

Abstract
The generation of human induced pluripotent stem cells (hiPSC) from somatic cells has enabled the possibility to provide patient-specific hiPSC for cell-based therapy, drug discovery, and other translational applications. Two major obstacles in using hiPSC for clinical application reside in the risk of genomic modification when they are derived with viral transgenes and risk of teratoma formation if undifferentiated cells are engrafted. In this study, we report the generation of “footprint-free” hiPSC-derived astrocytes. These are efficiently generated, have anatomical and physiological characteristics of fully differentiated astrocytes, maintain homing characteristics typical of stem cells, and do not give rise to teratomas when engrafted in the brain. Astrocytes can be obtained in sufficient numbers, aliquoted, frozen, thawed, and used when needed. Our results show the feasibility of differentiating astrocytes from “footprint-free” iPSC. These are suitable for clinical cell-based therapies as they can be induced from patients' specific cells, do not require viral vectors, and are fully differentiated. “Footprint-free” hiPSC-derived astrocytes represent a new potential source for therapeutic use for cell-based therapy, including treatment of high-grade human gliomas, and drug discovery.
Introduction
Human induced pluripotent stem cells (hiPSC) have the ability to undergo unlimited self-renewal and to differentiate into all cell types, including ectodermal lineage, such as neurons and astrocytes [1]. We and others have recently shown that astrocytes can be efficiently differentiated from hiPSC with characteristics similar to those differentiated from human embryonic stem cells (hESC) [2–4]. Astrocytes are native to the central nervous system, provide tropic and trophic support to neurons, and are highly secretory cells. These characteristics make them great tools for studying neurological diseases and/or to be used for translational studies assessing drug efficiency and/or as cell-based therapy.
To translate these results to the clinical setting, hiPSC-derived vectors with safe clinical criteria need to be developed. In particular, efforts have been focused toward generation of insertion-free or “footprint-free” iPSC to avoid the potential risk of insertional mutagenesis in humans [5]. Whereas viruses successfully demonstrated the feasibility of reprogramming somatic cells to a pluripotent stem cell stage [6–8], they also caused insertional mutagenesis by viral vector integration raising caution for human clinical applications. To address the inherent safety issues surrounding viral vectors, a number of vectors emerged in the attempt to generate “footprint-free” iPSC. Some of these included the use of nonintegrating adenoviral vectors, transient transfection of plasmids as well as episomal vectors [2,9–12]. However, the reprogramming efficiency was 100-fold lower with these vectors and the resulting iPSC colonies still had to be screened for residual integration of portions of these vectors into the host cell genome. To improve on the frequency of reprogramming and ensure the complete removal of all exogenous DNA without adversely affecting the already low reprogramming efficiency, other techniques to generate “footprint-free” iPSC were developed, including the RNA virus [Sendai virus (SeV)] [13,14] and modified RNA (modRNA) [15–17]. The former uses single SeV RNA infection leading to a robust iPSC colony generation after 14–21 days; the SeV RNA is lost from the iPSC cytoplasm between expansion passages 5–8. The latter uses synthetic modified mRNA. Both methodologies generate iPSC colonies with high efficiency and pose no risk to any type of accidental insertional mutagenesis [11,18].
Cell-based therapy can be used for a variety of diseases. In particular, diseases that shelter themselves from conventional treatments, such as chemotherapy, are in need for such therapy. Human high-grade gliomas (hHGG) are the most common primary brain tumors and remain a clinical challenge with an average life expectancy of 14 months after state of the art surgical, radiation, and chemotherapy treatments [19–23]. The infiltrative pattern of hHGG combined with the difficulty of chemotherapy to cross the blood–brain barrier is the main reason for treatment failure [24–26] and sparked interest in exploring cell-based therapy. Clinical and experimental data show that glioma invasion occurs during current cytotoxic therapies [27], highlighting the need of developing vectors that can infiltrate the brain while carrying proapoptotic genes. Stem cell-derived astrocytes are highly desirable vectors as they have the potential for maintaining migrating capacity and, therefore, could offer advantages over other delivery vectors in the treatment of brain tumors. We have published that mouse ESC (mESC)-derived astrocytes can be successfully used as cell-based gene therapy for treatment of experimental HGG [23,28]. To translate these results to the clinical setting, patient-specific iPSC derived without viral vectors, that is, “footprint-free”, are needed.
In this study, we tested the feasibility of differentiating astrocytes from two “footprint-free” hiPSC lines developed in our stem cell core facility. Our results show that we can differentiate a highly pure astrocytic population from “footprint-free” hiPSC having anatomical and physiological characteristics of fully differentiated astrocytes, maintaining in vivo migrating characteristics typical of naive astrocytes and not giving rise to teratomas when engrafted in the brain.
Materials and Methods
Generation and maintenance of hiPSC
modRNA-derived hiPSC were generated from adult human dermal fibroblasts (ScienCell Research Laboratories) reprogrammed using the mRNA Reprogramming Kit supplemented with the microRNA Booster Kit (Stemgent) according to the manufacturer's instructions. Briefly, 5×104 fibroblasts were plated on Matrigel-coated wells in Dulbecco's modified Eagle's medium (DMEM)/10% fetal calf serum (FCS) media containing B18R supplement (day −1). After 24 h, media were aspirated and fibroblasts were preincubated for 2–4 h with 2 mL of fresh mouse embryonic fibroblast (MEF)-conditioned media containing 4 ng/mL pluriton supplement and 300 ng/mL B18R supplement. Fibroblasts were transfected with 3.5 μL/well of miRNA in the Stemfect™ (Stemgent) reagent and transfection buffer (day 0). After 24 h, media were changed to fresh MEF-conditioned media supplemented with 4 ng/mL pluriton supplement and 300 ng/mL B18R. Cells were transfected with 1 μg/well of mRNA cocktail in the Stemfect transfection buffer (day 1). Day 1 procedure was repeated for the following 3 days. On day 5, the procedure from day 0 was repeated. From day 6 to 11, the procedure from day 1 was repeated. On day 12, media were changed to fresh MEF-conditioned pluriton media supplemented with 4 ng/mL pluriton supplement and 300 ng/mL B18R. Between days 14–16, Tra-1-60-positive colonies were picked and expanded on MEFs in the presence of hESC media or cultured feeder free in the presence of mTESR (Stemcells Technologies).
