according to 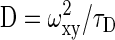 , whereas the transport coefficient is defined as
, whereas the transport coefficient is defined as 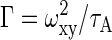 with D(τ) = Γ·τα-1.
with D(τ) = Γ·τα-1.
Fluorescence Recovery after Photobleaching
FRAP experiments were carried out on a Zeiss LSM 510 confocal microscope using an Plan-Neofluar 25× immersion objective and the 488 nm laser line for EGFP. Because of the high mobility of a subfraction of HP1 proteins, the pinhole was adjusted to increase the speed of image acquisition. Thirty images were taken before a bleach pulse of 50 ms time was performed using a strip across the whole nucleus with a length according to the size of the nucleus and a width of 1–2 μm. After the bleach pulse, at least 2000 images were taken, with a scan time of 63 ms per image. Quantitation of relative fluorescence intensity was done according to Chen and Huang (2001) using Excel software (Microsoft, Redmond, WA). FRAP curves were analyzed by nonlinear regression using Prism software version 4.0 (GraphPad Software Inc., San Diego, CA). Kinetic association models assuming one, two, or three individual protein populations according to Equation 4:
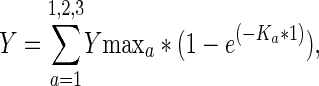 |
4 |
where Ka = association constant, Ymaxa = plateau value, and t = time, were tested. For all GFP-HP1 isoforms measured in interphase cells, best fits (according to R2 value and F-test significance) were obtained using the two-component model. Values of maximal recovery derived from nonlinear regression were used to calculate the proportion of the two differently mobile protein populations, whereas the very slow fraction was calculated by adding up the individual fractions to 100% (Christensen et al., 2002).
RESULTS
Expression of EGFP-HP1 Proteins in HEp-2 Cells
Stable HEp-2–derived cell lines, which constitutively express GFP-tagged HP1α, HP1β, HP1γ, and HP1γC59R, were generated. These cell lines were initially characterized by indirect immunofluorescence to analyze the distribution of the fusion proteins. In HEp-2 cells, endogenous and GFP-tagged HP1α, HP1β, and HP1γ localized to pericentromeric heterochromatin domains (Figure 1, A–C), as revealed by colocalization with centromeres (Supplementary Figure 1). All wild-type GFP isoforms were also detected diffusely throughout the euchromatic regions and some additional sites of the nucleoplasm that were significantly smaller in size than the large blocks of pericentromeric heterochromatin (Supplementary Figure 1). These distribution patterns are consistent with previous observations on HP1 localization in mammalian cell nuclei (Horsley et al., 1996; Nielsen et al., 1999; Minc et al., 2000; Gilbert et al., 2003). GFP-HP1γC59R showed a nuclear distribution pattern different to its wild-type counterpart. GFP-HP1γC59R encodes a mutant HP1γ peptide in which cysteine at position 59 is replaced by arginine. The mutant protein did not accumulate in heterochromatic regions, indicating that the point mutation abolished its recruitment to heterochromatin. Instead, it homogenously labeled the entire nucleoplasm and preferentially localized to 3–10 dot-like structures (Figure 1D). These structures were identified as promyelocytic leukemia nuclear bodies (PML NBs) in colocalization experiments (unpublished data), in line with the finding that an intact chromoshadow domain (aa 111–173) is sufficient to direct HP1γ to PML NBs when fused to GFP (Hayakawa et al., 2003).
Figure 1.

Characterization of GFP-tagged HP1 proteins in HEp-2 cells. HEp-2 cells stably expressing GFP-HP1α (A), HP1β (B), GFP-HP1γ (C), or GFP-HP1γC59R (D) were analyzed by indirect immunofluorescence and confocal microscopy. One confocal section displaying GFP signals (left) and DNA staining (right, DNA) is shown. (E), endogenous and GFP-tagged HP1 proteins were detected by Western blotting of protein lysates from GFP-HP1α, GFP-HP1β, GFP-HP1γ, or GFP-HP1γC59R-expressing cells (lanes 1–4, respectively). Nitrocellulose strips were probed with monoclonal antibodies against HP1α (lane 1), HP1β (lane 2), or HP1γ (lanes 3 and 4). (F) Coimmunoprecipitation analysis of GFP-HP1 fusion proteins. Upper panel: mononucleosome containing nuclear fractions (see Supplementary Figure 1) from transfected cells (lanes 1–4) and untransfected cells (lane 5) were used for immunoprecipitation with monoclonal anti-GFP antibody. Immunoprecipitates were subjected to SDS-PAGE and western blotting using antibody against lysine 9-methylated histone H3 (meK9-H3, upper panel). Lower panel: anti-GFP immunoprecipitates from soluble nuclear fractions were reacted with a human autoimmune serum recognizing all three HP1 isoforms (HP1α/β and HP1γ). (G, H) Nucleic acid binding abilities of GST-HP1γ fusion proteins. A 180 base pairs DNA fragment (G, DNA), or RNAs transcribed from the same DNA by T7 RNA polymerase (H, RNA) was incubated without (lanes 1 and 6) or with increasing amounts (100, 200, 300, and 400 nM) of GST-HP1γ (lanes 2–5) or GST-HP1γC59R (lanes 7–10) followed by EMSA.
Expression of GFP-HP1 fusion proteins was then analyzed by Western blotting. All four fusion constructs were expressed as full-length proteins (Figure 1E). Mammalian HP1 proteins bind to the methylated lysine 9 residue of histone H3 (meK9-H3) and form hetero-oligomers in vitro and in vivo (Huang et al., 1998; Wang et al., 2000; Zhao et al., 2000; Bannister et al., 2001, Lachner et al., 2001; Nielsen et al., 2001a). To verify that the GFP-tagged proteins in our transfected cells are capable of mediating such protein-protein interactions, soluble nuclear extracts from the respective cell lines were generated by micrococcal nuclease digest and subjected to coimmunoprecipitation analysis (Figure 1F). Anti-GFP antibody was used to immunoprecipitate GFP-HP1–containing complexes. A Western blot analysis of the immunoprecipitates revealed that each GFP isoform is associated with meK9-H3 in vivo (Figure 1F, top panel). Furthermore, the anti-GFP antibodies also coimmunoprecipitated endogenous HP1α, HP1β, and HP1γ, indicating their ability to form heterodimers (Figure 1F, bottom panel). The GFP-HP1γC59R mutant protein was not found to be associated with meK9-H3-containing nucleosomes but appeared to retain its ability to form complexes with endogenous HP1 proteins (Figure 1F, lane 4). GFP-tagged HP1α, HP1β, HP1γ, and endogenous HP1 proteins also showed an identical behavior in biochemical fractionation experiments (Supplementary Figure 2). Interestingly, although approximately half of the total amount of cellular HP1 was soluble in 0.3% NP40, a substantial fraction of all wild-type isoforms was found in the same polynucleosomal chromatin fraction that contained CENP-A, but not meK9-H3 (Supplementary Figure 2). This shows that a relatively insoluble subfraction of HP1 is tightly associated with densely packed heterochromatin in the absence of meK9-H3 containing nucleosomes.
Taken together, these results demonstrated that GFP-HP1 and endogenous HP1 proteins behave similarly with respect to nuclear localization, in vivo protein interactions, and biochemical properties. The inability of GFP-HP1γC59R to localize to heterochromatin regions (Figure 1D) is consistent with its absence from meK9-H3 containing nucleosomes (Figure 1F). Because the mutation in HP1γC59R lies within its nucleic acid binding domain (Sugimoto et al., 1996; Muchardt et al., 2002), we also performed DNA and RNA binding assays. These analyses demonstrated that wild-type HP1γ but not HP1γC59R is capable of binding to DNA and RNA in band shift assays (Figure 1, G and H, respectively), and suggest that the nucleic acid-binding activities are required to target HP1γ to heterochromatin.
