

Abstract
NMDA receptors are tetrameric complexes of GluN1, GluN2A-D, and GluN3A-B subunits and are involved in normal brain function and neurologic disorders. We identified a novel class of stereoselective pyrrolidinone (PYD) positive allosteric modulators for GluN2C-containing NMDA receptors, exemplified by methyl 4-(3-acetyl-4-hydroxy-1-[2-(2-methyl-1H-indol-3-yl)ethyl]-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate. Here we explore the site and mechanism of action of a prototypical analog, PYD-106, which at 30 μM does not alter responses of NMDA receptors containing GluN2A, GluN2B, and GluN2D and has no effect on AMPA [α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid] and kainate receptors. Coapplication of 50 μM PYD-106 with a maximally effective concentration of glutamate and glycine increases the response of GluN1/GluN2C NMDA receptors in HEK-293 cells to 221% of that obtained in the absence of PYD (taken as 100%). Evaluation of the concentration dependence of this enhancement revealed an EC50 value for PYD of 13 μM. PYD-106 increased opening frequency and open time of single channel currents activated by maximally effective concentrations of agonist but only had modest effects on glutamate and glycine EC50. PYD-106 selectively enhanced the responses of diheteromeric GluN1/GluN2C receptors but not triheteromeric GluN1/GluN2A/GluN2C receptors. Inclusion of residues encoded by GluN1-exon 5 attenuated the effects of PYD. Three GluN2C residues (Arg194, Ser470, Lys470), at which mutagenesis virtually eliminated PYD function, line a cavity at the interface of the ligand binding and the amino terminal domains in a homology model of GluN1/GluN2C built from crystallographic data on GluN1/GluN2B. We propose that this domain interface constitutes a new allosteric modulatory site on the NMDA receptor.
Introduction
N-Methyl-d-aspartate receptors (NMDARs) are ligand-gated cation channels that mediate a slow Ca2+-permeable component of excitatory synaptic transmission. NMDA receptors are involved in the development and normal function of the central nervous system (CNS). Dysfunction of NMDARs is associated with epilepsy, pain, depression, Parkinson’s disease, and schizophrenia, making these receptors an attractive therapeutic target (Hallett and Standaert, 2004; Kalia et al., 2008; Lisman et al., 2008; Preskorn et al., 2008; Wu and Zhuo, 2009; Endele et al., 2010; Traynelis et al., 2010; Balu and Coyle, 2011; Kostakis et al., 2011). The majority of NMDARs in the central nervous systems are heteromeric complexes formed by two GluN1 and two GluN2 subunits (Ulbrich and Isacoff, 2007), of which there are four subtypes (GluN2A-D) with temporal and spatial variation in expression (Watanabe et al., 1992; Ishii et al., 1993; Monyer et al., 1994). The development of GluN2-selective modulators provides a therapeutic opportunity to target NMDAR subtypes with anatomically restricted expression patterns, thereby minimizing potential side effects (Kalia et al., 2008; Ogden and Traynelis, 2011; Collingridge et al., 2013). Subunit-selective allosteric modulators exist for the GluN2A (TCN-201), GluN2B (ifenprodil), and GluN2C/GluN2D subunits (CIQ [3-chlorophenyl)(6,7-dimethoxy-1-[(4- methoxyphenoxy)methyl]-3,4-dihydroisoquinolin-2(1H)-yl)methanone], QNZ, UBP, DQP analogs) (Williams et al., 1993; Bettini et al., 2010; Mullasseril et al., 2010; Acker et al., 2011; Hansen and Traynelis, 2011; Costa et al., 2012; Hansen et al., 2012; Monaghan et al., 2012). However, no modulators to date have been able to distinguish between GluN2C and GluN2D subunits.
The GluN2C subunit is expressed in the cerebellum, amygdala, olfactory bulb, and retrosplenial cortex, as well as in thalamic, cortical, and hippocampal interneurons (Farrant et al., 1994; Monyer et al., 1994; Wenzel et al., 1997; Binshtok et al., 2006; Karavanova et al., 2007). Oligodendrocytes also express the GluN2C subunit (Karadottir et al., 2005; Salter and Fern, 2005; Micu et al., 2006). GluN2C-containing NMDARs have a lower sensitivity to voltage-dependent Mg2+ block, reduced Ca2+ permeability, and reduced conductance compared with GluN2A and GluN2B (Qian et al., 2005; Clarke and Johnson, 2006; Siegler Retchless et al., 2012). Behavioral studies evaluating the deletion of the GluN2C subunit suggest a possible role in working memory (Hillman et al., 2011). In addition, studies with the GluN2C/GluN2D-selective positive allosteric modulator CIQ suggest a possible role for enhancement of NMDAR function in emotional learning, working memory, and sensorimotor gating (Ogden et al., 2013; Suryavanshi et al., 2014). d-Cycloserine (DCS), an antibiotic treatment used for tuberculosis, acts as a partial agonist relative to glycine at GluN1/GluN2A, GluN1/GluN2B, and GluN1/GluN2D receptors. By contrast, a maximally effective concentration of DCS produces more current at GluN1/GluN2C receptors than the endogenous agonist glycine, leading to selective enhancement of GluN1/GluN2C receptors when DCS replaces glycine at the GluN1 agonist binding site (Sheinin et al., 2001; Dravid et al., 2010). NMDAR hypofunction has been suggested to underlie some aspects of schizophrenia (Krystal et al., 1994; Olney et al., 1999; Lisman, 2012), and DCS has shown positive results in schizophrenic patients, suggesting increased occupancy of the agonist binding site on GluN1 and thereby enhancement NMDAR function (Goff et al., 1995; Goff et al., 2008; Gottlieb et al., 2011). The ability of DCS to enhance the function of the GluN2C-containing receptors raises the possibility that GluN2C modulation may contribute to clinically relevant actions of DCS in schizophrenia (Norberg et al., 2008; Kaplan and Moore, 2011). Thus, better pharmacological tools are needed to evaluate the functional roles of GluN2C in neurologic diseases such as schizophrenia.
We describe here the mechanism of action and structural determinants for a GluN2C-selective class of compounds exemplified by PYD-106 (methyl 4-(3-acetyl-4-hydroxy-1-(2-(2-methyl-1H-indol-3-yl)ethyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate), an analog developed during the study of the structure-activity relationship around a pyrrolidinone identified from a high-throughput screen (Zimmerman et al., 2014). The PYD class of positive allosteric modulators is the only series that selectivity enhances the response to maximally effective concentrations of agonist for recombinant NMDARs containing two copies of the GluN2C subunit.
Materials and Methods
Molecular Biology.
GluN1, GluN2A (D13211), GluN2B (U11419), GluN2C (M91563), and GluN2D (L31611) cDNAs were provided by Drs. Heinemann (Salk Institute for Biologic Sciences, La Jolla, CA), Nakanishi (Kyoto University, Kyoto, Japan), and Seeburg (University of Heidelberg, Heidelberg, Germany). The GenBank accession numbers for GluN1-1a (hereafter GluN1) and splice variants GluN1-1b, -2a, -2b, -3a, -3b, -4a, -4b were U08261, U08263, U08262, U08264, U08265, U08266, U08267, U08268, respectively. Proteins are numbered starting with the initiating methionine as 1. The GluN2A-GluN2C chimeras and GluN2C point mutations were generated as previously described (Chen et al., 2008). Supplemental Table S1 lists the junctions for the chimeric receptors.
A GluN2C-2A C-terminal chimera was made from amino acids 1–851 of rat GluN2C and amino acids 841–1464 of rat GluN2A; these chimeric GluN2C-2A subunits will be referred to hereafter as GluN2C*. Constructs GluN2AC1, GluN2AC2, GluN2C*C1, and GluN2C*C2 were generated by adding a 23-residue synthetic linker followed by coiled-coil regions of the GABAB1 and GABAB2 receptor subunits (C1 and C2, respectively) plus a dilysine endoplasmic reticulum (ER) retention signal (KKTN) at the 3′ end of rat GluN2A and rat GluN2C*, as described previously (Hansen et al., 2014). Two point mutations were introduced into the agonist binding domains by site-directed mutagenesis using the Quikchange method and were GluN2A (R518K, T690I) and GluN2C* (R529K, T701I). All chimeric subunits and mutations were verified by DNA sequencing.
Two-Electrode Voltage Clamp Recordings.
Xenopus laevis oocytes were obtained from EcoCyte (Austin, TX) injected with cRNA created by in vitro transcription using the mMessage mMachine kits according to the manufacturer’s instructions (Life Technologies/Ambion, Grand Island, NY) as previously described (Hansen et al., 2013). Plasmids containing the genes for the GABAA (α1β2γ2s), GABAC (ρ1), glycine (α1), serotonin (5-HT3A), nicotinic acetylcholine receptor (α1β1δγ, α2β4, α4β3, α9α10), and purinergic (P2X2 rat, P2X2 human) receptors were provided by Drs. Heinemann (Salk), Weiss (Univ. of Texas, San Antonio), Papke (Univ. of Florida), and Hume (Univ. of Michigan); linearized; and used to synthesize cRNA for these receptors. One to four days after cRNA injection (5–15 ng), oocytes were recorded at room temperature under two-electrode voltage clamp (VHOLD −30 to −60 mV) in a solution containing (in mM) 90 NaCl, 1 KCl, 10 HEPES, 0.5 BaCl2, and 0.01 EDTA (pH 7.4). For triheteromeric receptor experiments, the cRNA was prepared at a ratio of 1:6:6 (GluN1:GluN2C1:GluN2C2), and approximately 5–10 ng of total cRNA was injected into each oocyte. Recordings were performed 3–4 days after injection. NMDAR currents were evoked by bath application of 50–100 μM glutamate and 30–100 μM glycine. Currents from the GluA1-4 and GluK1-2 receptors were evoked with 100 μM glutamate; GluK2 was incubated in 1 mg/ml concanavalin A for 10 minutes before recording. GluK2/GluK5 currents were evoked with 100 μM AMPA [α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid]. Currents were evoked for the following receptors using the agonist concentrations indicated: GABAC (1 μM GABA), GABAA (20 μM GABA), glycine α1 (50 μM glycine), 5-HT3A (1 μM serotonin), nicotinic acetylcholine α1β1δγ (1 μM acetylcholine), α3β4 (10 μM acetylcholine), α4β2 (10 μM acetylcholine), α9α10 (1 μM acetylcholine), α7 (300 μM acetylcholine), and the P2X2 receptors (9 μM ATP).
Patch-Clamp Recordings.
