

Abstract
Despite being extensively characterized structurally and biochemically, the functional role of histone deacetylase 8 (HDAC8) has remained largely obscure due in part to a lack of known cellular substrates. Herein, we describe an unbiased approach using chemical tools in conjunction with sophisticated proteomics methods to identify novel non-histone nuclear substrates of HDAC8, including the tumor suppressor ARID1A. These newly discovered substrates of HDAC8 are involved in diverse biological processes including mitosis, transcription, chromatin remodeling, and RNA splicing and may help guide therapeutic strategies that target the function of HDAC8.
Posttranslational acetylation of lysine residues is a highly conserved1 and important modification2 enabling the cellular calibration of protein function and/or stability resulting in effects ranging from cytoskeletal reorganization to changes in gene expression.3,4 Histone deacetylases (HDACs) play a key role in maintaining the balance of acetylation states by catalyzing the removal of acetyl groups from the ε-amino groups of acetylated lysine residues.4 As a result, these enzymes have become important therapeutic targets for a number of disease states including cancer5 and psychiatric illnesses.6 As their name implies, HDACs were thought to be primarily responsible for the deacetylation of histones; however, it has become apparent that a large number of non-histone proteins are substrates for these enzymes as well.2,7 The HDAC family comprises the NAD(+)-dependent sirtuins (class III) and the metal-dependent HDACs, which can be further divided into three classes (class I: HDACs 1, 2, 3, and 8, class II: HDACs 4, 5, 6, 7, 9, 10, and class IV: HDAC11) based on phylogenetic similarity,8 with class I being localized primarily in the nucleus and classes II and IV shuttling between the nucleus and the cytoplasm.4
Identification of the endogenous substrates of HDAC enzymes is a fundamental area of HDAC research, and this problem has been particularly acute for the class I enzyme HDAC8. Of all the HDACs, HDAC89 is arguably the best characterized structurally.10 It was the first human class I HDAC structure to be reported, and since then, over 25 additional structures bound to various classes of small molecule ligands and peptides have been disclosed (www.pdb.org).11 However, despite this knowledge, few of the enzyme’s natural substrates have been identified.11 To date, only two cellular substrates of HDAC8 have been identified, namely, the estrogen-related receptor alpha12 (ERR-α) and the structural maintenance of chromosome 313 (SMC3) protein, the latter of which plays a prominent role in Cornelia de Lange syndrome.13 It remains unclear which, if any, specific histone residues serve as viable substrates for this isoform. In terms of biological function, HDAC8 has been implicated in various cancers including neuroblastoma,14 urothelial,15 and breast cancer16 as well as in neural crest development.17 The HDAC8 substrates that mediate these effects are currently unknown.
To elucidate the cellular substrates and better define the biology of HDAC8, we undertook an unbiased, chemical biology approach that involved monitoring global acetylation and gene expression changes in a representative cell line following treatment with a known, potent, and highly selective small molecule inhibitor of HDAC8. Small molecule modulation coupled with mass spectrometry offers distinct advantages for the identification of acetylation substrates and specific lysine sites responsive to HDAC8 relative to protein knockdown, knockout, or pulldown approaches, including (1) deconvolution of catalytic versus scaffolding functions associated with HDACs,18 (2) temporal control, (3) increased resolution and sensitivity, and (4) the avoidance of complications associated with transient and/or metastable interactions and complexes. Therefore, we focused on using the highly selective and potent HDAC8 inhibitor PCI-3405119 as well as a suitably designed negative control compound to account for potential compound-driven off-target effects (Figure 1a,b). The inclusion of a negative control compound was particularly important, as PCI-34051 contains a metal-chelating hydroxamic acid group, and this motif has the potential to bind a variety of metalloenzymes. As such, we designed and synthesized BRD3811 (Figure 1a), a compound that retains the hydroxamic acid functionality and contains a minor structural modification to PCI-34051 (i.e., a single methyl group introduced ortho to the hydroxamic acid group) resulting in a 1,000-fold reduction in potency for inhibition of HDAC8 (Figure 1b). Consistent with this finding, molecular docking of PCI-34051 (Figure 1c) and BRD3811 (Figure 1e) into the active site of an HDAC8 crystal structure (PDB accession code 1T64) reveals that the methyl group of BRD3811 cannot be accommodated in the catalytic binding domain of HDAC8 while maintaining an optimal zinc chelation geometry.