FCS Reveals Two Differently Mobile Populations of HP1 in Euchromatin
To determine the dynamics of HP1 in euchromatin, the FCS laser beam was positioned in the least intensely stained regions of the nucleus, excluding nucleoli (Figure 2A, cross) and recording 10× 10-s measurement intervals. In all of the 10-s measurements the count rate displayed a mean fluorescence fluctuation trace dominated by 5 ± 3 pronounced spikes (Figure 2B, arrows). In living cells, such singular events during FCS measurements are caused by stable mobile structures containing many fluorescent molecules (Bacia et al., 2002; Gennerich and Schild, 2002). A likely explanation for the spikes in our measurements is the existence of large multiprotein complexes containing several, probably multimerized, GFP-HP1 molecules. The same results were obtained for GFP-HP1β, GFP-HP1γ, and GFP-HP1γC59R (unpublished data). Attempts to determine kinetic data from count trace rates with pronounced spikes or evaluation of the isolated spikes were not successful (unpublished data). However, prebleaching of the confocal volume with high laser intensity for 5 s diminished the appearance of the singular bright events in consecutive 10-s correlation intervals (Figure 2C), indicating that the remaining population now contains only uniformly labeled, highly mobile fluorescent molecules. The remaining fluctuation gave rise to autocorrelation, which revealed a single mobile fraction of GFP-HP1 molecules. Autocorrelation curves were recorded for all GFP-HP1 isoforms in stable cell lines after prebleaching of the confocal volume (Figure 2D). Autocorrelation functions (ACF) were fitted to one or two component models of free diffusion in three dimensions or to an anomalous diffusion model in three dimensions. In the case of nuclear GFP-HP1 proteins, fitting to the free diffusion models did not result in reproducible diffusion times. Instead, ACF of GFP-HP1 proteins recorded in euchromatin required fitting with an anomalous diffusion model for unambiguous interpretation (Figure 2E, red dotted line). To calculate the diffusion coefficient (D), 50–100 curves of at least 10 independent measurements were averaged. The diffusion coefficient of GFP-HP1α and GFP-HP1β was 0.6 ± 0.03 μm2 s-1, the diffusion coefficient of wild-type and mutant GFP-HP1γ was 0.7 ± 0.01 μm2 s-1. This shows that the highly mobile populations of all HP1 isoforms have similar kinetic properties within the nucleoplasm. Because GFP-HP1γC59R displayed similar count rate peaks during fluctuation measurements and had an identical diffusion coefficient compared with the wild type, we conclude that the mutation, although it causes loss of nucleic acid and meK9-H3 binding abilities (Figure 1, F–H), does not alter the intranuclear dynamics of the fast moving form of HP1γ in euchromatic regions of interphase cells. Because GFP-HP1γC59R is still able to interact with the other HP1 isoforms (Figure 1F), it is possible that the similar dynamic behavior stems from its dimerization with the other HP1 isoforms. As a control of our FCS measurements, we determined the kinetics of untagged GFP in the nucleoplasm (Figure 2D). As previously reported, FCS data of GFP dynamics in the nucleus can be fit to an anomalous diffusion model because this cellular compartment exhibits a low but significant degree of obstruction for monomeric GFP (Wachsmuth et al., 2000). Application of the anomalous diffusion model revealed a diffusion coefficient of D = 9.5 ± 0.4 μm2 s-1 for GFP (Figure 2F), consistent with published data (D = 8.7 ± 1.0 μm2 s-1, Wachsmuth et al., 2000). Thus, compared with GFP alone, the fast GFP-HP1 fraction is at least 15-fold less mobile in the nuclear compartment. As a further control, we compared the dynamics of GFP and GFP-HP1α in the cytoplasm. In both cases, autocorrelation curves could only be fitted with the one component three-dimensional diffusion model (unpublished data). Diffusion coefficients calculated from the fits were DGFP = 30 ± 2.7 μm2 s-1 compared with DGFP-HP1α = 26 ± 2 μm2 s-1 (Figure 2F), consistent with the assumption of free, unobstructed diffusion of both proteins within the cytoplasm.
Figure 2.

Two mobile populations of HP1 proteins in euchromatin. (A) LSM image of a HEp-2 cell stably expressing GFP-HP1α before FCS measurement. The cross indicates the position were the corresponding FCS measurement was performed. (B) Count rate trace of the FCS measurement shown in A. Pronounced spikes corresponding to large brightly stained HP1-containing structures with low mobility are indicated by arrows. (C) Count rate trace of FCS measured at the same position after a 5-s bleach pulse of the confocal volume. (D) Autocorrelation curves after amplitude normalization of GFP-HP1α (blue line), GFP-HP1β (red line), GFP-HP1γ (green line), GFP-HP1γC59R (gray line), and GFP alone (black dotted line) measured in euchromatic regions of the nucleus. (E) Fitting of the measured autocorrelation curve of HP1α (black line) with an anomalous diffusion model (red dotted line). (F) Diffusion coefficients of GFP proteins as determined by FCS in the nucleus and in the cytoplasm (cyto).
Taken together, our FCS measurements revealed at least two populations of HP1 proteins with different mobilities in the nucleoplasm of all cells analyzed, a highly mobile population of all HP1 isoforms with diffusion coefficients ranging between 0.6 and 0.7 (±0.03) μm2 s-1 and a second, much less mobile population detected as singular bright peaks during FCS count rate measurements. Because such spikes in count-rate measurements have been interpreted as large, slow moving stable structures or aggregates with many fluorophores (Bacia et al., 2002; Gennerich and Schild, 2002; Bacia and Schwille, 2003), we suggest that they correspond to large multiprotein complexes containing several HP1 molecules that are slowed down by multiple interactions within euchromatin.
FRAP: Mobile and Very Slow Populations of HP1 in Heterochromatin
To further probe the dynamic properties of GFP-HP1 proteins, we used FRAP. Stably transfected living cells were bleached by high-powered laser pulses in rectangular areas of the nucleus. Fluorescence recovery in the bleached area was recorded over time by sequential imaging scans (Figure 3A). Consistent with previous analyses (Cheutin et al., 2003; Festenstein et al., 2003) and our FCS results (see above), FRAP revealed that HP1 proteins are highly dynamic within the nucleoplasm outside the large HP1 domains (i.e., in euchromatin) and less dynamic in the larger heterochromatic domains (Figure 3, B and C). In euchromatin, the half time of fluorescence recovery (t1/2) of all GFP-HP1 isoforms was ∼1 s. However, GFP-HP1α and GFP-HP1β recovery reached 80% after ∼5 s, whereas t80 of GFP-HP1γ was much faster (∼2.5 s; Figure 3B). As already deduced from FCS measurements, the mobility of GFP-HP1γC59R was similar to wild-type GFP-HP1γ. Complete recovery of all HP1 isoforms was reached after 30 s, demonstrating the absence of an immobile fraction of HP1 proteins in euchromatin.
Figure 3.

FRAP analysis of GFP-HP1 proteins in HEp-2 cells. (A) FRAP analysis of a living cell expressing GFP-HP1α. A bar across the nucleus was bleached (indicated by brackets). Selected images of a time series of 2000 images are shown (Bar, 5 μm). Quantitative FRAP analysis for all HP1 isoforms was performed in euchromatin (B), and heterochromatin (C). (D) Comparison of quantitative FRAP of GFP-HP1α in euchromatin (blue line), and heterochromatin (red line) over a period of 30 s, or 120 s (inset). (E) Comparison of mobility of nuclear GFP alone (blue line) and GFP-HP1α in euchromatin (red line). The SD in all FRAP measurements was <5%. (F) Kinetic modeling of the FRAP data. Percentage and association half-time (t1/2) of each population was determined by nonlinear regression and fitting to a two-component model to FRAP curves.
The half-time of recovery of GFP-HP1 proteins in heterochromatin was almost three times slower than in euchromatin (Figure 3C). Recovery half-times were similar for all HP1 isoforms (t1/2 ≈ 3 s). In contrast to euchromatin, not all of the fluorescence signal was recovered in heterochromatin (Figure 3D). Recovery of all isoforms reached a plateau between 90 and 95% after 30 s and remained stable for several minutes (Figure 3D, inset, and unpublished data). This behavior indicates the presence of a very slow population of HP1 in heterochromatin. Quantitation of long time recovery curves consistently revealed that approximately 5 ± 3% of all HP1 isoforms are very slow in HEp-2 cells, suggesting the presence of a small fraction of more statically bound GFP-HP1 molecules in heterochromatin. A similar amount of very slow HP1 proteins was also detected in heterochromatin of mouse NIH3T3 cells (7 ± 4%) and freshly isolated human primary fibroblasts (5 ± 3%) after transient transfection of the GFP-HP1 constructs, and there was no difference in the amount of very slow fractions when transiently and stably transfected cells were compared (unpublished data). These observations indicate that the presence of a very slow fraction of HP1 in heterochromatin is a common feature of HP1 proteins in mammalian cells. As a control, the recovery kinetics of GFP-HP1α in euchromatin was analyzed in comparison to untagged nuclear GFP (Figure 3E). GFP-HP1α recovered much slower than untagged nuclear GFP reflecting chromatin interactions of HP1α.
Kinetic modeling of the FRAP data was used to obtain quantitative information about individually mobile fractions of GFP-HP1. Standard models assuming one, two, or three populations with different mobilities were tested (Kimura and Cook, 2001; Christensen et al., 2002). Nonlinear regression of the recovery values and curve fitting indicated with significance (p < 0.0001) that two different mobility states of HP1 contributed to the apparent FRAP kinetics in both euchromatin and heterochromatin. Models assuming one or three populations with different mobilities could not be fitted to FRAP curves or we obtained unrealistic recovery times of <50 ns and more than 3 h, respectively (unpublished data). Assumption of a two-component model is consistent with the FCS data, which indicated the presence of two mobile fractions of HP1 molecules (Figure 2). In euchromatin, ∼80% of all GFP-HP1 isoforms appeared to be highly mobile (recovery half-time t1/2 = 0.8–1.0 s), whereas ∼20% of the GFP-HP1 population was moving 10- to 15-fold slower than the highly mobile population (Figure 3F). In heterochromatin, similar kinetic populations of GFP-HP1 were determined, but the highly mobile population of GFP-HP1 was decreased to ∼50%, and the fraction of slow molecules increased to >40%, with a t1/2 of ∼10 s (Figure 3F). These observations indicate that euchromatin contains two differently mobile HP1 populations that are also present in heterochromatin.
