

Abstract
Tumors are composed of cancer cells but also a larger number of diverse stromal cells in the tumor microenvironment. Stromal cells provide essential supports to tumor pathophysiology but the distinct characteristics of their signaling networks are not usually considered in developing drugs to target tumors. This oversight potentially confounds proof-of-concept studies and increases drug development risks. Here, we show in established murine and human models of breast cancer how differential regulation of Akt by the small GTPase RhoB in cancer cells or stromal endothelial cells determines their dormancy versus outgrowth when angiogenesis becomes critical. In cancer cells in vitro or in vivo, RhoB functions as a tumor suppressor that restricts EGF receptor (EGFR) cell surface occupancy as well as Akt signaling. However, after activation of the angiogenic switch, RhoB functions as a tumor promoter by sustaining endothelial Akt signaling, growth, and survival of stromal endothelial cells that mediate tumor neoangiogenesis. Altogether, the positive impact of RhoB on angiogenesis and progression supercedes its negative impact in cancer cells themselves. Our findings elucidate the dominant positive role of RhoB in cancer. More generally, they illustrate how differential gene function effects on signaling pathways in the tumor stromal component can complicate the challenge of developing therapeutics to target cancer pathophysiology.
Introduction
Solid tumors are composed of neoplastic epithelial cells that exist in a microenvironment rich in resident fibroblasts, endothelial cells, pericytes, leukocytes, and extracellular matrix (ECM) proteins (1, 2). Both cancer cells and their surrounding stroma undergo dramatic alterations during the two main stages of tumor initiation and progression (3, 4). During initiation, a normal ductal epithelium with a quiescent stroma enters a hyperproliferative stage, with cells that harbor oncogenic mutations in genes promoting cell growth and survival (4). Initiation is mostly induced by the intrinsic properties of the epithelial cells themselves that facilitate transformation. The transformed cells subsequently progress to form a solid tumor that may ultimately acquire the ability to invade and metastasize to distant organs. Unlike initiation, progression is determined primarily by the surrounding stromal microenvironment. Cancer cells produce a range of growth factors and proteases that modify the adjacent stroma to form a permissive and supportive environment for tumor growth known as a reactive tumor stroma (5). One critical modification in the tumor stroma is the angiogenic switch in which proangiogenic molecules secreted by tumor cells recruit endothelial cells to the growing tumor to drive neovascularization. Thus, crosstalk between tumor cells and their surrounding stromal cells drives progression. Elucidating the character of the tumor-promoting signaling pathways that are present in both neoplastic cells and their stromal compartments is critical to define valid therapeutic targets, develop effective drugs and understand their mechanism of action.
RhoB is a member of the family of small GTPases that is distinguished from other Rho proteins by its subcellular localization in endosomes, Golgi-associated vesicles and the nucleus (6–8). In nontumor cells, RhoB controls the subcellular trafficking of important signaling molecules such as EGF receptor (EGFR), platelet-derived growth factor receptor (PDGFR), and Src between endosomes and other intracellular compartments (9–11). RhoB is also a sensor of cellular stress and is part of the immediate early response to EGF, TGF-β and Src activation (12). Thus, RhoB is considered a modifier of growth factor signals that are associated with cellular stresses coupled to neoplastic transformation (13). Alterations in RhoB expression have been detected in a number of human cancers. For example, RhoB expression is reduced significantly in invasive and poorly differentiated head and neck squamous cell carcinomas compared with normal epithelium (14). Similarly, expression of RhoB in lung and gastric cancers is diminished markedly compared to nonneoplastic tissues, and, moreover, overexpression of RhoB significantly inhibits the proliferation, migration, and invasion of gastric cancer cells in vitro (15, 16). Similar findings have been reported in ovarian adenocarcinomas (17). Cumulatively, these results support the concept that RhoB functions as a tumor suppressor or negative modifier gene in cancer (18).
In both tumor cells and stromal endothelial cells, RhoB function has been linked to the regulation of PI3K/Akt survival pathways (7, 19). PI3K/Akt signaling is crucial for tumor progression insofar as numerous genetic lesions have been discovered in Akt signaling components in human breast cancers and other solid tumors (20). In stromal endothelial cells, loss of RhoB decreases Akt phosphorylation and blocks its nuclear translocation (7). In tumor cells, regulation of the Akt signaling axis by RhoB controls invasion and migration (21, 22). However, a fully integrated view of how RhoB acts in vivo in tumor cells and stromal cells has yet to be developed. In this study, we show how RhoB differentially regulates the Akt pathway in neoplastic tumor cells versus stromal endothelial cells. Strikingly, the stromal effects of RhoB in the tumor vasculature override the effects of RhoB in tumor cells, such that the net in vivo effect is a marked reduction in the rate of tumorigenesis when RhoB is missing, challenging the prevailing view that this gene acts chiefly as a suppressor function in cancer.
Materials and Methods
Mice
Transgenic MMTV-PyT+/− males (Jackson Labs; ref. 23) were crossed with RhoB+/− females (12). Resulting double heterozygotes were crossed to obtain the required genotypes (MMTV-PyT+/−/RhoB+/+, MMTV-PyT+/−/RhoB+/−, MMTV-PyT+/−/RhoB−/−) in females. For mammary gland transplantations, NOD.CB17-Prkdcscid/J mice (NOD/SCID; Jackson labs) were crossed with 129/Sv/RhoB−/− mice. Resulting double heterozygotes were crossed to obtain Prkdcscid;Prkdcscid/RhoB−/− or Prkdcscid;Prkdcscid/RhoB+/− mice, which in turn were crossed to get Prkdcscid;Prkdcscid/RhoB+/−, Prkdcscid;Prkdcscid/RhoB−/− or Prkdcscid;Prkdcscid/RhoB+/+ mice. Transplantations were carried out with modifications of previous procedures (24). For orthotopic injection of tumor cells into severe combined immunodeficient (SCID) mice, 3 × 105 cells (MMTV-RhoB cells or MDA-MB-231) were embedded in Matrigel Matrix (BD Biosciences) and injected into the inguinal gland (no. 4). All tumors were measured with a caliper and total tumor volume was determined using the following formula: volume = (4/3)(π)(1/2 × smaller diameter)2 (1/2 × larger diameter). All studies were conducted in compliance with the Beth Israel Deaconess Medical Center or Lankenau Institute Institutional Animal Care and Use Committee guidelines (Boston, MA).
Human cells and shRNA
MDA-MB-231 cells obtained from American Type Culture Collection and authenticated by this established provider were cultured for less than 6 months after resuscitation in the laboratory in Dulbecco's modified Eagle's medium (DMEM) 4.5 g/L glucose containing 10% FBS. Proliferation assays were conducted using the MTT kit according to manufacturer's instructions (Roche). Migration assays were conducted using a Transwell assay for 4 hours (Corning). Colony formation in soft agar was conducted by resuspending 5 × 104 cells per well in culture medium containing 0.5% agar and 10% serum, and then plating onto a bottom layer of 1% agar in 6-well plates. 293T cells were used for production of short hairpin RNA (shRNA) purchased from The RNAi Consortium (TRC) at Broad Institute (Cambridge, MA) using recommended procedures (25). The targeting sequences of these constructs are listed in Supplementary Table S2.
Mouse primary tumor cells
Primary mouse tumor cells were isolated as described previously (26). Anchorage-independent cell growth was conducted as described on the nonadhesive substrate polyHEMA (Sigma; ref. 27). Cell proliferation was assessed using the BrdUrd assay from Roche. Three-dimensional (3D) morphogenesis assays in growth factor-reduced Matrigel (BD Biosciences) were conducted as previously described (28, 29).