Figure 1.
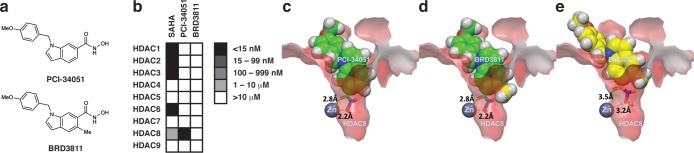
Chemical tools for studying HDAC8. (a) Chemical structures of the HDAC8 inhibitor PCI-34051 and the structurally related negative control compound BRD3811. (b) HDAC inhibitor potencies for PCI-34051, BRD3811, and the pan-inhibitor SAHA. Absolute potency values can be found in Supplementary Table 2. (c) PCI-34051 docked into a crystal structure of HDAC8 (PDB code 1T64). (d) Replacement of the ortho-hydrogen in the docked structure of PCI-34051 (c) with a methyl group. The methyl group protrudes from the enzyme pocket. (e) BRD3811 docked into a crystal structure of HDAC8 (PDB code 1T64).
Using these chemical tools, we compared the changes in global acetylation in a representative cell line known to express HDAC820 (i.e., MCF7) after treatment with each compound using Stable Isotope Labeling of Amino Acids in Cell Culture (SILAC)-based quantitative mass spectrometry (MS). Briefly, cells were grown in the presence of light, medium, or heavy arginine and lysine followed by treatment with either PCI-34051 (10 μM), BRD3811 (10 μM), or vehicle (DMSO) for 24 h (Figure 2a). Global acetylation profiling was completed by digesting cellular proteins with trypsin and enriching the acetylated peptides by immunoprecipitation using an antibody specific for acetylated lysine residues. Over two replicates, 1,360 acetylation sites were quantified using high-resolution MS (Supplementary Results, Supplementary Table 1).
Figure 2.

Identifying novel substrates of HDAC8. (a) Schematic of experimental design. (b) Acetylated proteins regulated by treatment with PCI-34051 as compared to DMSO or to (c) BRD3811 as the control. Each axis represents a single replicate and denotes log2-fold changes in acetylation with dashed lines indicating a 1.5-fold change in acetylation. Proteins that passed a p-value cutoff of ≤0.05 in both replicates and were not regulated by the negative control compound BRD3811 are highlighted in red. Insets show log2-fold changes in acetylation for select examples of replicate 1 and emphasize the relative lack of histone acetylation by comparison to SMC3 for each experiment. (d) Tables depicting acetylation sites regulated by more than 1.5-fold and passing p-value cutoffs of ≤0.05 in both replicates when PCI-34051 treatment was compared to DMSO or to (e) BRD3811 treatment as the control. (f) Steady state kinetic parameters (± standard error) for the deacetylation of synthetic acetylated peptides corresponding to a subset of identified HDAC8 substrates. Reactions were catalyzed by bacterially expressed human Zn-bound HDAC8 (see Methods). (g) Dependence of Zn-HDAC8-catalyzed deacetylation on the concentration of the ARID1A peptide. The Michaelis–Menten equation is fit to the data.
This approach enabled us to identify numerous protein sites whose acetylation increased by more than 1.5-fold in each of two replicates upon treatment with PCI-34051 relative to DMSO (Figure 2b). Of these, 7 passed a p-value cutoff of ≤0.05 (Figure 2b, red; Figure 2d) and were not regulated greater than 1.5-fold upon treatment with negative control compound BRD3811 relative to DMSO (Supplementary Table 1 and Supplementary Figure 1). Alternatively, a direct comparison of PCI-34051 treatment to BRD3811 treatment (Figure 2c) revealed 22 protein sites whose acetylation increased by more than 1.5-fold, with 7 passing a p-value cutoff of ≤0.05 in two replicates (Figure 2C, red; Figure 2e). From these data sets, we deemed 5 proteins (i.e., SMC3, RAI1, ZRANB2, NCOA3, and THRAP3) to be high-confidence substrates for HDAC8 as they were regulated by 1.5-fold or greater when PCI-34051 treatment was compared to both DMSO as well as to the negative control compound. Furthermore, ARID1A and SRSF5 were also considered candidate substrates for HDAC8 as they narrowly fell outside the bounds of our arbitrary cutoffs (i.e., 1.5-fold change and p-value ≤0.05) in only one of four experiments.