The presence of two fractions of all GFP-HP1 isoforms with different mobilities in all regions of the nucleus suggests free and unrestricted exchange of these proteins between heterochromatin and euchromatin. The decrease of the highly mobile and the increase of the amount of less mobile GFP-HP1 population in heterochromatin may be attributed to the existence of more HP1 binding sites in heterochromatin than in euchromatin or by transiently stable incorporation of HP1 into multiprotein complexes, or both. The difference also explains why the overall mobility of GFP-HP1 proteins in heterochromatin was ∼threefold less than in euchromatin.
The Very Slow HP1 Population Correlates with Chromatin Condensation and Transcriptional Activity
Previous FRAP analyses of resting mouse T cells revealed that ∼30% of HP1β molecules present in heterochromatic clusters are immobile, but when the T cells were activated, the pool of immobile HP1β in heterochromatin decreased to ∼10% (Festenstein et al., 2003). Because T-cell activation is accompanied by chromatin decondensation and widespread induction of gene expression (Smale and Fisher, 2002), these observations suggest that the amount of immobile HP1 proteins may be correlated with the state of chromatin condensation and/or transcriptional activity. To test this hypothesis, HEp-2 cells stably expressing HP1α were treated with the chromatin-decondensing drug trichostatin A (TSA; Yoshida et al., 1995) or with actinomycin D (ActD), which induces chromatin condensation (Gabbay and Wilson, 1978), and GFP-HP1 kinetics were determined by FRAP (Figure 4, A–E). Untreated cells showed typical GFP-HP1α distribution (Figure 4A), whereas in TSA-treated cells the protein was evenly distributed throughout the entire nucleoplasm (Figure 4B), concomittant with global heterochromatin decondensation (Figure 4B′). Actinomycin D treatment was monitored by visualization of nascent transcripts after in vivo Fluoro-UTP (Fl-U) incorporation (Kiesslich et al., 2002). Untreated GFP-HP1α-expressing cells showed strong nuclear Fl-U labeling, indicating high transcriptional activity (Figure 4C), whereas in ActD-treated cells transcription was almost abolished (Figure 4D′), and GFP-HP1α–containing heterochromatin clusters were increased size (Figure 4D). FRAP analysis of TSA-treated cells showed a more rapid exchange of GFP-HP1α compared with untreated cells, and recovery reached 100% in all regions of the nucleus after ∼20 s, thereby indicating free diffusion kinetics and absence of a very slow GFP-HP1α fraction in TSA-treated cells, respectively (Figure 4E). In contrast, chromatin condensation by treatment with ActD induced a decrease in GFP-HP1α mobility and fluorescence recovery was incomplete in both heterochromatin and euchromatin (Figure 4E). Similar kinetics were observed in GFP-HP1β and GFP-HP1γ-expressing HEp-2 cells (unpublished data). Quantification of the very slow population of all HP1 isoforms is shown in Figure 4F. Although very slow HP1 populations were not detected in euchromatin of transcriptionally active cells, up to a quarter of all GFP-HP1α molecules shifted from the highly mobile into a very slow mobile state in ActD–treated cells (Figure 4F, euchromatin). Similarly, the amount of the very slow fraction of all GFP-HP1 isoforms in heterochromatin increased six- to sevenfold (Figure 4F, heterochromatin). It should also be noted that the amounts of very slow fractions were isoform specific, with GFP-HP1α displaying the highest level of very slow populations (Figure 4F). A subpopulation of ∼5–10% of GFP-HP1–expressing cells were found, by light microscopy, to display typical features of apoptosis (chromatin condensation and membrane blebbing). FRAP analysis of GFP-HP1 in condensed chromatin of apoptotic cells revealed very slow fractions ranging between 50 and 60% (unpublished data), clearly supporting the induction of increased amounts of very slow HP1 molecules in condensed chromatin.
Figure 4.

Characterization of very slow HP1 populations. (A–D) Confocal images of GFP-HP1α expressing cells cultured in medium without drugs (A and C) or in the presence of trichostatin A (TSA) (B) or actinomycin D (ActD) (D). DNA staining of the cells shown in A and B demonstrates global chromatin decondensation in TSA-treated cells (B′) vs. untreated cells (A′). Fluorouridine incorporation assays were performed in control cells (C) and ActD-treated cells (D), and nascent RNA was visualized by immunofluorescence in the same cells (C′ and D′, respectively). (E) Quantitative FRAP analysis of GFP-HP1α in euchromatin (eu) and heterochromatin (hc) of control and drug-treated cells. (F) Very slow fractions as determined from FRAP for all HP1 isoforms in untreated, ActD, or TSA-treated HEp-2 cells. nd, not determined. (G) Confocal image of a GFP-HP1α–expressing cell. The black cross represents the FCS measuring focus positioned in a heterochromatin domain. (H) Decay in the FCS fluorescent trace measured at the cross of the cell shown in G. Heterochromatin-bound GFP-HP1α bleaches away quickly because of its low mobility or immobility (bleaching). (I) Stable association of a subfraction of HP1 with constitutive heterochromatin. Aliquots of whole cell extracts (WCE), cytoplasmic (C), nuclear (N), and micrococcal nuclease (MNase) fractions were subjected to 17.5% SDS-PAGE and analyzed by Coomassie staining (Coomassie) or Western blotting using specific antilysin 9-methylated histone H3 (meK9-H3), anticentromere protein A (CENP-A), anti-HP1α, and anti-GFP (GFP-HP1α, GFP-HP1γC59R) antibodies. For fractionation, nuclei were digested with 0.2, 1, or 5 U (u) of MNase and centrifuged to yield soluble supernatant (S) and insoluble pellet (P) fractions. Numbers on the left side of the gel indicate the position of marker proteins. Bars, 5 μm.
Taken together, these analyses demonstrate that the relatively small pool of very slow HP1 molecules in transcriptionally active and proliferating cells (5–6%) is dramatically increased (25–40%) when the cells undergo chromatin condensation and/or experience transcriptional inhibition.
The presence of a very slow population of GFP-HP1 was also probed by FCS. For this purpose, the FCS laser beam was positioned into the large, brightly fluorescent regions of GFP-HP1–expressing cells (Figure 4G, black cross). In contrast to euchromatin, fluctuation count rates derived from GFP-HP1α in constitutive heterochromatin displayed a strong decay of the fluorescent trace over time (Figure 4H). Such bleaching effects are usually observed if the chromophores exhibit a very low mobility or immobility and thus a long residence time in the FCS focus (Bacia et al., 2002; Bacia and Schwille, 2003). Consequently, the strong bleaching observed at low FCS laser intensities in heterochromatic regions of the nucleus indicates a very low mobility or immobility of a subfraction of GFP-HP1 molecules in heterochromatin. As expected, strong FCS bleaching effects were also observed in condensed chromatin of ActD–treated cells and apoptotic cells (unpublished data). Bleaching effects were never observed in FCS measurements of euchromatin-localized GFP-HP1 molecules (Figure 2). These observations are fully consistent with the FRAP data, which show complete (100%) fluorescence recovery to prebleach intensity of GFP-HP1 in euchromatin, but incomplete (90–95%) recovery in heterochromatin domains (Figure 3D). Taken together, these data unequivocally demonstrate the presence of very slow populations of HP1 proteins in the larger heterochromatin domains of living HEp-2 cells, which can dramatically increase under conditions of transcriptional inactivation and/or chromatin condensation. This result confirms that a fraction of constitutive heterochromatin is in a conformational state that harbors, in addition to very mobile HP1 proteins, a population of statically bound HP1 molecules with very low turnover rates.
Assuming that the mobility of nuclear proteins is correlated with their cellular solubility (i.e., binding strength to other nuclear structures), we performed chromatin fractionation assays on GFP-HP1–expressing HEp-2 cells (Figure 4I). Fractions enriched in euchromatin or heterochromatin can be prepared from intact nuclei subjected to digestion with increasing amounts of micrococcal nuclease (MNase; Huang and Garrard, 1989). This leads to supernatant (S) and pellet (P) fractions that each contain distinct monomeric or oligomeric populations of nucleosomes, depending on the chromatin accessibility of the MNase (Huang and Garrard, 1989; Supplementary Figure 2). As the MNase concentration increased, the S fractions were progressively enriched in core histones (Figure 4H, Coomassie). Western blot analysis confirmed that CENP-A, the histone H3 substitute in centromeric heterochromatin, was exclusively found in the “insoluble” pellet fractions (Figure 4I, CENP-A, lanes 5, 7, and 9) but not in “soluble” supernatant fractions (Figure 4I, CENP-A, lanes 4, 6, and 8). All pellet fractions also contained significant amounts of HP1α and GFP-HP1α (Figure 4I), indicating that a subfraction of HP1 molecules is as tightly bound to heterochromatin as CENP-A. We suggest that this small pool of HP1 protein corresponds to the very slow population of HP1 observed by FRAP and FCS. All other endogenous and GFP-tagged wild-type HP1 isoforms showed a similar biochemical fractionation behavior (Supplementary Figure 2C). As a further control, fractionation of GFP-HP1γC59R–expressing cells showed that the mutant protein was almost exclusively present in the soluble supernatant fractions (Figure 4I, GFP-HP1γC59R), consistent with its absence from heterochromatin (Figure 1D). The strength of the interaction between HP1 and chromatin was also analyzed by extracting HEp2 cell nuclei with increasing concentrations of NaCl. This revealed that a subfraction of all endogenous or GFP-tagged HP1 isoforms is still associated with the pellet fraction after extraction with 1 M NaCl, again indicating a strong interaction between a subpopulation of HP1 with chromatin (unpublished data).