SeV-derived hiPSC were generated from adult bone marrow cells (Reach Bio) and reprogrammed using the Sendai Virus Reprogramming Kit (Life Technologies) according to the manufacturer's instructions. Briefly, 2×105 actively growing fibroblasts were plated on gelatin-coated wells in DMEM/10% FCS media and infected overnight with the OCT4, SOX2, KLF4, and cMYC SeV at an MOI of 3. The next day media were changed and replaced with fresh DMEM/10% FCS media. Media were changed every other day until day 8. At day 8, cells were harvested and 5×104 or 1×105 cells were plated on MEFs in a 10-cm dish. The next day, DMEM/10% FCS media were gradually replaced by hESC media over the course of the next 2 days. From day 13 onward, hESC media were changed every other day until day 21. Between day 21 and 28, Tra-1-60-positive colonies were picked and expanded on MEFs in the presence of hESC media or cultured feeder free in the presence of mTESR (Stemcells Technologies).
Stem cell differentiation and cell culture
hESC H9 (WiCell; Research Institute) and hiPSC lines (SeV-derived iPSC line and modRNA-derived iPSC line) were differentiated following our previously published protocol [2]. Briefly, they were initially plated on Matrigel (BD Bioscience)-coated plates with mTeSR (Stemcells Technologies) in the presence of a Rock inhibitor (Stemgent). Embryoid bodies (EB) were generated by culturing clusters of cells in low attachment plates (Cornig Costar) in the presence of knockout serum replacement (Life Technologies). At day 6, EB were plated intact on a Matrigel-coated plate in the neurobasal medium containing N2 (Life Technologies) (2×), fibroblast growth factor-2 (FGF-2) (Life Technologies) (10 ng/mL), and epidermal growth factor (EGF; R&D Systems, Inc.) (20 ng/mL) for 3 days (day 6–9), followed by FGF+EGF+ciliary neurotrophic factor (CNTF; R&D Systems, Inc.) (20 ng/mL) for an additional 3 days (day 9–12), and finally, in FGF+CNTF (day 12–15) and in CNTF alone (day 15 onward). Neural tube-like rosettes were detached mechanically at day 15 of differentiation and cultured in the Matrigel-coated plate in the same medium. Noggin 500 ng/mL (R&D Systems, Inc.) was added to cultures for the first 12 days and SB431542 (Stemgent) (10 μM) was added from day 0 to 5 according to published protocols [29,30].
The hHGG cell line U87 (American Type Culture Collection) used for the in vitro and in vivo experiments was propagated in monolayer and cultured in DMEM, supplemented with 10% FCS (Corning, CellGro), 1% penicillin–streptomycin, and 1% l-glutamine (Gibco, Life Technologies) in a 37°C incubator supplemented with 5% CO2. Cells grew as an adherent monolayer and were maintained long term according to standard sterile cell culture techniques.
Immunocytochemistry and microscopy
Cells were fixed using 4% paraformaldehyde (PFA; Electron Microscopy) for 20 min, washed with phosphate-buffered saline (PBS), permeabilized using 0.5% Triton X in PBS, and blocked using 0.1% bovine serum albumin (BSA; Invitrogen), 10% goat (Chemicon Intl.), donkey (Jackson ImmunoResearch), or rabbit (Millipore) serum in PBS. Primary antibodies used for microscopy included Nestin (mouse, 1:200; R&D Systems, Inc.), glial fibrillary acid protein (GFAP) (rabbit 1:1,000; Millipore), A2B5 (mouse 1:200; Millipore), and Mushashi-1 (goat 1:200; Millipore). Secondary antibodies used were as follows: Alexa Fluor 488 (green)-conjugated anti-mouse (1:400; Life Technologies), Alexa Fluor 660-conjugated anti-goat (purple, 1:400; Life Technologies), and cy3-conjugated anti-rabbit IgG (red, for visualization of GFAP, 1:400; Millipore). Cells were counterstained with diamidophenylindole (DAPI; Sigma-Aldrich). Primary antibodies for flow cytometry included anti-human GFAP (hGFAP) rabbit polyclonal (1:200; Millipore) and anti-human A2B5 monoclonal (1:100; Millipore). Secondary antibodies used were Alexa Fluor 488-conjugated goat anti-rabbit and goat anti-mouse IgG (1:200; Life Technologies). Negative controls included isotype-specific primary serum and secondary antibodies.
Images of separate channels were acquired at 4×, 10×, and 20× magnification under fluorescence microscopy with a Leica DMI 4000B microscope and DFC340FX camera (Leica Microsystems) and an Olympus IX71 microscope and DP72 camera (Olympus America, Inc.) using the Leica Application suite software and the CellSens Standard software (Olympus) and exported to an imaging Adobe Photoshop software, where separate channel images were overlaid.
Physiologic iPSC-derived astrocyte characterization and glutamate assay
Aldolase c (ALDOC) and glutamate transporter 1 (GLT1) are considered physiologic markers as they are present only in fully functioning astrocytes capable of removing released glutamate from the synaptic cleft (GLT1) and performing glycolysis (ALDOC) [4]. For these experiments, quantitative real-time polymerase chain reaction (qRT-PCR) experiments were performed as summarized below.