Gratifyingly, our unbiased approach successfully identified SMC3, a known substrate of HDAC8,13 as being significantly regulated by treatment with the HDAC8-selective inhibitor and not BRD3811. Furthermore, we were able to demonstrate that HDAC8-mediated deacetylation occurs on K106 of SMC3, one of two sites known to be acetylated by the acetyltransferase ESCO1.21 Our coverage of acetylated proteins did not include ERR-α, the only other known cellular HDAC8 substrate, and therefore, we cannot verify its regulation by HDAC8 in MCF7 cells. Our coverage did include several histone proteins (Supplementary Table. 1), and we did not observe any significant changes in histone acetylation status upon treatment with PCI-34051 when compared to DMSO or BRD3811. Changes in H2A (H2AFX) and H1.3 (HIST1H1D) acetylation relative to SMC3 are shown for comparison (Figure 2b and c, insets).
To further validate the substrates identified in our proteomics experiments, we devised in vitro enzymatic deacetylation experiments using recombinant human HDAC8 and synthetic acetylated peptides (8–10 aa) based on the sequences of our candidate substrates. Our peptide design centered on the identified lysine (K) acetylation sites, ensuring that the regulated lysines were flanked on either side by several residues. These “artificial” substrates were incubated with HDAC8, and deacetylation was measured from the production of acetate using an enzyme-coupled assay to determine the steady state kinetic parameters (Figure 2f and Methods). Human Zn-HDAC8 catalyzed the deacetylation of all these synthetic peptides in vitro (Figure 2f), albeit with catalytic efficiencies (kcat/KM) ranging across 3 orders of magnitude. Four of the peptides assayed (i.e., NCOA3, ARID1A, CSRP2BP, and MLL2) have values of kcat/KM within a factor of 2 or higher than the peptide corresponding to SMC3, providing further evidence that these proteins are likely HDAC8 substrates. In particular, the peptide corresponding to ARID1A is the most efficient non-fluorophore conjugated peptide substrate of Zn-HDAC8 discovered to date (kcat/KM = 740 M–1 s–1) (Figure 2f,g).22 Consistent with previous work, peptides containing an aromatic residue adjacent to the target lysine (e.g., those corresponding to ARID1A and CSRP2BP) are preferred substrates of HDAC8.11 To further characterize the enzyme specificity of these synthetic substrates, we profiled the deacetylation activity of commercially available, human recombinant HDACs 1–9 (Supplementary Table 3). While multiple isozymes catalyzed deacetylation of all of the putative substrates, no single peptide was recognized by all of the HDACs tested, and none were uniquely deacetylated by HDAC8. However, the ARID1A peptide exhibited the largest kcat/KM values for HDAC8 and HDAC8’s closest homologue, HDAC3, with values of 2400 and 2500 M–1 s–1, respectively (Supplementary Table 3).8 Finally, we determined the deacetylase inhibitory activity of PCI-34051 toward the ARID1A peptide substrate using commercially available human HDAC8 and calculated a Ki value of 33 nM, demonstrating the ability of this molecule to inhibit HDAC8 in a manner consistent with our cell-based observations (Supplementary Figure 2).