Very Slow HP1α at Pericentromeric Heterochromatin During Mitosis
Constitutive heterochromatin such as pericentromeric DNA is a stable component of chromosomes throughout the cell cycle. One would therefore not expect alterations of HP1 mobility dependent on the cell cycle. To test this assumption, we determined HP1 dynamics in mitotic cells using FRAP and FCS. Similarly to endogenous HP1 (Minc et al., 1999), GFP-HP1 proteins in our cell lines redistributed dynamically in an isotype-specific manner. In prometaphase and metaphase, HP1α localized homogenously throughout the mitotic cytoplasm and accumulated at centromeres (Figure 5A), whereas HP1β and HP1γ were absent from prometaphase and metaphase chromosomes (unpublished data; Hayakawa et al., 2003). During anaphase and telophase, all HP1 isoforms relocalized to mitotic chromatin (Figure 5A, t; and unpublished data). These observations are consistent with previous reports on mitotic localization of endogenous HP1 (Minc et al., 1999) and GFP-HP1 (Hayakawa et al., 2003). Because only HP1α remained associated with pericentromeric heterochromatin throughout the cell cycle, we investigated this isoform in more detail during mitosis. At prometaphase, metaphase, and cytokinesis, GFP-HP1α within pericentromeric heterochromatin could be identified as distinct dot-shaped structures (Figure 5A, p, m, and c), whereas at anaphase and telophase these sites could not be identified because of the dynamic relocalization of cytoplasmic GFP-HP1α to the chromosomes (Figure 5, A and D, and unpublished data). This allowed us to perform FRAP on pericentromeric heterochromatin at distinct steps of mitosis (Figure 5C). Quantitation of FRAP data revealed that GFP-HP1α exchange at pericentromeric heterochromatin during prometaphase and metaphase, and cytokinesis showed kinetics similar to interphase heterochromatin (Figure 5C). Most importantly, fluorescence did not recover to prebleach levels. A very slow fraction of GFP-HP1α was observed at prometaphase (14 ± 5%), metaphase (15 ± 5%), and cytokinesis (8 ± 4%). These observations indicate that the very slow population of HP1α is maintained within constitutive heterochromatin during mitosis. As a control, recovery of GFP-HP1α in mitotic cytoplasm was significantly faster and reached 100% of the prebleach level after 10 s (Figure 5C).
Figure 5.

Dynamics of HP1 proteins in mitotic cells. (A) Confocal images of GFP-HP1α–expressing HEp-2 cells in prometaphase (p), metaphase (m), telophase (t), and cytokinesis (c). Arrows indicate regions that are shown as magnified images in insets. During cytokinesis, GFP-HP1α is also targeted to the midbody (arrow-head in c), consistent with previous observations (Sugimoto et al., 2001). (B) FRAP experiment of a GFP-HP1α–expressing HEp-2 cell at metaphase. a magnified view of the bleached area is shown in insets. (C) Quantitative FRAP of GFP-HP1α in mitotic cytoplasm of a metaphase cell, and in pericentromeric heterochromatin at different stages of mitosis. (D) FCS measurements within mitotic chromosomes (black cross) and in the mitotic cytoplasm (white cross) of GFP-HP1α–expressing cells in telophase. Bar, 5 μm. Count rate trace of FCS measurements in mitotic chromosomes (E) and mitotic cytoplasm (F) of the cell shown in D. (G) Autocorrelation curves of GFP-HP1α measured in mitotic chromosomes (solid line) and mitotic cytoplasm (dotted line). (H) Diffusion coefficients (D) from FCS analysis of all HP1 isoformes in telophase cells.
By late telophase, all GFP-HP1 isoforms had completely relocalized to chromatin (Figure 5D, and unpublished data), similar to endogenous HP1 (Minc et al., 1999). Employing FCS, we determined the diffusion coefficients of all HP1 isoforms within chromatin of telophase cells (Figure 5D-H). FRAP measurements were not possible in the mitotic cytoplasm because of the low fluorescence of the GFP signals within this compartment (Figure 5D). However, dynamics of all GFP-HP1 isoforms could easily be determined by FCS, demonstrating the power of this technique to measure the dynamics of fluorescent probes at very low concentrations. In contrast to interphase cells, the count rate trace of fluctuation measurements in diffusely stained areas of mitotic chromosomes and mitotic cytoplasm did not show single bright events, indicating the absence of large HP1-containing structures during mitosis (Figure 5, E and F, respectively). Initial photobleaching, however, was observed when the FCS laser beam was directed to the more intensely labeled HP1 chromatin domains, thereby confirming the presence of a very slow HP1 population within mitotic heterochromatin (unpublished data). Autocorrelation functions could be fitted to the anomalous three-dimensional diffusion model (Figure 5G) from which the diffusion coefficients of the GFP-HP1 proteins in mitosis were extracted (Figure 5H). Wild-type GFP-HP1 fusion proteins have D values between 0.3 and 0.4 μm2 s-1 in mitotic chromosomes, which is significantly slower (about twofold) than in interphase euchromatin (D = 0.6–0.7 μm2 s-1, Figure 2). The reduced mobility may be explained by the presence of more binding sites for HP1 per measuring volume within the more condensed mitotic chromosomes. It is interesting to note that all HP1 isoforms each have very similar diffusion coefficients (D ≈ 0.7 μm2 s-1) in interphasic euchromatin (Figure 2) and mitotic cytoplasm (Figure 4F). This suggests that the kinetics of the very mobile fraction of HP1 is not primarily governed by chromatin interactions. The GFP-HP1γC59R fusion protein showed no difference in kinetics between mitotic chromosomes and mitotic cytoplasm (D = 0.80 ± 0.04 and 0.75 ± 0.08 μm2 s-1, respectively), suggesting that the mutant is not able to interact with mitotic chromatin. Taken together, the presence of a very slow fraction of HP1α within pericentric chromatin during mitotis implies that the maintenance of constitutive heterochromatin is mediated by both, highly mobile and more stably associated HP1α.
DISCUSSION
An important function of chromatin is compartmentalization of individual portions of the genome into active and repressive states. This is achieved by the formation, maintenance, and propagation of euchromatin and heterochromatin, respectively. HP1 proteins have been implicated in constitutive heterochromatin and some forms of facultative heterochromatin (Cowell et al., 2002; Peters et al., 2002; Chadwick and Willard, 2003; Metzler-Guillemain et al., 2003). Kinetic microscopy in mammalian cells recently demonstrated a high turnover rate of HP1 proteins in heterochromatin, suggesting a model in which dynamic binding of HP1 to chromatin is the mechanism for the maintenance of stable heterochromatic domains (Cheutin et al., 2003; Festenstein et al., 2003). In the present study we have used fluorescence correlation spectroscopy and fluorescence recovery after photobleaching in combination to analyze the full spectrum of kinetic behavior of GFP-HP1 proteins in living mammalian cells.
Mobile HP1 Populations
All FCS recordings of GFP-HP1 in euchromatin showed frequent fluorescence peaks (spikes) dominating a stable mean fluorescence fluctuation, indicating at least two differently mobile populations of GFP-HP1 molecules within this nuclear compartment (Figure 2B). Singular bright events, alternatively designated “single transition events” in FCS count-trace rates (Gennerich and Schild, 2002) have been interpreted as large, strongly fluorescent mobile particles or aggregates passing through the FCS focus volume at random times (Bacia et al., 2002). A likely explanation for single transition events in our recordings is the existence of stable multiprotein complexes containing oligomers of GFP-HP1 molecules, which have been proposed to form in living cells (Huang et al., 1998; Wang et al., 2000; Nielsen et al., 2001a). Count-rate peaks were not detected in FCS measurements of mitotic cells, indicating that this HP1 population is only present in interphase.
