

Abstract
A new simple and practical protocol for scalable synthesis of a novel library of propargylated and PEGylated α-hydroxy acids toward the preparation of “clickable” polylactides was described. The overall synthesis starting from readily available propargyl alcohol, bromoacetaldehyde diethyl acetal, and OEGs or PEGs was developed as a convenient procedure with low cost and no need of column chromatographic purification. The terminal alkyne functionality survives from hydrolysis of the corresponding easily accessible cyanohydrin derivatives in methanolic sulfuric acid. Facile desymmetrization, monofunctionalization, and efficient chain-elongation coupling of OEGs further enable the incorporation of OEGs to α-hydroxy acids in a simple and efficient manner. At the end, synthesis of allyloxy lactic acid indicates that an alkene group is also compatible with the developed method.
Introduction
Polylactide (PLA), prepared from α-hydroxy acids, is considered to be one of the most promising polymer-based biomaterials because of its demonstrated excellent biocompatibility and biodegradability.1 Applications of PLA in the biomedical field include drug delivery vehicles,2,3 surgical sutures,4 tissue engineering scaffolds,5 and implants for bone fixation.6 While PLA has drawn increasing interests as a great biodegradable material, it is limited in scope due to its highly hydrophobic nature. The lack of functionality in backbones and side chains of PLA makes further tailoring of its physical and chemical properties very difficult. Therefore, to further modulate physical and chemical properties of PLA, such as hydrophilicity, via introducing a wide variety of functional groups to PLA is highly desirable.
Numerous α,ω-chain-end functionalized PLAs have been prepared via ring-opening polymerization (ROP) of lactides by judicious choice of initiating alcohols and further postpolymerization modification of ω-chain-end hydroxyl groups.7,8 Pendent functionalities along PLA backbones for functionalization of polymers provide great opportunities to tailor their physical and chemical properties.9 PLAs with appending terminal alkyne,10−13 azido,13 allyl,14 oligo(ethylene glycol) (OEG)/poly(ethylene glycol) (PEG),15−17 hydroxyl,18 and carboxyl19 functionalities have been prepared and functionalized by polymerization of functional lactide monomers and postpolymerization modification. Among various appending functionalities, terminal alkyne-functionalized PLA polymers have drawn increasing interests in scientific fields,10−12 since it allows facilely placing a broad range of pendant functional molecules onto polymers via convenient “click” reaction without backbone degradation.10 “Click” chemistry utilizing Cu(I)-catalyzed Huisgen 1,3-dipolar cycloaddition between azide and terminal alkyne is a highly reliable synthetic strategy because of its high selectivity, ease to perform, and broad functional group and reaction condition tolerance.20,21 Several novel biodegradable PLA materials have been prepared via “click” functionalization, such as hydrophilic PLA material,12 temperature responsive biodegradable PLA,10 a novel grafted PLA-drug conjugate,11 and novel redox-responsive PLA micelles.22 Besides its broad applications via alkyne-azide “click” reaction, terminal alkyne-functionalized PLA has recently found promising applications in radical-mediated thiol-yne “click” reaction.23 Thiol-yne “click” reaction incorporates two thiol units into one alkyne unit along the PLA backbone in one step at room temperature with high efficiency, selectivity and no need of potentially toxic catalysts. These studies clearly indicate that terminal alkyne-functionalized PLAs are of great importance in formation of novel biomaterials with unique physical or chemical properties via alkyne-azide or thiol-yne “click” reaction.
With continuous interest in the development of novel clickable PLA biomaterials and exploration of their promising biomedical applications, we designed a library of propargylated and PEGylated α-hydroxy acids (1, poly(ethylene glycol) monopropargyl ether lactic acids, Figure 1), essential precursors toward “clickable” PLAs. PEG or OEG is a biologically inert polymer that has been extensively used as spacers and linkers for targeted drug delivery systems.24 PEGylation plays an important role in the delivery of certain therapeutic agents and confers many beneficial properties, such as increased solubility, reduced interaction with serum and proteins, and thus significantly prolonged circulation times in the bloodstream.25 PEGylation also reduces or even eliminates antigenicity and immunogenicity.26 The biological effect of PEGylation has also been reported to be associated with molecular weights of PEG chains.27 We envision that PEGylation of terminal alkyne-functionalized α-hydroxy acids would benefit the ultimate PLAs with not only the “click” function but also the advantages of PEGylation, such as increased hydrophilicity,10,16,17 faster degradation rate, and protein fouling resistance.28,29 In addition, terminal alkyne functionality may endow this family of α-hydroxy acids many other advantages. The terminal alkyne group may polymerize to provide polyacetylene polymers with unique physical properties, including electrical conductivity, nonlinear optical properties, and magnetic properties.30,31 Furthermore, hyperbranched polymers,32−34 highly cross-linked polymer networks,35−38 and novel linear or comblike polymers39,40 may be readily prepared from these propargylated α-hydroxy acids via thiol-yne “click” polymerization.
Figure 1.

Structure of propargylated and PEGylated α-hydroxy acids (poly(ethylene glycol) monopropargyl ether lactic acids, 1a–g).
Synthesis of alkyne-functionalized lactide monomers has always been a limiting step in the preparation of “clickable” PLAs via ROP of corresponding lactide monomers. It mainly relies on dimerization of highly diluted terminal alkyne-functionalized α-hydroxy acids, which typically results in poor yield (Scheme 1).10−12,41,42 Three synthetic routes have been reported in the synthesis of terminal alkyne-functionalized α-hydroxy acids.10,13,22 However, they require using either specially prepared reagents via Passerini-type condensation13 or expensive and unstable compounds (propargyl bromide, ethyl glyoxylate, and Boc-l-tyrosine) via Reformatsky-type reaction or substitution reaction/deamination.10,22 In addition, column chromatography was also required to purify intermediates in the synthesis, which further limits their scalability and practicality. Hence, it is highly desirable to develop a simple protocol for rapid access to a library of terminal alkyne-functionalized α-hydroxy acids from convenient starting materials in high yield with no need of column chromatographic separation. This would allow for a sufficient supply of functional monomers for the ultimate preparation of “clickable” biodegradable PLAs.
Scheme 1. Ring Construction of Lactide Monomer via Dimerization of α-Hydroxy Acid.

Herein, we report a novel and general approach for the synthesis of a new library of propargylated and PEGylated α-hydroxy acids 1, which features the facile monofunctionalization and chain-elongation of OEGs and the convenient hydrolysis of easily accessible cyanohydrin derivatives. The key step involved in hydrolysis of a terminal alkyne-functionalized cyanohydrin to its corresponding α-hydroxy acids is rare in the literatures. By judicious choice of acid-catalyzed hydrolysis conditions, the developed process to propargylated and PEGylated α-hydroxy acids is easily and economically operational starting from propargyl alcohol, bromoacetaldehyde diethyl acetal (2), and OEGs/PEGs, since these starting materials are widely used in industry and can be supplied in large quantities at low prices. The synthesis was column chromatography free, which renders a scale-friendly process.
Results and Discussion
Synthesis of Propargyloxy Lactic Acid (1a) via Hydrolysis of a Cyanohydrin
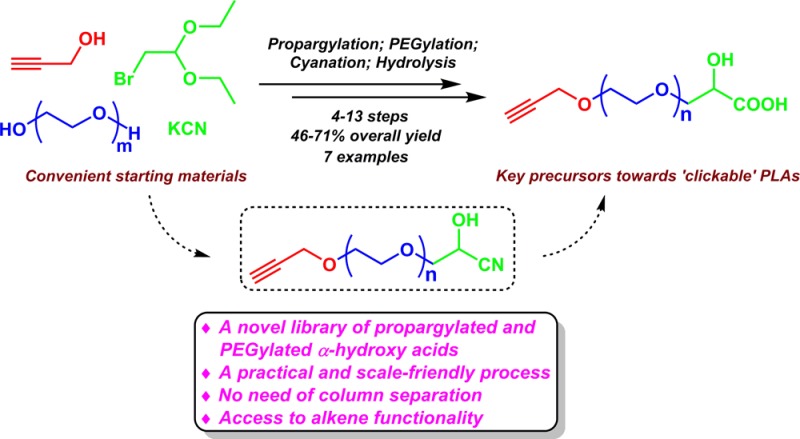
Propargyl alcohol contains two functional groups, a terminal alkyne and a hydroxy group. It is possible to employ its hydroxy group as a nucleophile under weak basic conditions without affecting the terminal alkyne group. Bromoacetaldehyde diethyl acetal (2) has a good leaving group (bromo group) and the protected aldehyde functionality, which can be later converted to a cyanohydrin. As shown in Scheme 2, an excess of propargyl alcohol was deprotonated by NaH in THF, and the resulting alkoxide nucleophilically attacked the bromo group of 2 to afford highly pure propargyloxyacetaldehyde diethyl acetal (3) as a pale red liquid in 91.8% yield without affecting the terminal alkyne group. The excess of propargyl alcohol was simply removed by washing a solution of 3 in CH2Cl2 with water because of its highly hydrophilic nature. Further purification of 3 via vacuum distillation can be completed, but is not necessary. The hydrolysis condition for converting diethyl acetal 3 to aldehyde 4 was optimized to be using a biphasic mixture of trifluoroacetic acid (TFA), water, and CH2Cl2 (1:1:4), which requires vigorous stirring to achieve quantitative conversion (Table 1, entry 8). However, p-toluenesulfonic acid (TsOH), HCl,43 H2SO4,44 and acetic acid (AcOH) were not effective in the deprotection of diethyl acetal 3 (Table 1, entries 1–6). The resulting aldehyde 4 was not isolated and directly used for the subsequent reaction.45−47
Scheme 2. Synthesis of Propargyloxy Lactic Acid (1a).