Mouse primary tumor endothelial cells
Tumors were collected in a petri dish, washed with cold Hank's balanced salt solution containing antibiotics, finely minced and digested in 0.2% Worthington type I collagenase for 30 minutes at 37° C. Digested tissue was filtered through a 100-micron cell strainer (BD Discovery Labware), centrifuged, and resuspended in PBS with 0.1% bovine serum albumin (BSA). The filtrate was incubated with magnetic beads (Invitrogen) conjugated to anti-mouse CD31 antibody (BD Pharmingen) for 15 minutes. The beads were washed vigorously 6 to 8 times in 0.1% BSA/PBS using the MPC magnet (Invitrogen) and plated on gelatin coated plates in high glucose DMEM supplemented with 25 mg Endothelial Cell Mitogen (Biomedical Technologies, Inc.), 100 μg/mL heparin, 20% FBS, and nonessential amino acids. At 80% confluence, cells were removed and incubated with beads conjugated to anti-mouse ICAM-2 (BD Pharmingen). Endothelial cell identity was confirmed by DiI-actylated Low Density Lipoprotein (LDL) fluorescence (Biomedical Technologies, Inc).
Whole-mount staining
Mammary glands were removed at 5 weeks of development and spread on a glass slide. After fixation for 4 hours in Carnoy's solution (10% glacial acetic acid, 30% chloroform, and 60% EtOH), the glands were rehydrated by washing sequentially in 70%, 50%, and 25% EtOH for 15 minutes each and then in H2O for 5 minutes. Each gland was stained overnight with carmine alum (Sigma), dehydrated in EtOH, cleared in xylene, and mounted in permamount (Fisher Scientific).
Immunoblot and immunoprecipitation
Western blot analysis was conducted using antibodies against phospho-Akt S473 (Cell signaling), total Akt (Cell signaling), phospho-EGFR Tyr1068 (Cell signaling), total EGFR (Cell signaling), and α-tubulin (Calbiochem). For immunoprecipitation, cell extracts were prepared in lysis buffer (150 mmol/L Tris, pH 7.5, 150 mmol/L NaCl, 1% NP-40 and 0.5% deoxycholic acid, 1 mmol/L sodium pyrophosphate, 200 mmol/L NaF, 50 nmol/L calyculin, and phosphatase/protease inhibitors). Immunoprecipitations were conducted with pAktS473 antibody (Cell Signaling) and a 50:50 mix of protein G and protein A Sepharose (GE), then analyzed by immunoblotting with antibody against Akt1, 2, or 3 (Cell Signaling Technology).
Immunostaining
Costaining of SMA (Sigma) and CD31 (Pharmingen) were conducted on 5-micron frozen sections using standard methods. Samples were mounted with Prolong Gold anti-fade mounting media containing 4′, 6 diamidino-2-phenylindole (DAPI; Invitrogen). Confocal images were taken using the Zeiss LSM510 confocal system and quantified using the Velocity software (Perkin Elmer).
In situ hybridization
In situ hybridization protocol has been previously detailed (30). Use of human tissue was approved by the Beth Israel Deaconess Medical Center Institutional Review Board.
Quantitative RT-PCR for gene expression
Tumor was collected in RNAlater solution (Ambion) overnight at 4°C for total RNA extraction using RNeasy Fibrous Tissue Mini Kit (Qiagen). cDNA were prepared from 0.8 μg total RNA using random hexaprimers as templates (Applied Biosystem kit). Quantitative real-time PCR (qRT-PCR) was carried out on an ABI Prism 7000 Sequence Detection System (Applied Biosystems). Cycling conditions were conducted as described previously (31). Multiple samples from independent tumors were run in triplicate. Primers are listed in Supplementary Table S1. VE-cadherin and glceraldehyde-3 phosphate dehydrogenase (GAPDH) were used as references for quantification of blood vessels RNA fraction and total RNA, respectively. The multigene transcriptional profiling method was used to determine mRNA copies per cell as described previously (32, 33).
Flow cytometry
Cells (5 × 105) were dissociated using 5 mmol/L EDTA, blocked in cold PBS with 3% BSA for 30 minutes, and incubated with IMC-ME1 antibody at 10 μg/mL for 1 hour on ice. After washing, cells were incubated with phycoerythrin-conjugated secondary antibody (Jackson Immunoresearch) for an additional 1 hour on ice, and then analyzed for IMC-ME1 surface binding using flow cytometry (Becton Dickinson FACSAria system). Data were analyzed using FlowJo (Tree Star, Inc.)
Results
RhoB is elevated in tumor blood vessels but less frequently expressed in tumor cells
There is considerable evidence that RhoB functions as a tumor suppressor in cancer cells where its expression correlates inversely with tumor aggressiveness (34). To begin to evaluate the overall contributions of RhoB during tumorigenesis in vivo, we investigated RhoB expression in tumor cells and the adjacent stroma of human breast tumor specimens using RNA in situ hybridization. Among the set of tumor specimens examined, 6 of 8 contained invasive ductal carcinoma with 2 of those 6 also containing ductal carcinoma in situ (DCIS). RhoB expression was detected in tumor cells in 4 of 6 of the invasive ductal carcinomas and 1 of 2 of the DCIS, whereas expression was lower in adjacent normal ductal epithelium. RhoB is expressed in the endothelial vasculature during neoangiogenesis in nonmalignant settings (7), so endothelial cells in the tumor and adjacent normal tissues were examined. Notably, all the breast tumor specimens showed elevated RhoB expression in tumor-associated blood vessels, with lower expression observed in blood vessels from adjacent normal tissue. Figure 1 shows antisense RNA hybridization images for adjacent normal tissue (Fig. 1A and B), tumor tissue and tumor-associated blood vessels (Fig. 1C and D), and sense RNA probe controls (Fig. 1E and F). These results suggested that RhoB expression was uniformly increased in tumor blood vessels. These findings agreed with a published dataset (35) of gene expression in 5 invasive ductal carcinoma and 5 invasive lobular carcinomas, where our bioinformatics analysis indicated that 8 of 10 breast tumor specimens displayed higher RhoB expression. Overall, it seemed that RhoB expression is commonly increased in the blood vessels of breast tumors, suggesting the hypothesis that RhoB may exert a role in the tumor vasculature that is distinct from its role in tumor cells.
Figure 1.
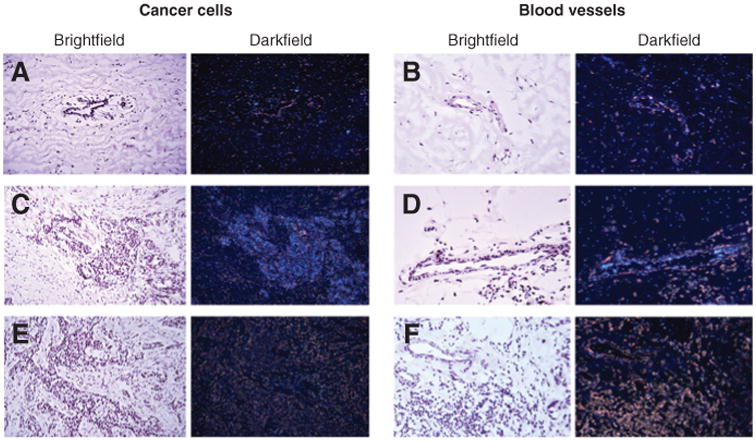
RhoB is expressed in blood vessels and cancer cells in human breast tumors. Antisense probe hybridized to noncancerous mammary duct (A), noncancerous blood vessel in adjacent normal tissue (B), ductal carcinoma (C), and blood vessels in tumor (D). The result shows less expression of RhoB in adjacent normal tissue compared with ductal carcinoma (A vs. C) and less in adjacent normal stromal blood vessels compared with blood vessels in the tumor (B vs. D). The control sense probe on tumor tissue (E) and tumor blood vessel (F) are also shown. Magnification 400×.
