

Abstract
DNA is a universal language encrypted with biological instruction for life. In higher organisms, the genetic information is preserved predominantly in an organized exon/intron structure. When a gene is expressed, the exons are spliced together to form the transcript for protein synthesis. We have developed a complexity reduction algorithm for sequence analysis (CRASA) that enables direct alignment of cDNA sequences to the genome. This method features a progressive data structure in hierarchical orders to facilitate a fast and efficient search mechanism. CRASA implementation was tested with already annotated genomic sequences in two benchmark data sets and compared with 15 annotation programs (10 ab initio and 5 homology-based approaches) against the EST database. By the use of layered noise filters, the complexity of CRASA-matched data was reduced exponentially. The results from the benchmark tests showed that CRASA annotation excelled in both the sensitivity and specificity categories. When CRASA was applied to the analysis of human Chromosomes 21 and 22, an additional 83 potential genes were identified. With its large-scale processing capability, CRASA can be used as a robust tool for genome annotation with high accuracy by matching the EST sequences precisely to the genomic sequences.
[Supplementary material is available online at http://www.genome.org and http://crasa.sinica.edu.tw/bioinformatics/Supplementary.htm.]
The entire human genome has been sequenced and annotated separately by Lander et al. (2001) and Venter et al. (2001). Altogether, 30,000 to 40,000 protein-coding genes were annotated from the genomic sequence. This number, roughly twice as many as in the worm or fly, deviates greatly from the earlier high estimates (Ewing and Green 2000; Liang et al. 2000; Roest Crollius et al. 2000). The exact gene number in the human genome remains to be determined by accurate annotation of the sequence data.
Genome annotation is based primarily on the ab initio and homology methods. The ab initio approach predicts genes directly from the genomic sequence using the computational properties of exons, introns, and other signature features without referencing the experimental data. Numerous ab initio prediction programs have been used extensively in genome annotation, including FGENESH (Solovyev et al. 1995; Salamov and Solovyev 2000), GeneID (Parra et al. 2000), GeneMark.hmm (Lukashin and Borodovsky 1998), GeneView (Milanesi et al. 1993), GENSCAN (Burge and Karlin 1997, 1998), Genie (Kulp et al. 1996; Reese et al. 2000), Grail (Xu et al. 1994), GrailEXP_Perceval (Hyatt et al. 2000), HMMgene (Krogh 1998, 2000), and MZEF (Zhang 1997).
The homology approach identifies genes with the aid of experimental data. This approach exploits sequence alignment between the genomic data and known cDNA or protein databases. Successful implementation of this method includes AAT (Huang et al. 1997), FGENESH+ and FGENESH++ (Salamov and Solovyev 2000), GAIA (Bailey et al. 1998), GeneBuilder (Milanesi et al. 1999), GenomeScan (Yeh et al. 2001), GrailEXP_Gawain (Hyatt et al. 2000), GeneWise (Birney and Durbin 2000), ICE (Pachter et al. 1999), and Procrustes (Gelfand et al. 1996; Sze and Pevzner 1997; Mironov et al. 1998). Among these programs FGENESH+ (and FGENESH++), GenomeScan, GeneWise, and Procrustes are combined tools of sequence homology and ab initio annotation.
Generally speaking, the ab initio approach tends to have a higher rate of false-positive predictions (overprediction) in annotating long genomic sequences with multiple genes (Dunham et al. 1999). The homology-based approaches demand high-performance computing and large storage space. Furthermore, these methods require extensive manual interventions to curate true gene prediction from large sets of matched data. The combination tools for sequence alignment and ab initio annotation, although highly accurate, are not robust in routine applications.
In this paper, we propose a new method, the Complexity Reduction Algorithm for Sequence Analysis (CRASA), for global alignment and annotation of the genomic sequence. The method finds the exact match between the cDNA data and genomic sequence; thus mapping the expressed genes directly to it. By using a set of filters, the enormous data complexity is reduced substantially. Thus, it provides an annotated framework of expressed genes in the genome.
The CRASA system restructures the cDNA data progressively into a pattern-based pyramidal data structure in hierarchical orders. The algorithm offers an automatic search of the entire database efficiently and is amicable to the implementation of parallel processing (see Methods). In this paper, CRASA was tested with two benchmark data sets, the SemiArtificial Genomic (SAG) sequences provided by Guigó et al. (2000) and the Real Genomic (RG) sequences generated ad hoc from GeneBank of NCBI (National Center of Biotechnolgy Information). In general, CRASA was capable of delivering annotation accuracy better than the other 15 programs tested in this study (see Results and Discussion).
The annotated human Chromosomes 21 (Hattori et al. 2000) and 22 (Dunham et al. 1999), although incomplete, are considered as standard benchmarks for genome annotation. In the benchmark test of human Chromosomes 21 and 22, CRASA's filters were able to remove the massive noise from matched hits, thus reducing the complexity of genome analysis. More significantly, our method identified 83 additional EST matches that were not annotated previously. These 83 matches were extracted and categorized into five classes.
Our results indicate that CRASA, with its capabilities of complexity reduction, progressive data transmission, and direct pattern match, is a robust and effective new method for genome annotation. The simplicity of program implementation allows for unlimited query size and parallel processing on multiple processors. It is well suited for annotating large genomic sequences.