Glutamate assay was carried out on iPSC-derived astrocytes at differentiation day 35. Human astrocytes (U87) and undifferentiated iPSC were used as control. Cells were homogenized in 100 μL of assay buffer and centrifugated at 13,000 g for 10 min to remove insoluble material, and spectrophotometry assay was carried out following the kit instruction (Abcam ab83389).
Reverse transcriptase polymerase chain reaction and quantitative PCR
To verify the lack of presence of the SeV in SeV-derived iPSC used in our experiments, we carried out a PCR using cDNA from day 8 SeV-infected cultures and SeV-derived iPSC after 25 passages with primers containing SeV genome sequences (SeV forward 5′ GGATCACTAGGT GATATCG AGC, SeV reverse 5′ ACCAGACAAGAGTTTAAG AGAT ATGTATC) (CytoTune-iPS reprogramming kit; Invitrogen).
Total RNA was extracted using a RNeasy kit (Qiagen) according to the manufacturer's instructions. All RNA preparations were spectrophotometrically quantified before reverse transcriptase (RT). The RT was performed on 1 μg of total RNA by using Super Script II (Invitrogen) and oligo-dT primers (Invitrogen) in a total volume of 20 μL.
qRT-PCRs were carried out on the Roche Light Cycler 480 in 384-well Optical Reaction Plates (Roche). The following program was applied: stage 1: 95°C for 1 min, stage 2: 95°C for 3 s and 60°C for 30 s, for 40 cycles. Additional dissociation curves of the products were created. The final reaction volume of 8 μL consisted of 4 μL of SYBR Green PCR Master Mix (Applied Biosystems), 0.25 μM of each primer (0.5 μL), 2 μL of cDNA (1:25 or 1:50 dilutions), and DNase/RNase-free water into the 8 μL volume. Each gene was analyzed in triplicate. Relative mRNA levels were calculated using the comparative delta Ct method and presented as a fold increase of the biological control (undifferentiated hESC or iPSC). mRNA levels of beta ACTIN genes were used as controls for normalization. All experiments done in triplicate; GLT1 and ALDOC in duplicate. The following primers were used: human Oct4 (hOCT4): forward 5′ F AACCTGGAGTTTGTGCCAGGGTTT, reverse 5′ R TGAACTTCACCTTCCCTCCAACCA; hGFAP: forward 5′ GTT GCT CCA GAC TGG GAC TG, reverse 5′ CTG CAA GCC CCA CCT AGA AG; human aldolase c (hALDOC): forward 5′ CTA ATT CAG CGG CTG CCT CC, reverse 5′ AAT AGA GAG CAG GGT CTG GGA; and human glutamate transporter (hGLT1): forward 5′ ATC CAG GCA ATC CCA AGC TCA AGA AC, reverse 5′ CGA AAG GTG ACA GGC AAA GTT CCA.
Magnetic-activated cell sorting
Glial-restricted progenitors from embryonic to adult human tissue have been immunomagnetically isolated according to the expression of A2B5. A2B5 is predominantly expressed in embryonic neural tissue and thus considered a marker for immature glial-committed precursors, defined as cells that give rise to glial types such as astrocytes and oligodentrocytes [31,32]. Cell plated on Matrigel were detached using 0.1 mM ethylenediaminetetraacetic acid (EDTA), resuspended in DMEM/10% FCS, and counted. The cell suspension was centrifuged at 300 g for 10 min, the supernatant was aspirated, and the pellet resuspended in 60 μL of magnetic-activated cell sorting (MACS) buffer per 107 total cells. After adding to this suspension 20 μL of FcR Blocking Reagent, cells were incubated for 10 min in the refrigerator (2–8°C). After 10 min, cells were incubated for 15 min in the refrigerator with 20 μL of Anti-A2B5 MicroBeads. Cells were washed twice and resuspended in 500 μL of buffer. Positive magnetic cell separation was performed using MiniMACS column and MACS Separator (Miltenyi Biotec).
Fluorescent-activated cell sorting
Cells were fixed using 4% PFA (Electron Microscopy) in a fluorescent-activated cell sorting (FACS) buffer, 0.1% BSA, 1×PBS, 10% fetal bovine serum (Atlanta Biologicals, Inc.), and 0.01% sodium azide (Sigma-Aldrich), PBS washed, permeabilized with 0.1% Triton X, and then washed with a 0.5% saponin buffer. A primary antibody was added overnight at 4°C, cells were then washed with the saponin buffer, then exposed to the secondary antibody, washed, and pellets were resuspended with the FACS buffer and analyzed with the FACScan cytometer (Beckton Dickinson) with CellQuest software (Beckton Dickinson). Primary antibodies included anti-hGFAP rabbit polyclonal (1:200; Millipore) and anti-human A2B5 monoclonal (1:100; Millipore). Secondary antibodies used were Alexa Fluor 488-conjugated goat anti-rabbit and goat anti-mouse IgG (1:200; Life Technologies). Negative controls included isotype-specific primary serum and secondary antibodies.
In vitro migration experiments
To assess the migratory capacity of hiPSC-derived astrocytes, transwell experiments were performed according to published protocols [28,33,34]. For these experiments, hESC- and hiPSC-derived astrocytes were plated in the top well (5×104 cells/well) of 8.0 μm pore size transwell culture dishes (BD Bioscience). In the bottom well, culture media or human malignant glioma cells (U87; 5×104 cells/well) (American Type Culture Collection) were added and incubated in DMEM+10% serum. At 48 and 72 h after plating, the migration of hiPSC-derived astrocytes was determined by fixing the membrane and staining the cells with hematoxylin and eosin (H&E). The number of hiPSC-derived astrocytes present in 10 random fields at 100× was quantified as the mean number of migrating cells. Experiments were done in triplicates.