HEK-293 cells (ATCC CRL-1573, hereafter HEK cells) were maintained in Dulbecco’s modified Eagle’s medium with GlutaMAX, 110 mg/ml sodium pyruvate, and 4.5 g/l glucose (Invitrogen, Grand Island, NY) supplemented with 10% dialyzed fetal bovine serum, 10 U/ml penicillin, and 10 μg/ml streptomycin. HEK cells were maintained in a 5% CO2 humidified 37°C incubator and transiently transfected using Fugene 6 with cDNAs encoding NMDAR subunits and green fluorescent protein (GFP) at a ratio of 1:1:5 for GluN1/GluN2A/GFP, 1:1:1 for GluN1/GluN2B/GFP, GluN1/GluN2C/GFP, and GluN1/GluN2D/GFP. Voltage clamp recordings were performed 12–36 hours after transfection (VHOLD −60 mV for whole cell, −80 mV for outside-out patches). The data were filtered at 8 kHz (8-pole Bessel filter, −3 dB) and digitized at 20–40 kHz. The extracellular solution consisted of (in mM) 150 NaCl, 10 HEPES, 3 KCl, 0.5 CaCl2, 0.01 EDTA, and 30 d-mannitol (pH 7.4 for whole cell and 8.0 for outside-out patches). The intracellular solution contained (in mM) 110 d-gluconate, 110 CsOH, 30 CsCl2, 5 HEPES, 4 NaCl, 0.5 CaCl2, 2 MgCl2, 5 BAPTA, 2 Na-ATP, and 0.3 Na-GTP; the pH was adjusted to 7.35 with CsOH. Rapid solution exchange was achieved using a two-barrel θ glass pipette controlled by a piezoelectric translator. The open tip junction currents had a 10–90% rise time of less than 1 ms. The solution exchange around a whole cell had a 10–90% rise time of 3.4 ± 0.3 ms (n = 9) as determined by exchanging the extracellular NaCl for KCl. Single channel recordings from outside-out patches were made in response to steady-state agonist application at pH 8.0. All recordings were performed at room temperature (23°C). The junction potential for HEK cell recording solution was +5.4 mV (Vance et al., 2011); single-channel chord conductance values were corrected for this value.
Recordings from outside-out patches were digitally filtered at 3–4 kHz (−3 dB) and idealized using time course fitting (SCAN; Dr. David Colquhoun, University College London; Colquhoun and Sigworth, 1995). An open resolution of 53 μs and shut resolution of 31 μs was imposed on the data, and open periods were combined for different conductance levels. Both open and closed duration histograms were fitted with the sum of 2–5 exponential functions using maximum likelihood methods. The open probability was calculated as the total time the patch was in an open state divided by the total time of the recording. Most outside-out patches contained multiple channels, because double and triple openings were occasionally observed. Multiple openings were excluded from analysis of channel dwell times, and thus the reported open probabilities are underestimates of the true open probability.
Molecular Modeling.
Amino acids are numbered with the initiating methionine set to 1. A protein sequence alignment of the different GluN2A-D sequences was generated with Muscle software (Edgar, 2004) using the GluN1-1a (i.e., GluN1) and GluN2B sequences obtained from the resolved GluN1/GluN2B crystal structure (PDB 4PE5). Five GluN1/GuN2C homology models were generated with Modeler 9v12 software (Sali and Blundell, 1993) using the GluN1/GluN2B crystal structure as template (PDB 4PE5; Karakas and Furukawa, 2014) No protein optimization was performed during model building. The models were subjected to protein quality analysis, and the model with the lowest discrete optimized protein energy score was selected. Side chain optimization and protonation state assignment was performed with the Protein Preparation Wizard modeling software (Sastry et al., 2013) and monitored by visual inspection. This was followed by energy minimization (heavy atom RMSD convergence of 0.3 Å; OPLS 2005 force field) to relieve the energetically unfavorable constraints. PYD-106 was prepared for docking using Ligprep (Schrödinger Release 2014-2: LigPrep, version 3.0, Schrödinger, New York). The docking grid was centered between the carbonyl oxygen of P428 and the Cβ-atom of S472. The diameter midpoint of the docked ligands was required to remain within a nested box (14 Å3) at the center of the grid. The extra precision (XP) scoring algorithm of Glide (Friesner et al., 2006) was used to obtain the best scoring poses during docking. PYD-106 was docked flexibly with docking poses being restricted to 10, followed by postdocking minimization (OPLS 2005 force field) with an energy threshold of 0.5 kcal/mol. A PDB file with PYD-106 docked into the homology model of GluN1/GluN2C is included as Supplemental Data).
Synthesis of Pyrrolidinone Analogs.
Compounds PYD-1 (methyl 4-(1-(2-(1H-indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate) and PYD-106 were prepared as previously described (see compounds in Zimmerman et al., 2014). Compounds were dissolved at 20 or 50 mM in 100% DMSO, and this stock was used for all solutions. Final DMSO concentrations were between 0.005 and 0.5% (vol/vol). Both compounds were soluble up to 100 µM, as determined by nephelometry. Compound purity was greater than 95%.
Statistics.
All results are presented as the mean ± standard error of the mean. Statistical significance was taken as P < 0.05 by t test or one-way analysis of variance (ANOVA; Tukey, Bonferroni, or Dunnett’s post hoc), as appropriate. For all tables and figures, n is the number of observations.
Results
Pyrrolidinones Are Selective Positive Allosteric Modulators of GluN1/GluN2C Receptors.
We studied the actions of the pyrrolidinone PYD-106 (Zimmerman et al., 2014) on recombinant GluN1/GluN2C receptor function. Figure 1 shows concentration-effect curves for glutamate in the absence and presence of PYD-106 as determined in Xenopus oocytes injected with GluN1/GluN2C mRNA. PYD-106 prominently enhanced the maximal fitted current response by over twofold. In addition, the glutamate EC50 was modestly increased by 70 μM PYD-106, being 0.72 ± 0.05 μM (n = 13, Hill slope 1.45 ± 0.03) in the absence and 1.17 ± 0.08 μM (n = 13, Hill slope 1.39 ± 0.02) in the presence of PYD-106 when studied in the same oocytes (P < 0.05; paired t test; 30 μM glycine present in all solutions). We also determined the glycine concentration-effect curve independently in the absence or presence of 70 μM PYD-106. Coapplication of PYD-106 with glycine modestly reduced the EC50 from 0.23 ± 0.004 μM (n = 6) in the absence to 0.16 ± 0.006 μM (n = 5) in the presence of PYD-106 (P < 0.05, unpaired t test; 100 μM glutamate present in all solutions). PYD-106 showed no agonist activity when applied alone, with the response amplitude being 1.1 ± 0.6% of control (n = 4). Coapplication of PYD-106 with either glutamate alone or glycine alone did not produce inward currents (n = 5). These data suggest that PYD-106 is a positive allosteric modulator that increases agonist efficacy and also reveal significant negative (glutamate) and positive (glycine) interactions with agonist potency.
Fig. 1.

PYD-106 enhances the maximal current response of GluN1/GluN2C receptors. (A) Concentration-effect curves for glutamate are shown recorded in the same oocyte with and without 70 μM PYD-106 (n = 13 oocytes); 30 μM glycine was present in all solutions. All data in PYD-106 are expressed as a percentage of the response to 10 μM glutamate without PYD-106. The smooth curve is a fit of the Hill equation to the data: Response (%) = Maximal Response/[1 + EC50/concentration)N], where EC50 is the concentration of agonist that produces half of the Maximal Response and N is the Hill slope. The fitted maximal response was increased by PYD-106 to 236%. (B) The concentration-effect curves were normalized to the responses at 10 μM glutamate for each and superimposed to more clearly illustrate the shift in glutamate EC50, which was increased by PYD-106 from 0.72 ± 0.05 μM (Hill slope 1.45 ± 0.03) in the absence of PYD-106 to 1.17 ± 0.08 μM (Hill slope 1.39 ± 0.02) in the presence of 70 μM PYD-106 (P < 0.05; t test).
We next evaluated the concentration-dependence of PYD-106 on recombinant NMDAR activated by maximally effective concentrations of glutamate (100 μM) and glycine (30 μM). The positive modulation produced by PYD-106 was both reversible and repeatable, with an EC50 value of 16 ± 0.5 μM and a Hill slope of 1.19 ± 0.03 in oocytes (n = 31, Fig. 2, A and B). Coapplication of 100 μM PYD-106 and saturating concentrations of glutamate (100 μM) and glycine (30 μM) increased the maximal current response to 204 ± 3.9% of control (the response in the absence of PYD-106 was taken as 100%, n = 31). At submaximal concentrations of glutamate (1 μM) and glycine (0.3 μM), the GluN1/GluN2C receptor response was increased to 223 ± 6.3% of control by 100 μM PYD-106, with an EC50 of 15 ± 1.9 μM (n = 5), which was not significantly different compared with the data obtained with saturating concentrations of glutamate and glycine (t test, P > 0.05).
Fig. 2.
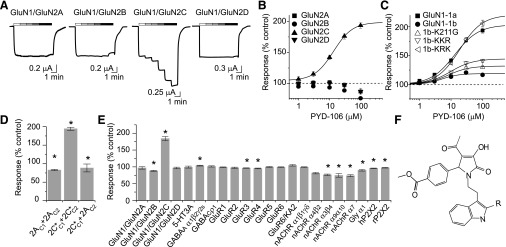
Subunit-selectivity of PYD-106. (A) Two-electrode voltage-clamp recordings are shown from Xenopus oocytes expressing recombinant NMDARs, which were activated by 100 μM glutamate and 30 μM glycine in the presence of PYD-106 (1, 3, 10, 30, 100 μM). (B) Composite concentration-effect curves are shown for PYD-106 at GluN1/GluN2A, GluN1/GluN2B, GluN1/GluN2C, and GluN1/GluN2D receptors. (C) PYD-106 is less effective for receptors expressing GluN1-exon5. GluN1-1b K211G and GluN1-1b K192G, K193G, R194G (1b-KKR) did not fully restore PYD modulation of GluN1/GluN2C receptors, whereas the triple mutation GluN1-1b K207G, R208G, K211G (1b-KRK) fully restored PYD-106 modulation. (D) The ratio of the response to maximally effective concentrations of glutamate (100 µM) and glycine (30 µM) with 50 µM PYD-106 compared with the maximal response in the absence of PYD (expressed as percent) was increased in Xenopus oocytes expressing diheteromeric GluN1/GluN2C receptors (2C*C1 + 2C*C2, see Materials and Methods). Maximal responses were slightly reduced in oocytes expressing diheteromeric GluN1/GluN2A receptors (2AC1 + 2AC2) and triheteromeric GluN1/GluN2A/GluN2C receptors (2AC1 + 2C*C2; *P < 0.001, n = 6-8 cells, one-way ANOVA, post hoc Tukey's test. (E) 30 μM PYD-106 was tested for off-target effects at wild-type NMDA, AMPA (GluA), kainate (GluK), GABA, nicotinic acetylcholine (nACh), glycine (Gly), and purinergic receptors (P2X), which were activated by their respective agonists (see Materials and Methods for concentrations). The ratio of the maximal current response in the presence and absence of PYD-106 is expressed as percent. Modest inhibition observed at some receptors was statistically significant. (F) The structure is shown for PYD-1(R=H) and PYD-106 (R=CH3).