RESULTS AND DISCUSSION
Principle of CRASA
In principle, CRASA is a homology-based approach for the alignment between the genomic and cDNA sequences in the databases. Unlike the other methods, CRASA implementation requires reconstructing a cDNA database with pattern-based CR processing and hashing (or indexing) the processed data with binary codes in a multilevel pyramidal structure. Thus, the cDNA data are organized hierarchically in coded bins to reduce the complexity by a factor of 16 at each level (Fig. 1; Methods). The original data are fully conserved under a new pyramidal scheme. One great advantage of using the reconfigured database is to reduce also the computational time complexity, when similarly processed genomic sequence is searched directly against cDNA data addressed by identical binary codes. It is an inherent nature of CRASA to perform parallel processing of genomic sequences without size limitation. We restructured the HGI database (The Institute for Genome Research, MD, USA) in CR pyramids up to the level 7 (detailed in Methods).
Figure 1.

The principle CRASA's pyramidal data structure. (A) An EST sequence with four gatc pattern sites indicated in boxes. (B) A gate CR pyramid with a three-level data structure. Each bin in the matrices from Level 1 to 3 is assigned with a binary code taken from both sides of the gate pattern base by base. Partitioning of Sites 1–3 from Level 1 to 3 is shown here to illustrate the progression of binary codes and data structure in a hierarchical order.
Because the expressed gene sequence represents merely 2% of the human genome, we anticipated a very high degree of noise in our database search. In the postprocessing of matched data, two main filters were installed to ascertain “true” hits: The matching sequence length is no less than 50 bp and the matched cDNA sequence is split into at least three colinear fragments (i.e., three possible exons). An example is given in Table 1 to illustrate the near exponential reduction of CRASA matches. It is clear that CRASA efficiently filters out a large amount of matched data with simple parameters.
Table 1.
The Performance of Complexity Reduction for CRASA
| No. of CRASA matches | |
| Matching length | Matched fragments |
| ≥20 bp | 367,359,815 |
| ≥30 bp | 37,801,885 |
| ≥40 bp | 4,717,948 |
| ≥50 bp | 835,533 |
| No. of ESTs (cach matched fragment ≥50 bp) | Matched ESTs |
| No. of fragments per EST ≥1 | 109,102 |
| No. of fragments per EST ≥2 | 65,817 |
| No. of fragments per EST ≥3 | 48,268 |
| After removing repetitive elements | 2880 |
| No. of patched fragments per EST ≥3 | 1681 |
| After removing the fragment in inconsistent order | 515 |
The query sequence is human Chromosome 21 (∼34 Mb).
CRASA was implemented by annotating the genomic sequence of two benchmark data sets and of the human Chromosomes 21 and 22 with the reconfigured HGI database. Its performance on a 16-CPU Linux cluster was robust and efficient, primarily because of its capability of parallel processing in hardware configuration and programming (data not shown). The filter sets greatly reduced the complexity of matched data analysis. Potentially, CRASA is a new global alignment method with high accuracy.
Alternative to the existing excellent EST-to-genome alignment methods, such as BLAT (Kent 2002), SSAHA (Ning et al. 2001), MegaBlast (Zhang et al. 2000), and sim-x (Chao et al. 1995, 1997), the CRASA algorithm defines a different model for viewing and searching data in the multileveled pyramidal structure. A pattern-associated binary code system is used to process and manage the database systematically and efficiently. In addition, it confers the flexibility of building a new data structure selectively high enough to decompose the original data complexity.
Accuracy Test of Genome Annotation
The accuracy of CRASA annotation results from two benchmark data sets and human Chromosomes 21 and 22 are presented and discussed in this section. The details of the CRASA system and test procedure are described in Methods.
We tested the CRASA system on two benchmark data sets, SAG and RG, with well-annotated gene locations. The results were compared with those obtained from 15 well-known annotation tools. The accuracy tests, Sensitivity (Sn and ME) and Specificity (Sp and WE) measurements at the exon level, are described briefly in Methods (Burset and Guigó 1996). The predicted exon is regarded as a correct one (i.e., true positive) only when the boundary on both sides is predicted correctly.
SAG Sequences
The first benchmark data set is SAG (SemiArtificial Genomic) sequences generalized by Guigó et al. (2000). Two different sets of SAG sequences were extracted according to protein similarity: strong versus moderate similarity (see Methods).
Figure 2 shows the comparison of CRASA results with those derived from GENSCAN, Procrustes, and GenWise reported early by Guigó et al. (2000). In their work, the SAG sequences were extracted and divided into two different sets, the strong similarity group of 17 genomic sequences (10−50 > BLASTX P-value > 10−∞) and the moderate similarity group of 26 sequences (10−6 > BLASTX P-value > 10−50), whereas the prediction accuracy of Procrustes and GenWise depends highly on protein similarity to the reference sequences. CRASA annotation was determined solely by direct sequence match between the SAG sequences and the EST database, HGI (Human Gene Index; see Methods). As shown in Figure 2, high accuracy of CRASA performance was observed in both protein similarity ranges, as it stays consistently above 80% at the exon level. Although GeneWise and Procrustes gave comparable results in the strong similarity group, CRASA outperformed these three tools in the moderate range. Gene prediction by GeneWise or Procrustes requires the input of a candidate homologous protein sequence for BLASTX search (Altschul et al. 1990, 1997) against the nonredundant (nr) protein database, in which most genes in SAG are represented (Guigó et al. 2000). The prediction accuracy of GeneWise and Procrustes may drop significantly as the cutoff of protein similarity relaxes (Yeh et al. 2001). On the contrary, our results indicate that CRASA is refractory to the arbitrary thresholds of protein similarity defined for the SAG subgroups.
Figure 2.

The exon-level accuracy of different annotation tools tested with the SAG data sets. The strong (left panel) and moderate (right panel) similarity groups contain 17 and 26 sequences, respectively. The Sn, Sp, and (Sn + Sp)/2 and measures of CRASA for the strong similarity data set are 0.88, 0.81, and 0.85, respectively, and for the moderate similarity dataset are 0.87, 0.79, and 0.83, respectively.