RhoB loss increases tumor initiation and tumor cell growth
To dissect the functional role of RhoB during breast cancer development, we used the well-established MMTV-PyT and MMTV-myc mouse models of spontaneous breast cancer (23). MMTV-PyT mice were interbred with RhoB null mice and breast tumorigenesis was evaluated in the backcrossed offspring. An evaluation of whole-mount staining of mammary fat pads revealed an increase in the number of early tumor lesions in RhoB−/− animals (average of 10 lesions per fat pad) compared with RhoB+/− animals (average of 0.5 lesions per fat pad; Fig. 2A (a and b)). We next isolated cells from the arising tumors and evaluated their growth in vitro. Three-dimensional growth of RhoB−/− tumor cells in Matrigel/collagen revealed an enhanced growth in acini compared with control RhoB cells+/− (Fig. 2B (a and b)). Similarly, RhoB−/− tumor cells survived and proliferated under anchorage-independent conditions, whereas RhoB+/− cells did not survive or grow in the same assay (Fig. 2C). The level of cell proliferation in RhoB−/− tumor cell cultures was assessed using BrdUrd analysis (Fig. 2D). The results indicated a much higher proliferation rate in RhoB−/− tumor cells compared with RhoB+/+ cells. Although both RhoB−/− and RhoB+/− tumor cells displayed increased proliferation at 72 hours, compared with 48 hours, RhoB−/− tumor cells displayed higher levels of proliferation at both time points.
Figure 2.
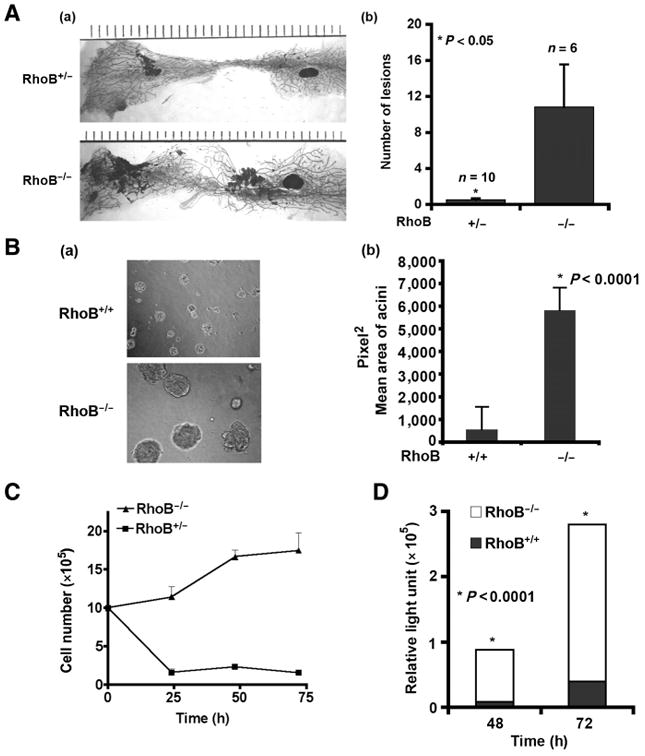
RhoB loss promotes the growth of breast tumor cells. A, mammary glands from 8-week-old mice are shown (a) and mean number of lesions graphed (b). n, the number of fat pads that were stained. B, 3D culture of primary tumor cells at 16 days (a). Acini size was quantified by ImageJ software (b). C, tumor cells counted from 0 to 72 hours on polyHEMA-coated dishes. D, BrdUrd incorporation for 48 hours of incubation for RhoB+/+ and RhoB−/− cells in 5% serum. See also Supplementary Fig. S1. P values were calculated with Prism4 by an unpaired 2-tailed test. Data are represented as mean ± SEM.
Similar findings were obtained in experiments in human MDA-MB-231 breast cancer cells where RhoB expression was inhibited by shRNA-mediated silencing (Fig. 3 and Supplementary Fig. S2A). RhoB downregulation elicited a marked phenotypic change in 3D Matrigel cultures such that highly branched and invasive structures were decreased and replaced with proliferative foci of tumor cells (Fig. 3A). Notably, this phenotypic change was associated with decreased invasion of the Matrigel matrix. In considering these characteristics in two-dimensional (2D) cell cultures, we found that RhoB-silenced cells were less migratory in Transwell assays (Fig. 3B) and more proliferative in MTT assays (Fig. 3C). The invasive phenotype seen in 3D Matrigel/collagen cell cultures were difficult to quantify in comparison with the loss-of-function phenotype, due to the differently shaped colonies observed, so this culture system was used to qualitatively evaluate the impact of RhoB. To quantify differences in 3D cell growth, we took advantage of agar 3D cell culture assays where invasion is inhibited, resulting in cell colonies that were similarly shaped and more readily compared in size (Fig. 3D (a and b)). In these cultures, we observed a significant increase in the size of colonies with loss of RhoB.
Figure 3.
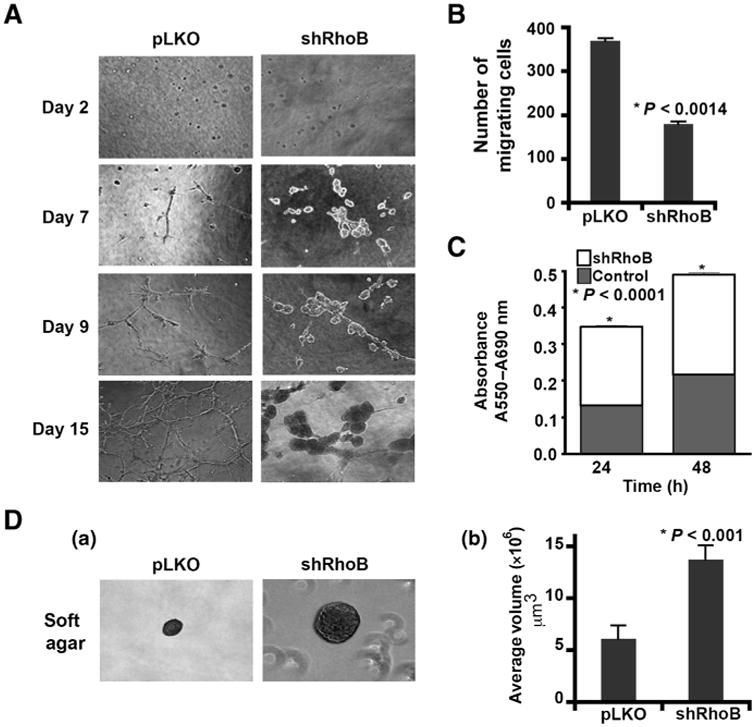
Loss of RhoB in human breast cancer cells promotes hyperproliferation phenotype. A, 3D growth of MDA-MB-231 RhoB silenced (shRhoB) or control (pLKO) breast cancer cells after 2 to 15 days. B, these cells were also assessed for Transwell migration after 4 hours. C, MTT proliferation/survival assays at 24 and 48 hours. D, growth in soft agar (a) with quantitation of 25 colonies per condition [volume = ((4/3)(pi)(rˆ3); (b)]. Data are represented as mean ± SEM. P values were calculated using an unpaired 2-tailed test. See also Supplementary Fig. S2.