Most of the substrates identified in our study are localized in the nucleus (Figure 2d,e) and include transcription factors as well as proteins intimately involved in epigenetic regulation, chromatin remodeling, or RNA splicing. Interestingly, histone proteins were not identified as substrates in our acetylome profiling experiments, and this fact was later confirmed in separate, targeted SILAC experiments designed to specifically monitor for histone acetylation changes (Supplementary Figure 3). These results are consistent with previous reports.23,24 Intrigued by the non-histone but primarily nuclear nature of the candidate substrates, we tested if HDAC8 inhibition could lead to changes in gene expression independent of changes in histone acetylation. To this end, we measured the expression changes in MCF7 cells of approximately 1,000 landmark genes (L1000) as a representative measure of genome-wide effects upon treatment with PCI-34051 or BRD3811 across the dose range of 0.04–10 μM (see Methods). We then selected the dose-responsive genes using the IsoGene package25 (http://CRAN.R-project.org/package=IsoGene). PCI-34051 altered the expression of significantly more genes (70 genes, Supplementary Table 4) than did BRD3811 (11 genes, Supplementary Table 5). While several genes, such as HMOX1, were differentially regulated by both PCI-34051 and BRD3811, the magnitude of the change was much greater for the PCI-34051 treatment.
In an attempt to connect the modulated transcripts to our candidate HDAC8 substrates, we searched for known biological pathway connections between our high-confidence HDAC8 substrates (Figure 2d,e) and the 70 genes dose-responsive to PCI-34051 treatment (Supplementary Table 4) using the Ingenuity IPA Knowledge Base (Figure 3a). No direct associations between HDAC8 and the newly identified substrates were found; however, both the known HDAC8 substrate SMC3 and the tumor suppressor p53 (TP53) were directly connected to HDAC8. Our acetylome coverage did not include p53, therefore we were unable to determine whether it can be directly modified by HDAC8. Several of the newly identified acetylation substrates were directly linked to genes differentially expressed upon inhibition of HDAC8. NCOA3, a protein known to positively regulate the expression of HMOX1, is one such example.26 Additionally, we found that 3 of the newly identified substrates (i.e., ARID1A, RAI1, and MLL2) were directly linked to the cell cycle regulator p21 (CDKN1A). This led us to speculate that the increased expression of p21 observed upon treatment with PCI-34051 could be driven in part by the acetylation changes of ARID1A. Alternatively, increased acetylation of RAI1 and/or MLL2 could influence the regulation of this important gene. Many HDAC inhibitors are known to cause the upregulation of p21, but until now the exact substrates responsible for mediating that effect have remained obscure.27 When MCF7 cells are treated with PCI-34051 over the dose range of 0.04–10 μM, a dose-dependent increase in the level of the p21 transcript is observed (Figure 3b). Conversely, BRD3811 increased the level of p21 transcript only at the highest dose tested (i.e., 10 μM) (Figure 3b). To further validate this finding, we examined changes in p21 protein levels after treatment with each compound, and PCI-34051 treatment (10 μM) increased p21 (visualized via Western blot) while BRD3811 (10 μM) treatment did not (Figure 3c,d).
Figure 3.

Pathway analysis of candidate HDAC8 substrates. (a) A connection network was generated with Ingenuity Pathways Analysis (IngenuitySystems, www.ingenuity.com), using HDAC8 and the proteins (Supplementary Table 1) and genes (Supplemenary Table 4) differentially acetylated and expressed, respectively, upon treatment with PCI-34051 as inputs. Only those proteins or genes with known connections to other proteins or genes in the pathway are shown. HDAC8 is highlighted in a blue ellipse, while red ellipses denote proteins differentially acetylated upon treatment with PCI-34051. The remaining nodes represent genes differentially expressed upon treatment with PCI-34051. Relationships indicated by lines in this graph are found by Ingenuity and can include protein–protein interactions, transcriptional regulation, co-expression, activation, binding, phosphorylation, inhibition, protein–DNA interactions, binding regulation, localization, molecular cleavage, and translocation. These relationships can be between two molecules (straight arrows) or between a molecule and itself (curved arrows), as in the case of self-phosphorylation, for example. (b) Treatment with PCI-34051 for 24 h results in a dose-dependent increase in p21 expression. (c) Representative Western blot after treatment of MCF7 cells with either PCI-34051 or BRD3811 at 10 μM for 48 h shows that PCI-34051, but not the negative control, induces an increase in p21 protein levels. (d) Quantitation of Western blot data from 4 independent experiments. The star denotes P ≤ 0.05, relative to DMSO, as determined by a one-way analysis of variance (ANOVA) utilizing a posthoc Dunnett’s multiple comparisons test.