Also different from the Procrustes and GenWise methods is that CRASA maps the expressed genes directly and exactly to genomic sequences. It does not require the setting of BLASTX P-value thresholds to select “optimal” candidate (or target) proteins for gene prediction. In the CRASA algorithm, finding the exact match between the expressed gene and the genomic sequence is compromised only by the quality of cDNAs or sequence variations without a priori selection of candidate genes.
RG Sequences
Because the intergenic sequences of SAG are artificial and most of these SAG sequences are longer than the length limitation to some annotation tools, we created a dataset of real genomic (RG) sequences (see Methods) randomly selected from GeneBank of NCBI. All the annotation tools tested on the RG data set below can be run directly on the Web sites and do not require any input of target sequences and P-value thresholds. For this accuracy test, 13 well-known annotation tools were included for comparative analysis: FGENESH, GeneID, GeneMark.hmm, Genie, GENSCAN, GenView, Grail II, GrailEXP–Perceval, HMMgene, MZEF (all ab initio approaches), AAT, GeneBuilder, and GrailEXP–Gawain (all homology-based approaches).
In addition to Sn and Sp, comparison of the accuracy measures also includes ME (the proportion of missing exons and actual exons) and WE (the proportion of predicted wrong exons and actual predicted exons). These accuracy measurement parameters are described in Methods. The annotation results of CRASA as well as the other 13 tools are illustrated at the exon level in Figure 3. The sensitivity (Sn and ME) and specificity (Sp and WE) values of CRASA clearly demonstrated a better performance than other annotation tools. The measures of Sn, Sp, ME, and WE of CRASA (0.92, 0.79, 0.05, and 0.1, respectively) are far better than those of the mean values of 13 other tools (i.e., 0.54, 0.47, 0.29, and 0.38, respectively). Also noted is the (Sn + Sp)/2 values of CRASA run on the RG (0.85), strong similarity (0.85), and moderate similarity SAG (0.83) data sets. Thus, CRASA annotation performed consistently under the present accuracy test condition.
Figure 3.

Comparison of the sensitivity and specificity of CRASA with the other 13 annotation tools. (A) The sensitivity (Sn, closed circle) and specificity (Sp, open circle) are computed from the annotation results of the RG data set by the indicated homology- and ab initio-based tools. (B) The exon-level accuracy in terms of missing exon (ME, closed circle) and wrong exon (WE, open circle) is calculated from the same results described in A. The mean values of Sn, Sp, ME, and WE of these 14 tools are, respectively, 0.54, 0.47, 0.29, and 0.38, whereas those of CRASA are 0.92, 0.79, 0.05, and 0.1.
Compared with the SAG data set, the greater exon density in RG is reflective of the fact that the average length of annotated sequences is at least five times smaller in size (Table 2). Unlike SAG, the RG data set contains the actual genomic sequences of multiple human genes and intergenic sequences. The accuracies reported here for GENSCAN (Sn = 0.67 and Sp = 0.48) and FGENESH (Sn = 0.68 andSp = 0.62) are comparable to the respective values of (Sn = 0.65 and Sp = 0.5) and (Sn = 0.68 and Sp = 0.66) in the analysis of the BRCA2 1.4-Mb region containing 20 verified genes of 168 exons (Couch et al. 1996; Salamov and Solovyev 2000). The BRAC2 contig of Chromosome 13 may also be considered as an ideal data set for testing annotation accuracy. Because the SAG sequences share the same property (Guigó et al. 2000), these two benchmark data sets used in this paper are valuable and reliable for practical evaluation and training of annotation tools.
Table 2.
Characteristics of the Benchmark Data Sets Used in This Study
| Variable | SAG (BLASTX similarity) | RG | |
| Strong | Moderate | ||
| No. of sequences | 17 | 26 | 20 |
| Mean sequence length (kb) | 164 | 174 | 29 |
| No. of genes | 64 | 93 | 29 |
| Mean gene length (bp) | 4496 | 4589 | 10,486 |
| No. of exons | 385 | 477 | 191 |
| Mean exon length (bp) | 197 | 181 | 201 |
| No. of exons/gene | 6.02 | 5.13 | 6.59 |
| No. of genes with one or two exons | 18 | 24 | 3 |
| Mean intron length (bp) | 659 | 886 | 1640 |
| No. of gene/Mb | 22.92 | 20.47 | 49.64 |
| Mean C + G % | 40.01 | 39.56 | 51.82 |
| Exon density (%) | 2.73 | 1.89 | 6.54 |
Exon density provides the percentage of nucleotides that occur in coding regions.
While testing the RG data set, we made several interesting observations. First, the homology-based approaches are generally more time-consuming than the ab initio methods. Secondly, as shown in Figure 3 for the ab inito approaches, the sensitivity (Sn and ME) is superior overall to the specificity (Sp and WE) except for MZEF. Thirdly, the sensitivity of the homology-based GeneBuilder is moderate, but its specificity is rather poor, compared with the high specificity of AAT (e.g., for the WE measure, AAT was second in the 14 tools tested). This result may reflect the difficulty in setting up suitable criteria to validate the potential EST matches by homology-based approaches.