We further confirmed our findings using the MMTV-myc mouse model of breast cancer where expression of c-Myc is under the control of the MMTV promoter/enhancer (36). Consistent with observations in the MMTV-PyT/RhoB−/− mice, loss of RhoB increased tumor incidence in MMTV-Myc/RhoB−/− mice (Supplementary Fig. S1A) such that the occurrence of tumor-free mice was more common in nullizygous mice (n = 34) than control heterozygous mice (n = 27; Supplementary Fig. S1A). In addition, there was a significant difference in tumor onset in virgin females between MMTV-myc/RhoB+/−(14.8%, n = 27) and MT-myc/RhoB−/− (26.5%, n = 34) mice (Supplementary Fig. S1B). The frequency of early ductal hyperplastic lesions in nullizygous virgin female mice (88.9%, n = 9) was significantly greater than heterozygous mice (11.1%, n = 9; Supplementary Fig. S1C, panels a and b). In vitro, there was increased growth of acini in 3D cultures that was evident with nullizygous tumor cells (Supplementary Fig. S1D). Finally, we found that MMTV-myc/RhoB−/− cells also grew relatively more aggressively under anchorage-independent conditions (Supplementary Fig. S1E). In summary, our findings in multiple mouse and human models supported the conclusion that RhoB acts primarily as a suppressor function in vivo during the initial stages of breast tumorigenesis.
RhoB loss enhances Akt signaling in breast tumor cells
In previous work, we showed that RhoB positively regulates Akt activity and signaling in nontumor endothelial cells (7). Yet studies in human tumor cells have implicated RhoB as a suppressor gene that downregulates growth factor-mediated Akt signaling (37). Here, we sought to determine whether differential alterations in Akt activity contributed to the observed tumor suppressor function of RhoB we documented in our breast cancer models. Increased levels of activated phospho-Akt isoform (pAkt) were detected in tumor tissues isolated from RhoB−/− mice, compared with control animals (Fig. 4A). Similarly, in tumor cells isolated from RhoB−/− tumors, we observed an increase in basal pAkt compared with cells isolated from wild-type animals, and this could be further increased by IGF-1 or EGF stimulation (Fig. 4B). Similar results were obtained in MDA-MB-231 and MCF7 cells, where shRNA-mediated or antisense-mediated knockdown of RhoB expression, respectively, enhanced levels of basal pAkt compared with control cells (Fig. 4C and Supplementary Figs. S2 and S3). To determine which Akt isoforms contributed to the increased level of total phosphorylated Akt and to the phenotypes produced by RhoB deficiency in vitro and in vivo, we conducted an immunoprecipitation analysis of pAkt-S473 followed by specific immunoblotting for Akt1, Akt2, or Akt3. In tumor cells from RhoB mice, as well as in MDA-MB-231 cells transduced with RhoB shRNA, we observed enhanced phosphorylation of S473 in all 3 Akt isoforms (Fig. 4D). In Akt knockdown experiments, we evaluated their individual contributions to tumor cell phenotypes by using shRNAs designed to selectively specifically silence each Akt isoform in RhoB+/+ and RhoB−/− mouse tumor cells (Supplementary Figs. S4 and S5). Paralleling the results in MDA-MB-231 cells, we found that silencing any one of the 3 Akt isoforms was sufficient to ameliorate the increased growth of RhoB−/− acini in 3D cultures (Fig. 4E and F). Quantification of the acini sizes confirmed an increase in the size of RhoB−/− acini (22426.81 Pixel2 vs. 9481.53 Pixel2) and revealed the significant reduction in size sustained after depletion of the Akt1, Akt2, or Akt3 isoforms (8688.84, 12208.94, and 12427.88 pixel2, respectively). Thus, we concluded that RhoB loss was sufficient to elevate the levels of phosphorylation in all Akt isoforms, suggesting effects on a redundant rather than nonredundant function of these isoforms that contributed to a more aggressive growth phenotype.
Figure 4.
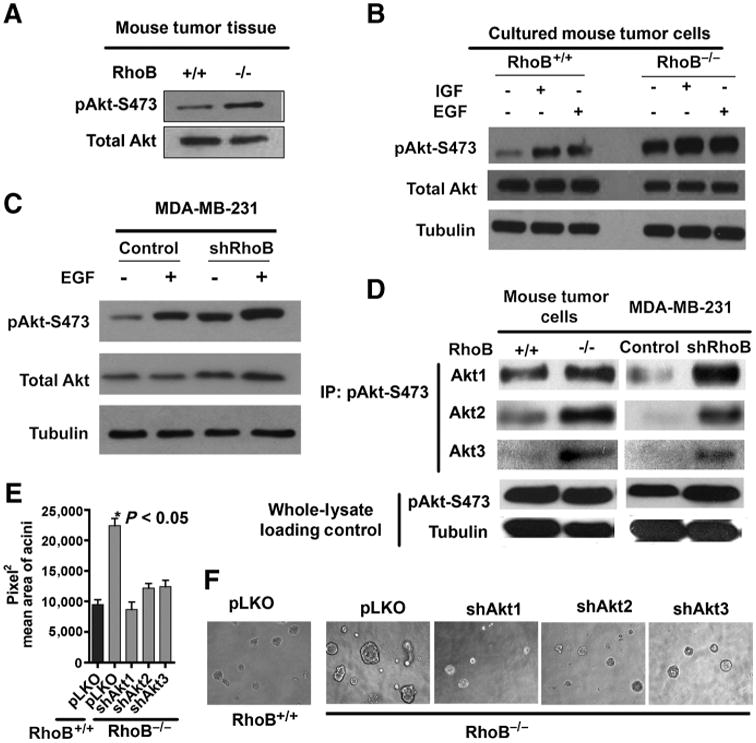
RhoB loss in breast tumor cells increases phospho-S473 on all Akt isoforms. A, Western blot analysis on tumor lysates. B, primary tumor cells starved and stimulated with 100 ng/mL insulin-like growth factor (IGF) or EGF for 10 minutes. C, human MDA-MB-231 starved and stimulated with EGF (20 ng/mL for 10 minutes). D, immunoprecipitation-Western blot analysis of phospho-S473 blotted for Akt isoforms 1, 2, and 3, with pAkt and tubulin Western blotting as lysate controls. E and F, RhoB+/+ and RhoB−/− mouse tumor cells silenced for the Akt isoforms indicated 3D Matrigel/collagen matrix after 16 days. Acini size was quantified using ImageJ and Prism4 software. See also Supplementary Figs. S2, S4, and S5.