To assess whether our findings extend beyond the context of a single cell type and to incorporate into our analysis orthogonal biological perturbations, we expanded our gene expression studies using L1000 into several cell lines representative of distinct tissue types: PC3 (prostate), HEPG2 (liver), HCC515 (lung), HA1E (kidney), A375 (skin), A549 (lung), and HT29 (colon). We then created a gene expression signature using the 1,000 landmark genes and compared PCI-34051 and BRD3811 treatments to the signatures of other bioactive perturbagens using the connectivity map (Cmap) database (www.broadinstitute.org/cmap/) as previously described (see Methods). We integrated the results of multiple independent Cmap queries using the cell lines highlighted above, and we observed that PCI-34051 treatment was highly correlated with the overexpression of p21 across multiple cell lines, whereas BRD3811 treatment was not (Supplementary Figure 4). In fact, p21 overexpression was the overexpression perturbation most highly correlated with PCI-34051 treatment, ranking in the 96th percentile on average (BRD3811 treatment did not correlate well with p21 overexpression, 66th percentile rank). It is quite attractive to speculate that some of the anticancer effects of the HDAC8 inhibitor PCI-3405119 are mediated in part by increasing p21 levels through these newly discovered substrates. These candidate substrates of HDAC8 may provide a more targeted approach toward specific cancers (or other diseases) driven by the dysregulation of proteins and/or genes within this HDAC8 network. In the case of ovarian clear cell carcinoma, mutations in ARID1A are found in almost half of all cases, and it has been demonstrated that in frame indel mutations fail to induce p21 expression through increased degradation in the nucleus or decreased promoter binding.28 Efforts toward defining the functional consequences of the change in acetylation of these proteins by HDAC8 are ongoing.
In conclusion, we have identified several novel substrates of HDAC8 by taking an unbiased approach coupling chemical tools with acetylome profiling. The proteins identified include the known HDAC8 substrate SMC3 but do not include histones. Furthermore, these candidate substrates were predominantly nuclear and involved in a diverse range of cellular functions including transcription and RNA splicing. We demonstrated through in vitro enzymatic assays as well as through gene and protein expression studies that inhibition of HDAC8 can affect acetylation status ultimately influencing the levels of downstream proteins. Our experimental design relied on using BRD3811, a negative control compound based on the structure of the potent and selective HDAC8 inhibitor PCI-34051. Our approach represents a general strategy that should prove useful in future studies aimed at the identification of the endogenous substrates of other members of the HDAC family of enzymes.
Methods
The details of the methods are described in the Supporting Information.
Acknowledgments
We thank M. Weïwer, R. Ratan, S. Sleiman, and A. Desouza for assistance and helpful discussions and L. Gaffney for her help with illustrations. This work was supported by the Stanley Medical Research Institute, the LINCS Program (Grant U54 HG006093 Large scale gene expression analysis of cellular states), the National Institutes of Health (Grants NIGMS GM40602 (C.A.F.), T32-GM-008353 (N.A.W.), and T32-GM-008597 (C.A.P.)), and the National Science Foundation (graduate fellowship DGE0718128 (N.A.W.)). This work was supported in part by the Broad Institute of MIT and Harvard and by grants from the US National Cancer Institute (U24CA160034, part of the Clinical Proteomics Tumor Analysis Consortium initiative, to S.A.C.) and National Heart, Lung, and Blood Institute (HHSN268201000033C and R01HL096738 to S.A.C.)