Annotation of Human Chromosomes 21 and 22 and Future Work
The annotated human Chromosomes 21 (Hattori et al. 2000) and 22 (Dunham et al. 1999) contain, respectively, 284 genes (127 known genes, 98 predicted or putative genes, and 59 pseudogenes) and 832 genes (339 known genes, 281 predicted or putative genes, and 212 pseudogenes). These annotated genes are classified into three sets in Table 3: (1) exon information not available at the time of study; (2) genes with 1 or 2 exons; and (3) genes analyzed by CRASA in this study. In the analyzed set, ∼83%, (130 + 305)/(177 + 347), of the genes (or 79% of the exons) were annotated by CRASA. Direct comparison of our results with Ensembl's annotated Chromosomes 21 and 22 (http://www.ensembl.org/Homo_sapiens/) showed that ∼17% of the annotated genes, mostly predictive, were missed by CRASA. The missing 47 + 42 = 89 genes (or 375 + 762 = 1137 exons) are either predicted or pseudogenes (i.e., Categories 2–5 defined by Hattori et al. 2000 at http://hgp.gsc.riken.go.jp/chr21/Genetable.html). Literally, CRASA is capable of identifying all the known genes (i.e., Category 1) reported by Hattori et al. (2000) and Dunham et al. (1999).
Table 3.
Results of CRASA for Human Chromosomes 21 and 22 Compared With the Annotated Genes
| Chr21 | Chr22 | |
| No. of annotated genes (including pseudogenes) | 284 | 832 |
| (1) No. of genes not available | 9 | 3 |
| (2) No. of genes with 1 or 2 exons | 98 | 482 |
| (3) No. of genes (exons) analyzed | 177 (1677) | 347 (3763) |
| (3.1) No. of genes (exons) annotated by CRASA | 130 (1302) | 305 (3001) |
| (3.2) No. of genes not annotated by CRASA | 47 | 42 |
| (3.2.1) No. of putative or predicted genes | 45 | 32 |
| (3.2.2) No. of nonfunctional pseudogenes | 2 | 10 |
| No. of additional genes annotated by CRASA | 46 | 37 |
| C1. Identical to known genes | 15 | 10 |
| C.1.1. Total length | 13 | 8 |
| C1.2. Partial length | 2 | 2 |
| C2. Similar to known genes | 4 | 6 |
| C2.1. Total length | 3 | 2 |
| C2.2. Partial length | 1 | 4 |
| C3. Not similar to any known genes | 16 | 11 |
| C4. Pseudogenes | 4 | 0 |
| C5. EST match only | 7 | 10 |
The genes that are not available means the mRNA sequences are not offered by the Consortium. The genes with 1 or 2 exons include 42 (or 341) functional genes and 56 (or 141) pseudogenes (processed or nonfunctional pseudogenes) on Chromosome 21 (or 22). The EST match only means the matched EST fragments are not open or the translated sequences <50 amino acids. The 1302 (or 3001) exons of Chromosome 21 (or 22) annotated by CRASA include 1145 (or 2556) exons both of whose boundaries are correctly matched and 157 (or 445) exons that are only partially matched to the actual exons.
From the CRASA-annotated results of Chromosome 21, we found that 46 ESTs had no matches to the genes of previous annotation (also termed the additional genes annotated by CRASA) and that 130 annotated genes were matched to 469 (i.e., 515 − 46) ESTs (see Tables 1 and 3). The latter may be attributed to data redundancy or splicing variants in the HGI database. It is therefore of great interest to study splicing variants with the CRASA algorithm in the future. More than 45,000 ESTs with at least three fragments matched to the Chromosome 21 contigs contain repetitive elements (Table 1). Although these ESTs were excluded from the present complexity reduction analysis, we believe that this observation deserves further attention in order to understand better the genome-wide transcription activities.
Of the 83 ESTs not matched to any annotated genes on Chromosomes 21 and 22 (Table 3), we searched the translated frames against the nr protein database of GenBank. Based on the search results, each EST was assigned to one of the following subcategories:
Category 1: Known Human Genes
Subcategory 1.1
The translated ESTs with 100% identity over essentially their total length to a known gene.
Subcategory 1.2
The translated ESTs with 100% identity over their partial length to a known gene.
Category 2: Similar to the Known Genes
Subcategory 2.1
The translated ESTs with similarities over essentially their total length to a known gene.
Subcategory 2.2
The translated ESTs with regional similarities to a known gene.
Category 3: The Translated ESTs With No Significant Similarity to Any Known Gene
Category 4: Pseudogenes
Category 5: EST Matches Only (Not Open)
All the CRASA-matched ESTs in Category 1 are genes verified only after the original annotation work was published (Dunham et al. 1999; Hattori et al. 2000). An additional 37 ESTs with an open translation frame in Categories 2 and 3 are potentially novel genes and need to be validated later. A detailed description of these 83 ESTs is listed in Supplementary Tables 4 and 5 (available online at http://www.genome.org and at http://crasa.sinica.edu.tw/bioinformatics/Supplementary.htm). It is true that new genes (10%–15% or more) can still be extracted from the presently incomplete EST databases.
In addition, the ab initio approaches remain viable and very useful when the query genome has no known homology of expressed information. As the ESTs continue to grow rapidly, homology-based approaches such as CRASA become more easy to annotate the genome with the expression information. In the present version, the single-exon (∼5%) and two-exon (∼14%) genes were excluded from the assessment of CRASA performance (Venter et al. 2001). It is well known that the human genome is littered with many processed pseudogenes and that the single-exon genes can be accurately predicted by the ab initio approaches. We intend to develop the next version of CRASA, capable of annotating the single- or two-exon genes as well.
METHODS
CRASA Approach
CRASA is implemented with two major components: reconstructing the cDNA database progressively to a multilevel pyramidal data structure in hierarchical orders (Fig. 4A) and annotating the genomic sequences (Fig. 4B). The latter includes pattern processing and matching to cDNA data in the corresponding pyramids. A stepwise description of CRASA annotation scheme is given below.
Figure 4.