Table 1. Optimization of Deprotection of 3 with Various Acids and Solvents.
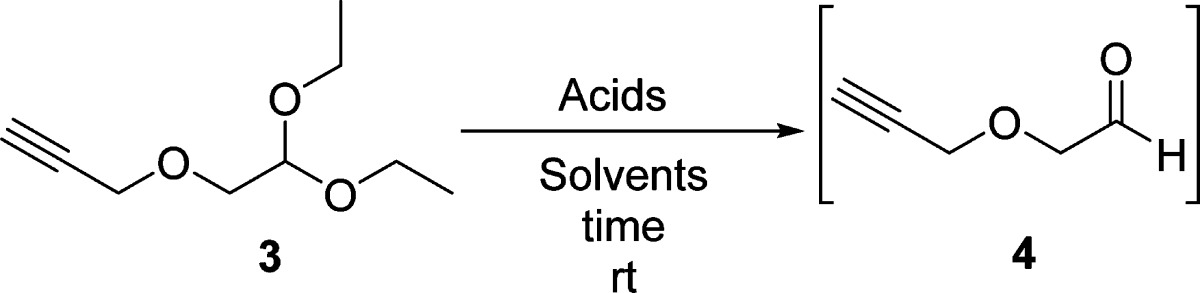
| entry | acid | solvent | time (h) | conversion (%)a |
|---|---|---|---|---|
| 1 | TsOH (10%) | H2O | 20 | 0 |
| 2 | TsOH (10%) | THF-H2O | 5 | <5 |
| 3 | HCl (2 M) | ether-H2O | 20 | 30 |
| 4 | HCl (2 M) | THF-H2O | 20 | 35 |
| 5 | H2SO4 (2 M) | AcOH-H2O (1:1)b | 36 | 40 |
| 6 | AcOH (25%)c | CH2Cl2-H2O (4:1)b | 36 | <10 |
| 7 | TFA (2%)c | CH2Cl2-H2O (4:1)b | 36 | <10 |
| 8 | TFA (16%)c | CH2Cl2-H2O (4:1)b | 12 | 100 |
Conversion was measured by 1H NMR.
Volume ratio.
Volume ratio to total solvent.
The cyanation of aldehyde 4 was performed under acidic conditions, and the resulting aqueous solution of 5 was extracted continuously with CH2Cl2 to provide a mixture of cyanohydrin 5 and acetic acid in 91.0% yield. Cyanohydrin 5 was subjected to acid-catalyzed hydrolysis without further purification to generate its α-hydroxy acid 1a (Table 2). The attempted hydrolysis of cyanohydrin 5 in aqueous HCl solution (2, 6, and 11 M) resulted in either nonhydrolyzed or decomposed starting materials. It is known that the terminal alkyne functionality can survive in refluxing 2 M H2SO4,48 so hydrolysis of 5 at various concentrations of H2SO4 and temperatures in a mixture of methanol and water was optimized (Table 2). The results showed that hydrolysis of 5 can proceed much faster at higher reaction temperature and higher concentration of H2SO4. However, high temperatures also resulted in slightly lower yields (Table 2, entries 5–8). The solvent ratio of MeOH and H2O did not affect the yields (Table 2, entries 7 and 8). The optimized conditions for hydrolysis of 5 were with 5–6 M H2SO4 for 24 h at 70 °C (Table 2, entries 3 and 4). The product consisting of a mixture of α-hydroxy acid (1a) and its methyl ester was continuously extracted with ether and hydrolyzed by aqueous NaOH (2 M) at room temperature. Since the sodium salt of 1a is not soluble in CH2Cl2 and cannot be extracted with CH2Cl2 from aqueous solution, all organic impurities, such as mineral oil from the first step, were simply removed via extracting the basic aqueous solution of 1a with CH2Cl2 in this step. This enables convenient synthesis of 1a with no need of column chromatographic separation. After acidifying the basic aqueous solution to pH < 1, the desired α-hydroxy acid 1a was obtained via continuous extraction as a slightly yellow solid with an overall yield of 71.2% (4 steps). The structure of propargylated α-hydroxy acid was verified by its 500 MHz 1H NMR spectrum (Figure S3, Supporting Information). The terminal alkyne segment of 1a was characterized by the triplet resonance for the terminal alkyne proton at δ 2.47 ppm and the doublet resonance for methylene protons at δ 4.22 ppm. The characteristic triplet resonance for the methine proton at δ 4.41 ppm clearly supports the α-hydroxy acid structure of 1a. HRMS spectrometry further confirmed the structure of 1a. This clearly indicates that terminal alkyne functionality is compatible with H2SO4-catalyzed cyanohydrin hydrolysis. It is worth to note that this procedure does not involve purification with column chromatography, which enables readily scaling up the synthesis of the desired library of propargylated and PEGylated α-hydroxy acids. Overall, it represents a simple and general process to provide sufficient quantities of terminal alkyne-functionalized α-hydroxy acid 1a with low cost and easily operational reaction conditions.
Table 2. Optimization of Hydrolysis Conditions of 5 to 1a at Various Concentrations of H2SO4, Temperatures, and Solvents.
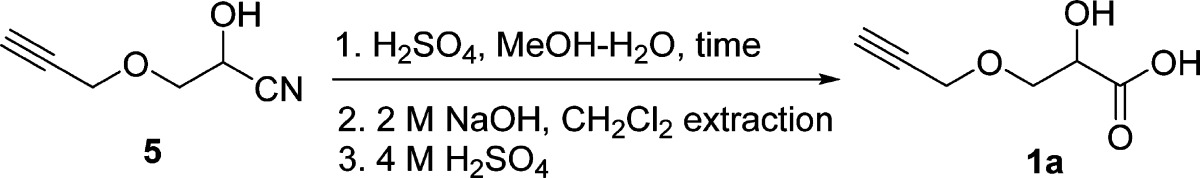
| entry | temperature (°C)a | MeOH/H2Ob | [H2SO4] (M) | time (h) | yield (1a) (%)c |
|---|---|---|---|---|---|
| 1 | 70 | 1:1 | 3.6 | 160 | 78 |
| 2 | 70 | 1:1 | 4 | 31 | 79 |
| 3 | 70 | 1:1 | 5 | 25 | 76d |
| 4 | 70 | 1:1 | 6 | 26 | 81 |
| 5 | 80 | 1:1 | 5 | 12 | 78 |
| 6 | 98 | 3:1 | 2 | 65 | 63 |
| 7 | 98 | 1:1 | 5 | 16 | 67 |
| 8 | 98 | 3:1 | 5 | 18 | 65 |
Temperature of heating source (oil bath).
Volume ratio.
Yield of 1a was measured by 1H NMR using an internal standard triphenylmethane after basic and acidic workup.
Isolated yield after basic and acidic workup, followed by continuous extraction with ether.