Supporting Information Available
Methods, Supplementary Figures 1–5, Supplementary Tables 1–5, and Supplementary References 26–38. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
D.E.O., N.D.U., J.P., J.J., S.A.C., X.L., N.A.W., C.A.F., F.F.W. and E.B.H. designed experiments. D.E.O., N.D.U., J.P., J.J., T.S., X.L., N.A.W., C.A.P., E.D.S., Y.Z., and J.P.G. performed experiments. D.E.O., N.D.U., J.P., J.J., S.A.C., T.N., A.S., N.A.W., C.A.F., F.F.W. and E.B.H. analyzed and interpreted the data. P.M. performed molecular docking computations. F.L. synthesized BRD3811. E.H. performed Ingenuity analysis. E.H. and D.B. performed gene expression dose-response analyses. The manuscript was written by D.E.O. with input from all authors.
The authors declare the following competing financial interest(s): E.B.H. is a consultant to KDAc Therapeutics.
The surname of contributing author Xiaodong Lu was incorrectly spelled in the version published August 11, 2014. This has been corrected and the revised version was re-posted on August 20, 2014.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Weinert B. T.; Wagner S. A.; Horn H.; Henriksen P.; Liu W. R.; Olsen J. V.; Jensen L. J.; Choudhary C. (2011) Proteome-wide mapping of the Drosophila acetylome demonstrates a high degree of conservation of lysine acetylation. Sci. Signaling 4, ra48. [DOI] [PubMed] [Google Scholar]
- Choudhary C.; Kumar C.; Gnad F.; Nielsen M. L.; Rehman M.; Walther T. C.; Olsen J. V.; Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. [DOI] [PubMed] [Google Scholar]
- Spange S.; Wagner T.; Heinzel T.; Krämer O. H. (2009) Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 41, 185–198. [DOI] [PubMed] [Google Scholar]
- Fass D. M. Kemp M. M., Schroeder F. A., Wagner F. F., Wang Q., Holson E. B. (2012) Histone Acetylation and Deacetylation, in Epigenetic Regulation and Epigenomics (Meyers R. A., Ed.), pp 515–561, Wiley-Blackwell, Weinheim. [Google Scholar]
- Acharya M. R.; Sparreboom A.; Venitz J.; Figg W. D. (2005) Rational development of histone deacetylase inhibitors as anticancer agents: a review. Mol. Pharmacol. 68, 917–932. [DOI] [PubMed] [Google Scholar]
- Gräff J.; Tsai L.-H. (2013) The potential of HDAC inhibitors as cognitive enhancers. Annu. Rev. Pharmacol. Toxicol. 53, 311–330. [DOI] [PubMed] [Google Scholar]
- Glozak M. A.; Sengupta N.; Zhang X.; Seto E. (2005) Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23. [DOI] [PubMed] [Google Scholar]
- Gregoretti I. V.; Lee Y.-M.; Goodson H. V. (2004) Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 338, 17–31. [DOI] [PubMed] [Google Scholar]
- Buggy J. J.; Sideris M. L.; Mak P.; Lorimer D. D.; McIntosh B.; Clark J. M. (2000) Cloning and characterization of a novel human histone deacetylase, HDAC8. Biochem. J. 350, 199–205. [PMC free article] [PubMed] [Google Scholar]
- Estiu G.; West N.; Mazitschek R.; Greenberg E.; Bradner J. E.; Wiest O. (2010) On the inhibition of histone deacetylase 8. Bioorg. Med. Chem. 18, 4103–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson N. A.; Pitcairn C. A.; Fierke C. A. (2012) HDAC8 substrates: Histones and beyond. Biopolymers 99, 112–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B. J.; Tremblay A. M.; Deblois G.; Sylvain-Drolet G.; Giguère V. (2010) An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol. Endocrinol. 24, 1349–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff M. A.; Bando M.; Nakato R.; Watrin E.; Itoh T.; Minamino M.; Saitoh K.; Komata M.; Katou Y.; Clark D.; Cole K. E.; De Baere E.; Decroos C.; Di Donato N.; Ernst S.; Francey L. J.; Gyftodimou Y.; Hirashima K.; Hullings M.; Ishikawa Y.; Jaulin C.; Kaur M.; Kiyono T.; Lombardi P. M.; Magnaghi-Jaulin L.; Mortier G. R.; Nozaki N.; Petersen M. B.; Seimiya H.; Siu V. M.; Suzuki Y.; Takagaki K.; Wilde J. J.; Willems P. J.; Prigent C.; Gillessen-Kaesbach G.; Christianson D. W.; Kaiser F. J.; Jackson L. G.; Hirota T.; Krantz I. D.; Shirahige K. (2012) HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 489, 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehme I.; Deubzer H. E.; Wegener D.; Pickert D.; Linke J. P.; Hero B.; Kopp-Schneider A.; Westermann F.; Ulrich S. M.; von Deimling A.; Fischer M.; Witt O. (2009) Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 15, 91–99. [DOI] [PubMed] [Google Scholar]