In vivo brain stereotactic engrafting and histology
Experimental protocols were in accordance with the National Institutes of Health Guidelines for Use of Live Animals and approved by our Institutional Animal Care and Use Committee (IACUC). Before engrafting, hiPSC-derived astrocytes (35 days differentiated) (∼5×105 cells in 5 μL of 1×PBS) were labeled with the vital dye PKH26 (Sigma) [35,36] and loaded in a microsyringe (Hamilton). Eight-week-old male nude mice (Taconic) were anesthetized, placed into a stereotactic frame (Stoelting), and underwent a stereotactic burr hole. The syringe needle was then positioned in the right striatum, using brain atlas coordinates [37] AP=−2 mm, LAT=3 mm; VERT=3 mm. An automatic micropump (Stoelting) was used for the engraftment injection 5 μm/60 s speed. For the experimental human brain tumor model, mice were first implanted with U87 cells, using the same techniques and stereotactic coordinates as reported above. Astrocyte engrafting was performed in the contralateral hemisphere using similar techniques 2 weeks thereafter.
Two weeks after hiPSC-derived astrocyte engrafting, animals were transcardially perfused with 4% PFA and after fixation and sucrose exposure were serially cut at 5 μm thickness, mounted on glass slides, and coverslipped using the cyanoacrylate ester glue. Slides were examined using a standard filter setup for TRIC (Olympus). Alternate slides were stained with H&E. Brains processed to rule out teratoma formation were serially coronally sliced, stained with H&E, and examined under light microscopy for the presence/absence of teratoma.
Statistical analysis
Statistical difference was assessed using the nonparametric Boostrap test [38], with significance set at P<0.005. Data are expressed and mean±standard error. Experiments were performed in triplicate unless otherwise specified.
Results
In vitro differentiation of “footprint-free” hiPSC into fully differentiated astrocytes
Two “footprint-free” hiPSC lines were differentiated and compared to a hESC line (H9) following our previous published protocol [2]. iPSC were initially picked on radiated MEFs and then maintained on MEFs or Matrigel. Passaging iPSC three times completely eliminate the presence of radiated MEFs. Colonies maintained on MEFs survived at a higher frequency than those on Matrigel. Nonetheless, the latter were used for differentiation to astrocytes.
After EBs were cultured for 5 days and plated at day 6 for differentiation, columnar neuroepithelial cells appeared at day 9 and formed typical neural tube-like rosettes at day 14. Within 3 weeks of differentiation, a progressive number of cells with distinctive astrocytic morphology, including stellate appearance and ultimately with tertiary highly branched processes, were found (Fig. 1A). Immunocytochemical characterization showed the presence of neural markers Musashi and Nestin in the neural rosettes and GFAP in differentiated astrocytes (Fig. 1B). Morphological and immunohistochemical appearance in the three cell lines did not vary at the different differentiation times.
FIG. 1.

Differentiation of astrocytes from “footprint-free” human induced pluripotent stem cells (hiPSC). (A) Phase-contrast microphotographs of human embryonic stem cells (hESC) (H9), modified RNA (modRNA)-, and Sendai virus (SeV)-derived iPSC during differentiation showing embryonic bodies at differentiation day 5 (first column), neural rosettes at day 14 (second column), neural cell differentiation at day 28 (third column), and fully differentiated astrocytes at day 35 (fourth column). (B) Immunofluorescence microphotographs of hESC (H9), modRNA-, and SeV-derived iPSC showing neural rosettes at differentiation day 14 expressing neural markers Nestin (green; Alexa 488), Musashi (purple; Alexa 660), and lack of glial fibrillary acid protein (GFAP) (red; cy3) (first column). Neural cells positive for Nestin (green) and GFAP (red) at differentiation day 28 (second column) and day 35 (third column). Scale bar: 100 μm. Nuclear stain: diamidophenylindole (DAPI).
qRT-PCR analysis confirmed the stemness nature of the undifferentiated hESC and “footprint-free” hiPSC by showing the presence of Oct4 at baseline, confirming stemness, followed by the increasing presence of GFAP and loss of Oct4 as differentiation progressed. Similarly, expression of physiologic astrocytic markers typical of fully differentiated astrocytes, GLT1 and ALDOC [4], increased in the late differentiation phases (Fig. 2). The glutamate assay showed no statistical difference in glutamate physiology between human astrocytes and iPSC-derived astrocytes, and a significant increase compared to undifferentiated iPSC (data not shown).
FIG. 2.

Loss of stemness and acquisition of differentiated astrocytic markers. Bar graph summarizing quantitative real-time polymerase chain reaction results in H9 (A), modRNA- (B), and SeV-derived cells (C), showing loss of Oct4 expression and increased expression of GFAP, glutamate transporter 1 (GLT1), and aldolase c (ALDOC) during differentiation. Data normalized to baseline (undifferentiated cells); mean±standard error (SE) of three experiments (see Materials and Methods).
Together, these results indicate that the transgene-free hiPSC lines can be successfully differentiated into astrocytic lineage with results similar to hESC. Since astrocytes can be differentiated with similar efficiency with both hiPSC lines, the modRNA line is best suited to be used for translational work as it does not require eight passages to lose the SeV needed for the SeV-iPSC line.
Purification of “footprint-free” hiPSC-derived astrocytes by MACS and lack of teratogenicity
To enhance the purity of the astrocytic population, 7 days after mechanical isolation of the rosettes, hiPSC were passed through a MACS column for positive cell sorting of A2B5+ cells [31]. This resulted in increased A2B5+ cells from 61% before MACS to 95% after MACS, in modRNA-derived ells (Fig. 3); similar results were found in the SeV-derived cell line (data not shown). After sorting, A2B5+ cells were plated again in Matrigel-coated plates in the neurobasal medium containing CNTF for 2 weeks. At differentiation day 28, GFAP+ cells measured by FACS analysis showed a 99% GFAP+ cell population (Fig. 4). The remaining cells were positive for Nestin and Olig2, neural markers, and negative for Oct4. As previously published, without MACS sorting, the GFAP+ cell population was 55% [2].