We subsequently tested the effect of PYD-106 at other recombinant ion channels expressed in oocytes. We used a concentration of PYD-106 (30 μM) that strongly enhanced the GluN1/GluN2C receptor response to maximally effective concentrations of glutamate and glycine. PYD-106 (30 μM) did not alter the amplitude for GluN1/GluN2A, GluN1/GluN2D, AMPA, kainate, GABAC, and 5-HT3A receptor-mediated currents in response to saturating agonist concentrations (Fig. 2E). Responses of GluN1/GluN2B, glycine α1, GABAA, and nicotinic acetylcholine receptors were inhibited to 73–88% of control by 30 μM PYD-106 (Fig. 2E, where the response in the absence of PYD is 100%). PYD-106 was also tested by the National Institute of Mental Health Psychoactive Drug Screening Program using a binding assay to assess interactions with G-protein–coupled receptors, transporters, and voltage-gated ion channels. Out of 42 proteins that were tested, 10 µM PYD-106 showed inhibition in excess of 50% of control for the kappa-opioid receptor, dopamine transporter, and the adrenergic α2C receptor (Supplemental Table S2). A subsequent experiment determined the binding affinity (Ki) for PYD-106 to be 6.1 μM for the kappa-opioid receptor; the Ki values for the α2C receptor and dopamine transporter were greater than 10 μM. These data suggest that PYD-106 selectively enhances the GluN1/GluN2C response within the glutamate receptor family but produces significant inhibition at multiple targets (including GluN2B at 100 μM).
Selectivity of Pyrrolidinones for GluN1 Splice Variants.
The GluN1 subunit RNA can be alternatively spliced with 8 different variants described (Hollmann et al., 1993). GluN1 alternative exon-5 encodes 21 amino acids, which are located near the amino terminal domain (ATD)–S1 interface of GluN1. GluN1-exon 5 is differentially expressed throughout the CNS (Laurie and Seeburg, 1994; Laurie et al., 1995), with expression in cerebellar granule neurons, which also express GluN2C (Akazawa et al., 1994; Prybylowski et al., 2000). Exon-5 has been shown to alter the effects of several NMDA receptor modulators including neurosteroids, extracellular protons, Zn2+, and polyamines, in addition to altering glutamate potency and the deactivation time course (Traynelis et al., 1995, 1998; Rumbaugh et al., 2000; Kostakis et al., 2011; Vance et al., 2012). We therefore evaluated the effects of RNA splicing of GluN1 on the activity of the positive allosteric modulator PYD-106. GluN1 subunits that contain exon-5 are referred to as “b” and those lacking exon-5 as “a”; different combinations of C-terminal splice variants that lack exon-21 and/or exon-22 are denoted by the suffix 2,3,4 (Hollmann et al., 1993). Comparison of PYD-106 activity (100 µM) at the different GluN1 splice variants coexpressed with GluN2C revealed significant differences in the maximal level of modulation compared with GluN1-1a/GluN2C (200 ± 5.6% of control, n = 16). Modulation was significantly reduced for GluN1-1b (125 ± 2.8%, n = 18), GluN1-2b (117 ± 0.6%, n = 7), GluN1-3a (173 ± 4.2%, n = 7), GluN1-3b (113 ± 0.4%, n = 9), and GluN1-4b (118 ± 0.9%, n = 10) splice variants (one-way ANOVA, Bonferroni’s post hoc, P < 0.05), whereas there were no significant differences for GluN1-2a (202 ± 4.5%, n = 6) and GluN1-4a (188 ± 3.4%, n = 6). That is, PYD-106 produces a stronger enhancement of the current response to maximal agonist concentrations for all GluN1 splice variants that lack exon-5 compared with those that contain exon-5. Concentration-effect curves demonstrated that the PYD-106 EC50 for enhancement of the maximal response of GluN1-1a (14 ± 1.1 μM, n = 25) was significantly different from the EC50 for GluN1-1b–containing receptors (5.6 ± 0.4 μM, n = 19, t test, P < 0.05; Fig. 2C).
To identify the structural determinants in the highly charged 21-amino acid segment encoded by exon-5 that reduced positive allosteric modulation by PYD-106, we first screened a series of triple charge neutralization mutations. The triple GluN1-1b exon-5 mutation K207G, R208G, K211G restored the effects of PYD-106, which enhanced the maximal current response to 206 ± 5.5% of control at 100 μM PYD-106 (EC50 of 21 ± 2.5 μM, n = 8, Fig. 1C). By contrast, the triple GluN1-1b mutant K192G, K193G, R194G only partially rescued modulation by 100 μM PYD-106 to 145 ± 3.2% of control (n = 4) with an EC50 of 9.6 ± 0.9 μM (n = 4) (Fig. 1C). Mutation of three negatively charged GluN1-1b residues encoded by exon-5 (E197A, D200A, D205A) did not restore the effects of 100 μM PYD-106 (116 ± 2.3% of control; n = 4).
A number of effects of exon-5 on allosteric modulators as well as the deactivation time course are controlled by Lys211 in exon-5 (Traynelis et al., 1995, 1998; Vance et al., 2012). We therefore assessed whether mutation of Lys211 to Gly or Arg restored the actions of PYD-106. The maximal current responses of GluN1-1b K211G and GluN1-1b K211R mutations were only modestly enhanced by 100 μM PYD-106 to 137 ± 2.3% (n = 9) and 129 ± 1.2% (n = 10), respectively (Fig. 2C, Supplemental Figure S1). The degree of modulation observed with GluN1-1b K211G was significantly different compared with PYD-106 modulation of wild-type GluN1-1b (t test, P < 0.05). The EC50 values for PYD-106 enhancement of the maximal response of GluN1-1b K211G and GluN1-1b K211R were 9.5 ± 0.9 μM (n = 3) and 9.0 ± 1.9 (n = 6) μM, respectively, which were not significantly different compared with GluN1-1a (P > 0.05). These data suggest that the structural determinants within exon-5 responsible for its effects on protons, polyamines, and Zn2+ are distinct from its effects on PYD-106.
Pyrrolidinone Activity on Triheteromeric NMDARs.
NMDARs can form diheteromeric complexes that contain one type of GluN2 subunit or triheteromeric receptors containing two different GluN2 subunits. For example, triheteromeric receptors containing GluN1/GluN2A/GluN2C have been reported to form functional receptors in cerebellar neurons (Chazot et al., 1994; Cathala et al., 2000; Lu et al., 2006). To determine whether PYD-106 enhanced the responses of triheteromeric receptors that contain a single copy of GluN2C, we adapted a recombinant expression system that promotes the surface expression of triheteromeric receptors and limits the surface expression of diheteromeric receptors (Hansen et al., 2014) to control expression of GluN1/GluN2A/GluN2C receptors. The GluN2C C terminus was replaced with that of GluN2A (referred to as GluN2C*), and heterodimeric coiled-coil regions with ER retention signals (C1 and C2) were added to the C terminus of both GluN2A and GluN2C* (hereafter named GluN2AC1, GluN2AC2, GluN2C*C1, and GluN2C*C2). NMDAR tetramers containing one C1-tagged GluN2 subunit and one C2-tagged GluN2 subunit are trafficked to the cell surface, whereas those complexes containing only a single C1 or C2 tag are retained in the ER (Hansen et al., 2014). The C-terminal domain of GluN2C was replaced with that of GluN2A because differences between the C-terminal domains could lead to differing trafficking patterns or hinder the C1-C2 interaction. To test how PYD-106 affects triheteromeric NMDAR responses, Xenopus oocytes were coinjected with GluN1/GluN2AC1/GluN2AC2, GluN1/GluN2C*C1/GluN2C*C2, or GluN1/GluN2AC1/GluN2C*C2, and receptors were activated with 100 µM glutamate and 30 µM glycine. PYD-106 application increased the maximal responses of GluN1/GluN2C*C1/GluN2C*C2 receptors to 212 ± 7.8% (n = 12) of control, whereas the responses of GluN1/GluN2AC1/GluN2AC2 and GluN1/GluN2AC1/GluN2C*C2 receptors were modestly inhibited by PYD-106 (86 ± 0.9% and 87 ± 2.2% of control, respectively, n = 13–14; Fig. 2D). To confirm that responses in oocytes expressing GluN1/GluN2AC1/GluN2C*C2 were mediated primarily by triheteromeric receptors, oocytes were coinjected as above with one double-mutated GluN2 that prevents glutamate binding, either GluN2C*C2 (R529K, T701I) or GluN2AC1 (R518K, T690I) (Laube et al., 1997; Hatton and Paoletti, 2005; Erreger et al., 2007; Hansen et al., 2014). The responses of oocytes coinjected with these mutated subunits to 100 μM glutamate and 30 μM glycine were 8.6 ± 1.6% (n = 15, GluN2AC1 escape) and 1.8 ± 1.1% (n = 12, GluN2CC1 escape) of GluN1/GluN2AC1/GluN2C*C2 responses, indicating that approximately 90% of the NMDAR current was mediated by triheteromeric receptors. Together, these data suggest that PYD-106 selectively enhances diheteromeric GluN1/GluN2C receptors.
We subsequently tested 18 additional PYD analogs that were active at diheteromeric receptors containing GluN2C to determine whether the requirement of two GluN2C subunits was a feature of the entire class of PYD compounds. None of the PYD analogs with activity at diheteromeric GluN1/GluN2C receptors enhanced the responses of triheteromeric GluN1/GluN2A/GluN2C* receptors, suggesting that the ability to distinguish between the GluN2 composition of the receptors was a property of this class of modulator (Supplemental Table S3).
Mechanism of Action of Pyrrolidinones in GluN2C Modulation.
