

Abstract
In pancreatic β-cells, KATP channels consisting of Kir6.2 and SUR1 couple cell metabolism to membrane excitability and regulate insulin secretion. Sulfonylureas, insulin secretagogues used to treat type II diabetes, inhibit KATP channel activity primarily by abolishing the stimulatory effect of MgADP endowed by SUR1. In addition, sulfonylureas have been shown to function as pharmacological chaperones to correct channel biogenesis and trafficking defects. Recently, we reported that carbamazepine, an anticonvulsant known to inhibit voltage-gated sodium channels, has profound effects on KATP channels. Like sulfonylureas, carbamazepine corrects trafficking defects in channels bearing mutations in the first transmembrane domain of SUR1. Moreover, carbamazepine inhibits the activity of KATP channels such that rescued mutant channels are unable to open when the intracellular ATP/ADP ratio is lowered by metabolic inhibition. Here, we investigated the mechanism by which carbamazepine inhibits KATP channel activity. We show that carbamazepine specifically blocks channel response to MgADP. This gating effect resembles that of sulfonylureas. Our results reveal striking similarities between carbamazepine and sulfonylureas in their effects on KATP channel biogenesis and gating and suggest that the 2 classes of drugs may act via a converging mechanism.
Keywords: KATP channel, gating, sulfonylurea receptor 1, sulfonylureas, pharmacological chaperones
Introduction
ATP-sensitive potassium (KATP) channels are hetero-octameric complexes of 4 pore-forming inwardly rectifying potassium channel subunits, Kir6.1 or Kir6.2, and 4 regulatory sulfonylurea receptor subunits, SUR1 or SUR2.1 Collectively, KATP channels function as molecular sensors to convert metabolic signals into changes in membrane potential; they are important for a wide range of physiological functions such as protection of cardiac and neuronal cells from ischemic injuries, control of vascular tone, and regulation of hormone secretion.2 Dysfunction of KATP channels underlies a number of human diseases, including congenital hyperinsulinism, neonatal diabetes, DEND syndrome (Developmental delay, Epilepsy and Neonatal Diabetes), dilated cardiomyopathy, and Cantu syndrome.3-8
In pancreatic β-cells, KATP channels composed of SUR1 and Kir6.2 play a key role in glucose-stimulated insulin secretion.3,9 KATP channels are gated by intracellular nucleotides. ATP interacts with the pore subunit Kir6.2 to inhibit channel activity in a non-hydrolysis dependent manner; whereas MgATP and MgADP interact with the nucleotide binding domains (NBDs) of SUR1 to stimulate channel activity.2,10,11 Regulation of SUR1 is thought to occur via hydrolysis of MgATP to MgADP, which induces a conformational change of the NBDs to open the Kir6.2 pore; an increase in MgADP concentration has a potent stimulatory effect by stabilizing the NBDs in the post-hydrolytic conformation.11 Thus, channel activity reflects the balance between nucleotide inhibition and Mg-nucleotide stimulation.2,3 When blood glucose levels rise, the ATP/ADP ratio in β-cells increases to shift the balance toward channel closure, leading to membrane depolarization, opening of voltage-gated Ca2+ channels, and insulin secretion. As blood glucose levels fall, the ATP/ADP ratio decreases to favor KATP channel opening by MgADP and stop insulin secretion. Mutations that render channels less sensitive to ATP inhibition, or more sensitive to MgADP stimulation, cause neonatal diabetes and in some cases also DEND syndrome.3,12-14 By contrast, mutations which render channels unresponsive to the stimulatory effect of MgADP result in loss of channel function and are frequently found in patients with congenital hyperinsulinism, which is characterized by persistent insulin secretion despite life-threatening hypoglycemia.15,16 Aside from gating defects, channel biogenesis and trafficking defects which prevent expression of functional channels in the β-cell plasma membrane are also major causes of congenital hyperinsulinism.17
Sulfonylureas such as tolbutamide and glibenclamide inhibit KATP channel activity to stimulate insulin secretion and are thus effective in treating type II diabetes as well as some cases of neonatal diabetes/DEND syndrome caused by hyperactive KATP channels.3 The inhibitory effect of sulfonylureas on KATP channel activity is largely attributed to an inhibition of channel response to MgADP.18 In addition to inhibiting channel activity, sulfonylureas have been shown to act as KATP channel pharmacological chaperones and correct trafficking defects caused by a subset of mutations in SUR1, specifically those in the first transmembrane domain TMD0.19,20 We have previously shown that mutant channels rescued to the cell surface by the high-affinity sulfonylurea glibenclamide are unable to open in response to metabolic inhibition in intact cells as assessed by 86Rb+ efflux assays.19 Closer examination by inside-out patch-clamp recording revealed that the rescued channels failed to respond to MgADP, likely because the drug remained bound to the channel even after extensive washout.19 However, a lower affinity sulfonylurea tolbutamide could be washed out from rescued surface channels to recover channel response to MgADP and to allow channels to open upon metabolic inhibition in 86Rb+ efflux assays.19,20