- Niegisch G.; Knievel J.; Koch A.; Hader C.; Fischer U.; Albers P.; Schulz W. A. (2013) Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers. Urol. Oncol.: Semin. Orig. Invest. 31, 1770–1779. [DOI] [PubMed] [Google Scholar]
- Park S. Y.; Jun J. A.; Jeong K. J.; Heo H. J.; Sohn J. S.; Lee H. Y.; Park C. G.; Kang J. (2011) Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol. Rep. 25, 1677–1681. [DOI] [PubMed] [Google Scholar]
- Haberland M.; Mokalled M. H.; Montgomery R. L.; Olson E. N. (2009) Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 23, 1625–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You S.-H.; Lim H. W.; Sun Z.; Broache M.; Won K. J.; Lazar M. A. (2013) Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat. Struct. Mol. Biol. 20, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian A.; Ramos J.; Luo W.; Sirisawad M.; Verner E.; Buggy J. J. (2008) A novel deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 22, 1026–1034. [DOI] [PubMed] [Google Scholar]
- Ververis K.; Karagiannis T. C. (2012) An atlas of histone deacetylase expression in breast cancer: fluorescence methodology for comparative semi-quantitative analysis. Am. J. Transl. Res. 4, 24–43. [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Shi X.; Li Y.; Kim B. L.; Jia J.; Huang Z.; Yang T.; Fu X.; Jung S. Y.; Wang Y.; Zhang P.; Kim S. T.; Pan X.; Qin J. (2008) Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol. Cell 31, 143–151. [DOI] [PubMed] [Google Scholar]
- Madsen A. S.; Olsen C. A. (2012) Profiling of substrates for zinc-dependent lysine deacylase enzymes: HDAC3 exhibits decrotonylase activity in vitro. Angew. Chem., Int. Ed. 51, 9083–9087. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Ota Y.; Masaki R.; Bando M.; Gotoh A.; Itoh Y.; Tsumoto H.; Tatum P. R.; Mizukami T.; Nakagawa H.; Iida S.; Ueda R.; Shirahige K.; Miyata N. (2012) Rapid discovery of highly potent and selective inhibitors of histone deacetylase 8 using click chemistry to generate candidate libraries. J. Med. Chem. 55, 9562–9575. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Muto N.; Bando M.; Itoh Y.; Maski A.; Ri M.; Ota Y.; Nakagawa H.; Iida S.; Shirahige K.; Miyata N. (2014) Design, synthesis, and biological activity of NCC149 derivatives as histone deacetylase 8-selective inhibitors. ChemMedChem. 9, 657–664. [DOI] [PubMed] [Google Scholar]
- Pramana S.; Lin D.; Haldermans P.; Shkedy Z.; Verbeke T.; Göhlmann H.; De Bondt A.; Talloen W.; Bijnens L. (2010) IsoGene: An R package for analyzing dose-response studies in microarray experiments. R J. 2, 5–12. [Google Scholar]
- Kim J. H.; Yu S.; Chen J. D.; Kong A. N. (2013) The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains. Oncogene 32, 514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z.; Feng D.; Fang B.; Mullican S. E.; You S. H.; Lim H. W.; Everett L. J.; Nabel C. S.; Li Y.; Selvakumaran V.; Won K. J.; Lazar M. A. (2013) Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol. Cell 52, 769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan B.; Gao M.; Wu C.-H.; Wang T.-L.; Shih l.-M. (2012) Functional analysis of in-frame indel ARID1A mutations reveals new regulatory mechanisms of its tumor suppressor functions. Neoplasia 14, 986–993A. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.