We evaluated the voltage-dependence of PYD-106 positive allosteric modulation to assess whether there are interactions with either the pore or process of ion permeation. Evaluation of the current-voltage curve showed that the reversal potential of GluN1/GluN2C receptor responses in oocytes was identical in the absence (−6.6 ± 1.9 mV, n = 4) and presence (−6.7 ± 1.9 mV, n = 4) of 100 μM PYD-106. Modulation was voltage independent, being 195 ± 4.2% of control at −40 mV and 236 ± 44% of control at +40 mV (n = 4, P > 0.05, t test). The EC50 value and degree of modulation by PYD-106 were compared in the absence and presence of 1 mM Mg2+, which exerts a voltage-dependent block of the channel. The EC50 for PYD-106 modulation in the absence of Mg2+ was 17 ± 1.2 μM (n = 7), only modestly different from the EC50 observed in the presence of 1 mM Mg2+ (23 ± 1.2 μM, n = 7, P < 0.05, t test). These data suggest that the actions of PYD-106 are largely voltage independent.
To gain more insight into the functional mechanism by which PYD-106 enhances GluN1/GluN2C responses, we evaluated the time course of macroscopic current responses recorded from NMDARs transiently expressed in HEK cells. PYD-106 increased the GluN1/GluN2C whole cell current responses to maximally effective concentrations of glutamate (100 μM) and glycine (30 μM) with an EC50 of 13 ± 1.0 μM and a Hill slope of 1.30 ± 0.04 (n = 10, Fig. 3, A and D). Selective modulation of GluN2C-containing NMDARs activated by a maximally effective concentration of glutamate and glycine was observed upon addition of 50 µM PYD-106 (224 ± 4.5% of control, n = 6) compared with weak inhibition of GluN1/GluN2A (88 ± 2.7% of control, n = 5), GluN1/GluN2B (81 ± 1.2% of control, n = 6), and GluN1/GluN2D (81 ± 1.0% of control, n = 6) NMDARs expressed in HEK cells. After rapid removal of glutamate in the presence of 50 μM PYD-106, the time constants for a dual exponential function fitted to the current deactivation time course (τfast = 67 ± 17 ms, %fast = 83 ± 3%, τslow = 260 ± 32 ms, n = 5) were not detectably different from those observed in the absence of PYD-106 (τfast = 62 ± 15 ms %fast = 74 ± 10%, τslow = 295 ± 2 ms, Fig. 3, B and C, n = 5, paired t test, P > 0.05). There was no significant effect on the relative proportion of the two components. However, τweighted was slightly faster in the presence of PYD-106 (τweighted = 204 ± 9 ms) compared with control (τweighted = 255 ± 8 ms, n = 5, paired t test, P < 0.05), perhaps reflecting a combined effect of slightly faster and less prominent slow component in PYD-106.
Fig. 3.
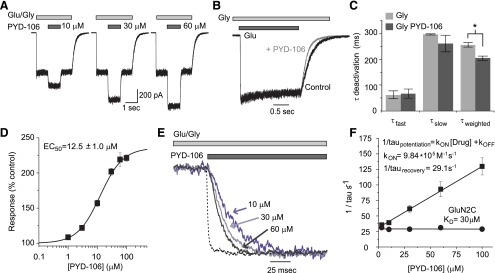
Mechanism of action of PYD modulation. (A) Whole-cell recordings of HEK cells expressing GluN1/GluN2C receptors activated with 100 μM glutamate and 30 μM glycine (Gly) showed concentration-dependent modulation by PYD-106. (B) Normalized current responses are superimposed to show the deactivation time course after removal of glutamate in the absence (black trace) and presence (gray trace) of PYD-106. (C) The deactivation time constant was fit with a dual exponential function. The time constants describing the independent fast and slow component were not significantly different in the presence or absence of PYD-106 (n = 5, t test, P > 0.05), although the mean τ weighted by the relative amplitude of each τ was significantly faster in the presence of PYD-106 (n = 5, t test, P < 0.05). (D) Composite concentration-effect curve for PYD-106 enhancement of maximal response in HEK cells transiently expressing the GluN1/GluN2C receptor. (E) Expansion of the onset of PYD-106 enhancement of the response at 10, 30, and 60 μM illustrates the normalized concentration-dependent time course. The dashed line is the fluid exchange rate around the cell, determined by switching between 150 mM NaCl with 150 mM KCl. Exchange time around a cell had a τ of 2.3 ± 0.3 ms with a 10–90% rise time of 3.4 ± 0.3 ms (n = 5), 3.8-fold faster than the onset of current increase at 100 μM PYD-106. (F) τONSET and τRECOVERY were used to estimate the kon and koff, and KD for PYD-106 was determined as koff /kon. The linear relationship between concentration and 1/τONSET suggests that the compounds act directly on the receptor rather than, for example, through a multistep intracellular signaling pathway.
The time course for the onset of PYD-106 enhancement of GluN1/GluN2C was rapid (τONSET = 9.0 ± 1.6 ms at 100 μM, n = 6–9) and could be well described by a single exponential function (Fig. 3E). We determined the upper limit of the solution exchange time around the cell by measuring the leak current during exchange of NaCl with KCl (Mott et al., 2001; Erreger and Traynelis, 2005). The time constant describing solution exchange was 2.3 ± 0.3 ms, fourfold faster than the onset of PYD-106 effects at 100 μM (Fig. 3E). This indicates that solution exchange was not a limiting step in assessing the association (kon) and dissociation (koff) rates of PYD-106. The reciprocal of the time constant for modulation of PYD-106 was linearly related to the concentration, with a slope corresponding to kon of 9.84 × 105 M-1⋅s−1. The dissociation rate koff was estimated from koff = 1/τrecovery to be 32 second−1, which was similar to that determined from the intercept of the relationship between concentration and 1/τONSET (koff-intercept = 29 ± 0.6 second−1). The dissociation constant (KD) for PYD-106 was estimated from koff /kon to be 30 μM (n = 6–9, Fig. 3F).
We subsequently evaluated the single channel mechanism of PYD-106 positive allosteric modulation of GluN1/GluN2C-containing NMDAR responses by analyzing individual single channel openings recorded from excised outside-out patches (Fig. 4). Outside-out patches containing rat recombinant GluN1/GluN2C receptors were activated by 100 μM glutamate and 30 μM glycine in the absence and presence of 100 μM PYD-106. Because all patches contained more than one active channel, data analysis was restricted to openings of individual channels. Two sublevels for the unitary currents were observed under both the control conditions (2.58 ± 0.11 and 3.31 ± 0.16 pA) as well as in the presence of PYD-106 (2.40 ± 0.25 and 3.30 ± 0.16 pA; Table 1). The chord conductance levels in the absence and presence of drug were not significantly different (Table 1; n = 4, P < 0.05, paired t test). The product of the number of channels in the patch and open probability (n Po) under control conditions (0.032 ± 0.015) was significantly increased in the presence of 100 µM PYD-106 (0.067±0.024, n = 4, P < 0.05, paired t test). The observed increase can be attributed partly to a significant increase in the mean open time (0.35 ± 0.07 to 0.48 ± 0.07 ms) in the presence of drug (n = 4, P < 0.05, paired t test). This change in mean open time reflects a shift toward longer duration openings, rather than a change in the time constants describing the dual component open time histogram (Fig. 4B, Table 1). These data suggest that PYD-106 stabilizes the open state of the receptor. Opening frequency was 0.086 ± 0.045 Hz in control and 0.159 ± 0.069 Hz in the presence of PYD-106 (n = 4, Table 1).
Fig. 4.

Representative excised outside-out patch recordings of HEK293 cells expressing the GluN1/GluN2C NMDAR activated by 100 μM glutamate and 30 μM glycine in the absence and presence of 100 μM PYD-106. The highlighted region is expanded below demonstrating the open (o) and closed (c) levels of the recordings. (B) The composite distributions of all the open and closed periods from four patches for the control (black) and for PYD-106 (gray) were fitted with two (open times) and five (shut times) exponential components (Table 1).
TABLE 1.
Effects of PYD-106 on single channel properties
All values are mean ± S.E.M., All patches contained 2 or more channels (n = 4 patches). Intervals either adjacent to or containing a double opening were omitted from analysis. nPo is the product of the number of channels in the patch and the open probability for an individual channel, which is the probability of at least one channel being open.
| Control | + 100 μM PYD-106 | |
|---|---|---|
| nPo | 0.032 ± 0.015 | 0.067 ± 0.024* |
| nPo (% control) | 100 | 257 ± 45%* |
| Opening frequency (Hz) | 0.086 ± 0.045 | 0.159 ± 0.069 |
| Opening frequency (% control) | 100 | 217 ± 34%* |
| Mean shut time | 24.3 ± 10.6 | 10.0 ± 3.2 |
| Mean open time | 0.35 ± 0.07 | 0.48 ± 0.07* |
| Open τ 1, ms (%) | 0.19 ± 0.07 (39) | 0.25 ± 0.11 (42) |
| Open τ 2, ms (%) | 0.46 ± 0.10 (61) | 0.65 ± 0.16 (58) |
| Amplitude 1 (pA) | 2.58 ± 0.11 | 2.40 ± 0.25 |
| Amplitude 2 (pA) | 3.31 ± 0.16 | 3.30 ± 0.16 |
| Conductance 1 (pS) | 32.3 ± 1.4 | 29.9 ± 3.1 |
| Conductance 2 (pS) | 41.4 ± 2.0 | 41.3 ± 2.0 |
P < 0.05, paired t test.
Interaction of Pyrrolidinones with Known Modulator Sites.
We recently described three new classes of nonselective allosteric modulators, which also act on the GluN1/GluN2C receptor, exemplified by CIQ (Mullasseril et al., 2010), QNZ-46 [(E)-4-[6-methoxy-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl]-benzoic acid] (Mosley et al., 2010), and DQP-1105 [4-[5-(4-bromophenyl)-3-(6-methyl-2-oxo-4- phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoic acid] (Acker et al., 2011). CIQ enhances GluN1/GluN2C receptor function through actions at the GluN2 pre-M1/M1 helices (Ogden and Traynelis, 2013), whereas QNZ-46 and DQP-1105 inhibit GluN1/GluN2C function through actions that apparently involve residues within the GluN2 S2 region of the ligand binding domain (Acker et al., 2011; Hansen and Traynelis, 2011). To assess whether PYD-106 might interact with either of these two sites or downstream mechanisms, we tested if positive allosteric modulation by PYD-106 would still be observed in the presence of maximally effective concentrations of CIQ, QNZ-46, or DQP-1105 on oocytes expressing the GluN1/GluN2C receptor. If the mechanisms of allosteric modulation for PYD and CIQ are independent, the effects of CIQ (8 μM) or PYD-106 (100 μM) measured individually should add to give a combined enhancement of the maximal response of 318% of control. The measured modulation by coapplication of CIQ and PYD-106 was 340 ± 14%, which was not significantly different from the estimated modulation of 318% (one-way ANOVA), suggesting that CIQ and PYD-106 do not compete for the same molecular determinants or share downstream mechanisms of action (Supplemental Figure S2). To further evaluate the possibility that CIQ and PYD-106 might share a portion of their respective binding sites, we mutated T578I in the GluN2C M1 domain, which is equivalent to the GluN2D T592I mutation that eliminates CIQ modulation (Mullasseril et al., 2010). We found that the mutation GluN2C(T578I) eliminates the actions of CIQ on the GluN1/GluN2C receptor (104 ± 2.0%, n = 4) but has no effect on PYD-1 (100 μM) enhancement of the maximal response (249 ± 11.3% of control, n = 4, P < 0.05).