An overview of the CRASA system. (A) The construction of CR pyramids. (B) CRASA annotation of the genomic sequences, which includes the pattern matching, pattern sites processing, and data filtering.
Construction of a Pyramidal Data Structure
The main property of a pyramidal structure is multiresolution (or progressive) transmission, which has been applied quite extensively to other research areas of the computing sciences. In multiresolution transmission, the original data are viewed globally by partial transmission or processing only. By selecting different resolutions, massive data can be treated dynamically, adaptively, and efficiently. The pyramidal data structure used here represents a simple application to processing sequence information. Within the pyramid the higher the level is, the finer the data resolution is achieved. We therefore constructed different pyramid levels to filter the noise and to patch the matched exons.
A virtual gate pyramid with three levels of complexity reduction (CR) is illustrated in Figure 1. An EST sequence is scanned base by base from the 5′ to the 3′ end. Each 4-base string is grouped as a pattern. In total, there are 44 = 256 possible patterns in the CRASA system. The pyramid is constructed by scanning the right and left neighbors of the identified pattern (Fig. 1B, “gatc”) one base by one base as binary codes added to each level.
Suppose that the gatc pattern is processed as (Fig. 1A): Four gatc patterns are found in the sequence. We then construct the gatc pyramid by scanning the neighbors of each gatc pattern. For the first level of the gate pyramid, the left and right neighbors of the first three gatc patterns are all g and c, and the binary code of the fourth pattern is c and g. Hence, we define the gatc Sites 1–3 as g/c and the Site 4 as c/g at Level 1 of the data pyramid. Each site at Level 1 is thus 1 + 4 + 1 = 6 bp in length. The related location information of each site including the position and EST accession number (e.g., HGI_6.0 contains >388,000 ESTs) is recorded in the corresponding coordinate (bin) of Level 1 of a 4 × 4 matrix (Fig. 1B). In the given example, Sites 1–3 are recorded in the g/c bin, whereas Site 4 is in the c/g bin. Similarly, the location information of these sites is recorded individually as binary codes at Levels 2 (16 × 16 matrix) and 3 (64 × 64 matrix) in the gatc pyramid. Level l can be regarded as a 4l × 4l matrix with 42l bins. As illustrated in Figure 1B, Sites 1–3 are indistinguishable at Level 1 for sharing the same binary code. However, they are addressed in three separate bins with different binary codes at Level 3. Conversely, data complexity is reduced by a factor of 16 from one level to the next within a pyramidal structure. Also noted is the data reversibility and inheritability between levels.
Extending from the three-level-pyramid skeleton shown in Figure 1B, a massive amount of sequence data can be processed and maintained systematically by CRASA. The depth of levels to be constructed in a CR pyramid is often dictated by the size of databases. For the present study, we constructed merely the 3rd and 7th levels in our system. The entire EST database (HGI_6.0) is 1-base-shift scanned, and the location information of each pattern site is addressed to the corresponding bin in a 4-base patterned pyramid. Each bin includes the corresponding EST accession number and the location of 4-base pattern sites for each EST. For saturated reconfiguration, a total of 256 pyramids is constructed for the HGI database to minimize the effect of sequence quality. It is apparent that a higher order of progressive level may reduce the complexity further, however, at the expense of longer construction time and greater data storage space.
Annotating the Genomic Sequences
Pattern Matching
The querying genomic sequence was similarly processed by CRASA and mapped to the corresponding HGI pyramids and bins. At the 7th level, each bin contains pattern sites of 18 bases in length (7 + 4 + 7 bases). By direct mapping, the matched 18-bp pattern sites between the query and EST sequences were identified. The entire process of pattern matching is likely to be very fast, because the time complexity is O(m) if the query length is m bp.
Pattern Sites Assembly
After pattern matching, the 18-bp pattern sites of matched ESTs are sorted along with the corresponding positions on the query sequence and assembled into the longest and nonoverlapping fragments. Throughout our system, the time cost is binding to the sorting process. If the total number of the matched 18-bp pattern sites is k, then the time complexity is O(k log k). Presumably, k becomes larger as the query length increases.
Filtering of Matched ESTs
To assess the efficiency of the filtering process, we analyzed the ESTs matched to the query sequences from human Chromosome 21. Table 1 shows that the number of matched fragments is reduced about 1 log for every 10-bp increment of matching length (e.g., from 3.7 × 108 [length ≥ 20 bp] to 8.4 × 105 [length ≥ 50 bp]). Only matched fragments ≥50 bp are considered for further analysis. It is known that <20% of human genes have one or two exons and that the average number of exons per gene is 7.8 (Venter et al. 2001). To further reduce the analysis complexity, ESTs with one or two split fragments matched to query sequences were removed in this study. As indicated in Table 1, the number of matched ESTs was reduced from 109,102 to 48,268. Although the definition of a gene is better represented by the interrupted colinear genomic fragments, it is possible that an EST with one or two fragments matched to a genome query is derived from a true gene, because the 3′ cDNA sequences are overrepresented in the databases. Additionally, matched ESTs with repetitive elements present in the genomic sequences, as shown in Figure 5A–C, are filtered out to further reduce the complexity substantially.
Figure 5.

Four possible scenarios of ESTs matched to a genomic sequence by CRASA. (A) Multiple ESTs match to the same region of genomic sequence. (B) Several internal segments of an EST match to the same region of a genomic sequence. (C) One segment within an EST matches to several regions of a genomic sequence. (D) The potential coding region of a genomic sequence matches colinearly to an EST in split segments and the processes of small fragments matching and gap patching. Step 1: EST_7 has three segments matched to the genomic sequence, which are at least 50 bp in length. Step 2: Small fragment matching: the small matched fragments (10–49 bp) are shown (open box). Step 3: Gap patching with the genomic sequence: While Gap 1 stays independently as a match, Gap 2, corrected by the gap-patching rule, is patched contiguously to the matched fragment downstream.