FIG. 3.
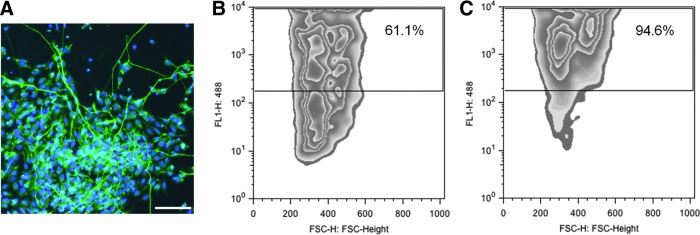
A2B5-positive cell sorting increases astrocytic purification. (A) Immunofluorescence microphotograph of A2B5+ modRNA iPSC-derived cells at differentiation day 21. Flow cytometry analysis of the same cells before (B) and after (C) magnetic-activated cell sorting (MACS) showing an increase of the A2B5+ cell population from 61% before MACS to 95% after MACS. Scale bar: 50 μm. Nuclear stain: DAPI.
FIG. 4.

hiPSC-derived fully differentiated astrocytes do not result in teratoma formation. (A) Flow cytometry analysis of modRNA hiPSC-derived astrocytes after MACS at differentiation day 28, showing a highly pure astrocytic population 99% GFAP positive. (B) PCR confirming the lack of Oct4 and presence of GFAP at differentiation day 28, in modRNA-derived cells. Microphotographs of fully differentiated SeV hiPSC-derived astrocytes with phase-contrast microscopy (C) and after immunocytochemistry for GFAP (D) showing typical stellate astrocytic processes and GFAP positivity. Scale bar: 50 μm. Nuclear stain: DAPI.
To confirm the lack of teratogenicity of our hiPSC-derived astrocytes, they were stereotactically implanted in the mice striatum before (n=4) and after MACS (n=10); brains were examined 3 months thereafter. Teratomas were found in all four mice engrafted with hiPSC-derived astrocytes at differentiation day 28 without MACS and not found in those engrafted with cells after MACS. These results corroborate the evidence that positive cell sorting eliminates the presence of undifferentiated cells and, consequently, the potential risk of teratoma formation when used in vivo.
hiPSC-derived astrocytes maintain migration characteristics in vitro and in vivo
The migration characteristics of stem cells, stem cell-derived astrocytes, and neural progenitor cells (NPC) in the brain have been reported [39,40]. To confirm that hiESC-derived astrocytes exhibit similar migration properties, we performed in vitro transwell migration assays. As shown in Fig. 5, hiPSC-derived astrocytes had in vitro migratory capacity. This had a threefold increase in the presence of human glioma cells U87, indicating the migrating and tropism capacity of hiPSC-derived astrocytes for human brain tumor cells 72 h after exposure to glioma cells.
FIG. 5.

hiPSC-derived astrocytes maintain stem cell-like migration characteristics in vitro. (A) Microphotograph of migrated modRNA- and SeV-derived astrocytes (pink) without (control: ctr) and with U87 tumor cells at the bottom of the well 72 h after plating. (B) Bar graphs representation of migration fold change, normalized to controls. In the presence of U87 tumor cells, hiPSC-derived astrocytes maintain homing characteristics, migrating toward the tumor cells with a greater than threefold increase in their absence. Cell stain: H&E, hematoxylin and eosin; mean±SE of three experiments; *P<0.05; scale bar: 50 μm.
To assess the migration characteristics of hiPSC-derived astrocytes in vivo, these were grafted into the striatum of mice with and without experimental brain tumors; brains were studied 2 weeks after engrafting. hiPSC-derived astrocytes were found to have migrated away from the engraftment side within 2 weeks. They were present in the cortex and gray matter equally distributed between the two hemispheres. In the presence of an experimental brain tumor, hiPSC were found to cluster around and within the tumor with very few astrocytes scattered within the brain (Fig. 6).
FIG. 6.

hiPSC-derived astrocytes maintain migration characteristics in vivo. Microphotographs of “footprint-free” hiPSC-derived astrocytes labeled with the vital stain PKH26 before engrafting on the hemocytometer phase contrast (A), fluorescence (B), after engrafting in the normal mouse brain (C, D) and after engrafting in the mouse brain in presence of tumor (F, G) and control (I, J). (E, H, K) are diagrams of coronal mouse brain sections showing the hiPSC-derived astrocyte engraftment site (S) and tumor location (T). The box in each diagram shows the location of the area shown on light microscopy (C, F, I) and fluorescence (D, G, J). After engrafting, astrocytes (arrows) migrated through the normal brain (D). In the presence of a brain tumor (G), they clustered within and around the tumor (arrowheads); this was not observed in the control animal (J). Scale bar: 200 μm.
These results confirm the migrating characteristics of the hiPSC-derived astrocytes in the presence of a brain tumor, with characteristics similar to those described for other stem cell-derived astrocytes or NPC [41].
Discussion
Human stem cells, including hESC and hiPSC, represent important cell resources and hold high promise for disease modeling, cell-based therapies, and drug and pharmaceutical applications [7,42]. hiPSC offer significant advantages over ESC, including lack of ethical concerns and the possibility to be patient specific. The latter is particularly important in translational clinical application as it avoids the need for immunotherapy [43]. Current limitations in the translational use of hiPSC include the potential of teratogenesis in the presence of undifferentiated cells and the need for viral induction with inherent risk of insertional mutagenesis. Mesenchymal stem cells have been associated in some studies with tumor progression in breast metastasis [44] and enhanced growth of brain tumor glioblastoma cells [45]. Other stem cells are also considered for similar applications, such us NPC; however, they have significant limitations, including ethical concerns, as they need to be harvested in the fetus, difficulty of harvesting as they reside in the subventricular zone, need for immunotherapy as they cannot be patient specific, and potential for teratogenesis as they are not fully differentiated.