A similar approach was used to estimate the inhibition of QNZ-46 or DQP-1105 on oocytes when coapplied with PYD-106. The response when QNZ-46 (20 μM) or DQP-1105 (20 μM) were coapplied with PYD-106 (100 μM) as a percent of control should be 108% and 46% if these compounds do not interact. Consistent with this idea, we observed 108 ± 2.3% and 39 ± 3.5% modulation compared with control for QNZ-46 or DQP-1105 in the presence of PYD-106, respectively (P > 0.05 for both, one-way ANOVA with Tukey’s post hoc test). Together, these data suggest that the structural determinants of PYD positive allosteric modulation are distinct from the elements required for CIQ, QNZ-46, and DQP-1105 modulation (Supplemental Figure S2).
Only one other ligand, DCS, is known to show selectivity in its actions as a coagonist for NMDARs at the glycine site for GluN1/ GluN2C over GluN1/GluN2A, GluN1/GluN2B, and GluN1/GluN2D (Sheinin et al., 2001; Dravid et al., 2010). A maximally effective concentration of DCS (100 μM) induces more current at GluN1/GluN2C than a maximally effective concentration of glycine (IDCS/IGLYCINE = 180 ± 4.9%, n = 6). By contrast, a maximally effective concentration of DCS produces a smaller response than glycine at GluN1/GluN2A, GluN1/GluN2B, GluN1/GluN2D receptors (Sheinin et al., 2001). We recently described structural determinants of the subunit-selective actions of DCS at the dimer interface between GluN1 and GluN2C ligand binding domains (Dravid et al., 2010). To assess whether DCS and PYD share either common structural determinants or similar mechanisms downstream of binding, we determined whether the actions of the PYD analogs and DCS at GluN1/GluN2C were additive. In this experiment, PYD-106 (100 μM) applied in the presence of both glutamate (100 μM) and DCS (100 μM, a saturating concentration) increased the maximal response to 172 ± 8.5% (n = 6). This reflects only a slight decrease of PYD-106 modulation in DCS compared with glycine evaluated in the same oocytes (207 ± 5.7% of control, n = 6, paired t test, P < 0.05). Thus, the actions of PYD-106 and DCS are largely additive, suggesting they do not share an overlapping site or mechanism of action.
Molecular Determinants of Pyrrolidinones.
We constructed a series of GluN2A-GluN2C chimeric subunits to identify the regions of GluN2C necessary for pyrrolidinone modulation (Supplemental Table S1). For this extensive mutagenesis screen, we used a close analog of PYD-106 that can be more readily synthesized (PYD-1, EC50 19 ± 1.1 μM), which increased the maximal response to 231 ± 10% of control (n = 14). Xenopus laevis oocytes were used to express and test whether the chimeric receptors show altered responses to 100 μM PYD-1 compared with wild-type receptors. Figure 5A shows that chimeras GluN2A (2C ATD), GluN2A (2C ATD-L0), and GluN2A (2C L0-S2) did not transfer modulation of PYD-1 to the GluN2A subunit. By contrast, the gain-of-function GluN2A(2C ATD-L0-S1) chimera was sensitive to PYD-1 (151 ± 2.2% of control, n = 8), suggesting that the ATD, L0, and S1 domains are required for PYD-1 modulation. To determine if all three regions are necessary, potential loss-of-function chimeras were generated individually by substituting the ATD, L0, and S1 of the GluN2A subunit into GluN2C. Replacement of the GluN2C ATD or S1 individually with the corresponding region from GluN2A eliminated modulation of GluN2C by PYD-1. Substitution of GluN2A L0 into GluN2C significantly decreased modulation (Fig. 5B). These data suggest that the ATD and S1 domains of GluN2C-containing receptors might harbor a binding pocket for PYD-1.
Fig. 5.
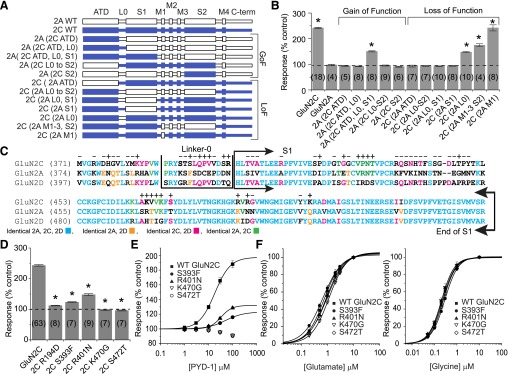
Structural determinants of PYD modulation. (A) Diagram of the gain-of-function (GoF) and loss-of-function (LoF) chimeric GluN2 subunits that were evaluated. (B) The change in the response to 100 μM glutamate and 30 μM glycine by 100 μM PYD-1 is shown as a percent of the control response (taken as 100%). The data from the chimera subunits suggest that the ATD, L0, and S1 regions of the GluN2C subunit are required for PYD modulation (*P < 0.05 compared with GluN2A, ANOVA, Dunnett’s post hoc, number of oocytes in parentheses). (C) Sequence alignment of the GluN2C, GluN2A, and GluN2D subunits illustrates the identity between the three subunits. A total of 61 mutations were constructed in the ATD, L0, and S1 regions, and the effects of each mutation on PYD-1 modulation is indicated by a “+” (P < 0.05, ANOVA) or “−” (not statistically significant). All tested mutations except for R194D, E341D, G342N, and F345L are indicated. (D) The modulation by 100 μM PYD-1 of the response in oocytes to 100 μM glutamate and 30 μM glycine is shown as a percent of control for wild-type (WT) GluN1/GluN2C and GluN2C mutants that showed the strongest effects (P < 0.05 against wild-type GluN2C, ANOVA, post hoc Dunnett’s). Table 2 gives the degree of PYD-1 modulation observed for all mutations. (E) PYD-1 concentration-effect curves show that S393F and R401N decreased the maximum modulation and PYD-1 EC50, whereas K470G and S472T mutations caused a complete loss of modulation by PYD-1 at all concentrations tested. Glutamate (F) and glycine (G) concentration-effect curves for these same mutations show minimal or no changes in EC50 values.
To identify the residues necessary for modulation, 61 GluN2C point mutations were constructed in the ATD, L0, and S1 domains of the GluN2C subunit (Table 2). Sequence alignment of the GluN2A/C/D subunits was used to identify residues that were candidates for mutation on the basis of unique identity in GluN2C or a position near or within L0 (i.e., the ATD-S1 linker) (Fig. 5C). Point mutations S393F, R401N, and K467S significantly decreased modulation by 50 μM PYD-1 (123 ± 1.5% of control, 148 ± 3.2% and 164 ± 2.2%, respectively, ANOVA, P < 0.05, n = 7–11). Interestingly, mutations K470G and S472T eliminated all effects of PYD-1. Several additional mutations (Q395D, V397E, V468R, V469I) significantly enhanced the effects of PYD-1, which increased the maximal responses up to 301–324% (n = 4–8, ANOVA, P < 0.0001, Table 2). Evaluation of the PYD-1 concentration-effect curves showed that the S393F and R401N mutations decreased the potency to 90 ± 9.5 and 36 ± 0.7 μM and decreased the enhancement of the maximal current response to 117 ± 1.2 and 136 ± 1.5%, respectively.
TABLE 2.
Structural determinants of PYD modulation
Response as a percent of control are shown for 100 μM PYD-1 coapplied with 100 μM glutamate and 30 μM glycine.
| GluN2C Mutation | ITEST/ICONTROL | n | GluN2C Mutation | ITEST/ICONTROL | n |
|---|---|---|---|---|---|
| % | % | ||||
| Wild type | 247 ± 2.3 | 63 | P428R# | 208 ± 6.0* | 8 |
| R194D# | 112 ± 1.1* | 8 | N429D# | 182 ± 8.0* | 5 |
| E341D | 217 ± 4.7 | 4 | T430S# | 203 ± 3.2* | 7 |
| G342N | 259 ± 4.2 | 4 | R435K | 244 ± 3.8 | 5 |
| F345L | 262 ± 1.4 | 3 | Q436F | 244 ± 8.1 | 6 |
| M371V | 263 ± 5.4 | 7 | S437V | 274 ± 16 | 4 |
| R374S | 267 ± 5.7 | 6 | N438F | 244 ± 9.5 | 5 |
| D376E | 239 ± 6.1 | 8 | H439I | 258 ± 3.9 | 8 |
| H377N | 276 ± 7.9* | 6 | T440N | 242 ± 4.7 | 3 |
| G378Q | 238 ± 7.2 | 6 | F441N | 250 ± 4.0 | 5 |
| V379T | 229 ± 9.9 | 6 | S443T | 262 ± 6.7 | 6 |
| Y381S | 263 ± 11.2 | 6 | G444N | 238 ± 7.7 | 8 |
| M382L | 242 ± 5.4 | 4 | D445N | 259 ± 6.2 | 6 |
| K383R | 217 ± 6.9* | 6 | L446E | 242 ± 6.5 | 6 |
| S391K | 277 ± 2.8 | 3 | T447G | 283 ± 6.9* | 6 |
| T392S | 273 ± 7.1 | 7 | P448M | 254 ± 6.3 | 4 |
| S393F | 123 ± 1.5* | 7 | Y449N | 246 ± 10.0 | 5 |
| L394S | 252 ± 4.9 | 6 | A466S# | 184 ± 4.3* | 6 |
| Q395D | 314 ± 9.4* | 6 | K467S# | 164 ± 2.2* | 1 |
| P396C | 215 ± 8.8* | 5 | V468R | 324 ± 8.6* | 6 |
| V397E | 301 ± 11.1* | 8 | V469I | 322 ± 4.3* | 4 |
| V398P | 257 ± 3.4 | 7 | K470G# | 97 ± 1.3* | 7 |
| S400D | 194 ± 4.9* | 12 | S472T# | 96 ± 1.6* | 7 |
| R401N# | 147 ± 3.2* | 9 | R486K | 275 ± 4.9* | 8 |
| T404S | 233 ± 8.2 | 8 | R488N | 276 ± 5.3* | 6 |
| V405I | 255 ± 4.5 | 8 | G489N | 257 ± 3.1 | 5 |
| A406V | 262 ± 3.1 | 8 | Y499V | 265 ± 3.3 | 7 |
| R411A | 238 ± 6.6 | 6 | K501Q | 281 ± 3.6* | 8 |
| S418D | 253 ± 8.2 | 7 | K470D# | 89 ± 2.0* | 8 |
| P419I | 190 ± 3.5* | 10 | K470T# | 102 ± 4.0* | 8 |
| G422L | 237 ± 4.2 | 8 | K470W# | 102 ± 1.3* | 6 |
| G424E# | 189 ± 2.8* | 5 | S472A# | 134 ± 2.8* | 7 |
| G425T | 240 ± 2.5 | 8 | S472R# | 90 ± 3.1* | 7 |
| V427I# | 294 ± 5.2* | 6 | S472E# | 98 ± 2.2* | 6 |
P < 0.05 by ANOVA, Bonferroni post hoc; #Residues that form part of the proposed PYD-106 binding pocket and have a significant effect on PYD-106 modulation.