Small Fragment Matching
Because of the quality of cDNA sequences in the EST database, matched fragments shorter than 50 bases may be interrupted and excluded from the annotation process. A default value of 10 bases is thus set to recover small fragments. These 10-bp to 49-bp fragments are patched to the neighboring matches at Level 3 of CR pyramids (step 2 in Fig. 5D).
Gap Patching
A gap-patching rule is defined below to determine if the gap between successive EST fragments can be patched by the high-quality genomic sequence.
Gap-patching Rule:
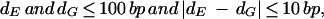 |
where dE and dG stand for the position differences of two successive fragments, respectively, on the EST and the corresponding query sequence. As in step 2 of Figure 5D, Gap 2 is patched with its genomic sequence, whereas Gap 1 is not by the gap-patching rule (step 3 in Fig. 5D). In this case EST_7 is matched to the query genomic sequence in four fragments.
After gap patching, 1681 ESTs are matched to Chromosome 21 queries (Table 1). One additional filter is installed to remove ESTs with matched fragments in a physical order inconsistent to the genomic sequence. The overall performance of CRASA annotation for Chromosome 21 in complexity reduction is >99.5%, as the number of matched ESTs is reduced from the original 109,102 to 515 (Table 1). Finally, the standard signatures of a gene, such as the initiation/stop codons and splicing signals, are used to determine the exact exon boundaries and coding region in the matched fragments.
Annotation Tools Tested in This Study
In this study 15 presently well-known annotation tools were tested along with CRASA, which include the ab initio (or statistic-based) and homology-based approaches. All these tools tested are the newly updated versions. Their Web sites are listed below.
Ab Initio Approaches
FGENESH: http://www.softberry.com/nucleo.html.
GeneID (v.1): http://www1.imim.es/geneid.html.
GeneMark.hmm (v. 2.2a): http://genemark.biology.gatech.edu/GeneMark/hum.cgi.
GENSCAN: http://genes.mit.edu/GENSCAN.html.
Grail II (Gene Recognition and Assembly Internet Link v.1.3): http://compbio.ornl.gov/Grail-1.3/.
GrailEXP-Perceval (v.3.0): http://grail.lsd.ornl.gov/grailexp/.
HMMgene (v.1.1): http://www.cbs.dtu.dk/services/HMMgene/.
Homology-Based Approaches
11. AAT (Analysis and Annotation Tool): http://genome.cs.mtu.edu/aat.html.
12. GeneBuilder: http://l25.itba.mi.cnr.it/∼webgene/genebuilder.html.
13. GeneWise (or Wise2, v.2.1.20): http://www.sanger.ac.uk/Software/Wise2/.
14. GrailEXP – Gawain (v3.0): http://grail.lsd.ornl.gov/grailexp/.
15. PROCRUSTES (v.4.0): http://www-hto.usc.edu/software/procrustes/qpn.html.
In our tests, the parameters used were the default values defined by the host sites except for GeneWise and PROCRUSTES (references in Guigó et al. 2000) and AAT (using the HGI instead of the UniGene database).
cDNA Database
The cDNA database used here is the HGI (Human Gene Index) version 6.0 (184 Mb for 388,006 sequences), which was kindly provided by TIGR (The Institute for Genomic Research). Compared with the recent UniGene version (102 Mb for 96,109 sequences) of NCBI (National Center for Biotechnology Information), HGI 6.0 appears to contain more expressed gene information. Interested readers may obtain a licensing agreement on the HGI database at http://www.tigr.org/tigr-scripts/license/new.pl?genre=gi. The CR pyramids will be updated continually as the new version of HGI is released.
Benchmark Data Sets
Genomic sequences in the SAG and RG data sets were used to evaluate the annotation tools in this paper. The SAG (semiartificial genomic) sequences were generated and offered generously by Guigó et al. (2000). In the SAG data set, a set of annotated gene sequences was arbitrarily placed in the background of random intergenic DNAs and the length was generated artificially by normal distribution. For testing the accuracy of gene prediction, two separate groups with strong and moderate sequence similarity were extracted from the SAG sequences. Each gene in the strong similarity group has a BLASTX P-value < 10−50), whereas the BLASTX P-value of the moderate similarity sequences is between 10−50 and 10−6.
We have also created a set of RG (real genomic) sequences selected randomly from GenBank. In the RG data set, each sequence contains annotated gene(s) and “real” intergenic sequence. Because of the limitation on query size for some annotation tools (e.g., 50 kb for GeneBuilder and 200 kb for MZEF), each sequence in the RG set is no longer than 50 kb. Table 2 lists the general features of these three benchmark data sets used for the evaluation of annotation tools in this study.
Accuracy Evaluation
To determine the performance of exon-based alignment by CRASA annotation, all the tested tools were evaluated for accuracy at the exon level. The standardized measures for accuracy evaluation used in this paper were defined previously by Burset and Guigó (1996), and are described briefly below.
Sensitivity
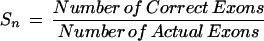 |
and
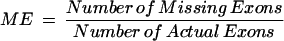 |
Specificity
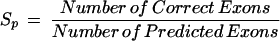 |
and
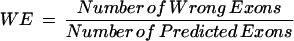 |
For Sn and Sp, the larger the values are, the more accurate the annotation tool is. On the contrary, the smaller the values of ME and WE are, the more accurate the annotation tool is. Measures of annotation results are computed at the exon level for all the query sequences. A correct exon is scored only when both ends of its boundary are annotated correctly.