Recent advances in hiPSC induction focused on addressing the need to avoid using retroviral and lentiviral vectors, the first reprogramming vectors used to induce iPSC. Whereas these vectors successfully demonstrated the feasibility of this technology to reprogram somatic cells to a pluripotent stem cell stage [6,7], they also caused insertional mutagenesis by viral vector integration raising caution for human clinical applications. To address the inherent safety issues surrounding viral vectors, a number of vectors emerged in the attempt to generate “footprint-free” iPSC. Some of these included the use of nonintegrating adenoviral vectors, transient transfection of plasmids, as well as episomal vectors [9–12]. However, the reprogramming efficiency was 100-fold lower with these vectors and the resulting iPSC colonies still had to be screened for residual integration of portions of these vectors into the host cell genome. To improve on the frequency of reprogramming and ensure the complete removal of all exogenous DNA without adversely affecting the already low reprogramming efficiency, other techniques to generate “footprint-free” iPSC were developed, including the RNA virus (SeV) [13,14] and modRNA [15–17]. The former uses sequential RNA transfection while blocking the interferon response, leads to robust iPSC colonies, and results in loss of the SeV after eight passages; the latter uses synthetic modRNA. In our study, we show that we can successfully differentiate astrocytes from “footprint-free” hiPSC with the same ease and characteristics of astrocytes differentiated from hESC or “nonfootprint-free” iPSC, as previously published [2]. The lack of need for passages to lose the SeV makes the modRNA-derived iPSC line very desirable for clinical studies.
Differentiation of astrocytes from hiPSC has been reported before by us and others [2,46]. In the previous work, however, the differentiated cells were not a pure astrocytic population, therefore raising the question of safety if considered for translational use. Additionally, in previous studies, cells were not maintained in serum-free and feeder-free conditions. Exposure to animal products using media containing serum and feeders might further increase the risk of nonhuman pathogen transmission, further limiting the use of cells obtained using those conditions for clinical use. Thus, serum-free and feeder-free conditions are necessary before considering using them for clinical application. An additional advantage of deriving cells under a feeder-free and serum-free condition is that the variability among hiPSC is minimized compared to when derived using serum and/or feeders [13,47]. In the current study, we showed that we can obtain astrocytes from hiPSC expanded and maintained in serum-free and feeder-free conditions.
Experimental allotransplantation of undifferentiated ESC and iPSC into the brain or other tissues results in teratoma formation [48–52]. This very serious risk has hindered the use of hiPSC-derived cells from clinical trials. In this study, we used positive cell sorting with MACS showing a highly purified cell population, 99% GFAP positive, with lack of OCT4 and the absence of teratoma formation in long-term experiments.
A number of studies observed the migration of various implanted stem cells and stem cell-derived cells into lesions of the brain and indicated that possibly stem cells are targeted by inflammatory chemotactic factors and cytokines extracted from ischemic brain tissue or surrounding brain tumors [53–55]. Similarly, native brain astrocytes have migration characteristics elicited by a wide variety of CNS pathologies, including trauma, stroke, and tumors [56]. These were first described in the early 1990s and recently substantiated by identifying molecular pathways at the basis of migration [57–59]. Our results show that hiPSC-derived astrocytes maintain migration capacity in vitro and after brain engraftment. Additionally, we showed that they maintain migration capacity and migrate toward and within brain tumors. These results corroborate our previous results with mESC [42,60] and highlight the importance of considering them as promising vectors for cell-based gene therapy for adjuvant therapy of brain tumors or other neurological disorders.
In summary, our work reports an efficient and defined method to differentiate astrocytes from “footprint-free” hiPSC. These are highly purified, do not result in teratoma formation after allotransplantation, and maintain migration characteristics after engraftment. Astrocytes can be obtained in generous numbers, aliquoted, frozen, thawed, and used when needed. These results suggest that astrocytes derived with this methodology can be used for clinical applications of cell-based therapies and drug discoveries.
Acknowledgment
The authors thank Dr. Stephan Cosenza for offering us the use of the fluorescence microscope Olympus IX71.
Author Disclosure Statement
The authors indicate no potential conflicts of interest.