Synthesis of Oligo(ethylene glycol) Monopropargyl Ether Lactic Acid (1b–e) via Hydrolysis of Corresponding Cyanohydrins
The synthesis of oligo(ethylene glycol) monopropargyl ether lactic acids (1b–e) starts from their corresponding OEGs with 1–4 glycol units, all of which are inexpensive and commercially available. The synthetic strategy is based on the H2SO4-catalyzed hydrolysis of propargylated and PEGylated cyanohydrin derivatives prepared from efficient coupling of monotritylated OEGs, propargyl alcohol, and 2. As shown in Scheme 3, OEGs 6b–e were desymmetrized and monoprotected with trityl chloride to afford a mixture of monotritylated OEGs 7b–e and ditritylated OEGs as white solids (7b and 7c) or pale red viscous liquids (7d and 7e) in high yields (>92%) without further purification.49,50 Byproducts ditritylated OEGs in 7b–e were not removed, and they or their derivatives were carried over until final purification of 1b–e, since it was anticipated that they could be easily removed in their derivatized forms (OEG dipropargyl ethers) while purifying final α-hydroxy acids 1b–e.
Scheme 3. Synthesis of Oligo(ethylene glycol) Monopropargyl Ether Lactic Acids 1b–e.

Direct incorporation of a propargyl group to 7b–e is not plausible due to the fact that deprotection of the trityl group will employ a Pd-catalyzed hydrogenolysis and due to possible side reactions of terminal alkyne when excess NaH is used in subsequent steps. Thus, 7c–e were first coupled with 2 quantitatively to provide 9c–e without further purification. However, the coupling between monotrityl ether (7b) and 2 only proceeded with 66% conversion after refluxing for 48 h, probably due to the low solubility of alkoxide of 7b, and this would dramatically lower the yield of 1b and complicate the purification process. This problem was solved by using commerically available ethylene glycol monobenzyl ether (8) as starting material to afford pure 9b. It is worth to note that the excess of 2 could be simply removed while drying 9c–e under vacuum at 40 °C, which simplifies the following purification step and enables column-free isolation. The selective deprotection of trityl and benzyl groups of 9b–e via Pd-catalyzed hydrogenolysis in both THF and CH2Cl2 was not effective or required very high catalyst loading (10 mol % of Pd/C) and long reaction time to achieve high conversion.50 To bypass this problem, ethanol was selected as the solvent51 for selective hydrogenolysis with 2 mol % of Pd/C catalyst with 100% conversion without affecting the protected aldehyde. Notably, the recycled catalyst Pd/C is still active and can be reused after hydrogenolysis in ethanol. The resulting alcohols52 were then carefully tosylated with tosyl chloride to provide intermediates 10b–e50 quantitatively, which were nucleophilically substituted with propargyl alcohol to afford terminal alkyne-functionalized acetals 11b–e. The desired propargylated cyanohydrins 12b–e were obtained via deprotection and cyanation of intermediates 11b–e, followed by continuous extraction. Cyanohydrins 12b–e were hydrolyzed in methanolic H2SO4 (6 M), and all organic compounds including the desired α-hydroxy acids 1b–e were extracted with CH2Cl2 and treated with aqueous NaOH (2M) to hydrolyze methyl esters of 1b–e. The resulting basic aqueous solution was diluted and extracted with CH2Cl2 to remove all nonionic organic impurities, such as oligo(ethylene glycol) dipropargyl ethers from ditrityl protected OEGs and mineral oil from NaH. After being acidified with 4 M HCl, pure propargylated and PEGylated α-hydroxy acids 1b–e were obtained via continuous extraction with CH2Cl2 as dark red viscous liquids in 40–60% yields. These results indicate that the process to propargylated and PEGylated α-hydroxy acids 1b–e is economical, practical, and scale-friendly.
Synthesis of Octa(ethylene glycol) Monopropargyl Ether Lactic Acid (1f)
OEGs with up to four glycol units are available commerically in large quantities with low prices, and thus, one can afford to waste these inexpensive diols during the desymmetrization and monofunctionalization. However, when extending the number of repeating units of OEGs to greater than four, it is not acceptable to start with OEGs (n > 4) in the synthesis of the corresponding α-hydroxy acid due to their extremely high cost. Therefore, it is highly desirable to synthesize these extended bifunctional OEGs (n > 4) with an efficient route not involving complicated purifications, such as column chromatographic separations. Using an inexpensive THP protection/deprotection strategy, we have developed a simple procedure to produce longer well-defined bifunctional octa(ethylene glycol) (n = 8). Introducing orthogonal protecting/functional groups to two shorter oligo(ethylene glycol)s and then coupling of the two building blocks furnished the facile synthesis of propargylated and PEGylated α-hydroxy acid 1f.53
The synthesis of building block monobenzylated tetraethylene glycol (13) started from inexpensive and commercially available tetraethylene glycol (6e) (Scheme 4).53 OEG 6e was monoprotected with dihydropyran (DHP) to afford a mixture of mono-THP ether 14 and bis-THP ether 15 (8:1). This mixture was benzylated with benzyl bromide to provide a mixture of 15 and heterobifunctional 16 (∼1:8) in 92.8% yield. THP protection of 15 and 16 was completely cleaved via acetal exchange in MeOH under acidic conditions, and the resulting THP ether of methanol was readily removed in vacuo because of their low boiling points.54 The highly hydrophilic OEG 6e from THP deprotection of 15 was simply removed by washing a solution of crude product 13 in CH2Cl2 with aqueous NaCl solution to afford pure building block 13 in 92.0% yield. The overall procedure described here allows for scalable monofunctionalization of OEGs and the synthesis of pure building block 13 in a simple and efficient manner.
Scheme 4. Synthesis of Monobenzylated Tetraethylene Glycol (13).

Similar to the synthesis of 13, building block monotetrahydropyranyl tetraethylene glycol monotosylate (17) was prepared as shown in Scheme 5. A mixture of mono-THP ether 14 and bis-THP ether 15 (8:1) was tosylated with tosyl chloride to give a mixture of 15 and heterobifunctional 17 (∼1:8) in quantitative yield. After removal of the THP protecting group and side products, pure monotosylate 18 was obtained and further protected with DHP in anhydrous THF to provide pure heterobifunctional 17 in 98.3% yield.
Scheme 5. Synthesis of Monotetrahydropyranyl Tetraethylene Glycol Monotosylate (17).

The synthesis of α-hydroxy acid 1f via efficient OEG chain extension using two short building blocks 13 and 17 is illustrated in Scheme 6. Monobenzylated tetraethylene glycol (13) was deprotonated by NaH and coupled with 17 (1.1 equiv) via Williamson ether synthesis under optimized conditions to provide chain-extended heterobifunctional 19, which can be easily purified because of negligible side products. Analytically pure octa(ethylene glycol) monobenzyl ether (20) was obtained in 90% yield after deprotection of the THP group in MeOH. The coupling of 20 with 2 and subsequent hydrogenolysis and tosylation provided compound 21, which was coupled with propargyl alcohol to afford propargylated intermediate 22 in 90% yield. After deprotection of diethyl acetal, cyanation, and hydrolysis, intermediate 22 was converted to the desired α-hydroxy acid 1f in 65% yield.
Scheme 6. Synthesis of Octa(ethylene glycol) Monopropargyl Ether Lactic Acid (1f).

Synthesis of Poly(ethylene glycol) Monopropargyl Ether Lactic Acid (1g)
The THP protection/deprotection method (Schemes 4 and 5) used in preparing building blocks 13 and 17 makes it ideal for the synthesis of propargylated and PEGylated α-hydroxy acids because of the inexpensive DHP source, easy installation, facile deprotection, and ready removal from the reaction mixture after THP deprotection. As shown in Scheme 7, PEG 24 (n ≈ 23) was monoprotected with DHP to give a mixture of mono-THP ether 25 and bis-THP ether 26 (∼7:1) in 85% yield after washing with water to remove excess 24. The minor bis-THP ether 26 was intended to be carried over during the synthesis and removed as its derivative 30 at the end. Coupling of 25 with 2, followed by deprotection of THP ethers and tosylation of the resulting alcohols, provided monotosylate intermediate 27 with 28 as a minor product (92% total yield). The monotosylate 27 was propargylated with propargyl alcohol via Williamson ether synthesis to give 29 in 86% yield, which was converted to cyanohydrin 31 (95% yield) via acetal hydrolysis and cyanation. Similar to the preparation of 1f, intermediate 31 was hydrolyzed in 6 M H2SO4, and the resulting mixture was extracted with CH2Cl2 continuously and concentrated. After treatment with aqueous 2 M NaOH solution, the resulting mixture was extracted with hexane/CH2Cl2 (1:1) twice to remove nonionic impurities, such as mineral oil and derivative 30. The use of CH2Cl2 as extraction solvent in this step would result in dramatic loss of 1g because of the increased solubility of the sodium salt of 1g in CH2Cl2. α-Hydroxy acid 1g was obtained as a white waxlike solid in 74% yield via acidification of the above basic solution, continuous extraction with CH2Cl2, and washing the CH2Cl2 solution of 1g with 1 M HCl. The successful synthesis of 1g was confirmed by 1H NMR (Figure S28, Supporting Information) and further with HRMS (Figure S29 and Table S1, Supporting Information). NMR spectroscopy shows the incorporation of both α-hydroxy acid and terminal alkyne segments as end groups of 1g. Monomodal molecular weight distribution in HRMS indicates that there is only one set of end groups for polymeric 1g, and all detected mass peaks ([M – H]−) separated by one ethylene glycol unit of m/z 44 match well with the calculated exact mass of 1g ([M – H]−) (Table S1). These results present a simple and practical procedure to prepare propargylated and PEGylated α-hydroxy acids. The convenience of the THP protection/deprotection strategy to selective monofunctionalize PEG and no need of precious Pd catalyst and pressurized reaction vessels make it superior to the trityl/benzyl-involved process (1b–e) in the synthesis of PEGylated α-hydroxy acids.