Deliberate prebleaching of the confocal volume for 5 s eliminated count-rate peaks. The remaining fluorescence fluctuation revealed a highly mobile HP1 population with diffusion coefficients ranging between D = 0.6 ± 0.03 μm2 s-1 for HP1α and HP1β, and D = 0.7 ± 0.01 μm2 s-1 for HP1γ. Nuclear proteins with similar kinetics within the nucleoplasm include the rRNA processing protein fibrillarin (D = 0.53 μm2 s-1), splicing factor SF2/ASF (D = 0.24 μm2 s-1) and nucleosomal binding protein HMG-17 (D = 0.45 μm2 s-1; Phair and Misteli, 2000). FRAP analyses confirmed that in euchromatin HP1 is divided into a highly mobile (t1/2 ≈ 1 s) and a less mobile (t1/2 ≈ 10 s) fraction (Figure 3F). Kinetic modeling of FRAP data (Figure 3F) yields the minimum number of two populations required to fit the data, and more populations may actually exist. However, combining our FRAP and FCS data, it is plausible to assume that the highly mobile and less mobile fractions determined by FRAP correspond to the highly mobile fraction (D = 0.6–0.7 μm2 s-1) and the presumed multiprotein complexes detected by FCS count rate peaks, respectively. The less mobile fraction may indeed be composed of a heterogeneity of diffusing species exhibiting a distribution of mobilities with a mean recovery halftime ranging between 10 and 15 s.
FRAP in heterochromatin revealed similar populations of HP1 mobilities, indicating unrestricted passage of both the fast fraction and the less mobile complexes throughout the entire nuclear space. The different relative amounts of each fraction in euchromatin (80% fast, 20% slow) and heterochromatin (50% fast, 40% slow) suggest that these populations are interconvertible. Alternatively, assuming a slow exchange of HP1 between the highly mobile pool and the complexes, the increase of the amount of slow HP1-containing complexes may be attributable to a higher number of binding sites in heterochromatin.
Mobile Oligomeric HP1 Complexes
The presence of large, slow-moving complexes containing self-associated HP1 molecules is not surprising because such complexes have been postulated based on the ability of HP1 to form higher-order multimeric complexes that could be relevant to heterochromatin formation (Orlando and Paro, 1995; Platero et al., 1995; Le Douarin et al., 1996; Cowell and Austin, 1997; Ye et al., 1997; Brasher et al., 2000; Smothers and Henikoff, 2000; Wang et al., 2000; Nielsen at al., 2001a). Interestingly, extracts from maternally loaded cells of early Drosophila embryos were found to contain three distinct HP1-containing oligomeric species. The predominant species represented a protein complex with a molecular weight of 39 kDa (HP1 dimers), whereas ∼10% of the HP1 population migrated as 290 and 720 kDa complexes (Huang et al., 1998). The formation of multimeric complexes appears to be a common feature among chromodomain proteins because stable high-molecular-weight complexes (2–5 × 106 Da) containing Pc-G proteins were identified in Drosophila embryonic nuclear extracts (Franke et al., 1992). Despite the lack of biochemical evidence for similar complexes in mammalian cells, the large HP1 complexes in Drosophila embryos demonstrate the intrinsic ability of HP1 to form stable complexes in vivo. Furthermore, bacterially purified HP1 can oligomerize in vitro to form complexes ranging in size between 158 and 443 kDa (Wang et al., 2000; Festenstein et al., 2003). Multimeric HP1-containing complexes may interact with each other in a cooperative manner to extend, stabilize, and/or cross-link heterochromatic DNA (Zhao et al., 2000). On the nucleosome level, cooperativity may be enhanced by the interaction of multimeric HP1 with several meK9-H3 residues, the histone fold domain of H3, histone H1, histone H4, DNA, and RNA (Zhao et al., 2000; Nielsen et al., 2001a; Peters et al., 2001; Polioudaki et al., 2001; Maison et al., 2002; Muchardt et al., 2002; Meehan et al., 2003). It is therefore conceivable that the oligomeric HP1 complexes play an important role in the formation and maintenance of heterochromatin. The HP1 multiprotein complexes may contain additional proteins such as subunits of the origin recognition complex (Pak et al., 1997), members of the TIF1/KAP family (Nielsen et al., 1999; Lechner et al., 2000; Schultz et al., 2002), or CAF I (Murzina et al., 1999). Such complexes may have individual functions, i.e., silencing of individual genes residing in euchromatin by establishing small microcosms of heterochromatin in promoter regions (Ayyanathan et al., 2003, Fahrner and Baylin et al., 2003).
Mobile and Very Slow HP1 Populations in Constitutive Heterochromatin of Mammalian Cells
Consistent with recent observations (Cheutin et al., 2003; Festenstein et al., 2003), the highly mobile population of HP1 suggests dynamic binding to chromatin and supports a model in which continuous HP1 exchange is instrumental to allow competition with various other chromatin-regulating factors on the level of nucleosomes (Cheutin et al., 2003). Although the model of maintenance of heterochromatin domains by dynamic HP1 binding is compatible with the dynamic nature of facultative heterochromatin and HP1-mediated silencing of gene promoters in euchromatin (Fahrner and Baylin, 2003), it may not be fully applicable to constitutive heterochromatin. In this respect, an important conclusion from our work is that interphasic pericentromeric heterochromatin (but not euchromatin) of all cell types analyzed contained very slow HP1 molecules.
Quantitation by FRAP revealed that, depending on the cell type, 5 ± 3 to 7 ± 4% of all HP1 isoforms remained very slow in pericentromeric heterochromatin of proliferating and transcriptionally active HEp-2 cells. The dramatic increase of this very slow fraction of HP1 in transcriptionally inhibited cells in both, euchromatin (5–6%) and heterochromatin (25–40%; Figure 4F) suggests a functional link between HP1 mobility and the chromatin condensation status of a given cell. This interpretation is fully consistent with the observation that activated, proliferating mouse T cells contain in their constitutive heterochromatin ∼10% of immobile HP1β, whereas nonproliferating, quiescent T cells contain ∼30% immobile HP1β (Festenstein et al., 2003). The very slow fraction of HP1 proteins in HEp-2 cells was confirmed by the observation of a strong initial decay of the fluorescence fluctuation trace in heterochromatin during FCS measurements (Figure 4H). This decay is caused by photobleaching effects in which molecules in the observation volume are photochemically destroyed because they are bleached before exiting the volume because of their relative immobility (Bacia and Schwille, 2003; Haustein and Schwille, 2003; Hink et al., 2003). Finally, the presence of such a stable HP1 fraction was also suggested by our biochemical fractionation analysis (Figure 4I).
Very Slow HP1α within Pericentromeric Heterochromatin in Mitosis
Previous life cell analyses have revealed that, although the majority of HP1α diffuses into the cytoplasm, some populations are retained in the centromeric heterochromatin region throughout mitosis (Sugimoto et al., 2001). In this report we show that this pericentromeric heterochromatin contains significant amounts of very slow GFP HP1α (Figure 5C), strongly suggesting that a stably heterochromatin-associated fraction of this HP1 isoform may be essential for maintaining the heterochromatin status of centromere DNA during mitosis. Compared with constitutive heterochromatin of interphase cells, the amount of very slow HP1α increased two- to threefold in pericentromeric heterochromatin of mitotic cells, either reflecting increased chromatin condensation at centromeres or functional upregulation of this HP1α population. Interestingly, HP1β and HP1γ are absent from pericentromeric heterochromatin until anaphase (Minc et al., 1999; Hayakawa et al., 2003; and our own unpublished data). This differential behavior of HP1 isoforms may be regulated by subtle differences in primary sequence and by posttranslational modifications, such as the recently reported isotype-specific hyperphosphorylation of HP1α during mitosis (Minc et al., 1999).
Contribution of Very Slow HP1 to Heterochromatin Maintenance
A minor fraction of very slow HP1 in constitutive heterochromatin could be indicative of a stable structural network within this chromatin compartment that could serve as a binding interface for mobile populations of HP1. Thus, although high HP1 mobility appears to be essential, more stably associated HP1 molecules and stable oligomeric HP1 complexes seem to have an important structural function in maintaining constitutive heterochromatin integrity. This model is supported by our (and previous) findings (Festenstein et al., 2003) that the amount of very slow or immobile HP1 is several-fold increased in transcriptionally inactive cells or during chromatin condensation (Figure 4F). Very slow HP1 molecules may be tightly DNA-bound due to certain higher-order conformational states of chromatin fibers in constitutive heterochromatin (Gilbert and Allan, 2001), by cross-linking of nucleosomes through HP1 dimerization, and by cooperative chromatin binding of higher-order multimeric HP1 complexes (Zhao et al., 2000; Nielsen et al., 2001a). We therefore propose to extend the current model of dynamic heterochromatin maintenance (Cheutin et al., 2003; Festenstein et al., 2003) in that highly mobile (“freely” diffusing), mobile oligomeric complexes as well as very slow populations of HP1 contribute to the integrity of constitutive heterochromatin in mammalian cells (Figure 6). To further unravel the mechanism of heterochromatin formation and maintenance, future studies should address the question whether other heterochromatin components exhibit similar dynamics as HP1 in living cells.
Figure 6.