RhoB loss delays breast tumor progression
Having established that RhoB loss promoted the initiation and early growth of breast tumors, we went on to assess its effects on tumor angiogenesis and progression after activation of the angiogenic switch. Strikingly, the greater number of early lesions in RhoB-deficient mice did not convert at later times to a greater tumor burden. Specifically, we found that the appearance of palpable tumors was markedly delayed in MMTV-PyT and MMTV-myc mice when RhoB was deleted (Fig. 5A and data not shown). In addition, at the endpoint of the experiment, RhoB-deficient mice displayed small, slow growing tumors, whereas the tumor burden of RhoB-expressing animals was much larger (Fig. 5B). Because RhoB was deleted in both the tumor and stromal compartments, we reasoned that it may exert different competing biologic functions in these compartments, as hinted by the opposite directions of pAkt regulation we had observed in tumor cells versus endothelial cells. Indeed, by establishing that RhoB loss retards angiogenic sprouting in normal tissues, a previous study had hinted that tumor neoangiogenesis might also be similarly impaired (7). In support of this likelihood, tumors stained with CD31 to mark endothelial cells and α smooth muscle actin (SMA) to mark pericytes revealed a general deficiency in the tumor vascular network in RhoB−/− animals consistent with a defect in sprouting angiogenesis (Fig. 5C). Furthermore, whole tumor mRNA analysis revealed significantly lower levels of endothelial-specific VE-cadherin mRNA in RhoB-deficient mice, another indication of fewer endothelial cells in the tumor tissue (Fig. 5D). We isolated endothelial cells from tumors growing in RhoB −/− and RhoB +/+ mice to assess pS473 levels in Akt expressed in tumor endothelial cells (Fig. 5E). As expected, a dosedependent decrease was documented in pAkt that was associated with allelic losses in RhoB +/+, RhoB+/−, and RhoB−/− tumor endothelial cells. There were no dramatic changes in the expression of Akt isoforms detected at the RNA level in endothelial cells taken from RhoB−/− mouse lung (Fig. 5F). In summary, we concluded that a loss of Akt phosphorylation rather than expression in endothelial cells may contribute to the angiogenic failure in RhoB-null tumors.
Figure 5.
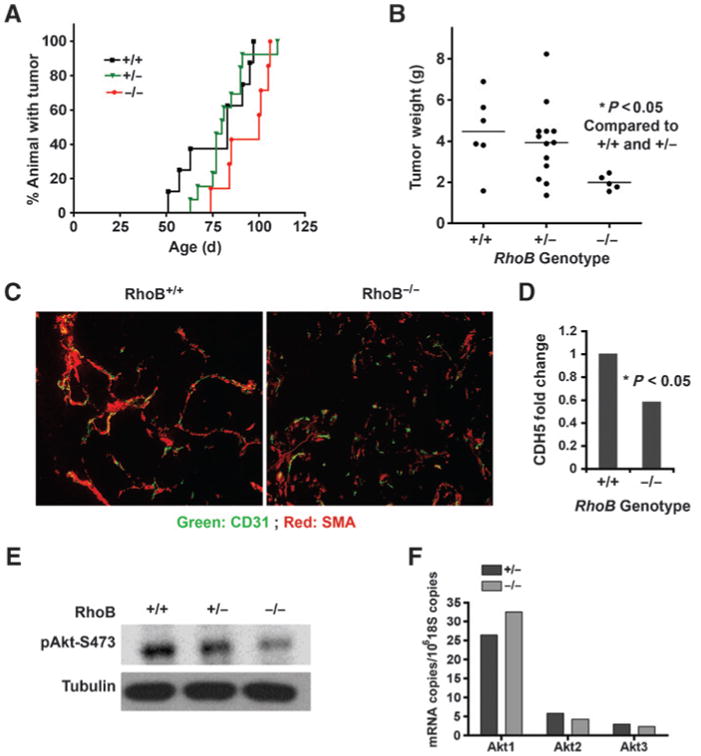
RhoB deficiency delays tumor growth and reduces blood vessel density. A, the percentage of animals with palpable tumor in RhoB+/+, +/−, and −/− tumors over time. B, total tumor weight at day 110 for all genotypes. C, representative endothelial (CD31, green) and pericyte (SMA, red) staining of vasculature are shown at magnification 200×. D, vascular content was quantified by real-time PCR of the endothelial-specific gene ve-cadherin (CDH5) relative to GAPDH. E, primary RhoB+/+, +/−, and −/− tumor endothelial cells analyzed by immunoblotting for pAkt (S473). F, qRT-PCR quantitation of Akt isoforms in endothelial cells normalized to 1 million copies of 18S RNA.
Benefits of RhoB loss to tumor cell growth are subordinate to angiogenic limitations in the tumor stromal compartment
Given the differential functions of RhoB seen on tumor cells in vitro where loss of RhoB increased cell growth contrasted with the slowed overall tumor grown in the RhoB-null MMTV-PyT mice, we hypothesized that the impact of RhoB on stromal functions may be dominant to its tumor cell autonomous functions. To examine this hypothesis, we conducted reciprocal transplants of tumor and host RhoB genotypes. We observed consistent results regardless of whether minced tumor pieces or primary tumor cells grown in culture were used as orthotopic transplants. We first compared the growth RhoB tumor grafted into RhoB+/+ nullizygous or heterozygous hosts. Notably, tumor growth was restricted in nullizygous hosts, establishing that the RhoB−/− stroma was sufficient to enforce a restriction in tumor growth (Fig. 6A). Specifically, there was a delay in the appearance of palpable tumors in RhoB−/− stromal microenvironment leading to a reduction in overall tumor size at 35 and 50 days after implantation. This difference diminished by approximately 80 days after implantation, suggesting the imposition of a selection by the microenvironment for the emergence of properties that could overcome the antiangiogenic barrier that the microenvironment initially enforced (Fig. 6A). A breakdown in the rate of tumor growth at early time points (0–50 days) versus later time points (60–80 days) showed that the barrier conferred by the RhoB−/− stroma is active against the initial phases of growth rather than the later stages of progression. Therefore, we interpreted the delay as one that could trigger the angiogenic switch, as once the tumor vasculature was established in the RhoB−/− hosts, tumor growth proceeded similarly to that observed in wildtype hosts. Overall, these findings are consistent with previous work defining the critical role of RhoB in sprouting, the earliest and first steps in neoangiogenesis (7).
Figure 6.
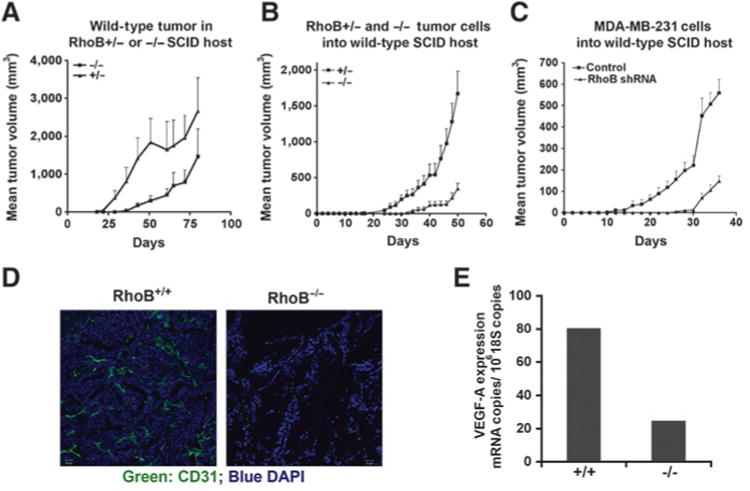
Positive impact of RhoB on tumor angiogenesis supercedes its negative impact in cancer cells. A, minced RhoB+/+ tumors were orthotopically transplanted into RhoB+/− or RhoB−/− SCID mouse hosts (n = 10). Labels indicate the genotype of the recipient host. B, tumor cells isolated from RhoB+/− or RhoB−/− mice in Matrigel were similarly orthotopically transplanted into wild-type SCID mice (n = 10). Labels indicate the genotype of the donor tumor cells. C, human MDA-MB-231 cells were implanted orthotopically into wild-type SCID mice (n = 10). Labels indicate the genotype of the donor tumor cells. D, tumors from B above were stained with anti-CD31 to assess the microvascular density. Representative images are shown. E, mRNA expression levels for murine VEGF-A in RhoB+/− and RhoB−/− endothelial cells isolated from tumors were assessed by qRT-PCR.