Scheme 7. Synthesis of Poly(ethylene glycol) Monopropargyl Ether Lactic Acid (1g).

Synthesis of Allyloxy Lactic Acid (1h) via Hydrolysis of Cyanohydrin
Alkene-functionalized polymers have also attracted much attention due to their wide applications via efficient thiol-ene click reaction.55 We are delighted to find that our synthetic route to propargylated and PEGylated α-hydroxy acids described above could also be applied successfully to the preparation of allyloxy lactic acid (1h). As outlined in Scheme 8, the synthesis of allyloxy lactic acid (1h) was prepared following a similar procedure to propargyloxy lactic acid (1a, Scheme 2) using allyl alcohol and bromoacetaldehyde 2 as starting materials. The coupling of 2 and allyl alcohol gave 32 in 76.1% yield after vacuum distillation. After hydrolysis of aldehyde 32 under acidic conditions, cyanohydrin 33 was obtained quantitatively as a pale red liquid (containing some acetic acid) and used directly for subsequent reaction without further purification. Following hydrolysis under acidic conditions, basic treatment with aqueous NaOH solution, and removal of organic impurities, allyloxy lactic acid (1h) was extracted from the acidic aqueous solution in 71.8% yield. These results indicate that the sensitive alkene functionality is well tolerated under the reaction conditions and the developed process can be extended to prepare alkene-functionalized α-hydroxy acids as well.
Scheme 8. Synthesis of Allyloxy Lactic Acid (1h).

Conclusion
We have developed a convenient and practical protocol for scalable synthesis of a new library of propargylated and PEGylated α-hydroxy acids toward “clickable” PLAs via hydrolysis of easily accessible cyanohydrin derivatives. Starting from readily available propargyl alcohol and bromoacetaldehyde diethyl acetal, terminal alkyne-functionalized cyanohydrin 5 was prepared and hydrolyzed to afford propargylated α-hydroxy acid 1a in good overall yield. The terminal alkyne is compatible with H2SO4-catalyzed hydrolysis of cyanohydrin during the synthesis. The incorporation of OEGs or PEGs as spacers between terminal alkyne and α-hydroxy acid units resulted in a library of propargylated and PEGylated α-hydroxy acids 1b–g. The facile synthesis of propargylated and PEGylated α-hydroxy acids 1b–g features efficient desymmetrization, monofunctionalization, and chain-elongation of OEGs or PEGs with no need of column chromatographic separation. The alkene functionality is also well tolerated under the reaction conditions in the synthesis of allyloxy lactic acid. The overall synthesis requires inexpensive chemicals and does not involve column chromatography, which indicates its high efficiency and low cost in supplying large quantities of propargylated and PEGylated α-hydroxy acids to prepare “clickable” PLAs.
Experimental Section
General Information
Propargyl alcohol, allyl alcohol, bromoacetaldehyde diethyl acetal, oilgo(ethylene glycol) (OEG 6b–e, n = 1, 2, 3, 4), poly(ethylene glycol) (PEG 24, average Mn = 1000), ethylene glycol monobenzyl ether (8), dihydropyran, sodium hydride, trityl chloride, and palladium on activated charcoal (10%) were purchased from commercial sources and used as received. THF was refluxed over sodium/benzophenone and distilled before use. All other reagents and solvents were ACS grade and used as received unless specified. The progress of reactions was monitored by 1H NMR.
Characterization
Melting points were measured on a melting point apparatus. FT-IR spectra were taken with an FT-IR spectrometer. High-resolution mass spectra (HRMS) were taken on an ESI-TOF mass spectrometer. 1H NMR and 13C NMR were recorded on a 300, 500, or 600 MHz instrument in CDCl3 unless otherwise noted. CDCl3 was used as the internal standard for both 1H NMR (δ = 7.24) and 13C NMR (δ = 77.0). Data for 1H NMR are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, bs = broad signal, bs m = broad signal multiplet), integration, and coupling constant. Data for 13C NMR are reported in terms of chemical shift (δ ppm).
Synthesis of Propargyloxyacetaldehyde Diethyl Acetal (3)
Propargyl alcohol (134 mL, 2.25 mol) in THF (200 mL) was added to NaH (97 g, 60% dispersion in mineral oil, 2.4 mol) in THF (1.1 L) dropwise over 1 h at 0 °C, and the resulting reaction mixture was stirred for another 1 h at rt. Bromoacetaldehyde (2) (234 mL, 1.50 mol) and NaI (2.0 g, 13 mmol) in THF (100 mL) was added to the mixture prepared above, and the resulting suspension was refluxed for 4 days to achieve 96% conversion. The suspension was filtered, and the resultant filtrate was concentrated and redissolved in CH2Cl2 (500 mL). The CH2Cl2 layer was washed with aqueous NaCl solution (1 × 200 mL), dried over anhydrous sodium sulfate, concentrated, and dried under vacuum to afford 3 as a pale red liquid (237 g, 91.80%). 1H NMR (500 MHz, CDCl3) δ 4.62 (t, 1H, J = 5 Hz), 4.19 (d, 2H, J = 2.5 Hz), 3.62–3.72 (m, 2H), 3.50–3.58 (m, 4H), 2.40 (t, 1H, J = 2.5 Hz), 1.19 (t, 6H, J = 10 Hz); 13C NMR (125 MHz, CDCl3) δ 100.8, 79.5, 74.6, 70.1, 62.2, 58.6, 15.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C9H17O3 173.1178; Found 173.1174.
Synthesis of Propargyloxy Lactic Acid (1a)
Diethyl acetal 3 (222 g, 1.29 mol) was added to a mixture of TFA (230 mL), water (230 mL), and CH2Cl2 (920 mL) dropwise, and the mixture was stirred vigorously for 3 days until full conversion of diethyl acetal 3 to aldehyde 4. CH2Cl2 was removed, and acetic acid (450 mL) was added. The resulting mixture was cooled in an ice bath, followed by dropwise addition of KCN (256 g, 3.90 mol) in water (420 mL), and the resultant mixture was stirred for 18 h until 1H NMR showed no aldehyde left. The resulting aqueous solution was extracted continuously with CH2Cl2, and the organic layer was concentrated in vacuo to provide a mixture of acetic acid and propargylated cyanohydrin 5 (644 g, 91.0% yield estimated from 1H NMR), which was used for the subsequent step without further purification, 1H NMR (500 MHz, CDCl3) δ 4.62 (t, 1H, J = 5 Hz), 4.27 (d, 2H, J = 2.5 Hz), 3.76–3.84 (m, 2H), 2.50 (t, 1H, J = 2.5 Hz). This mixture was cooled in an ice bath, followed by the addition of concentrated H2SO4 (450 mL), methanol (150 mL), and H2O (100 mL) (final concentration of H2SO4 was ∼6 M). The resulting solution was stirred for 24 h in a 70 °C oil bath and was continuously extracted with ether to give a dark red liquid after removal of acetic acid. This dark red liquid was treated with aqueous NaOH solution (2 M) for 24 h and extracted with CH2Cl2 to remove all organic impurities, such as mineral oil. The basic solution was acidified with 4 M H2SO4 and continuously extracted with ether to afford the desired α-hydroxy acid (1a) as a slight yellow solid (144 g, 85.2%). mp 58–61 °C. 1H NMR (500 MHz, CDCl3) δ 4.41 (t, 1H, J = 4 Hz), 4.22 (d, 2H, J = 2.5 Hz), 3.83–3.92 (m, 2H), 2.47 (t, 1H, J = 2.5 Hz); 13C NMR (125 MHz, CDCl3) δ 176.12, 78.6, 75.4, 70.6, 70.2, 58.9; IR 3286, 2933, 2122, 1735, 1358, 1203, 1129, 1104, 1044; HRMS (ESI-TOF) m/z: [M – H]− Calcd for C6H7O4 143.0343; found 143.0344.