The glutamate EC50 for wild-type GluN1/GluN2C receptors is 0.75 ± 0.08 (n = 5), which is not significantly different from that determined for the GluN2C mutations S393F (0.68 ± 0.08 μM, n = 3), R401N (0.90 ± 0.05 μM, n = 7), and S472T (1.0 ± 0.1 μM, n = 8) but is modestly different from K470G (1.2 ± 0.1 μM, n = 7, P < 0.05, ANOVA, Dunnett’s post hoc, Fig. 5F). The glycine EC50 for wild-type GluN1/GluN2C receptors is 0.23 ± 0.01 (n = 8), which is not significantly different from S393F (0.29 ± 0.01 μM, n = 3), R401N (0.21 ± 0.01 μM, n = 7), K470G (0.22 ± 0.03 μM, n = 6), or S472T (0.26 ± 0.02 μM, n = 6, ANOVA, post hoc Dunnett’s; Fig. 5F). Thus, these mutations do not cause substantive changes to agonist potency or receptor function.
Because K470G and S472T eliminated the effects of PYD analogs, we substituted additional residues at these positions to further evaluate the effects of side chain composition, size, polarity, and hydrogen bonding capability. The GluN2C mutations K470D, K470T, K470W, S472R, and S472E all blocked PYD allosteric modulation completely (Table 2). Only one mutation (S472A) permitted minimal modulation (134 ± 2.8% of control, n = 6–8), suggesting that these two residues are critically positioned to control the actions of PYD analogs.
We subsequently evaluated this region in a homology model of GluN1/GluN2C built from the GluN1/GluN2B crystal structure, which is thought to represent an inhibitory conformational state of the receptor (see Supplemental Data) (Karakas and Furukawa, 2014; Lee et al., 2014). This suggests the structure of the modeled GluN1/GluN2C receptor may also represent a similar inactive conformation. The modeled structure shows extensive interactions between the ATD and the S1 region of the ligand binding domain, which our mutagenesis data suggest harbors the PYD binding site (Fig. 6, A and B). Inspection of the structure between the ATD and S1 portion of the ligand binding domain revealed a small cavity of sufficient volume to act as a binding site for PYD analogs. A total of 35 residues form a pocket located between the ATD (R2) and ligand binding domains (LBD)(S1) (Karakas and Furukawa, 2014), which shows 43% sequence identity between the GluN2A-D subunits. Interestingly, 10 of 24 residues (44%, Table 2) that were found to markedly alter the modulation by PYD analogs in the mutagenesis screen formed part of the lining of this pocket (Fig. 6C). Five additional mutations with significant effects resided in the nearby ATD-S1 linker within 12–14 Å of the cavity. These data support the idea that this cavity is involved in the actions of PYD.
Fig. 6.

Site of action of PYD positive allosteric modulators. (A and B) A homology model of the GluN1/GluN2C receptor was based on the GluN1-1a/GluN2B crystal structure (see Supplemental Data) (Karakas and Furukawa, 2014; see Materials and Methods). (C) A depiction of residues in close proximity to the proposed PYD binding pocket of GluN2C residing between the ATD and LBD. Residues in blue and green were identified using a mutational screen. Blue-colored residues represent mutations that significantly decreased PYD modulation of the maximal current response. Residues colored green increased modulation by PYD of the maximal current response. The residue colored red was selected from PYD binding poses to test; mutations at this site significantly decrease the effect of PYD. (D) A docking pose of PYD-106 in the binding pocket is shown. Panels (C) and (D) are viewed form the same angle. Protein Data Bank (PDB): homology model of the GluN2C subunit based on the GluN2B crystal structure (see Karakas and Furukawa, 2014).
To test whether PYD modulators can fit within this newly identified pocket, we docked PYD-106 into this site on the GluN2C subunit (Fig. 6D). The resulting docking pose was then used to identify additional residues within the pocket that could be central to the binding of PYD-106. The docked pose suggested that the mutation GluN2C R194D should perturb PYD-106 binding, and thus we examined the effects of this mutation on PYD106 modulation. We observed that GluN1/GluN2C R194D nearly eliminated the actions of PYD-106 (112 ± 1.1%, n = 8). These data further support the identification of a binding pocket in the homology model of GluN2C based on the crystal structure of the GluN2B subunit (Karakas and Furukawa, 2014; Lee et al., 2014).
Discussion
We describe here the site and mechanism of action of the first class of selective positive allosteric modulators of GluN2C-containing NMDARs, which do not enhance the maximal response at GluN2A, GluN2B, or GluN2D. We also show this class of modulator has the unique ability to sense the composition of the tetrameric assembly, acting on receptors with two copies of GluN2C but not receptors with a single copy of GluN2C. Furthermore, the compound series shows minimal activity when residues encoded by the alternative exon-5 are included in the GluN1 ATD. The subunit selectivity of this class of compounds suggests it should be useful in identifying NMDARs that lack GluN1 exon-5 and contain two copies of GluN2C throughout the CNS.
Recently, several new NMDAR modulators with novel candidate binding sites were identified (Ogden and Traynelis, 2011; Monaghan et al., 2012). Among these, the GluN2C/D potentiator CIQ and related tetrahydroisoquinolines were proposed to interact with the extracellular end of the M1 transmembrane domain and pre-M1 cuff helix (Ogden et al., 2013). The GluN2C/D inhibitors QNZ46 and the DQP series appear to involve residues in the GluN2 S2 region of the agonist binding domain (Acker et al., 2011; Hansen and Traynelis, 2011). TCN-201 binds within the dimer interface between GluN1 and GluN2A agonist binding domains (Hansen et al., 2012). In addition, ifenprodil is a GluN2B-selective negative allosteric modulator that binds at the GluN1-GluN2B ATD heterodimer interface (Karakas and Furukawa, 2014). Several classes of positive and negative UBP ligands appear to interact with the ligand binding domain (Costa et al., 2010). The neurosteroid pregnanolone sulfate inhibits all NMDARs, with the highest potency for GluN2C- and GluN2D-containing receptors (Malayev et al., 2002; Petrovic et al., 2005). By contrast, pregnenolone sulfate shows positive allosteric modulation of GluN2A- and GluN2B-containing NMDARs and negative allosteric modulation of GluN2C- and GluN2D-containing NMDARs (Malayev et al., 2002). Pregnenolone sulfate enhancement of GluN2B receptor function has been suggested to involve helices J/K in the S2 portion of the ligand binding domain in addition to residues near the M4 transmembrane helix (Jang et al., 2004). Negative allosteric modulation by pregnanolone sulfate has been suggested to involve the S2 region of the ligand binding domain (Petrovic et al., 2005). Thus, the molecular determinants of both positive and negative allosteric regulation of NMDAR function by neurosteroids are distinct from regions of the receptor proposed to interact with PYD (e.g., Fig. 6). In addition, our data suggest that the selectivity and structural determinants for the PYD class of NMDAR modulators are distinct from other known modulators.
The potential site for PYD function at the S1-ATD interface has several interesting features. For example, the ATD-S1 linker region is only weakly conserved among GluN2 subunits, and not previously known as a site of action for any modulator active within the glutamate receptor family. The site clearly exists in three crystal structures independently solved (Karakas and Furukawa, 2014; Lee et al., 2014) and may well be a feature of the subunit family, raising the possibility that new classes of ligands may act at this pocket in other GluN2 subunits. The relatively fast time course for the onset and recovery from positive allosteric modulation suggests that the extracellular binding site for PYD is readily accessible. Moreover, the region between the ATD and LBD is well positioned to influence a wide range of channel properties including agonist EC50, deactivation time course and open probability (Gielen et al., 2009; Yuan et al., 2009; Hansen et al., 2013).
Within this new potential site, our data suggest specific roles for several of the residues lining the pocket. The interactions described here are those proposed for the R enantiomer of PYD-106, because its docking pose shows a more consistent correlation with the mutational data than the S enantiomer (not shown). Whereas the two enantiomers show significantly different biologic activities (Zimmerman et al., 2014), the absolute configuration of the most active enantiomer has yet to be determined. Our models show that Ser472 (GluN2C) forms a hydrogen bond with His402, which in turn hydrogen bonds with Asp474, perhaps stabilizing the ATD and LBD interface. Moreover, these interactions may allow hydrogen bond formation between adjacent residues Tyr473 and Ile475 and the acetyl and methyl ester moieties of PYD-106, respectively. The backbone of three residues at which mutations perturb PYD-106 modulation (Pro428, Asn429, and Thr430) form part of the binding pocket of the indole moiety in PYD-106. The latter is predicted to form a hydrogen bond with Pro428, which is retained in models of GluN2C P428R, a result that may explain the modest reduction in potency observed with this mutation. The similar modest decrease in modulation for GluN2C N429D might be explained by the loss of hydrogen bonding with Asp460 of the LBD. GluN2C A466S also reduces PYD modulation modestly, perhaps by influencing the orientation of the phenol ring of Tyr473. Interestingly, one of the 10 GluN2C mutations lining the pocket (V427I) increases the degree of modulation, which we speculate may alter the interaction between the ATD and LBD due to steric effects of the larger Ile side chain. The loss of PYD modulation by mutating Lys470 could be due to a loss of its interaction with Asp220 (ATD), which we suggest may stabilize the ATD and LBD interface. It will be important in the future to determine the absolute configuration of the more active enantiomer of PYD-106 as well as details of its binding interactions within the ATD-LBD domain interface of GluN2C.