We further addressed this question by conducting a reciprocal experiment in which RhoB−/− tumors were grown in wild-type hosts. In this case, RhoB−/− mouse tumor cells and RhoB-silenced MDA-MB-231 human breast cancer cells both exhibited a delay in progression, even in a RhoB+/+ microenvironment, despite having exhibited enhanced growth in cell culture (Fig. 6B and C). Thus, it was clear that the effects of RhoB on tumor growth could not be explained simply by its cell-autonomous contribution in endothelial cells. Instead, the results implied that the loss of RhoB in cancer cells conferred a nonautonomous effect(s) on the stromal compartment that was sufficient to restrain tumor growth. Indeed, an examination of the vascular bed in these tumors revealed a marked decrease in CD31 staining showing a reduction in neoangiogenesis (Fig. 6D). This result was not surprising because the secretion of proangiogenic signals by tumor cells is known to be essential to recruit endothelial cells to the tumor mass and to vascularize it (4). In assessing the likelihood that RhoB−/− tumors may have a deficiency in such signals, we documented a significant decrease in the expression of VEGF-A that is pivotal to tumor neoangiogenesis (Fig. 6E). In profiling gene expression in nullizygous and control wild-type breast tumor tissues, and in cells derived from those tumors, we also documented a reproducible decrease in expression of PDGF-β and thrombospondin1 (THBS1) and a reproducible increase in stem cell factor 1 (SCF1), the c-KIT ligand, in cells where RhoB was deleted (Supplementary Table S3). To evaluate whether increased Akt activity could explain the downstream gene expression changes, we also profiled gene expression in tumor cells silenced for Akt1, Akt2, or Akt3. Unlike the general impact of each Akt isoform on 3D tumor growth, only SCF1 seemed to be altered by Akt2 silencing in RhoB−/− cells. In summary, we concluded that the delays in tumor progression caused by RhoB deficiency reflected both direct and indirect effects on tumor angiogenesis, involving both Akt-dependent and Akt-independent pathways.
RhoB deficiency in breast tumor cells increases expression of EGFR
RhoB localizes to endosomes and regulates endocytic trafficking of the EGFR (10, 38). A reduction in endocytic removal of active ligand-receptor complex from the cell surface is the chief process through which signal transduction by tyrosine kinase receptors is amplified (39). For these reasons, we investigated whether loss of RhoB affected the level of EGFR on the cell surface in our breast cancer models, as one way to stimulate Akt signaling. Immunoblot analysis showed an increased steady-state level of EGFR in mammary tumor cells isolated from RhoB−/− mice that were maintained in full growth media in 2D cell cultures (Fig. 7A). The enhancement to EGFR expression occurred at the steady-state protein level, as we did not see significant changes at the mRNA level (Fig. 7B). However, as the immunoblot analysis did not indicate where the increase in protein levels occurred, we conducted fluorescence-activated cell sorting (FACS) analysis to examine the levels of EGFR expression on the cell surface. We observed that RhoB loss increased the cell surface expression level of EGFR in both mouse tumor cells and MDA-MB-231 cells (Fig. 7C and Supplementary Fig. S6). Together, these results indicated that the increased levels of EGFR caused by RhoB loss were also reflected by an increase at the cell surface, most likely reflecting the defect in intracellular trafficking produced by RhoB loss described previously (10).
Figure 7.
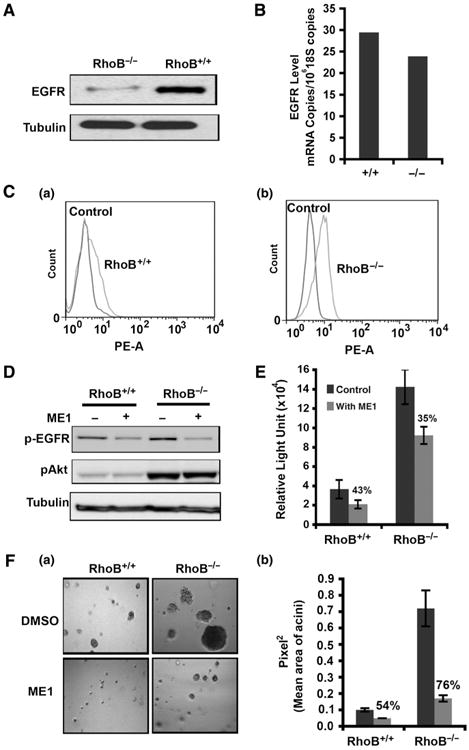
Loss of RhoB in mouse tumor cells increases EGFR cell surface levels. A, Western blot for EGFR in RhoB+/+ and RhoB−/− mouse tumor cells. B, qRT-PCR of EGFR mRNA. C, FACS analysis of EGFR on the cell surface of mouse tumor cells. D, p-EGFR (p-Try1068) and p-Akt (p-S473) after treatment with EGFR-neutralizing antibody ME-1. E, proliferation of mouse tumor cells via BrdUrd incorporation after 72-hour treatment with ME1 in 5% FBS. F, 3D growth in Matrigel culture after ME1 blockade of EGFR signaling (a). The average size of the acini (b). Data are represented as mean ± SEM. P values were calculated using an unpaired 2-tailed test.
To determine whether the increased accumulation of EGFR on the cell surface contributed to the enhancement of Akt activation and proliferation rate caused by RhoB loss, we examined the effects of a ligand blocking antibody (ME1) that specifically recognizes murine EGFR. Western blot analysis showed a higher level of pEGFR-Tyr 1068 in RhoB−/− cells (Fig. 7D). Although ME1 addition to cell cultures reduced the level of pEGFR-Tyr 1068 in both RhoB− + − and RhoB−/− cells, there was no effect on the levels of pAkt-S473. Cell proliferation assays based on BrdUrd incorporation or growth in 3D culture confirmed that ME1 addition could proportionately reduce proliferation of both RhoB+/+ and RhoB−/− mouse tumor cells (Fig. 7E and F). Therefore, while pEGFR elevation could activate Akt signaling and cell proliferation, and RhoB loss could elevate the levels of pEGFR, it was evident that pEGFR elevation was not the sole mechanism through which RhoB modulated the Akt pathway.
Discussion
The stress-inducible RhoB gene is frequently attenuated in tumor cells with most studies finding an association between its reduced expression and increased tumor aggressiveness (15-17). The identity of specific modifiers that modulate RhoB expression during cancer development remain largely undefined. Loss of heterozygosity (LOH) in the human RhoB gene has been reported to correlate with poorer outcomes in lung cancer (40). Transcriptional variations have been linked to epigenetic modifications of the promoter, with polymorphisms in this region implicated in the heterogeneity of RhoB expression in human populations as well as mechanisms for cancerrelated silencing events that impact chemotherapeutic responses (41–43). Our present study provides new insights into the functional consequences of such changes in various well-established models of breast cancer.
Our investigations of early tumor lesions in these models support previous studies that indicate RhoB attenuation is sufficient to increase tumor aggressiveness (12). However, the more expanded scope of our investigations into how RhoB affects tumor progression challenge the generally held interpretation of a solely cell autonomous function that acts as a pivotal regulator of overall tumor growth. Indeed, while suppression of cell growth by RhoB was critical during the early stages of cell transformation in mouse models, we found that later in progression it exerted a distinct cell nonautonomous impact on the stromal microenvironment that was dominant to its cell autonomous impact. This perspective gained from murine models was confirmed in studies of the highly invasive and metastatic human MDA-MB-231 breast cells, where we found that RhoB silencing heightened the proliferative phenotype, in support of studies of RhoB in other types of human cancer cells (34). Because small-molecule–based therapeutic interventions necessarily target both tumor and stroma, in the many types of human tumor cells where RhoB is attenuated, the primary benefit of inhibiting its primary Akt effector signals would be to block stromal activities that are essential to support angiogenesis. In broader terms, our findings illustrate how a putative suppressor function thought to be solely cancer cell-centric in nature may also be cell nonautonomous and tumor promoting in nature, as a result of crosstalk with tumor stromal components that dominate tumor growth, such as the case with RhoB and its Akt effector pathways in the tumor vasculature.