General Procedure for Synthesis of Oligo(ethylene glycol) Monopropargyl Ether Lactic Acids (1b–e)
General Procedure for Synthesis of Oligo(ethylene glycol) Monotrityl Ethers (7b–e)
A 500 mL three-neck round-bottom flask was charged with oligo(ethylene glycol) (6b–e, 3.0 mol) and pyridine (30 mL), and the resulting mixture was heated to 55 °C. Powdered trityl chloride (42 g, 0.15 mol) was added, and the reaction mixture was vigorously stirred for 18 h at 55 °C. The suspension was mixed with water (1000 mL) and extracted with toluene (5 × 100 mL). The combined organic layer was washed with 0.3 M HCl (2 × 600 mL), aqueous NaOH (1 × 600 mL), and water (3 × 600 mL), dried over anhydrous sodium sulfate, and concentrated to give 7b–e as white solids or viscous liquids containing their corresponding ditrityl ethers as minor byproducts. These mixtures were used directly for the next reaction without further purification.
Ethylene Glycol Monotrityl Ether (7b)
A white solid (5% ditrityl ether), 42 g, 92%, mp 95–98 °C. 1H NMR (300 MHz, CDCl3) δ 7.47–7.54 (m, 6H), 7.18–7.40 (m, 9H), 3.76–3.86 (bs m, 2H), 3.28–3.35 (m, 2H), 2.03 (bs, 1H, −OH).
Diethylene Glycol Monotrityl Ether (7c)
A white solid (8% ditrityl ether), 48 g, 92%, mp 108–110 °C. 1H NMR (500 MHz, CDCl3) δ 7.48–7.55 (m, 6H), 7.30 (m, 9H), 3.62–3.81 (m, 6H), 3.28–3.34 (m, 2H), 2.06 (bs, 1H, −OH).
Triethylene Glycol Monotrityl Ether (7d)
A viscous liquid (4% ditrityl ether), 57 g, 97%. 1H NMR (300 MHz, CDCl3) δ 7.47–7.55 (m, 6H), 7.19–7.38 (m, 9H), 3.64–3.82 (m, 10H), 3.27–3.34 (m, 2H).
Tetraethylene Glycol Monotrityl Ether (7e)
A viscous liquid (9% ditrityl ether), 62 g, 95%. 1H NMR (300 MHz, CDCl3) δ 7.47–7.55 (m, 6H), 7.18–7.40 (m, 9H), 3.60–3.80 (m, 14H), 3.29 (t, 2H, J = 6 Hz), 2.50 (bs, 1H, −OH).
General Procedure for Synthesis of 9b–e
Alcohol intermediates 7c–e (containing their corresponding ditrityl ethers) and ethylene glycol monobenzyl ether (8) (1.00 equiv) were deprotonated by NaH (1.35 equiv) in THF at 0 °C. The resulting reaction mixtures were stirred for 12 h at room temperature, followed by dropwise addition of a solution of 2 (1.25 equiv) in THF at 0 °C. The reaction mixtures were refluxed for 3–6 days until all alcohol intermediates were consumed (monitored by 1H NMR). The resulting suspensions were filtered, and the filtrates were concentrated, redissolved in CH2Cl2, and washed with aqueous NaCl solution. The organic layer was dried over anhydrous sodium sulfate, concentrated, and dried at 55 °C in vacuo to afford the desired products 9b–e (containing the corresponding ditrityl ethers).
9b
From 8 (60.8 g, 0.400 mol). A pale red liquid (96 g, 91%). 1H NMR (600 MHz, CDCl3) δ 7.20–7.27 (m, 5H), 4.62 (t, 1H, J = 6 Hz), 4.54 (s, 2H), 3.71–3.63 (m, 4H), 3.58–3.63 (m, 2H), 3.50–3.57 (m, 4H), 1.19 (t, 6H, J = 6 Hz); 13C NMR (150 MHz, CDCl3) δ 138.2, 128.2, 127.6, 127.4, 101.1, 73.1, 71.88, 70.86, 69.4, 62.2, 15.3.
9c
From 7c (138 g, containing 0.350 mol of 7c). A pale red liquid, 177 g, 98.3%. 1H NMR (500 MHz, CDCl3) δ 7.46–7.54 (m, 6H), 7.20–7.36 (m, 9H), 4.67 (t, 1H, J = 5 Hz), 3.54–3.80 (m, 12H), 3.27 (t, 2H, J = 5 Hz), 1.23 (t, 6H, J = 7 Hz).
9d
From 7d (190 g, containing 0.440 mol of 7d). A dark red liquid, 229 g, 90.8%. 1H NMR (500 MHz, CDCl3) δ 7.45–7.55 (m, 6H), 7.20–7.37 (m, 9H), 4.66 (t, 1H, J = 5 Hz), 3.54–3.79 (m, 16H), 3.25 (t, 2H, J = 5 Hz), 1.24 (t, 6H, J = 7 Hz).
9e
From 7e (236 g, containing 0.420 mol of 7d). A light brown liquid, 276 g, 96.5%. 1H NMR (500 MHz, CDCl3) δ 7.47–7.54 (m, 6H), 7.21–7.36 (m, 9H), 4.68 (t, 1H, J = 5 Hz), 3.52–3.79 (m, 20H), 3.30 (t, 2H, J = 5 Hz), 1.28 (t, 6H, J = 7 Hz).
General Procedure for Synthesis of Propargylated Intermediates 11b–e
A high-pressure autoclave reactor with a glass insert was charged with benzylate 9b or tritylates 9c–e, 10% Pd/C (2 mol %), and absolute ethanol (150 mL). Hydrogenolysis was carried out with effective stirring for 48 h at room temperature under 1400 psi of H2. The reaction was stopped upon completion. Triphenylmethane and catalyst (which can be reused) were removed via filtration. The filtrate was concentrated in vacuo carefully to give a mixture of desired alcohols, triphenylmethane or toluene and ethanol (<5%), which were used without further purification. These alcohol mixtures prepared above and TsCl (1.5 equiv) were dissolved in THF, and the resulting solutions were cooled in an ice bath, followed by addition of aqueous KOH solution (4 equiv) to provide the desired tosylates 10b–e quantitatively as light yellow liquids.50 These tosylates were applied to the next step directly without further purification. The propargylation of 10b–e with propargyl alcohol was carried out as described for 3 to afford propargylated intermediates 11b–e with high yields (containing byproducts from impurities), and these propargylated intermediates were used directly for the next step without purification
11b
A light yellow liquid, 35 g, 95%. 1H NMR (600 MHz, CDCl3) δ 4.62 (t, 1H, J = 6 Hz), 4.18 (d, 2H, J = 2.4 Hz), 3.65–3.71 (m, 6H), 3.50–3.58 (m, 4H), 2.40 (t, 1H, J = 2.4 Hz), 1.19 (t, 6H, J = 6 Hz).
11c
A pale red liquid, 48 g, 92%. 1H NMR (600 MHz, CDCl3) δ 4.59 (t, 1H, J = 6 Hz), 4.16 (d, 2H, J = 2.4 Hz), 3.57–3.68 (m, 10H), 3.42–3.56 (m, 4H), 2.38 (t, 1H, J = 2.4 Hz), 1.17 (t, 6H, J = 6 Hz).
11d
A pale red liquid, 62 g, 94%. 1H NMR (600 MHz, CDCl3) δ 4.62 (t, 1H, J = 6 Hz), 4.19 (d, 2H, J = 2.4 Hz), 3.60–3.73 (m, 14H), 3.48–3.58 (m, 4H), 2.41 (t, 1H, J = 2.4 Hz), 1.20 (t, 6H, J = 6 Hz).