In terms of mechanism, single channel analysis of the GluN1/GluN2C shows that PYD-106 increases the open probability and mean open time. This suggests that the compound increases receptor-mediated currents and agonist efficacy in part by stabilization of the open state, which prolongs the channel open periods. Enhancement of receptor open frequency and open duration by molecules binding to the ATD-LBD interface is consistent with emerging understanding of control of receptor function by the ATD (Hansen et al., 2010; Zhu et al., 2013). The strong influence that the ATD has on NMDAR gating can also now be understood through the extensive contacts between the ATD and ligand binding domain, as observed in the NMDAR crystal structure (Karakas and Furukawa, 2014; Lee et al., 2014). It is intriguing that the PYD modulators that act at this site require two GluN2C subunits to be present in the receptor complex. It remains unclear whether this reflects the need for modification of the receptor conformation driven by the binding of molecules at two sites across the region of twofold symmetry, or whether the nature of the pocket itself is perturbed when a GluN2 subunit other than GluN2C is present in the receptor complex. This observation emphasizes the need to consider the actions of allosteric modulators on triheteromeric receptors and provides a new precedent illustrating the potential for new pharmacological probes that sense receptor stoichiometry. Such probes could become useful tools for evaluating receptor composition. For example, we predict that PYD will not be active at cerebellar granule neurons, which express GluN1 exon-5 (Laurie and Seeburg, 1994; Laurie et al., 1995) in addition to GluN1/GluN2A/GluN2C triheteromeric receptors (Chazot et al., 1994; Cathala et al., 2000; Lu et al., 2006). However, this probe could be used to assess subunit stoichiometry of receptors containing GluN2C in other neurons that lack GluN1 exon-5. In summary, the identification of the site and mechanism of action of this series at the ATD-LBD interface should spur new work evaluating whether this site in other NMDAR subunits can bind to allosteric modulators and provides new tools with which to study neuronal NMDAR subunit composition.
Supplementary Material
Acknowledgments
The authors thank the Custom Cloning Core Facility at Emory University for constructing a subset of the chimeras and point mutations reported used in this study and Phuong Le, Jing Zhang, and Anel Tankovic for excellent technical assistance.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ANOVA
analysis of variance
- ATD
amino terminal domain
- CIQ
3-chlorophenyl)(6,7-dimethoxy-1-[(4- methoxyphenoxy)methyl]-3,4-dihydroisoquinolin-2(1H)-yl)methanone
- CNS
central nervous system
- DCS
d-cycloserine
- DQP-1105
4-[5-(4-bromophenyl)-3-(6-methyl-2-oxo-4- phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoic acid
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- HEK
human embryonic kidney cell
- LBD
ligand-binding domain
- NMDA
N-methyl-d-aspartate
- PDB
Protein Data Bank
- PYD
pyrrolidinone
- PYD-1
methyl 4-(1-(2-(1H-indol-3-yl)ethyl)-3-acetyl-4-hydroxy-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate
- PYD-106
methyl 4-(3-acetyl-4-hydroxy-1-(2-(2-methyl-1H-indol-3-yl)ethyl)-5-oxo-2,5-dihydro-1H-pyrrol-2-yl)benzoate
- QNZ-46
(E)-4-[6-methoxy-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl]-benzoic acid
- SQRT
square root
- TMD
transmembrane domain
Authorship Contributions
Participated in research design: Khatri, Burger, Swanger, Hansen, Zimmerman, Karakas, Liotta, Furukawa, Snyder, Traynelis.
Conducted experiments: Khatri, Burger, Hansen, Swanger, Zimmerman, Karakas.
Contributed new reagents or analytic tools: Hansen, Zimmerman, Karakas, Liotta, Furukawa.
Performed data analysis: Khatri, Burger, Snyder, Traynelis.
Wrote or contributed to the writing of the manuscript: Khatri, Burger, Swanger, Hansen, Zimmerman, Karakas, Liotta, Furukawa, Snyder, Traynelis.
Footnotes
This work was supported by the National Institutes of Health National Institute of Neurologic Disorders and Stroke [Grants F32-NS078873, R01-NS065371], National Institute of Mental Health [Grant R21-MH094525], National Institute of General Medicine [Grants P20-GM103546, R01-GM105730], and the Michael J Fox Foundation.
Several of the authors (D.C.L., J.P.S., S.F.T., S.Z.) are coinventors on Emory University-owned intellectual property, board members (D.C.L.), or paid consultants for companies developing NMDAR modulators (S.F.T., K.B.H.).
 This article has supplemental material available at mol.aspetjournals.org.
This article has supplemental material available at mol.aspetjournals.org.
References
- Acker TM, Yuan H, Hansen KB, Vance KM, Ogden KK, Jensen HS, Burger PB, Mullasseril P, Snyder JP, Liotta DC, et al. (2011) Mechanism for noncompetitive inhibition by novel GluN2C/D N-methyl-D-aspartate receptor subunit-selective modulators. Mol Pharmacol 80:782–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. (1994) Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J Comp Neurol 347:150–160 [DOI] [PubMed] [Google Scholar]
- Balu DT, Coyle JT. (2011) Neuroplasticity signaling pathways linked to the pathophysiology of schizophrenia. Neurosci Biobehav Rev 35:848–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettini E, Sava A, Griffante C, Carignani C, Buson A, Capelli AM, Negri M, Andreetta F, Senar-Sancho SA, Guiral L, et al. (2010) Identification and characterization of novel NMDA receptor antagonists selective for NR2A- over NR2B-containing receptors. J Pharmacol Exp Ther 335:636–644 [DOI] [PubMed] [Google Scholar]
- Binshtok AM, Fleidervish IA, Sprengel R, Gutnick MJ. (2006) NMDA receptors in layer 4 spiny stellate cells of the mouse barrel cortex contain the NR2C subunit. J Neurosci 26:708–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathala L, Misra C, Cull-Candy S. (2000) Developmental profile of the changing properties of NMDA receptors at cerebellar mossy fiber-granule cell synapses. J Neurosci 20:5899–5905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazot PL, Coleman SK, Cik M, Stephenson FA. (1994) Molecular characterization of N-methyl-D-aspartate receptors expressed in mammalian cells yields evidence for the coexistence of three subunit types within a discrete receptor molecule. J Biol Chem 269:24403–24409 [PubMed] [Google Scholar]
- Chen PE, Geballe MT, Katz E, Erreger K, Livesey MR, O’Toole KK, Le P, Lee CJ, Snyder JP, Traynelis SF, et al. (2008) Modulation of glycine potency in rat recombinant NMDA receptors containing chimeric NR2A/2D subunits expressed in Xenopus laevis oocytes. J Physiol 586:227–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RJ, Johnson JW. (2006) NMDA receptor NR2 subunit dependence of the slow component of magnesium unblock. J Neurosci 26:5825–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Volianskis A, Bannister N, France G, Hanna L, Mercier M, Tidball P, Fang G, Irvine MW, Costa BM, et al. (2013) The NMDA receptor as a target for cognitive enhancement. Neuropharmacology 64:13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sigworth F. (1995). Fitting In: Sakmann B, Neher E (eds). Single-channel Recordings, edn. New York, NY: Plenum Press. pp 483–587 [Google Scholar]
- Costa BM, Irvine MW, Fang G, Eaves RJ, Mayo-Martin MB, Laube B, Jane DE, Monaghan DT. (2012) Structure-activity relationships for allosteric NMDA receptor inhibitors based on 2-naphthoic acid. Neuropharmacology 62:1730–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa BM, Irvine MW, Fang G, Eaves RJ, Mayo-Martin MB, Skifter DA, Jane DE, Monaghan DT. (2010) A novel family of negative and positive allosteric modulators of NMDA receptors. J Pharmacol Exp Ther 335:614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravid SM, Burger PB, Prakash A, Geballe MT, Yadav R, Le P, Vellano K, Snyder JP, Traynelis SF. (2010) Structural determinants of D-cycloserine efficacy at the NR1/NR2C NMDA receptors. J Neurosci 30:2741–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113 DOI: 10.1186/1471-2105-5-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, Milh M, Kortüm F, Fritsch A, Pientka FK, et al. (2010) Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet 42:1021–1026 [DOI] [PubMed] [Google Scholar]
- Erreger K, Geballe MT, Kristensen A, Chen PE, Hansen KB, Lee CJ, Yuan H, Le P, Lyuboslavsky PN, Micale N, et al. (2007) Subunit-specific agonist activity at NR2A-, NR2B-, NR2C-, and NR2D-containing N-methyl-D-aspartate glutamate receptors. Mol Pharmacol 72:907–920 [DOI] [PubMed] [Google Scholar]
- Erreger K, Traynelis SF. (2005) Allosteric interaction between zinc and glutamate binding domains on NR2A causes desensitization of NMDA receptors. J Physiol 569:381–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Feldmeyer D, Takahashi T, Cull-Candy SG. (1994) NMDA-receptor channel diversity in the developing cerebellum. Nature 368:335–339 [DOI] [PubMed] [Google Scholar]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49:6177–6196 [DOI] [PubMed] [Google Scholar]
- Gielen M, Siegler Retchless B, Mony L, Johnson JW, Paoletti P. (2009) Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature 459:703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DC, Cather C, Gottlieb JD, Evins AE, Walsh J, Raeke L, Otto MW, Schoenfeld D, Green MF. (2008) Once-weekly D-cycloserine effects on negative symptoms and cognition in schizophrenia: an exploratory study. Schizophr Res 106:320–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DC, Tsai G, Manoach DS, Coyle JT. (1995) Dose-finding trial of D-cycloserine added to neuroleptics for negative symptoms in schizophrenia. Am J Psychiatry 152:1213–1215 [DOI] [PubMed] [Google Scholar]
- Gottlieb JD, Cather C, Shanahan M, Creedon T, Macklin EA, Goff DC. (2011) D-cycloserine facilitation of cognitive behavioral therapy for delusions in schizophrenia. Schizophr Res 131:69–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett PJ, Standaert DG. (2004) Rationale for and use of NMDA receptor antagonists in Parkinson’s disease. Pharmacol Ther 102:155–174 [DOI] [PubMed] [Google Scholar]