Our work deepens the concept that the early response stressactivated protein RhoB is a pivotal modifier of Akt signaling in cancer pathobiology. In epithelial cancer cells, we learned that silencing individual Akt isoforms was sufficient to ablate the proliferative benefits conferred by RhoB loss. Thus, RhoB seemed to affect the redundant functions rather than nonredundant functions of Akt isoforms, perhaps by affecting regulators common to each. In endothelial stromal cells, we observed decreased phosphorylated Akt, consistent with earlier results in retinal endothelial cells where loss of RhoB also reduced levels of Akt activation (7). While we do not yet fully understand the basis by which RhoB modulates Akt activity, this work sets a precedent for differential responses to stress pathways between tumor cells and endothelial cells or other cell types in tumors.
An additional general implication of our work is it reveals a fundamental molecular difference in the wiring of signaling pathways in endothelium and cancer cells, which heretofore have been generally assumed to be similar in organization and functional output. This core distinction likely has its roots in the differential response of the endothelium to metabolic stresses such as glucose or oxygen deprivation, which promote growth and angiogenesis in opposition to the cell stasis or autophagic responses triggered by these conditions in cancer cells or other nonendothelial cells. This distinction speaks to the core function of the vasculature in providing access to nutrients by expansion in times of nutrient deprivation.
In establishing a critical function for RhoB in tumor angiogenesis, our work points to future work in determining how this small GTPase influences responses to hypoxia and glucose deprivation, stresses of central relevance to cancer pathophysiology that other recent studies suggest RhoB may control (44, 45). Our findings reinforce the concept that genetic validation of a candidate target in the tumor stroma is critical to monitor, as effects at this level may dominate the desired effects on tumor cells and alter interpretations of what constitutes a suitable therapeutic candidate. In summary, we have shown that RhoB differentially influences tumorigenesis in tumor cells and vascular cells, showing that the latter effects are dominant in terms of disease pathobiology. The unexpected differential phenotype of in vivo tumor progression versus in vitro transformation assays and 3D growth underscores the importance of exploring gene function in a full in vivo context that includes the tumor microenvironment, when contemplating therapeutic intervention.
Supplementary Material
Acknowledgments
The authors thank Janice Nagy and Christopher V. Carmen for their critical review of the paper. In addition, the authors received support from the core facilities at BIDMC and ImClone Systems including Ying Chen and Ying Wang for bioinformatics, LayHong Ang and Yi Zheng for Histology, Shou-ching Shih and Dan Li for qRT-PCR, and Qi Xue for in vivo support.
Grant Support: This work was supported by grants from Susan G. Komen for the Cure (to S. Kazerounian), NIH R01 HL071049 and CA0109086 (to L.E. Benjamin) and NIH R01 CA100123 and CA082222 (to G.C. Prendergast), and P01 CA092644 (to A. Toker and L.E. Benjamin) and K99CA157945 (to Y.R. Chin).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: A. Toker is employed by ASBMB Today as Chair of the Board and by Biochemical Journal as Vice Chair. No potential conflicts of interest were disclosed by other authors.
The Editor-in-Chief of Cancer Research (G.C. Prendergast) is an author of this article. In keeping with the AACR's Editorial Policy, the paper was peer reviewed and a member of the AACR's Publications Committee rendered the decision concerning acceptability.
Authors' Contributions: Conception and design: S. Kazerounian, M. Huang, D. Udayakumar, A. Bravo-Nuevo, A. Toker, G.C. Prendergast, L.E. Benjamin
Development of methodology: S. Kazerounian, M. Huang, Y.R. Chin, D. Udayakumar, N. Zheng
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): S. Kazerounian, D. Gerald, M. Huang, Y.R. Chin, D. Udayakumar, R.K. O'Donnell, C. Perruzzi, L. Mangiante, J. Pourat, S. McNamara, J.B. DuHadaway
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): S. Kazerounian, D. Gerald, M. Huang, Y.R. Chin, R.K. O'Donnell, S. Shechter, S. McNamara, O.N. Kocher, L.F. Brown, A. Toker, L.E. Benjamin
Writing, review, and/or revision of the manuscript: S. Kazerounian, D. Gerald, M. Huang, R.K. O'Donnell, C. Perruzzi, A. Bravo-Nuevo, A. Toker, G.C. Prendergast, L.E. Benjamin
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): S. Kazerounian, D. Gerald, M. Huang, C. Perruzzi, L. Mangiante, J. Pourat, T.L. Phung, S. Shechter, O.N. Kocher, L.E. Benjamin
Study supervision: S. Kazerounian, A. Toker, L.E. Benjamin, G.C. Prendergast
References
- 1.Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res. 2010;316:1324–31. doi: 10.1016/j.yexcr.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 2.McAllister SS, Weinberg RA. Tumor-host interactions: a far-reaching relationship. J Clin Oncol. 2010;28:4022–8. doi: 10.1200/JCO.2010.28.4257. [DOI] [PubMed] [Google Scholar]
- 3.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–7. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–64. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 5.Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–49. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 6.Michaelson D, Silletti J, Murphy G, D'Eustachio P, Rush M, Philips MR. Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J Cell Biol. 2001;152:111–26. doi: 10.1083/jcb.152.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adini I, Rabinovitz I, Sun JF, Prendergast GC, Benjamin LE. RhoB controls Akt trafficking and stage-specific survival of endothelial cells during vascular development. Genes Dev. 2003;17:2721–32. doi: 10.1101/gad.1134603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wheeler AP, Ridley AJ. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res. 2004;301:43–9. doi: 10.1016/j.yexcr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 9.Sandilands E, Brunton VG, Frame MC. The membrane targeting and spatial activation of Src, Yes and Fyn is influenced by palmitoylation and distinct RhoB/RhoD endosome requirements. J Cell Sci. 2007;120:2555–64. doi: 10.1242/jcs.003657. [DOI] [PubMed] [Google Scholar]
- 10.Gampel A, Parker PJ, Mellor H. Regulation of epidermal growth factor receptor traffic by the small GTPase rhoB. Curr Biol. 1999;9:955–8. doi: 10.1016/s0960-9822(99)80422-9. [DOI] [PubMed] [Google Scholar]