Three HP1 populations with different mobilities in mammalian cell nuclei. The diagram summarizes the kinetic data of HP1 obtained by FCS and FRAP. Euchromatin (EU) and heterochromatin (HC) contain two differently mobile HP1 populations, a highly mobile fraction with a FRAP recovery half-time (t1/2) of ∼1 s (D: diffusion coefficient), and a less mobile with a t1/2 of ∼10 s. In addition, there is a minor fraction of very slow HP1 molecules of 5–7% present in constitutive heterochromatin.
Supplementary Material
Acknowledgments
We thank Tom Misteli and Thierry Cheutin for critically reading the manuscript, Hiroyoshi Ariga and Howard Worman who kindly provided HP1 cDNAs, Anna von Mikecz for anti-HP1 autoimmune serum, Marianne Koch and Sabine Ohndorf for excellent technical assistance. L.S. received a fellowship from the “EXIST-HighTEPP” grant provided by the Bundesministerium für Bildung und Forschung.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E03-11-0827. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E03-11-0827.
Online version of this article contains supporting material.
Online version is available at www.molbiolcell.org.
References
- Avner, P., and Heard, E. (2001). X-chromosome inactivation: counting, choice and initiation. Nat. Rev. Genet. 2, 59-67. [DOI] [PubMed] [Google Scholar]
- Ayyanathan, K., Lechner, M.S., Bell, P., Maul, G.G., Schultz, D.C., Yamada, Y., Tanaka, K., Torigoe, K. and Rauscher, F.J., 3rd. (2003). Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: a mammalian cell culture model of gene variegation. Genes Dev. 17, 1855-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacia, K., Majoul, I.V., and Schwille, P. (2002). Probing the endocytic pathway in live cells using dual-color fluorescence cross-correlation analysis. Biophys. J. 83, 1184-1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacia, K., and Schwille, P. (2003). A dynamic view of cellular processes by in vivo fluorescence auto- and cross-correlation spectroscopy. Methods 29, 74-85. [DOI] [PubMed] [Google Scholar]
- Bannister, A.J., Zegerman, P., Partridge, J.F., Miska, E.A., Thomas, J.O., Allshire, R.C., and Kouzarides, T. (2001). Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120-124. [DOI] [PubMed] [Google Scholar]
- Bernard, P., Maure, J.F., Partridge, J.F., Genier, S., Javerzat, J.P., and Allshire, R.C. (2001). Requirement of heterochromatin for cohesion at centromeres. Science 294, 2539-2542. [DOI] [PubMed] [Google Scholar]
- Brasher, S.V. et al. (2000). The structure of mouse HP1 suggests a unique mode of single peptide recognition by the shadow chromo domain dimer. EMBO J. 19, 1587-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli, G. (2002). Chromatin as a eukaryotic template of genetic information. Curr. Opin. Cell Biol. 14, 269-278. [DOI] [PubMed] [Google Scholar]
- Chadwick, B.P., and Willard, H.F. (2003). Chromatin of the Barr body: histone and non-histone proteins associated with or excluded from the inactive X chromosome. Hum. Mol. Genet. 12, 2167-2178. [DOI] [PubMed] [Google Scholar]
- Chen, D., and Huang, S. (2001). Nucleolar components involved in ribosome biogenesis cycle between the nucleolus and nucleoplasm in interphase cells. J. Cell Biol. 153, 169-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheutin, T., McNairn, A.J., Jenuwein, T., Gilbert, D.M., Singh, P.B., and Misteli, T. (2003). Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science 299, 721-725. [DOI] [PubMed] [Google Scholar]
- Christensen, M.O., Barthelmes, H.U., Feineis, S., Knudsen, B.R., Andersen, A.H., Boege, F., and Mielke, C. (2002). Changes in mobility account for camptothecin-induced subnuclear relocation of topoisomerase I. J. Biol. Chem. 277, 15661-15665. [DOI] [PubMed] [Google Scholar]
- Cohen, D.E., and Lee, J.T. (2002). X-chromosome inactivation and the search for chromosome-wide silencers. Curr. Opin. Genet. Dev. 12, 219-224. [DOI] [PubMed] [Google Scholar]
- Cowell, I.G. et al. (2002). Heterochromatin, HP1 and methylation at lysine 9 of histone H3 in animals. Chromosoma 111, 22-36. [DOI] [PubMed] [Google Scholar]
- Cowell, I.G., and Austin, C.A. (1997). Self-association of chromo domain peptides. Biochim. Biophys. Acta 1337, 198-206. [DOI] [PubMed] [Google Scholar]
- Cowieson, N.P., Partridge, J.F., Allshire, R.C., and McLaughlin, P.J. (2000). Dimerisation of a chromo shadow domain and distinctions from the chromodomain as revealed by structural analysis. Curr. Biol. 10, 517-525. [DOI] [PubMed] [Google Scholar]
- Dillon, N., and Festenstein, R. (2002). Unravelling heterochromatin: competition between positive and negative factors regulates accessibility. Trends Genet. 18, 252-258. [DOI] [PubMed] [Google Scholar]
- Eissenberg, J.C., and Elgin, S.C. (2000). The HP1 protein family: getting a grip on chromatin. Curr. Opin. Genet. Dev. 10, 204-210. [DOI] [PubMed] [Google Scholar]
- Ekwall, K., Javerzat, J.P., Lorentz, A., Schmidt, H., Cranston, G., and Allshire, R. (1995). The chromodomain protein Swi 6, a key component at fission yeast centromeres. Science 269, 1429-1431. [DOI] [PubMed] [Google Scholar]
- Elson, E.L. (2001). Fluorescence correlation spectroscopy measures molecular transport in cells. Traffic 2, 789-796. [DOI] [PubMed] [Google Scholar]
- Fahrner, J.A., and Baylin, S.B. (2003). Heterochromatin: stable and unstable invasions at home and abroad. Genes Dev. 17, 1805-1812. [DOI] [PubMed] [Google Scholar]
- Festenstein, R., Pagakis, S.N., Hiragami, K., Lyon, D., Verreault, A., Sekkali, B., and Kioussis, D. (2003). Modulation of heterochromatin protein 1 dynamics in primary Mammalian cells. Science 299, 719-721. [DOI] [PubMed] [Google Scholar]
- Franke, A., DeCamillis, M., Zink, D., Cheng, N., Brock, H.W., and Paro, R. (1992). Polycomb and polyhomeotic are constituents of a multimeric protein complex in chromatin of Drosophila melanogaster. EMBO J. 11, 2941-2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbay, E.J., and Wilson, W.D. (1978). Intercalating agents as probes of chromatin structure. Methods Cell Biol. 18, 351-384. [DOI] [PubMed] [Google Scholar]
- Gennerich, A., and Schild, D. (2002). Anisotropic diffusion in mitral cell dendrites revealed by fluorescence correlation spectroscopy. Biophys. J. 83, 510-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, N., and Allan, J. (2001). Distinctive higher-order chromatin structure at mammalian centromeres. Proc. Natl. Acad. Sci. USA 98, 11949-11954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, N., Boyle, S., Sutherland, H., de Las Heras, J., Allan, J., Jenuwein, T., and Bickmore, W.A. (2003). Formation of facultative heterochromatin in the absence of HP1. EMBO J. 22, 5540-5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greil, F., van der Kraan, I., Delrow, J., Smothers, J.F., de Wit, E., Bussemaker, H.J., van Driel, R., Henikoff, S., and van Steensel, B. (2003). Distinct HP1 and Su(var)3–9 complexes bind to sets of developmentally coexpressed genes depending on chromosomal location. Genes Dev. 17, 2825-2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal, S.I., and Elgin, S.C. (2002). Heterochromatin: new possibilities for the inheritance of structure. Curr. Opin. Genet. Dev. 12, 178-187. [DOI] [PubMed] [Google Scholar]
- Grewal, S.I., and Moazed, D. (2003). Heterochromatin and epigenetic control of gene expression. Science 301, 798-802. [DOI] [PubMed] [Google Scholar]
- Hager, G.L., Elbi, C., and Becker, M. (2002). Protein dynamics in the nuclear compartment. Curr. Opin. Genet. Dev. 12, 137-141. [DOI] [PubMed] [Google Scholar]
- Hall, I.M., Noma, K., and Grewal, S.I. (2003). RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast. Proc. Natl. Acad. Sci. USA 100, 193-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, I.M., Shankaranarayana, G.D., Noma, K., Ayoub, N., Cohen, A., and Grewal, S.I. (2002). Establishment and maintenance of a heterochromatin domain. Science 297, 2232-2237. [DOI] [PubMed] [Google Scholar]
- Haustein, E., and Schwille, P. (2003). Ultrasensitive investigations of biological systems by fluorescence correlation spectroscopy. Methods 29, 153-166. [DOI] [PubMed] [Google Scholar]