11e
A light brown liquid, 81 g, 90%. 1H NMR (600 MHz, CDCl3) δ 4.64 (t, 1H, J = 6 Hz), 4.20 (d, 2H, J = 2.4 Hz), 3.62–3.74 (m, 18H), 3.52–3.61 (m, 4H), 2.43 (t, 1H, J = 2.4 Hz), 1.22 (t, 6H, J = 6 Hz).
General Procedure for Synthesis of Oligo(ethylene glycol) Monopropargyl Ether Lactic Acids (1b–e)
Propargylated and PEGylated α-hydroxy acids (1b–e) were prepared following a similar procedure as that described in the synthesis of 1a from diethyl acetal 3, except for the continuous extraction of intermediates or desired products with CH2Cl2. The resulting dark solutions of 1b–e were continuously extracted with CH2Cl2, and the obtained organic layer was concentrated in vacuo to remove acetic acid. The resultant acid mixtures were treated with aqueous 2 M NaOH solution for 24 h at room temperature, followed by dilution with 1 M NaOH solution. The resulting solution was extracted with CH2Cl2 to remove all organic impurities, such as oligo(ethylene glycol) dipropargyl ethers, propargyl ethyl ether, and mineral oil. After the resulting solution was acidified with 4 M HCl, the desired α-hydroxy acids 1b–e were obtained via continuous extraction with CH2Cl2 and concentrated under vacuum as dark red liquids. α-Hydroxy acids (1d and 1e) were further purified by washing their solutions in CH2Cl2 with 1 M HCl (2 × 100 mL).
1b
A dark red liquid, 20 g, 66%. 1H NMR (500 MHz, CDCl3) δ 4.35 (t, 1H, J = 5 Hz), 4.19 (d, 2H, J = 2.5 Hz), 3.77–3.89 (dq, 2H, J = 4.5 and 10.5 Hz), 3.67–3.75 (m, 4H), 2.44 (t, 1H, J = 2.5 Hz); 13C NMR (125 MHz, CDCl3) δ 174.5, 79.1, 75.01, 72.1, 70.8, 70.1, 68.9, 58.4; IR 3286, 2922, 2878, 2122, 1746, 1446, 1355, 1248, 1203, 1110, 1038; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C8H13O5 189.0763; found 189.0754.
1c
A dark red liquid, 60 g, 67%. 1H NMR (500 MHz, CDCl3) δ 4.34 (t, 1H, J = 5 Hz), 4.19 (d, 2H, J = 2.5 Hz), 3.77–3.88 (dq, 2H, J = 4.5 and 10.5 Hz), 3.60–3.73 (m, 8H), 2.44 (t, 1H, J = 2.5 Hz); 13C NMR (125 MHz, CDCl3) δ 174.1, 79.3, 74.9, 72.3, 71.0, 70.4, 70.3, 70.2, 69.0, 58.4; IR 3269, 2916, 2878, 2117, 1746, 1460, 1352, 1253, 1201, 1104, 1035; HRMS (ESI-TOF) m/z: [M – H]− Calcd for C10H15O6 231.0869; found 231.0868.
1d
A dark red liquid, 39 g, 59%. 1H NMR (500 MHz, CDCl3) δ 4.3 (t, 1H, J = 5 Hz), 4.17 (d, 2H, J = 2.5 Hz), 3.78–3.89 (dq, 2H, J = 4.5 and 10.5 Hz), 3.58–3.70 (m, 12H), 2.41 (t, 1H, J = 2.5 Hz); 13C NMR (125 MHz, CDCl3) δ 174.3, 79.3, 74.75, 72.3, 70.9, 70.43, 70.40, 70.3, 70.1, 68.9, 58.2; IR 3264, 2911, 2878, 2117, 1746, 1457, 1358, 1250, 1203, 1104; HRMS (ESI-TOF) m/z: [M – H]− Calcd for C12H19O7 275.1131; found 275.1129.
1e
A dark red liquid, 48 g, 56%. 1H NMR (500 MHz, CDCl3) δ 4.34 (t, 1H, J = 5 Hz), 4.20 (d, 2H, J = 2.5 Hz), 3.79–3.90 (dq,, 2H, J = 4.5 and 10.5 Hz), 3.60–3.79 (m, 16H), 2.44 (t, 1H, J = 2.5 Hz); 13C NMR (125 MHz, CDCl3) δ 173.9, 79.4, 74.6, 72.3, 71.0, 70.52, 70.47, 70.46, 70.45, 70.24, 70.22, 70.1, 68.9, 58.3; IR 3258, 2916, 2878, 2122, 1749, 1457, 1355, 1297, 1250, 1107; HRMS (ESI-TOF) m/z: [M – H]− Calcd for C14H23O8 319.1393; found 319.1390.
Synthesis of Octa(ethylene glycol) Monopropargyl Ether Lactic Acid (1f)
Intermediates monobenzylated tetraethylene glycol (13), tetraethylene glycol monotosylate (18), and monotetrahydropyranyl tetraethylene glycol monotosylate (17) were prepared according to our previous report.53
Monobenzylated Tetraethylene Glycol (13)53,56
A slightly yellow liquid, 67.2 g, 72.1% (3 steps). 1H NMR (500 MHz, CDCl3) δ 7.22–7.38 (m, 5H), 4.55 (s, 2H), 3.61–3.71 (m, 14H), 3.57–3.60 (m, 2H), 2.34–68 (bs, 1H, −OH); 13C NMR (125 MHz, CDCl3) δ 138.2, 128.3, 127.7, 127.5, 73.2, 72.4, 70.63 (bs and tall peak), 70.60, 70.58, 70.4, 69.4, 61.7.
Tetraethylene Glycol Monotosylate (18)53
A light yellow liquid, 200 g, 97.0%. 1H NMR (500 MHz, CDCl3) δ 7.80 (d, 2H, J = 10 Hz), 7.36 (d, 2H, J = 10 Hz), 4.16 (t, 2H, J = 5 Hz), 3.53–3.75 (m, 14H), 2.49 (br., 1H, −OH), 2.45 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 144.8, 132.9, 129.9, 127.9, 72.4, 70.7, 70.6, 70.4, 70.3, 69.2, 68.7, 61.7, 21.6.
Monotetrahydropyranyl Tetraethylene Glycol Monotosylate (17)53,57
A dark red liquid, 244 g, 79.8% (4 steps). 1H NMR (500 MHz, CDCl3) δ 7.77 (d, 2H, J = 8 Hz), 7.31 (d, 2H, J = 8 Hz), 4.60 (t, 1H, J = 3.5 Hz), 4.14 (t, 2H, J = 5 Hz), 3.79–3.88 (m, 2H), 3.52–3.70 (m, 13H), 3.43–3.51 (m, 1H), 2.42 (s, 3H), 1.74–1.86 (m, 1H), 1.64–1.73 (m, 1H), 1.42–1.61 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 144.7, 133.1, 129.8, 128.0, 99.0, 70.8, 70.7, 70.6, 70.5 (bs and tall peak), 69.2, 68.7, 66.6, 62.2, 30.6, 25.4, 21.6, 19.5.
Intermediates monotetrahydropyranyl octa(ethylene glycol) monobenzyl ether (19) and monobenzylated octa(ethylene glycol) (20) were prepared according to our previous report.53
Monotetrahydropyranyl Octa(ethylene glycol) Monobenzyl Ether (19)
A reddish viscous liquid, 135 g, 99.3%. 1H NMR (500 MHz, CDCl3) δ 7.20–7.37 (m, 5H), 4.60 (t, 1H, J = 3.5 Hz), 4.53 (s, 2H), 3.79–3.87 (m, 2H), 3.54–3.70 (m, 31H), 3.42–3.50 (m, 1H), 1.74–1.86 (m, 1H), 1.64–1.73 (m, 1H), 1.43–1.61 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 138.3, 128.3, 127.7, 127.5, 98.9, 73.2, 70.5–70.6 (bs m and tall peaks), 69.4, 66.6, 62.1, 30.5, 25.4, 19.4.
Octa(ethylene glycol) Monobenzyl Ether (20)56,58
A pale viscous liquid, 111 g, 90.0%. 1H NMR (500 MHz, CDCl3) δ 7.22–7.38 (m, 5H), 4.54 (s, 2H), 3.52–3.72 (m, 32H), 2.55 (t, 1H, J = 5 Hz, −OH); 13C NMR (125 MHz, CDCl3) δ 138.3, 128.3, 127.7, 127.5, 73.2, 72.5, 70.5–70.6 (bs and tall peaks.), 70.3, 69.4, 61.7.