Program Implementation
Two main CRASA programs, the construction of CR pyramids and the annotation processes, were written in Fortran, which allow parallel processing in a distributed memory computing system. The users' interface and the control scripts for program execution were written in Python. These programs are compiled (using Portland Group's PGF77 and MPICH) and executed on a 16-node Linux PC cluster. Each node has a 1 AMD K7 900 MHz processor and 512 Mb of RAM.
Practically, it takes ∼6 h to construct and 2.5 Gb of HD space to store the HGI database (version 6.0) at the 7th level of 256 CR pyramids. The time and storage requirements are dependent on the database size and the level depth in the pyramid. It is clear that the construction of CR pyramids is a preprocessing of the cDNA database. On the other hand, the execution performance (EP) for CRASA annotation is a function of the number of PC nodes in a Linux cluster (EP ≅ 2 log2 z − 2 kb/sec, where z is the used number of nodes). For instance, annotating the 34-Mb human Chromosome 21 sequences by CRASA takes ∼3 h of runtime for both DNA strands.
Data and Program Availability
Both the strong similarity and moderate similarity data sets of SAG sequences are at http://www1.imim.es/databases/gpecal2000/ (Guigó et al. 2000). The RG sequences and the related information, including the source code of CRASA, are available from http://crasa.sinica.edu.tw/bioinformatics/bioinformatics.html. The Web-based package of CRASA is presently in preparation.
WEB SITE REFERENCES
http://crasa.sinica.edu.tw/bioinformatics/bioinformatics.html; source code of CRASA, RG sequences, and related information.
http://crasa.sinica.edu.tw/bioinformatics/Supplementary.htm; Supplementary material available.
http://hgp.gsc.riken.go.jp/chr21/Genetable.html; Category 1 genes.
http://www.ensembl.org/Homo_sapiens/; Ensembl's annotated Chromosomes 21 and 22.
http://www.tigr.org/tigr-scripts/license/new.pl?genre=gi; HGI database.
http://www1.imim.es/databases/gpecal2000/; strong similarity and moderate similarity data sets of SAG sequences.
Acknowledgments
This work was supported by the National Science Council, Republic of China, under contract NSC 89-2316-B-001-021 and a Post-doctoral Researcher Award of the National Health Research Institutes. We thank Roderic Guigó for providing the SAG data sets and TIGR for the HGI database. We are also thankful to all the providers of annotation tools on the Web sites. This work was made possible by the public availability of the annotated human genome databases. We are grateful to Rudy Chen for his enthusiasm for this project.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL lychang@ibms.sinica.edu.tw; FAX 886-2-27858594.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.313703.
REFERENCES
- 1.Altschul S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. [DOI] [PubMed] [Google Scholar]
- 2.Altschul S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailey L.C., Fischer, S., Schug, J., Crabtree, J., Gibson, M., and Overton, G.C. 1998. GAIA: Framework annotation of genomic sequence. Genome Res. 8: 234-250. [DOI] [PubMed] [Google Scholar]
- 4.Birney E. and Durbin, R. 2000. Using GeneWise in the Drosophila annotation experiment. Genome Res. 10: 547-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burge C.B. and Karlin, S. 1997. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268: 78-94. [DOI] [PubMed] [Google Scholar]
- 6.___, 1998. Finding the genes in genomic DNA. Curr. Opin. Struct. Biol. 8: 346-354. [DOI] [PubMed] [Google Scholar]
- 7.Burset M. and Guigó, R. 1996. Evaluation of gene structure prediction programs. Genomics 34: 353-367. [DOI] [PubMed] [Google Scholar]
- 8.Chao K.M., Zhang, J., Ostell, J., and Miller, W. 1995. A local alignment tool for very long DNA sequences. Comput. Appl. Biosci. 11: 147-153. [DOI] [PubMed] [Google Scholar]
- 9.___, 1997. A tool for aligning very similar DNA sequences. Comput. Appl. Biosci. 13: 75-80. [DOI] [PubMed] [Google Scholar]
- 10.Couch F.J., Rommens, J.M., Neuhausen, S.L., Couch, E.J., Rommens, J.M., Neuhausen, S.L., Belanger, C., Dumont, M., Abel, K., Bell, R., et al. 1996. Generation of an integrated transcription map of the BRCA2 region on chromosome 13q12–q13. Genomics 36: 86-99. [DOI] [PubMed] [Google Scholar]
- 11.Dunham I., Hunt, A.R., Collins, J.E., Bruskiewich, R., Beare, D.M., Clamp, M., Smink, L.J., Ainscough, R., Almeida, J.P., et al. 1999. The DNA sequence of human Chromosome 22. Nature 402: 489-495. [DOI] [PubMed] [Google Scholar]
- 12.Ewing B. and Green, P. 2000. Analysis of expressed sequence tags indicates 35,000 human genes. Nat. Genet. 25: 232-234. [DOI] [PubMed] [Google Scholar]
- 13.Gelfand M.S., Mironov, A.A., and Pevzner, P.A. 1996. Gene recognition via spliced sequence alignment. Proc. Natl. Acad Sci. 93: 9061-9066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guigó R., Agarwal, P., Abril, J.F., Burset, M., and Fickett, J.W. 2000. An assessment of gene prediction accuracy in large DNA sequences. Genome Res. 10: 1631-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hattori M., Fujiyama, A., Taylor, T.D., Watanabe, H., Yada, T., Park, H.-S., Toyoda, A., Ishii, K., Totoki, Y., Choi, D.-K., et al. 2000. The DNA sequence of human Chromosome 21. Nature 405: 311-319. [DOI] [PubMed] [Google Scholar]
- 16.Huang X., Adams, M.D., Zhou, H., and Kerlavage, A.R. 1997. A tool for analyzing and annotating genomic sequences. Genomics 46: 37-45. [DOI] [PubMed] [Google Scholar]
- 17.Hyatt, D., Snoddy, J., Schmoyer, D., Chen, G., Fischer, K., Parang, M., Vokler, I., Petrov, S., Locascio, P., Olman, V., et al. 2000. Improved analysis and annotation tools for whole-genome computational annotation and analysis: GRAIL-EXP genome analysis toolkit and related analysis tools. Genome Sequencing & Biology Meeting, May..