In a recent study, we identified carbamazepine as a novel KATP channel ‘corrector’ able to rescue trafficking defects caused by the same set of TMD0 mutations rescued by sulfonylureas.21 Interestingly, functional analysis of a SUR1-TMD0 trafficking mutant, F27S, showed that channels rescued to the cell surface by carbamazepine also failed to open upon metabolic inhibition.21 However, mutant channel activity gradually recovered as carbamazepine was washed out, with activity near the level observed in wild-type (WT) channels after 90 min washout.21 In this study, we investigated how carbamazepine affects KATP channel gating to prevent rescued channels from opening in metabolically stressed cells. We show that carbamazepine, like sulfonylureas, specifically suppresses channel response to MgADP such that channels are unable to open when the intracellular ATP/ADP was lowered by metabolic inhibition.
Results
The ability of KATP channels to respond to metabolic signals is critically dependent on their response to ATP and MgADP. We therefore determined the response of carbamazepine-rescued F27S mutant channels to ATP and ADP by inside-out patch-clamp recording. First, to verify that the mutation itself does not alter channel-gating properties, we recorded mutant channels from cells not treated with the pharmacological corrector carbamazepine. Note although the F27S mutation severely hinders channel trafficking to the cell surface, sufficient currents (~10% of averaged WT channel current amplitude) can be detected in a small number of cells to allow analysis of ATP and MgADP sensitivity. Compared with WT channels, mutant F27S channels exhibited comparable sensitivity to ATP inhibition and MgADP stimulation (Fig. 1), indicating that the F27S mutation per se does not have a significant effect on channel response to intracellular nucleotides.

Figure 1. The F27S mutation in SUR1 does not alter KATP channel gating property. (A) Representative recordings testing MgADP responses of WT and F27S channels expressed in COSm6 cells without overnight carbamazepine treatment. (B) Quantification of the MgADP response. Currents in 0.1 mM ATP or 0.1 mM ATP plus 0.25 mM ADP were expressed as percent currents of those observed in K-INT. There is no statistical significance in MgADP response between F27S and WT channels.
Next, we recorded F27S mutant channels from cells treated overnight with carbamazepine. As expected, carbamazepine markedly increased the current amplitudes detected in isolated membrane patches indicating increased mutant channel expression at the cell surface. However, although these channels displayed normal ATP sensitivity they showed greatly reduced MgADP response (Fig. 2) compared with channels from cells not treated with carbamazepine (Fig. 1). As the MgADP stimulatory effect is crucial for KATP channels to open upon glucose deprivation,15,16 this explains why carbamazepine-rescued F27S mutant channels were unable to open upon metabolic inhibition, as observed in our previous study.21 Interestingly, washout of carbamazepine by incubating cells in fresh media without the drug for 2 h prior to recording recovered the channel’s response to MgADP, suggesting carbamazepine impairs MgADP response. This prompted us to test whether carbamazepine has the same effect on WT channels. As observed in the F27S mutant, WT channels recorded from cells treated overnight with carbamazepine also had little MgADP response; upon washout for more than 2 h, the MgADP response recovered to nearly the extent seen in channels not previously exposed to carbamazepine (Fig. 2). These results show that carbamazepine disrupts the ability of channels to respond to MgADP. In addition to the physiological activator MgATP/MgADP, KATP channels can be activated pharmacologically by the potassium channel opener diazoxide via SUR1 in a Mg-nucleotide dependent manner. We found that both WT and F27S mutant channels from cells treated overnight with carbamazepine also exhibited greatly reduced response to diazoxide; and again, after washout, diazoxide response recovered to the level comparable to that reported previously for WT channels (Fig. 3).