Propargylated intermediate 22 was synthesized from 20 following the procedure for the synthesis of 11b–e from 9b–e; an amber liquid, 101 g, 74.0% (4 steps). 1H NMR (600 MHz, CDCl3) δ 4.60 (t, 1H, J = 6 Hz), 4.17 (d, 2H, J = 2.4 Hz), 3.59–3.68 (m, 34H), 3.47–3.57 (m, 4H), 2.41 (t, 1H, J = 2.4 Hz), 1.18 (t, 6H, J = 2.4 Hz); 13C NMR (150 MHz, CDCl3) δ 101.2, 79.7, 74.5, 71.9, 70.9, 70.5–70.6 (bs m and tall peaks), 70.4, 69.1, 62.3, 58.4, 15.3. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C25H48NaO11 547.3094; found 547.3112.
Octa(ethylene glycol) monopropargyl ether lactic acid (1f) was synthesized following the procedure for the synthesis of 1d–e from 11d–e. A light yellow viscous liquid, 58 g, 65%. 1H NMR (500 MHz, CDCl3) δ 4.29 (t, 1H, J = 5 Hz), 4.17 (d, 2H, J = 2.5 Hz), 3.79–3.91 (dq, 2H, J = 4 and 10.5 Hz), 3.53–3.70 (m, 32H), 2.41 (t, 1H, J = 2.5 Hz); 13C NMR (125 MHz, CDCl3) δ 173.6, 79.6, 74.5, 72.4, 71.0, 70.3–70.6 (bs m and tall peaks), 70.2, 69.1, 58.4; IR 3241, 2878, 2117, 1746, 1457, 1352, 1297, 1250, 1107; HRMS (ESI-TOF) m/z: [M – H]− Calcd for C22H39O12 495.2442; found 495.2437.
Synthesis of Poly(ethylene glycol) Monopropargyl Ether Lactic Acid (1g, n ≈ 23)
PEG 24 (Mn ≈ 1000, n ≈ 23, 250 g, ∼0.250 mol) and TsOH (1.0 g, 5.0 mmol) were dissolved in CH2Cl2 (3 L), followed by dropwise addition of DHP (11.5 mL, 0.125 mol) at room temeprature. The resulting reaction mixture was stirred for 4 h at rt. Half of the solvent was evaporated, and the resulting mixture was washed with H2O (60 × 500 mL), dried over anhydrous sodium sulfate, and concentrated to afford a mixture of 25 and 26 (∼7:1) as a white solid, 106 g, 84.8%. 1H NMR (500 MHz, CDCl3) δ 4.59 (t, 2H, J = 4 Hz), 3.78–3.86 (m, 2H), 3.40–3.76 (m, 90H), 1.73–1.84 (m, 1H), 1.63–1.71 (m, 1H), 1.40–1.60 (m, 4H). This mixture (106 g, ∼0.100 mol) was converted to intermediate 29 with 30 as minor byproduct via alkylation with 2, transacetalization with ethanol,53 tosylation, and propargylation as described for the synthesis of 22. A light yellow solid, 89.3 g, 79.1% (4 steps). 1H NMR (600 MHz, CDCl3) δ 4.55 (t, 1H, J = 5.4 Hz), 4.12 (d, 2H, J = 2.4 Hz), 3.43–3.70 (m, 101H), 2.38 (t, 1H, J = 2.4 Hz), 1.13 (t, 6H, J = 7.2 Hz).
Poly(ethylene glycol) monopropargyl ether lactic acid (1g, n ≈ 23) was prepared using a modified procedure for the synthesis of 1f from 22 as discussed above. A solvent mixture of CH2Cl2 and hexane (1:1, 2 × 100 mL) was used to extract a basic solution of 1g to remove organic impurities. Product 1g was obtained as a white wax solid, 63.2 g, 74.4% (3 steps). 1H NMR (500 MHz, CDCl3) δ 4.29 (t, 1H, J = 4 Hz), 4.18 (d, 2H, J = 2.5 Hz), 3.74–3.86 (dq, 2H, J = 4 and 10 Hz), 3.56–3.72 (m, 113H), 2.41 (t, 1H, J = 2.5 Hz); 13C NMR (125 MHz, CDCl3) δ 173.4, 79.7, 74.5, 72.4, 71.1, 70.5–70.6 (bs m and tall peaks), 70.44, 70.41, 70.3, 70.2, 69.1, 58.4; IR 3252, 2872, 2111, 1749, 1463, 1355, 1300, 1248, 1112.
Synthesis of Allyloxy Lactic Aicd (1h)
α-Hydroxy acid 1h was prepared using the same procedure as described for the synthesis of propargyloxy lactic acid (1a).
Allyloxyacetaldehyde Diethyl Acetal (32)
A colorless liquid, 46 °C/2 Torr, 198 g, 76.1%. 1H NMR (600 MHz, CDCl3) δ 5.80–5.89 (m, 1H), 5.18–5.25 (m, 1H), 5.08–5.14 (m, 1H), 4.58 (t, 1H, J = 6 Hz), 3.98 (d, 2H, J = 6 Hz), 3.60–68 (m, 2H), 3.47–3.55 (m, 2H), 3.42 (d, 2H, J = 6 Hz), 1.16 (t, 6H, J = 6 Hz); 13C NMR (150 MHz, CDCl3) δ 134.5, 117.2, 101.1, 72.3, 70.6, 62.1, 15.2; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C9H19O3 175.1334; found 175.1329.
Allyloxy Cyanohydrin (33)
Crude product without further purification, 145 g, 100%. 1H NMR (500 MHz, CDCl3) δ 5.82–5.93 (m, 1H), 5.21–5.33 (m, 2H), 4.56 (t, 1H, J = 5 Hz), 4.10 (dt, 2H, J = 1.5 and 5 Hz), 3.64–3.72 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 133.3, 118.5, 118.2, 72.7, 70.4, 60.7.
Allyloxy Lactic Aicd (1h)
Light yellow liquid, 120 g, 71.8%. 1H NMR (500 MHz, CDCl3) δ 5.80–5.91 (m, 1H), 5.16–5.32 (m, 2H) 4.37 (t, 1H, J = 5 Hz), 4.00–4.09 (m, 2H), 3.70–3.81 (dq, 2H, J = 5 and 10 Hz); 13C NMR (125 MHz, CDCl3) δ 176.0, 133.7, 118.7, 118.0, 72.6, 70.8, 70.2; IR 3407, 3093, 2977, 2933, 2872, 2500–3500 (br.), 1744, 1424, 1352, 1206, 1123, 1041; HRMS (ESI-TOF) m/z: [M – H]− Calcd for C6H9O4 145.0511; found 145.0499.
Acknowledgments
This manuscript is dedicated in memory of my advisor, Prof. Gregory L. Baker, who passed away unexpectedly while this paper was under preparation. The authors gratefully acknowledge the invaluable discussions, assistance, and guidance from Prof. William D. Wulff at Michigan State University during the preparation of this manuscript. This work was funded by NIH 5RC2ES018756-02.