- Hayakawa, T., Haraguchi, T., Masumoto, H., and Hiraoka, Y. (2003). Cell cycle behavior of human HP1 subtypes: distinct molecular domains of HP1 are required for their centromeric localization during interphase and metaphase. J. Cell Sci. 116, 3327-3338. [DOI] [PubMed] [Google Scholar]
- Hemmerich, P., Stoyan, T., Wieland, G., Koch, M., Lechner, J., and Diekmann, S. (2000). Interaction of yeast kinetochore proteins with centromere-protein/transcription factor Cbf1. Proc. Natl. Acad. Sci. USA 97, 12583-12588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, S.T., Huang, S., Heikal, A.A., and Webb, W.W. (2002). Biological and chemical applications of fluorescence correlation spectroscopy: a review. Biochemistry 41, 697-705. [DOI] [PubMed] [Google Scholar]
- Heun, P., Taddei, A., and Gasser, S.M. (2001). From snapshots to moving pictures: new perspectives on nuclear organization. Trends Cell Biol. 11, 519-525. [DOI] [PubMed] [Google Scholar]
- Hink, M.A., Borst, J.W., and Visser, A.J. (2003). Fluorescence correlation spectroscopy of GFP fusion proteins in living plant cells. Methods Enzymol. 361, 93-112. [DOI] [PubMed] [Google Scholar]
- Hoppe, G.J., Tanny, J.C., Rudner, A.D., Gerber, S.A., Danaie, S., Gygi, S.P., and Moazed, D. (2002). Steps in assembly of silent chromatin in yeast: Sir3-independent binding of a Sir2/Sir4 complex to silencers and role for Sir2-dependent deacetylation. Mol. Cell. Biol. 22, 4167-4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsley, D., Hutchings, A., Butcher, G.W., and Singh, P.B. (1996). M32, a murine homologue of Drosophila heterochromatin protein 1 (HP1), localises to euchromatin within interphase nuclei and is largely excluded from constitutive heterochromatin. Cytogenet. Cell. Genet. 73, 308-311. [DOI] [PubMed] [Google Scholar]
- Houtsmuller, A.B., and Vermeulen, W. (2001). Macromolecular dynamics in living cell nuclei revealed by fluorescence redistribution after photobleaching. Histochem. Cell. Biol. 115, 13-21. [DOI] [PubMed] [Google Scholar]
- Huang, D.W., Fanti, L., Pak, D.T., Botchan, M.R., Pimpinelli, S., and Kellum, R. (1998). Distinct cytoplasmic and nuclear fractions of Drosophila heterochromatin protein 1, their phosphorylation levels and associations with origin recognition complex proteins. J. Cell Biol. 142, 307-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S.Y., and Garrard, W.T. (1989). Electrophoretic analyses of nucleosomes and other protein-DNA complexes. Methods Enzymol. 170, 116-142. [DOI] [PubMed] [Google Scholar]
- Hwang, K.K., Eissenberg, J.C., and Worman, H.J. (2001). Transcriptional repression of euchromatic genes by Drosophila heterochromatin protein 1 and histone modifiers. Proc. Natl. Acad. Sci. USA 98, 11423-11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, S.A., Taverna, S.D., Zhang, Y., Briggs, S.D., Li, J., Eissenberg, J.C., Allis, C.D., and Khorasanizadeh, S. (2001). Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J. 20, 5232-5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James, T.C., Eissenberg, J.C., Craig, C., Dietrich, V., Hobson, A., and Elgin, S.C. (1989). Distribution patterns of HP1, a heterochromatin-associated nonhistone chromosomal protein of Drosophila. Eur. J. Cell Biol. 50, 170-180. [PubMed] [Google Scholar]
- Jenuwein, T., and Allis, C.D. (2001). Translating the histone code. Science 293, 1074-1080. [DOI] [PubMed] [Google Scholar]
- Jones, D.O., Cowell, I.G., and Singh, P.B. (2000). Mammalian chromodomain proteins: their role in genome organisation and expression. Bioessays 22, 124-137. [DOI] [PubMed] [Google Scholar]
- Kellum, R., Raff, J.W., and Alberts, B.M. (1995). Heterochromatin protein 1 distribution during development and during the cell cycle in Drosophila embryos. J. Cell Sci. 108, 1407-1418. [DOI] [PubMed] [Google Scholar]
- Kiesslich, A., von Mikecz, A., and Hemmerich, P. (2002). Cell cycle-dependent association of PML bodies with sites of active transcription in nuclei of mammalian cells. J. Struct. Biol. 140, 167-179. [DOI] [PubMed] [Google Scholar]
- Kimura, H., and Cook, P.R. (2001). Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 153, 1341-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachner, M., O'Carroll, D., Rea, S., Mechtler, K., and Jenuwein, T. (2001). Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116-120. [DOI] [PubMed] [Google Scholar]
- Lamond, A.I., and Earnshaw, W.C. (1998). Structure and function in the nucleus. Science 280, 547-553. [DOI] [PubMed] [Google Scholar]
- Le Douarin, B., Nielsen, A.L., Garnier, J.M., Ichinose, H., Jeanmougin, F., Losson, R., and Chambon, P. (1996). A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. EMBO J. 15, 6701-6715. [PMC free article] [PubMed] [Google Scholar]
- Lechner, M.S., Begg, G.E., Speicher, D.W. and Rauscher, F.J., 3rd. (2000). Molecular determinants for targeting heterochromatin protein 1-mediated gene silencing: direct chromoshadow domain-KAP-1 corepressor interaction is essential. Mol. Cell. Biol. 20, 6449-6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y., Kirschmann, D.A., and Wallrath, L.L. (2002). Does heterochromatin protein 1 always follow code? Proc. Natl. Acad. Sci. USA 99(Suppl 4), 16462-16469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., Snapp, E., and Kenworthy, A. (2001). Studying protein dynamics in living cells. Nat. Rev. Mol. Cell. Biol. 2, 444-456. [DOI] [PubMed] [Google Scholar]
- Locke, J., Kotarski, M.A., and Tartof, K.D. (1988). Dosage-dependent modifiers of position effect variegation in Drosophila and a mass action model that explains their effect. Genetics 120, 181-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, K., Vega-Palas, M.A., and Grunstein, M. (2002). Rap1-Sir4 binding independent of other Sir, y Nospacekuqq, or histone interactions initiates the assembly of telomeric heterochromatin in yeast. Genes Dev. 16, 1528-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magde, D., Elson, E.L., and Webb W.W. (1974) Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers 13, 29-61. [DOI] [PubMed] [Google Scholar]
- Maison, C., Bailly, D., Peters, A.H., Quivy, J.P., Roche, D., Taddei, A., Lachner, M., Jenuwein, T., and Almouzni, G. (2002). Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat. Genet. 30, 329-334. [DOI] [PubMed] [Google Scholar]
- Meehan, R.R., Kao, C.F., and Pennings, S. (2003). HP1 binding to native chromatin in vitro is determined by the hinge region and not by the chromodomain. EMBO J. 22, 3164-3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzler-Guillemain, C., Luciani, J., Depetris, D., Guichaoua, M.R., and Mattei, M.G. (2003). HP1beta and HP1gamma, but not HP1alpha, decorate the entire XY body during human male meiosis. Chromosome Res. 11, 73-81. [DOI] [PubMed] [Google Scholar]
- Minc, E., Allory, Y., Worman, H.J., Courvalin, J.C., and Buendia, B. (1999). Localization and phosphorylation of HP1 proteins during the cell cycle in mammalian cells. Chromosoma 108, 220-234. [DOI] [PubMed] [Google Scholar]
- Minc, E., Courvalin, J.C., and Buendia, B. (2000). HP1gamma associates with euchromatin and heterochromatin in mammalian nuclei and chromosomes. Cytogenet. Cell. Genet. 90, 279-284. [DOI] [PubMed] [Google Scholar]
- Misteli, T. (2001). Protein dynamics: implications for nuclear architecture and gene expression. Science 291, 843-847. [DOI] [PubMed] [Google Scholar]
- Muchardt, C., Guilleme, M., Seeler, J.S., Trouche, D., Dejean, A., and Yaniv, M. (2002). Coordinated methyl and RNA binding is required for heterochromatin localization of mammalian HP1alpha. EMBO Rep. 3, 975-981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzina, N., Verreault, A., Laue, E., and Stillman, B. (1999). Heterochromatin dynamics in mouse cells: interaction between chromatin assembly factor 1 and HP1 proteins. Mol. Cell 4, 529-540. [DOI] [PubMed] [Google Scholar]
- Nakayama, J., Rice, J.C., Strahl, B.D., Allis, C.D., and Grewal, S.I. (2001). Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292, 110-113. [DOI] [PubMed] [Google Scholar]
- Nielsen, A.L., Ortiz, J.A., You, J., Oulad-Abdelghani, M., Khechumian, R., Gansmuller, A., Chambon, P., and Losson, R. (1999). Interaction with members of the heterochromatin protein 1 (HP1) family and histone deacetylation are differentially involved in transcriptional silencing by members of the TIF1 family. EMBO J. 18, 6385-6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, A.L., Oulad-Abdelghani, M., Ortiz, J.A., Remboutsika, E., Chambon, P., and Losson, R. (2001a). Heterochromatin formation in mammalian cells: interaction between histones and HP1 proteins. Mol. Cell 7, 729-739. [DOI] [PubMed] [Google Scholar]