- 18.Kent W.J. 2002. BLAT—The BLAST-like alignment tool. Genome Res. 12: 656-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krogh A., et al. 1998. An introduction to hidden Markov models for biological sequences. In Computational methods in molecular biology (ed. S.L. Salzberg), Chapter 4, pp. 45–63. Elsevier, Amsterdam.
- 20.___, 2000. Using database matches with HMMGene for automated gene detection in Drosophila. Genome Res. 10: 523-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lander E.S. and Waterman, M.S., 1995. Calculating the secrets of life Chapter 3. National Academy Press, Washington, DC.
- 22.Lander E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- 23.Liang F., Holt, I., Pertea, G., Karamycheva, S., Salzberg, S.L., and Quackenbush, J. 2000. Gene Index analysis of the human genome estimates approximately 120,000 genes. Nat. Genet. 25: 239-240. [DOI] [PubMed] [Google Scholar]
- 24.Lukashin A.V. and Borodovsky, M. 1998. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 264: 1107-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milanesi L., Kolchanov, N.A., Rogozin, I.B., Ischenko, I.V., Kel, A.E., Orlov, Yu.L., Ponomarenko, M.P., Vezzoni, P., et al. 1993. GenView: A computing tool for protein-coding regions prediction in nucleotide sequences. In Proceedings of the Second International Conference on Bioinformatics, Supercomputing and Complex Genome Analysis (ed. H.A. Lim), pp. 573–588. World Scientific Publishing, Singapore.
- 26.Milanesi L., D'Angelo, D., and Rogozin, I.B. 1999. GeneBuilder: Interactive in silico prediction of gene structure. Bioinformatics 15: 612-621. [DOI] [PubMed] [Google Scholar]
- 27.Mironov A.A., Roytberg, M.A., Pevzner, P.A., and Gelfand, M.S. 1998. Performance-guarantee gene predictions via spliced alignment. Genomics 51: 332-339. [DOI] [PubMed] [Google Scholar]
- 28.Ning Z., Cox, A.J., and Mullikin, J.C. 2001. SSAHA: A fast search method for large DNA databases. Genome Res. 11: 1725-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pachter L., Batzoglou, S., Spitkovsky, V.I., Banks, E., Lander, E.S., Kleitman, D.J., and Berger, B. 1999. A dictionary-based approach for gene annotation. J. Comput. Biol. 6: 419-430. [DOI] [PubMed] [Google Scholar]
- 30.Parra G., Blanco, E., and Guigó, R. 2000. GeneID in Drosophila. Genome Res. 10: 511-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reese M.G., Eeckman, F.H., Kulp, D., and Haussler, D., 1997. Improved splice site detection in Genie. In Proceedings of the First Annual International Conference on Computational Molecular Biology (RECOMB) 1997, Santa Fe, NM. ACM Press, New York, NY. [DOI] [PubMed]
- 32.Reese M.G., Kulp, D., Tammana, H., and Haussler, D. 2000. Genie—Gene finding in Drosophila melanogaster. Genome Res. 10: 529-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roest Crollius H., Jaillon, O., Bernot, A., Dasilva, C., Bouneau, L., Fischer, C., Fizames, C., Wincker, P., Brottier, P., Quetier, F., et al. 2000. Estimate of human gene number provided by genome-wide analysis using Tetraodon nigroviridis DNA sequence. Nat. Genet. 25: 235-238. [DOI] [PubMed] [Google Scholar]
- 34.Salamov A.A. and Solovyev, V.V. 2000. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10: 516-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solovyev V.V., Salamov, A.A., and Lawrence, C.B. 1995. Identification of human gene structure using linear discriminant functions and dynamic programming. Proc. Int. Conf. Intell. Syst. Mol. Biol. 3: 367-375. [PubMed] [Google Scholar]
- 36.Sze S.H. and Pevzner, P.A. 1997. Las Vegas algorithms for gene recognition: Suboptimal and error-tolerant spliced alignment. J. Comput. Biol. 4: 297-309. [DOI] [PubMed] [Google Scholar]
- 37.Venter J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291: 1304-1351. [DOI] [PubMed] [Google Scholar]
- 38.Xu Y. and Uberbacher, E., 1996. Gene prediction by pattern recognition and homology search. In Proceedings of the Fourth International Conference on Intelligent Systems for Molecular Biology, pp. 241–251. AAAI Press, June. [PubMed]
- 39.Xu Y., Mural, R., Shah, M., and Uberbacher, E. 1994. Recognizing exons in genomic sequence using GRAIL II. Genet. Eng. (NY) 16: 241-253. [PubMed] [Google Scholar]
- 40.Yeh R.-F., Lim, L.P., and Burge, C.B. 2001. Computational interface of homologous gene structures in the human genome. Genome Res. 11: 803-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang M.Q. 1997. Identification of protein coding regions in the human genome by quadratic discriminant analysis. Proc. Natl. Acad. Sci. 94: 565-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Z., Schwartz, S., Wagner, L., and Miller, W. 2000. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7: 203-214. [DOI] [PubMed] [Google Scholar]