- Hansen KB, Furukawa H, Traynelis SF. (2010) Control of assembly and function of glutamate receptors by the amino-terminal domain. Mol Pharmacol 78:535–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Traynelis SF. (2012) Subunit-selective allosteric inhibition of glycine binding to NMDA receptors. J Neurosci 32:6197–6208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Yuan H, Traynelis SF. (2014) Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron 81:1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Tajima N, Risgaard R, Perszyk RE, Jørgensen L, Vance KM, Ogden KK, Clausen RP, Furukawa H, Traynelis SF. (2013) Structural determinants of agonist efficacy at the glutamate binding site of N-methyl-D-aspartate receptors. Mol Pharmacol 84:114–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Traynelis SF. (2011) Structural and mechanistic determinants of a novel site for noncompetitive inhibition of GluN2D-containing NMDA receptors. J Neurosci 31:3650–3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton CJ, Paoletti P. (2005) Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron 46:261–274 [DOI] [PubMed] [Google Scholar]
- Hillman BG, Gupta SC, Stairs DJ, Buonanno A, Dravid SM. (2011) Behavioral analysis of NR2C knockout mouse reveals deficit in acquisition of conditioned fear and working memory. Neurobiol Learn Mem 95:404–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Boulter J, Maron C, Beasley L, Sullivan J, Pecht G, Heinemann S. (1993) Zinc potentiates agonist-induced currents at certain splice variants of the NMDA receptor. Neuron 10:943–954 [DOI] [PubMed] [Google Scholar]
- Ishii T, Moriyoshi K, Sugihara H, Sakurada K, Kadotani H, Yokoi M, Akazawa C, Shigemoto R, Mizuno N, Masu M, et al. (1993) Molecular characterization of the family of the N-methyl-D-aspartate receptor subunits. J Biol Chem 268:2836–2843 [PubMed] [Google Scholar]
- Jang MK, Mierke DF, Russek SJ, Farb DH. (2004) A steroid modulatory domain on NR2B controls N-methyl-D-aspartate receptor proton sensitivity. Proc Natl Acad Sci USA 101:8198–8203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia LV, Kalia SK, Salter MW. (2008) NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol 7:742–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan GB, Moore KA. (2011) The use of cognitive enhancers in animal models of fear extinction. Pharmacol Biochem Behav 99:217–228 [DOI] [PubMed] [Google Scholar]
- Káradóttir R, Cavelier P, Bergersen LH, Attwell D. (2005) NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 438:1162–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Furukawa H. (2014) Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 344:992–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karavanova I, Vasudevan K, Cheng J, Buonanno A. (2007) Novel regional and developmental NMDA receptor expression patterns uncovered in NR2C subunit-beta-galactosidase knock-in mice. Mol Cell Neurosci 34:468–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostakis E, Jang MK, Russek SJ, Gibbs TT, Farb DH. (2011) A steroid modulatory domain in NR2A collaborates with NR1 exon-5 to control NMDAR modulation by pregnenolone sulfate and protons. J Neurochem 119:486–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51:199–214 [DOI] [PubMed] [Google Scholar]
- Laube B, Hirai H, Sturgess M, Betz H, Kuhse J. (1997) Molecular determinants of agonist discrimination by NMDA receptor subunits: analysis of the glutamate binding site on the NR2B subunit. Neuron 18:493–503 [DOI] [PubMed] [Google Scholar]
- Laurie DJ, Putzke J, Zieglgänsberger W, Seeburg PH, Tölle TR. (1995) The distribution of splice variants of the NMDAR1 subunit mRNA in adult rat brain. Brain Res Mol Brain Res 32:94–108 [DOI] [PubMed] [Google Scholar]
- Laurie DJ, Seeburg PH. (1994) Regional and developmental heterogeneity in splicing of the rat brain NMDAR1 mRNA. J Neurosci 14:3180–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Lü W, Michel JC, Goehring A, Du J, Song X, Gouaux E. (2014) NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 511:191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J. (2012) Excitation, inhibition, local oscillations, or large-scale loops: what causes the symptoms of schizophrenia? Curr Opin Neurobiol 22:537–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. (2008) Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci 31:234–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Fu Z, Karavanov I, Yasuda RP, Wolfe BB, Buonanno A, Vicini S. (2006) NMDA receptor subtypes at autaptic synapses of cerebellar granule neurons. J Neurophysiol 96:2282–2294 [DOI] [PubMed] [Google Scholar]
- Malayev A, Gibbs TT, Farb DH. (2002) Inhibition of the NMDA response by pregnenolone sulphate reveals subtype selective modulation of NMDA receptors by sulphated steroids. Br J Pharmacol 135:901–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, McRory JE, Rehak R, et al. (2006) NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 439:988–992 [DOI] [PubMed] [Google Scholar]
- Monaghan DT, Irvine MW, Costa BM, Fang G, Jane DE. (2012) Pharmacological modulation of NMDA receptor activity and the advent of negative and positive allosteric modulators. Neurochem Int 61:581–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. (1994) Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12:529–540 [DOI] [PubMed] [Google Scholar]
- Mosley CA, Acker TM, Hansen KB, Mullasseril P, Andersen KT, Le P, Vellano KM, Bräuner-Osborne H, Liotta DC, Traynelis SF. (2010) Quinazolin-4-one derivatives: A novel class of noncompetitive NR2C/D subunit-selective N-methyl-D-aspartate receptor antagonists. J Med Chem 53:5476–5490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott DD, Erreger K, Banke TG, Traynelis SF. (2001) Open probability of homomeric murine 5-HT3A serotonin receptors depends on subunit occupancy. J Physiol 535:427–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullasseril P, Hansen KB, Vance KM, Ogden KK, Yuan H, Kurtkaya NL, Santangelo R, Orr AG, Le P, Vellano KM, et al. (2010) A subunit-selective potentiator of NR2C- and NR2D-containing NMDA receptors. Nat Commun 1:90 DOI: 10.1038/ncomms1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberg MM, Krystal JH, Tolin DF. (2008) A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry 63:1118–1126 [DOI] [PubMed] [Google Scholar]
- Ogden KK, Traynelis SF. (2011) New advances in NMDA receptor pharmacology. Trends Pharmacol Sci 32:726–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden KK, Traynelis SF. (2013) Contribution of the M1 transmembrane helix and pre-M1 region to positive allosteric modulation and gating of N-methyl-D-aspartate receptors. Mol Pharmacol 83:1045–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Newcomer JW, Farber NB. (1999) NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res 33:523–533 [DOI] [PubMed] [Google Scholar]
- Petrovic M, Sedlacek M, Horak M, Chodounska H, Vyklický L., Jr (2005) 20-oxo-5beta-pregnan-3alpha-yl sulfate is a use-dependent NMDA receptor inhibitor. J Neurosci 25:8439–8450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. (2008) An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol 28:631–637 [DOI] [PubMed] [Google Scholar]
- Prybylowski K, Rumbaugh G, Wolfe BB, Vicini S. (2000) Increased exon 5 expression alters extrasynaptic NMDA receptors in cerebellar neurons. J Neurochem 75:1140–1146 [DOI] [PubMed] [Google Scholar]
- Qian A, Buller AL, Johnson JW. (2005) NR2 subunit-dependence of NMDA receptor channel block by external Mg2+. J Physiol 562:319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh G, Prybylowski K, Wang JF, Vicini S. (2000) Exon 5 and spermine regulate deactivation of NMDA receptor subtypes. J Neurophysiol 83:1300–1306 [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815 [DOI] [PubMed] [Google Scholar]
- Salter MG, Fern R. (2005) NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 438:1167–1171 [DOI] [PubMed] [Google Scholar]
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27:221–234 [DOI] [PubMed] [Google Scholar]
- Sheinin A, Shavit S, Benveniste M. (2001) Subunit specificity and mechanism of action of NMDA partial agonist D-cycloserine. Neuropharmacology 41:151–158 [DOI] [PubMed] [Google Scholar]
- Siegler Retchless B, Gao W, Johnson JW. (2012). A single GluN2 subunit residue controls NMDA receptor channel properties via intersubunit interaction. Nat Neurosci 15: 406–413, S1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryavanshi PS, Ugale RR, Yilmazer-Hanke D, Stairs DJ, Dravid SM. (2014). GluN2C/GluN2D subunit-selective NMDA receptor potentiator CIQ reverses MK-801-induced impairment in prepulse inhibition and working memory in Y-maze test in mice. Br J Pharmacol 171(3):799–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Burgess MF, Zheng F, Lyuboslavsky P, Powers JL. (1998) Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J Neurosci 18:6163–6175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Hartley M, Heinemann SF. (1995) Control of proton sensitivity of the NMDA receptor by RNA splicing and polyamines. Science 268:873–876 [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich MH, Isacoff EY. (2007) Subunit counting in membrane-bound proteins. Nat Methods 4:319–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance KM, Hansen KB, Traynelis SF. (2012) GluN1 splice variant control of GluN1/GluN2D NMDA receptors. J Physiol 590:3857–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance KM, Simorowski N, Traynelis SF, Furukawa H. (2011) Ligand-specific deactivation time course of GluN1/GluN2D NMDA receptors. Nat Commun 2:294 DOI: 10.1038/ncomms1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Inoue Y, Sakimura K, Mishina M. (1992) Developmental changes in distribution of NMDA receptor channel subunit mRNAs. Neuroreport 3:1138–1140 [DOI] [PubMed] [Google Scholar]
- Wenzel A, Fritschy JM, Mohler H, Benke D. (1997) NMDA receptor heterogeneity during postnatal development of the rat brain: differential expression of the NR2A, NR2B, and NR2C subunit proteins. J Neurochem 68:469–478 [DOI] [PubMed] [Google Scholar]
- Williams K, Russell SL, Shen YM, Molinoff PB. (1993) Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron 10:267–278 [DOI] [PubMed] [Google Scholar]
- Wu LJ, Zhuo M. (2009) Targeting the NMDA receptor subunit NR2B for the treatment of neuropathic pain. Neurotherapeutics 6:693–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF. (2009) Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J Neurosci 29:12045–12058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stroebel D, Yao CA, Taly A, Paoletti P. (2013) Allosteric signaling and dynamics of the clamshell-like NMDA receptor GluN1 N-terminal domain. Nat Struct Mol Biol 20:477–485 [DOI] [PubMed] [Google Scholar]
- Zimmerman SS, Khatri A, Garnier-Amblard EC, Mullasseril P, Kurtkaya NL, Gyoneva S, Hansen KB, Traynelis SF, Liotta DC. (2014) Design, synthesis, and structure-activity relationship of a novel series of GluN2C-selective potentiators. J Med Chem 57:2334–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.