- Nielsen, P.R., Nietlispach, D., Mott, H.R., Callaghan, J., Bannister, A., Kouzarides, T., Murzin, A.G., Murzina, N.V., and Laue, E.D. (2002). Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature 416, 103-107. [DOI] [PubMed] [Google Scholar]
- Nielsen, S.J. et al. (2001b). Rb targets histone H3 methylation and HP1 to promoters. Nature 412, 561-565. [DOI] [PubMed] [Google Scholar]
- Orlando, V., and Paro, R. (1995). Chromatin multiprotein complexes involved in the maintenance of transcription patterns. Curr. Opin. Genet. Dev. 5, 174-179. [DOI] [PubMed] [Google Scholar]
- Pak, D.T., Pflumm, M., Chesnokov, I., Huang, D.W., Kellum, R., Marr, J., Romanowski, P., and Botchan, M.R. (1997). Association of the origin recognition complex with heterochromatin and HP1 in higher eukaryotes. Cell 91, 311-323. [DOI] [PubMed] [Google Scholar]
- Pal-Bhadra, M., Leibovitch, B.A., Gandhi, S.G., Rao, M., Bhadra, U., Birchler, J.A., and Elgin, S.C. (2004). Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science 303, 669-672. [DOI] [PubMed] [Google Scholar]
- Peters, A.H., Mermoud, J.E., O'Carroll, D., Pagani, M., Schweizer, D., Brockdorff, N., and Jenuwein, T. (2002). Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat. Genet. 30, 77-80. [DOI] [PubMed] [Google Scholar]
- Peters, A.H. et al. (2001). Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107, 323-337. [DOI] [PubMed] [Google Scholar]
- Phair, R.D., and Misteli, T. (2000). High mobility of proteins in the mammalian cell nucleus. Nature 404, 604-609. [DOI] [PubMed] [Google Scholar]
- Piacentini, L., Fanti, L., Berloco, M., Perrini, B., and Pimpinelli, S. (2003). Heterochromatin protein 1 (HP1) is associated with induced gene expression in Drosophila euchromatin. J. Cell Biol. 161, 707-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platero, J.S., Hartnett, T., and Eissenberg, J.C. (1995). Functional analysis of the chromo domain of HP1. EMBO J. 14, 3977-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polioudaki, H., Kourmouli, N., Drosou, V., Bakou, A., Theodoropoulos, P.A., Singh, P.B., Giannakouros, T., and Georgatos, S.D. (2001). Histones H3/H4 form a tight complex with the inner nuclear membrane protein LBR and heterochromatin protein 1. EMBO Rep. 2, 920-925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea, S. et al. (2000). Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593-599. [DOI] [PubMed] [Google Scholar]
- Remboutsika, E., Lutz, Y., Gansmuller, A., Vonesch, J.L., Losson, R., and Chambon, P. (1999). The putative nuclear receptor mediator TIF1alpha is tightly associated with euchromatin. J. Cell Sci. 112, 1671-1683. [DOI] [PubMed] [Google Scholar]
- Richards, E.J., and Elgin, S.C. (2002). Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell 108, 489-500. [DOI] [PubMed] [Google Scholar]
- Rigler, R., Ü. Mets, J. Widengren, and P. Kask. (1993). Fluorescence correlation spectroscopy with high count rate and low background: analysis of translational diffusion. Eur. Biophys. J. 22, 169-175. [Google Scholar]
- Rigler, R., Widengren, J. and Mets, and Ü. (1992) Interaction and kinetics of single molecules as observed by fluorescence correlation spectroscopy. In: Fluorescence Spectroscopy, ed. O.S.E. Wolfbeis, Berlin: Springer, 13-24.
- Rusche, L.N., Kirchmaier, A.L., and Rine, J. (2002). Ordered nucleation and spreading of silenced chromatin in Saccharomyces cerevisiae. Mol. Biol. Cell 13, 2207-2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton, M.J. (2001). Anomalous subdiffusion in fluorescence photobleaching recovery: a Monte Carlo study. Biophys. J. 81, 2226-2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, D.C., Ayyanathan, K., Negorev, D., Maul, G.G. and Rauscher, F.J., 3rd. (2002). SETDB 1, a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 16, 919-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwille, P. (2001). Fluorescence correlation spectroscopy and its potential for intracellular applications. Cell. Biochem. Biophys. 34, 383-408. [DOI] [PubMed] [Google Scholar]
- Schwille, P., Korlach, J., and Webb, W.W. (1999). Fluorescence correlation spectroscopy with single-molecule sensitivity on cell and model membranes. Cytometry 36, 176-182. [DOI] [PubMed] [Google Scholar]
- Shankaranarayana, G.D., Motamedi, M.R., Moazed, D., and Grewal, S.I. (2003). Sir2 regulates histone H3 lysine 9 methylation and heterochromatin assembly in fission yeast. Curr. Biol. 13, 1240-1246. [DOI] [PubMed] [Google Scholar]
- Smale, S.T. and Fisher, A.G. (2002). Chromatin structure and gene regulation in the immune system. Annu. Rev. Immunol. 20, 427-462. Epub 2001: Oct 2004. [DOI] [PubMed] [Google Scholar]
- Smith, D.B., and Johnson, K.S. (1988). Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67, 31-40. [DOI] [PubMed] [Google Scholar]
- Smothers, J.F., and Henikoff, S. (2000). The HP1 chromo shadow domain binds a consensus peptide pentamer. Curr. Biol. 10, 27-30. [DOI] [PubMed] [Google Scholar]
- Strahl, B.D., and Allis, C.D. (2000). The language of covalent histone modifications. Nature 403, 41-45. [DOI] [PubMed] [Google Scholar]
- Sugimoto, K., Yamada, T., Muro, Y., and Himeno, M. (1996). Human homolog of Drosophila heterochromatin-associated protein 1 (HP1) is a DNA-binding protein which possesses a DNA-binding motif with weak similarity to that of human centromere protein C (CENP-C). J. Biochem. (Tokyo) 120, 153-159. [DOI] [PubMed] [Google Scholar]
- Sugimoto, K., Tasaka, H., and Dotsu, M. (2001). Molecular behaviour in living mitotic cells of human centromere heterochromatin protein HP1 alpha ectopically expressed as a fusion to red fluorescent protein. Cell Struct. Funct. 26, 705-718. [DOI] [PubMed] [Google Scholar]
- Sun, F.L., Cuaycong, M.H., Craig, C.A., Wallrath, L.L., Locke, J., and Elgin, S.C. (2000). The fourth chromosome of Drosophila melanogaster: interspersed euchromatic and heterochromatic domains. Proc. Natl. Acad. Sci. USA 97, 5340-5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, N.L. (1991). Fluorescence correlation spectroscopy. In: Topics in Fluorescence Spectroscopy, vol. 1, ed. J.R., Lakowicz, New York: Plenum, 337-378. [Google Scholar]
- von Mikecz, A., Zhang, S., Montminy, M., Tan, E.M., and Hemmerich, P. (2000). CREB-binding protein (CBP)/p300 and RNA polymerase II colocalize in transcriptionally active domains in the nucleus. J. Cell Biol. 150, 265-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachsmuth, M., Waldeck, W., and Langowski, J. (2000). Anomalous diffusion of fluorescent probes inside living cell nuclei investigated by spatially-resolved fluorescence correlation spectroscopy. J. Mol. Biol. 298, 677-689. [DOI] [PubMed] [Google Scholar]
- Wallrath, L.L., and Elgin, S.C. (1995). Position effect variegation in Drosophila is associated with an altered chromatin structure. Genes Dev. 9, 1263-1277. [DOI] [PubMed] [Google Scholar]
- Wang, G., Ma, A., Chow, C.M., Horsley, D., Brown, N.R., Cowell, I.G., and Singh, P.B. (2000). Conservation of heterochromatin protein 1 function. Mol. Cell. Biol. 20, 6970-6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wreggett, K.A., Hill, F., James, P.S., Hutchings, A., Butcher, G.W., and Singh, P.B. (1994). A mammalian homologue of Drosophila heterochromatin protein 1 (HP1) is a component of constitutive heterochromatin. Cytogenet. Cell. Genet. 66, 99-103. [DOI] [PubMed] [Google Scholar]
- Ye, Q., Callebaut, I., Pezhman, A., Courvalin, J.C., and Worman, H.J. (1997). Domain-specific interactions of human HP1-type chromodomain proteins and inner nuclear membrane protein LBR. J. Biol. Chem. 272, 14983-14989. [DOI] [PubMed] [Google Scholar]
- Yoshida, M., Horinouchi, S., and Beppu, T. (1995). Trichostatin A and trapoxin: novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays 423-430. [DOI] [PubMed]
- Zhao, T., Heyduk, T., Allis, C.D., and Eissenberg, J.C. (2000). Heterochromatin protein 1 binds to nucleosomes and DNA in vitro. J. Biol. Chem. 275, 28332-28338. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.