- 11.Huang M, Duhadaway JB, Prendergast GC, Laury-Kleintop LD. RhoB regulates PDGFR-beta trafficking and signaling in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2007;27:2597–605. doi: 10.1161/ATVBAHA.107.154211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu AX, Rane N, Liu JP, Prendergast GC. RhoB is dispensable for mouse development, but it modifies susceptibility to tumor formation as well as cell adhesion and growth factor signaling in transformed cells. Mol Cell Biol. 2001;21:6906–12. doi: 10.1128/MCB.21.20.6906-6912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu A, Cerniglia GJ, Bernhard EJ, Prendergast GC. RhoB is required to mediate apoptosis in neoplastically transformed cells after DNA damage. Proc Natl Acad Sci U S A. 2001;98:6192–7. doi: 10.1073/pnas.111137198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adnane J, Muro-Cacho C, Mathews L, Sebti SM, Munoz-Antonia T. Suppression of rho B expression in invasive carcinoma from head and neck cancer patients. Clin Cancer Res. 2002;8:2225–32. [PubMed] [Google Scholar]
- 15.Zhou J, Zhu Y, Zhang G, Liu N, Sun L, Liu M, et al. A distinct role of RhoB in gastric cancer suppression. Int J Cancer. 2011;128:1057–68. doi: 10.1002/ijc.25445. [DOI] [PubMed] [Google Scholar]
- 16.Mazieres J, Antonia T, Daste G, Muro-Cacho C, Berchery D, Tillement V, et al. Loss of RhoB expression in human lung cancer progression. Clin Cancer Res. 2004;10:2742–50. doi: 10.1158/1078-0432.ccr-03-0149. [DOI] [PubMed] [Google Scholar]
- 17.Couderc B, Pradines A, Rafii A, Golzio M, Deviers A, Allal C, et al. In vivo restoration of RhoB expression leads to ovarian tumor regression. Cancer Gene Ther. 2008;15:456–64. doi: 10.1038/cgt.2008.12. [DOI] [PubMed] [Google Scholar]
- 18.Prendergast GC. Actin' up: RhoB in cancer and apoptosis. Nat Rev Cancer. 2001;1:162–8. doi: 10.1038/35101096. [DOI] [PubMed] [Google Scholar]
- 19.Liu A, Prendergast GC. Geranylgeranylated RhoB is sufficient to mediate tissue-specific suppression of Akt kinase activity by farnesyl-transferase inhibitors. FEBS Lett. 2000;481:205–8. doi: 10.1016/s0014-5793(00)02003-2. [DOI] [PubMed] [Google Scholar]
- 20.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–91. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee YC, Cheng TH, Lee JS, Chen JH, Liao YC, Fong Y, et al. Nobiletin, a citrus flavonoid, suppresses invasion and migration involving FAK/PI3K/Akt and small GTPase signals in human gastric adenocarcinoma AGS cells. Mol Cell Biochem. 2011;347:103–15. doi: 10.1007/s11010-010-0618-z. [DOI] [PubMed] [Google Scholar]
- 22.Bousquet E, Mazieres J, Privat M, Rizzati V, Casanova A, Ledoux A, et al. Loss of RhoB expression promotes migration and invasion of human bronchial cells via activation of AKT1. Cancer Res. 2009;69:6092–9. doi: 10.1158/0008-5472.CAN-08-4147. [DOI] [PubMed] [Google Scholar]
- 23.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–61. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ip MM, Asch BB, editors. editors Methods in Mammary Gland Biology and Breast Cancer Research. New York: Kluwer Academic/Plenum Publishers; 2000. [Google Scholar]
- 25.http://www.broad.mit.edu/rnai/trc/lib.
- 26.Lanari C, Luthy I, Lamb CA, Fabris V, Pagano E, Helguero LA, et al. Five novel hormone-responsive cell lines derived from murine mammary ductal carcinomas: in vivo and in vitro effects of estrogens and progestins. Cancer Res. 2001;61:293–302. [PubMed] [Google Scholar]
- 27.Lebowitz PF, Sakamuro D, Prendergast GC. Farnesyl transferase inhibitors induce apoptosis of Ras-transformed cells denied substratum attachment. Cancer Res. 1997;57:708–13. [PubMed] [Google Scholar]
- 28.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–68. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 29.Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3:785–92. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ffrench-Constant C, Van de Water L, Dvorak HF, Hynes RO. Reappearance of an embryonic pattern of fibronectin splicing during wound healing in the adult rat. J Cell Biol. 1989;109:903–14. doi: 10.1083/jcb.109.2.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shih SC, Zukauskas A, Li D, Liu G, Ang LH, Nagy JA, et al. The L6 protein TM4SF1 is critical for endothelial cell function and tumor angiogenesis. Cancer Res. 2009;69:3272–7. doi: 10.1158/0008-5472.CAN-08-4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aerts JL, Gonzales MI, Topalian SL. Selection of appropriate control genes to assess expression of tumor antigens using real-time RT-PCR. Biotechniques. 2004;36:84–6. doi: 10.2144/04361ST04. [DOI] [PubMed] [Google Scholar]
- 33.Hollenhorst PC, Jones DA, Graves BJ. Expression profiles frame the promoter specificity dilemma of the ETS family of transcription factors. Nucleic Acids Res. 2004;32:5693–702. doi: 10.1093/nar/gkh906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang M, Prendergast GC. RhoB in cancer suppression. Histol Histopathol. 2006;21:213–8. doi: 10.14670/HH-21.213. [DOI] [PubMed] [Google Scholar]
- 35.Turashvili G, Bouchal J, Baumforth K, Wei W, Dziechciarkova M, Ehrmann J, et al. Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer. 2007;7:55. doi: 10.1186/1471-2407-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell. 1987;49:465–75. doi: 10.1016/0092-8674(87)90449-1. [DOI] [PubMed] [Google Scholar]
- 37.Chen Z, Sun J, Pradines A, Favre G, Adnane J, Sebti SM. Both farnesylated and geranylgeranylated RhoB inhibit malignant transformation and suppress human tumor growth in nude mice. J Biol Chem. 2000;275:17974–8. doi: 10.1074/jbc.C000145200. [DOI] [PubMed] [Google Scholar]
- 38.Ellis S, Mellor H. Regulation of endocytic traffic by rho family GTPases. Trends Cell Biol. 2000;10:85–8. doi: 10.1016/s0962-8924(99)01710-9. [DOI] [PubMed] [Google Scholar]
- 39.Waterman H, Yarden Y. Molecular mechanisms underlying endocytosis and sorting of ErbB receptor tyrosine kinases. FEBS Lett. 2001;490:142–52. doi: 10.1016/s0014-5793(01)02117-2. [DOI] [PubMed] [Google Scholar]
- 40.Sato N, Fukui T, Taniguchi T, Yokoyama T, Kondo M, Nagasaka T, et al. RhoB is frequently downregulated in non-small-cell lung cancer and resides in the 2p24 homozygous deletion region of a lung cancer cell line. Int J Cancer. 2007;120:543–51. doi: 10.1002/ijc.22328. [DOI] [PubMed] [Google Scholar]
- 41.Fritz G, Kaina B. Transcriptional activation of the small GTPase gene rhoB by genotoxic stress is regulated via a CCAAT element. Nucleic Acids Res. 2001;29:792–8. doi: 10.1093/nar/29.3.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tovar D, Faye JC, Favre G. Cloning of the human RHOB gene promoter: characterization of a VNTR sequence that affects transcriptional activity. Genomics. 2003;81:525–30. doi: 10.1016/s0888-7543(03)00044-2. [DOI] [PubMed] [Google Scholar]
- 43.Mazieres J, Tovar D, He B, Nieto-Acosta J, Marty-Detraves C, Clanet C, et al. Epigenetic regulation of RhoB loss of expression in lung cancer. BMC Cancer. 2007;7:220. doi: 10.1186/1471-2407-7-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skuli N, Monferran S, Delmas C, Lajoie-Mazenc I, Favre G, Toulas C, et al. Activation of RhoB by hypoxia controls hypoxia-inducible factor-1alpha stabilization through glycogen synthase kinase-3 in U87 glioblastoma cells. Cancer Res. 2006;66:482–9. doi: 10.1158/0008-5472.CAN-05-2299. [DOI] [PubMed] [Google Scholar]
- 45.Bravo-Nuevo A, Sugimoto H, Iyer S, Fallon Z, Lucas JM, Kazerounian S, et al. RhoB loss prevents streptozotocin-induced diabetes and ameliorates diabetic complications in mice. Am J Pathol. 2011;178:245–52. doi: 10.1016/j.ajpath.2010.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.