References
- 1.Bergstrom T. and Forsberg-Nilsson K. (2012). Neural stem cells: brain building blocks and beyond. Ups J Med Sci 117:132–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Emdad L, D'Souza SL, Kothari HP, Qadeer ZA. and Germano IM. (2012). Efficient differentiation of human embryonic and induced pluripotent stem cells into functional astrocytes. Stem Cells Dev 21:404–410 [DOI] [PubMed] [Google Scholar]
- 3.Hu BY, Weick JP. and Yu J. (2010). Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci U S A 107:4335–4340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaltouki A, Peng J, Liu Q, Rao MS. and Zenget X. (2013). Efficient generation of astrocytes from human pluripotent stem cells in defined conditions. Stem Cells 31:941–952 [DOI] [PubMed] [Google Scholar]
- 5.Okano H, Nakamura M, Yoshida K, Okada Y, Tsuji O, Nori S, Ikeda E, Yamanaka S. and Miura K. (2013). Steps toward safe cell therapy using induced pluripotent stem cells. Circ Res 112:523–533 [DOI] [PubMed] [Google Scholar]
- 6.Takahashi K. and Yamanaka S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676 [DOI] [PubMed] [Google Scholar]
- 7.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K. and Yamanaka S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872 [DOI] [PubMed] [Google Scholar]
- 8.Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II. and Thomson JA. (2009). Human induced pluripotent stem cells free of vector and transgene sequences. Science 324:797–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stadtfeld M, Nagaya M, Utikal J, Weir G. and Hochedlinger K. (2008). Induced pluripotent stem cells generated without viral integration. Science 322:945–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tucker BA, Anfinson KR, Mullins RF, Stone EM. and Young MJ. (2013). Use of synthetic xeno-free culture substrate for inducing pluripotent stem cells induction and retinal differentiation. Stem Cells Transl Med 2:16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920 [DOI] [PubMed] [Google Scholar]
- 12.Zhou H, Wu S, Joo JY, Zhu S, Han DW, Lin T, Trauger S, Bien G, Yao S, et al. (2009). Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 4:381–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Macarthur CC, Fontes A, Ravinder N, Kuninger D, Kaur J, Bailey M, Taliana A, Vemuri MC. and Lieu PT. (2012). Generation of human-induced pluripotent stem cells by a non integrating RNA Sendai virus vector in feeder-free or xeno-free conditions. Stem Cells Int 2012:564–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fusaki N, Ban H, Nishiyama A, Saeki K. and Hasegawa M. (2009). Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci 85:348–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, Ebina W, Mandal PK, Smith ZD, et al. (2010). Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7:618–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warren L, Ni Y, Wang J. and Guo X. (2012). Feeder-free derivation of human induced pluripotent stem cells with messenger RNA. Sci Rep 2:657–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mandal PK. and Rossi DJ. (2013). Reprogramming human fibroblasts to pluripotency using modified mRNA. Nat Protoc 8:568–582 [DOI] [PubMed] [Google Scholar]
- 18.Okita K, Nakagawa M, Hyenjong H, Ichisaka T. and Yamanaka S. (2008). Generation of mouse induced pluripotent stem cells without viral vectors. Science 322:949–953 [DOI] [PubMed] [Google Scholar]
- 19.Benveniste R. and Germano IM. (2003). Evaluation of factors predicting accurate resection of high-grade gliomas using frameless image-guided stereotactic guidance. Neurosurg Focus 14:1–4 [DOI] [PubMed] [Google Scholar]
- 20.Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinski Y, Stroup NE, Kruchko C. and Barnholtz-Sloan JS. (2013). CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol 15Suppl 2:ii1–ii56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Germano IM, Ito M, Cho K, Hoshino T, Davis RL. and Wilson CB. (1989). Correlation of histopathological features and proliferative potential of individual gliomas. J Neurosurg 70:701–706 [DOI] [PubMed] [Google Scholar]
- 22.Germano IM, Fable J, Gultekin SH. and Silvers A. (2003). Adenovirus/herpes simplex-thymidine kinase/gancyclovir complex: preliminary results of a phase I trial in patients with recurrent malignant gliomas. J Neurooncol 65:279–289 [DOI] [PubMed] [Google Scholar]
- 23.Germano IM, Uzzaman M, Benveniste RJ, Zaurova M. and Keller G. (2006). Apoptosis in human glioblastoma cells produced using embryonic stem cell-derived astrocytes expressing tumor necrosis factor-related apoptosis-inducing ligand. J Neurosurg 105:88–95 [DOI] [PubMed] [Google Scholar]
- 24.Kanu OO, Mehta A, Di C, Lin N, Bortoff K, Bigner DD, Yan H. and Adamson DC. (2009). Glioblastoma multiforme: a review of therapeutic targets. Expert Opin Ther Targets 13:701–718 [DOI] [PubMed] [Google Scholar]
- 25.Schiffer D, Giordana MT, Germano IM. and Mauro A. (1986). Anaplasia and heterogeneity of GFAP expression in gliomas. Tumori 72:163–170 [DOI] [PubMed] [Google Scholar]
- 26.Stupp R, Mason W, van den Bent M, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, et al. (2005). Radiotherpay plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996 [DOI] [PubMed] [Google Scholar]
- 27.Adamson C, Kanu OO, Mehta AI, Di C, Li N, Mattox AK. and Bigner DD. (2009). Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin Investig Drugs 18:1061–1083 [DOI] [PubMed] [Google Scholar]
- 28.Germano IM, Emdad L, Qadeer ZA, Binello E. and Uzzman M. (2010). Embryonic stem cell (ESC)-mediated transgene delivery induces growth suppression, apoptosis and radiosensitization, and overcomes temozolomide resistance in malignant gliomas. Cancer Gene Ther 17:664–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang SC, Wernig M, Duncan ID, Brstle O. and Thomson JA. (2001). In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol 19:1129–1133 [DOI] [PubMed] [Google Scholar]
- 30.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M. and Studer L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27:275–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dietrich J, et al. (2002). Characterization of A2B5+ glial precursor cells from cryopreserved human fetal brain progenitor cells. Glia 40:65–77 [DOI] [PubMed] [Google Scholar]