Supporting Information Available
NMR spectra for all pure compounds and HRMS data for 1g. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Status
∥ Prof. Gregory L. Baker passed away on October 18, 2012, unexpectedly.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Albertsson A. C.; Varma I. K. Biomacromolecules 2003, 4, 1466–1486. [DOI] [PubMed] [Google Scholar]
- Jiang W.; Gupta R. K.; Deshpande M. C.; Schwendeman S. P. Adv. Drug Delivery Rev. 2005, 57, 391–410. [DOI] [PubMed] [Google Scholar]
- Rancan F.; Papakostas D.; Hadam S.; Hackbarth S.; Delair T.; Primard C.; Verrier B.; Sterry W.; Blume-Peytavi U.; Vogt A. Pharm. Res. 2009, 26, 2027–2036. [DOI] [PubMed] [Google Scholar]
- Dechy-Cabaret O.; Martin-Vaca B.; Bourissou D. Chem. Rev. 2004, 104, 6147–6176. [DOI] [PubMed] [Google Scholar]
- Pan Z.; Ding J. Interface Focus 2012, 2, 366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L. E.; Ignatius A. A.; Rehm K. E.; Scholz C. Biomaterials 1996, 17, 1621–1626. [DOI] [PubMed] [Google Scholar]
- Mizutani M.; Arnold S. C.; Matsuda T. Biomacromolecules 2002, 3, 668–675. [DOI] [PubMed] [Google Scholar]
- Stanford M. J.; Dove A. P. Macromolecules 2009, 42, 141–147. [Google Scholar]
- Yu Y.; Zou J.; Cheng C. Polym. Chem. 2014, 10.1039/C4PY00667D. [DOI] [Google Scholar]
- Jiang X.; Vogel E. B.; Smith M. R. III; Baker G. L. Macromolecules 2008, 41, 1937–1944. [Google Scholar]
- Yu Y.; Zou J.; Yu L.; Ji W.; Li Y.; Law W.-C.; Cheng C. Macromolecules 2011, 44, 4793–4800. [Google Scholar]
- Coumes F.; Darcos V.; Domurado D.; Li S.; Coudane J. Polym. Chem. 2013, 4, 3705–3713. [Google Scholar]
- Rubinshtein M.; James C. R.; Young J. L.; Ma Y. J.; Kobayashi Y.; Gianneschi N. C.; Yang J. Org. Lett. 2010, 12, 3560–3563. [DOI] [PubMed] [Google Scholar]
- Mecerreyes D.; Miller R. D.; Hedrick J. L.; Detrembleur C.; Jerome R. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 870–875. [Google Scholar]
- Jiang X.; Smith M. R. III; Baker G. L. Macromolecules 2008, 41, 318–324. [Google Scholar]
- Jiang X.; Vogel E. B.; Smith M. R.; Baker G. L. J. Polym. Sci., Part A: Polym. Chem. 2007, 45, 5227–5236. [Google Scholar]
- Castillo J. A.; Borchmann D. E.; Cheng A. Y.; Wang Y.; Hu C.; García A. J.; Weck M. Macromolecules 2012, 45, 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noga D. E.; Petrie T. A.; Kumar A.; Weck M.; García A. J.; Collard D. M. Biomacromolecules 2008, 9, 2056–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y.; Shirotani K.; Yamane H.; Kitao T. Macromolecules 1988, 21, 3338–3340. [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- Yuan Z.; Kuang G.-C.; Clark R. J.; Zhu L. Org. Lett. 2012, 14, 2590–2593. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Yin L.; Tu C.; Song Z.; Zhang Y.; Xu Y.; Tong R.; Zhou Q.; Ren J.; Cheng J. ACS Macro Lett. 2013, 2, 40–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Yin L.; Xu Y.; Tong R.; Lu Y.; Ren J.; Cheng J. Biomacromolecules 2012, 13, 3456–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B.; Desai A.; Tang S.; Thomas T. P.; Baker J. R. Org. Lett. 2010, 12, 1384–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plank C.; Mechtler K.; Szoka F. S. Jr.; Wanger E. Hum. Gene Ther. 1996, 7, 1437–1446. [DOI] [PubMed] [Google Scholar]
- Veronese F. M.; Pasut G. Drug Discovery Today 2005, 10, 1451–1458. [DOI] [PubMed] [Google Scholar]
- Peterson H.; Fechner P. M.; Martin A. L.; Kunath K.; Stolnik S.; Roberts C. J.; Davies M. C.; Kissel T. Bioconjugate Chem. 2002, 13, 845–854. [DOI] [PubMed] [Google Scholar]
- Dalsin J. L.; Lin L.; Tosatti S.; Vörös J.; Textor M.; Messersmith P. B. Langmuir 2005, 21, 640–646. [DOI] [PubMed] [Google Scholar]
- Banerjee I.; Pangule R. C.; Kane R. S. Adv. Mater. 2011, 23, 690–718. [DOI] [PubMed] [Google Scholar]
- Shiotsuki M.; Sanda F.; Masuda T. Polym. Chem. 2011, 2, 1044–1058. [Google Scholar]
- Masuda T. J. Polym. Sci., Part A: Polym. Chem. 2007, 45, 165–180. [Google Scholar]
- Konkolewicz D.; Gray-Wealeb A.; Perrier S. J. Am. Chem. Soc. 2009, 131, 18075–18077. [DOI] [PubMed] [Google Scholar]
- Konkolewicz D.; Gray-Wealeb A.; Perrier S. Chem. Commun. 2011, 47, 239–241. [DOI] [PubMed] [Google Scholar]
- Barbey R.; Perrier S. ACS Macro Lett. 2013, 2, 366–370. [DOI] [PubMed] [Google Scholar]
- Fairbanks B. D.; Sims E. A.; Anseth K. S.; Bowman C. N. Macromolecules 2010, 43, 4113–4119. [Google Scholar]
- Chan J. W.; Zhou H.; Hoyle C. E.; Lowe A. B. Chem. Mater. 2009, 21, 1579–1585. [Google Scholar]
- Chan J. W.; Shin J.; Hoyle C. E.; Bowman C. N.; Lowe A. B. Macromolecules 2010, 43, 4937–4942. [Google Scholar]
- Fairbanks B. D.; Scott T. F.; Kloxin C. J.; Anseth K. S.; Bowman C. N. Macromolecules 2009, 42, 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Türünç O.; Meier M. A. R. J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 1689–1695. [Google Scholar]
- Han J.; Zheng Y.; Zhao B.; Li S.; Zhang Y.; Gao C. Sci. Rep. 2014, 4, 4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portis A. M.; Carballo G.; Baker G. L.; Chan C.; Walton S. P. Microsc. Res. Tech. 2010, 73, 878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Chen C.-K.; Law W.-C.; Weinheimer E.; Sengupta S.; Prasad P. N.; Cheng C. Biomacromolecules 2014, 15, 524–532. [DOI] [PubMed] [Google Scholar]
- Compernolle F.; Toppet S. J. Heterocycl. Chem. 1986, 23, 541–544. [Google Scholar]
- Bitan G.; Muller D.; Kasher R.; Gluhov E. V.; Gilon C. J. Chem. Soc., Perkin Trans. 1 1997, 1501–1510. [Google Scholar]
- Nishida H.; Miyazaki Y.; Mukaihira T.; Saitoh F.; Fukui M.; Harada K.; Itoh M.; Muraoka A.; Matsusue T.; Okamoto A.; Hosaka Y.; Matsumoto M.; Ohnishi S.; Mochizuki H. Chem. Pharm. Bull. 2002, 50, 1187–1194. [DOI] [PubMed] [Google Scholar]
- Liu C. F.; Tam J. P. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 6584–6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vugts D. J.; Aktas H.; Al-Mafraji K.; de Kanter F. J. J.; Ruijter E.; Groen M. B.; Orru R. V. A. Eur. J. Org. Chem. 2008, 1336–1339. [Google Scholar]
- Zhang Q.; Ren H.; Baker G. L. Beilstein J. Org. Chem. 2014, 10, 1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzo A. D.; Vigo A.; Donzelli G. Makromol. Chem. 1989, 190, 2457–2461. [Google Scholar]
- Chen Y.; Baker G. L. J. Org. Chem. 1999, 64, 6870–6873. [DOI] [PubMed] [Google Scholar]
- Mirrington R. N.; Schmalzl K. J. J. Org. Chem. 1972, 37, 2877–2881. [Google Scholar]
- It is worth mentioning that the resulting alcohols could not be completely dried under vacuum to prevent inter- or intramolecular transacetalization. Moreover, those alcohols contaminated by inter- or intramolecular transacetalization can be rapidly recovered by stirring their solutions in ethanol under nitrogen (100 psi) if inter- or intramolecular transacetalization takes place.
- Zhang Q.; Ren H.; Baker G. L. Tetrahedron Lett. 2014, 55, 3384–3386. [Google Scholar]
- Abe K.; Sato T.; Nakamura N.; Sakan T. Chem. Lett. 1977, 6, 817–818. [Google Scholar]
- Lowe A. B. Polym. Chem. 2010, 1, 17–36. [Google Scholar]
- French A. C.; Thompson A. L.; Davis B. G. Angew. Chem., Int. Ed. 2009, 48, 1248–1252. [DOI] [PubMed] [Google Scholar]
- Asakawa M.; Ashton P. R.; Balzani V.; Credi A.; Mattersteig G.; Matthews O. A.; Montalti M.; Spencer N.; Stoddart J. F.; Venturi M. Chem.—Eur. J. 1997, 3, 1992–1996. [Google Scholar]
- Li Y.; Thapa B.; Zhang H.; Li X.; Yu F.; Jeong E.-K.; Yang Z.; Jiang Z.-X. Tetrahedron 2013, 69, 9586–9590. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.