- 32.Seidenfaden R, Desoeuvre A, Bosio A, Virard I. and Cremer H. (2006). Glial conversion of SVZderived committed neuronal precursors after ectopic grafting into the adult brain. Mol Cell Neurosci 32:187–198 [DOI] [PubMed] [Google Scholar]
- 33.Heese O, Disko A, Zirkel D, Westphal M. and Lamszus K. (2005). Neural stem cell migration toward gliomas in vitro. Neuro Oncol 7:476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, Chen J, Hentschel S, Vecil G, et al. (2005). Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res 65:3307–3318 [DOI] [PubMed] [Google Scholar]
- 35.Poon RY. (2002). PKH fluorescent cell linker dyes. In: In Living Color: Flow Cytometry and Cell Sorting Protocols. Diamond RA, De Maggio S, eds. Springer, New York. pp 302–352 [Google Scholar]
- 36.Bin Z, Xiaolei S, Xiulan L, Qiang Y, Yanjun L, Yang Z, Bing L. and Xinlong M. (2013). Imprroved preparation of a cellular nerve scaffold and application of PKH26 fluorescent labeling combined with in vivo fluorescent imaging system in nerve tissue engineering. Neurosci Lett 556:52–57 [DOI] [PubMed] [Google Scholar]
- 37.Franklin KBJ. and Paxinos G. (2010). The Mouse Brain in Stereotaxic Coordinates, Second Edition. Academic Press, New York [Google Scholar]
- 38.Mooney CZ. and Duval RD. (1993). Bootstrapping: A Nonparametric Approach to Statistical Inference. Sage, Newbury Park, CA [Google Scholar]
- 39.Tabernero A, Orfao A. and Medina JM. (1996). Astrocyte differentiation in primary culture followed by flow cytometry. Neurosci Res 24:131–138 [DOI] [PubMed] [Google Scholar]
- 40.Krencik R. and Zhang SC. (2011). Directed differentiation of functional astroglial subtypes from human pluripotent stem cells. Nat Protoc 6:1710–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benveniste RJ, Keller G. and Germano I. (2005). Embryonic stem cell-derived astrocytes expressing drug-inducible transgenes: differentiation and transplantion into the mouse brain. J Neurosurg 103:115–123 [DOI] [PubMed] [Google Scholar]
- 42.Uzzaman M, Keller G. and Germano IM. (2009). In vivo gene delivery by embryonic-stem-cell-derived astrocytes for malignant gliomas. Neuro Oncol 11:102–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cao J, Li X, Zhang C, Yu H. and Zhao T. (2013). Cells derived from iPSC can be immunogenic—yes or no?. Protein Cell 5:1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R. and Weinberg RA. (2007). Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449:557–563 [DOI] [PubMed] [Google Scholar]
- 45.Akimoto K, Kimura K, Nagano M, Takano S, To'a Salazar G, Yamashita T. and Ohneda O. (2013). Umbilical cord blood-derived mesenchymal stem cells inhibit, but adipose tissue-derived mesenchymal stem cells promote, glioblastoma multiforme proliferation. Stem Cells Dev 22:1370–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan T, Liao W, Feng NH, Lou YL, Niu X, Zhang AJ, Wang Y. and Deng ZF. (2013). Human induced pluripotent stem cell-derived neural stem cells survive, migrate, differentiate, and improve neurological function in a rat model of middle cerebral artery occlusion. Stem Cell Res Ther 4:73–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung HC, Lin RC, Logan GJ, Alexander IE, Sachdev PS. and Sidhu KS. (2012). Human induced pluripotent stem cell derived under feeder-free conditions display unique cell cycle and DNA replication gene profiles. Stem Cells Dev 21:206–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramirez MA, Pericuesta E, Fernandez-Gonzalez R, Pintado B. and Gutiérrez-Adán A. (2007). Inadvertent presence of pluripotent cells in monolayers derived from differentiated embryoid bodies. Int J Dev Biol 51:397–407 [DOI] [PubMed] [Google Scholar]
- 49.Fu W, Wang SJ, Zhou GD, Liu W, Cao Y. and Zhang WJ. (2012). Residual undifferentiated cells during differentiation of induced pluripotent stem cells in vitro and in vivo. Stem Cells Dev 21:521–529 [DOI] [PubMed] [Google Scholar]
- 50.Liu Z, Tang Y, Lü S, Zhou J, Du Z, Duan C, Li Z. and Wang C. (2013). The tumorigensis of iPSC and their differentiated derivates. J Cell Mol Med 17:782–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Erdo F, Buhrle C, Blunk J, Hoehn M, Xia Y, Fleischmann B, Föcking M, Küstermann E, Kolossov E, et al. (2003). Host-dependent tumorigenesis of embryonic stem cell transplantation in experimental stroke. J Cereb Blood Flow Metab 23:780–785 [DOI] [PubMed] [Google Scholar]
- 52.Wakitani S, Takaoka K, Hattori T, Miyazawa N, Iwanaga T, Takeda S, Watanabe TK. and Tanigami A. (2003). Embryonic stem cells injected into the mouse knee joint form teratomas and subsequently destroy the joint. Rheumatology 42:162–165 [DOI] [PubMed] [Google Scholar]
- 53.Wang L, Li Y, Chen X, Chen J, Gautam SC, Xu Y. and Chopp M. (2002). MCP-1, MIP-1,IL-8 and ischemic cerebral tissue enhance human bone marrow stromal cell migration in interface culture. Hematology 7:113–117 [DOI] [PubMed] [Google Scholar]
- 54.Zhang H, Vutskits L, Pepper MS. and Kiss JZ. (2003). VEGF is a chemoattractant for FGF-2-stimulated neural progenitors. J Cell Biol 163:1375–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shinojima N, Hossain A, Takezaki T, Fueyo J, Gumin J, Gao F, Nwajei F, Marini FC, Andreeff M, Kuratsu J. and Lang FF. (2013). TGF-β mediates homing of bone marrow-derived human mesenchymal stem cells to glioma stem cells. Cancer Res 73:2333–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buffo A, Rolando C. and Ceruti S. (2010). Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential. Biochem Pharmacol 79:77–89 [DOI] [PubMed] [Google Scholar]
- 57.Dahl D, Mansourour H. and Bignami A. (1991). Astrocytes colonize dorsal root ganglia transplanted unto rat brain. Brain Res Bull 27:169–173 [DOI] [PubMed] [Google Scholar]
- 58.Kang W. and Hebert JM. (2011). Signaling pathways in reactive astrocytes, a genetic perspective. Mol Neurobiol 43:147–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ho KW, Lambert WS. and Calkins DJ. (2014). Activation of the TRPV1 cation channel contributes to stress-induced astrocyte migration. Glia [Epub ahead of print]; DOI: 10.1002/glia.22691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Germano IM. and Binello E. (2009). Gene therapy as an adjuvant treatment for malignant gliomas: from bench to bedside. J Neurooncol 93:79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]