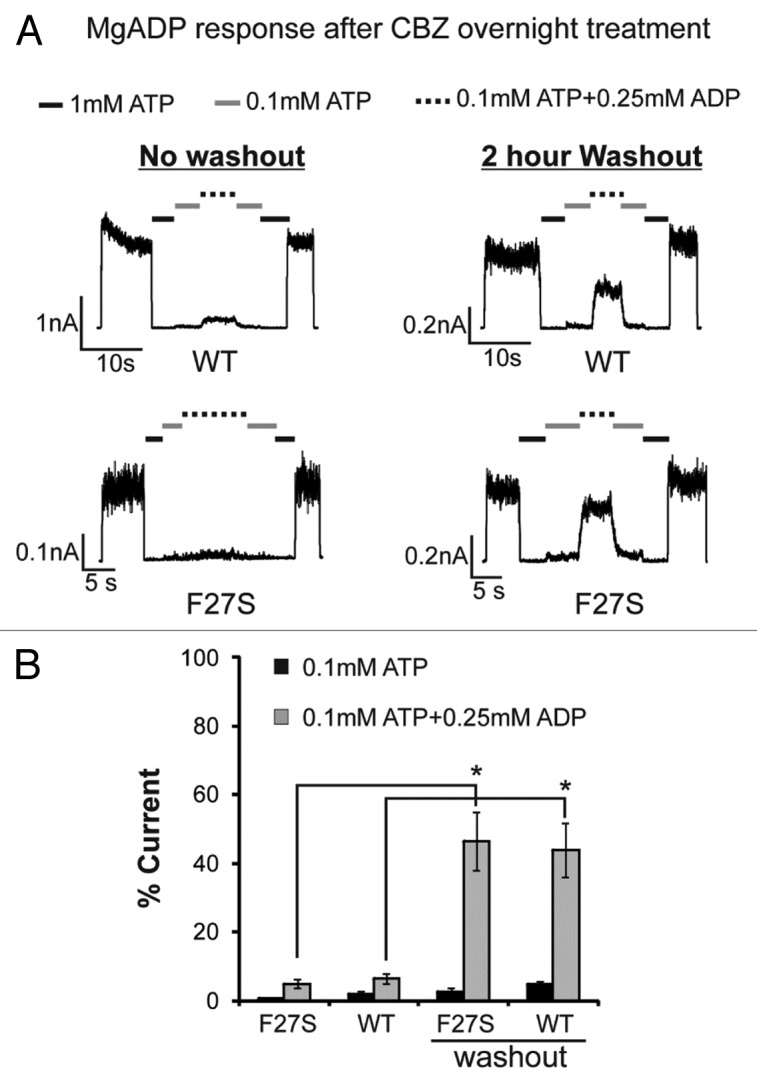
Figure 2. Channels from cells treated overnight with carbamazepine show diminished response to MgADP, an effect that is reversed after > 2-h washout of the drug. (A) Representative inside-out patch-clamp recordings testing MgADP responses of WT and F27S channels expressed in COSm6 cells treated overnight with 10 µM carbamazepine with (right) or without (left) 2 h of washout prior to recording. (B) Quantification of MgADP response. Currents in 0.1mM ATP or 0.1mM ATP plus 0.25mM ADP were expressed as percent of those observed in K-INT. Asterisks indicate statistical significance in MgADP response between no washout and > 2-h of washout of carbamazepine prior to recording (P < 0.05, n = 3) by 2-tailed, unpaired Student t test.
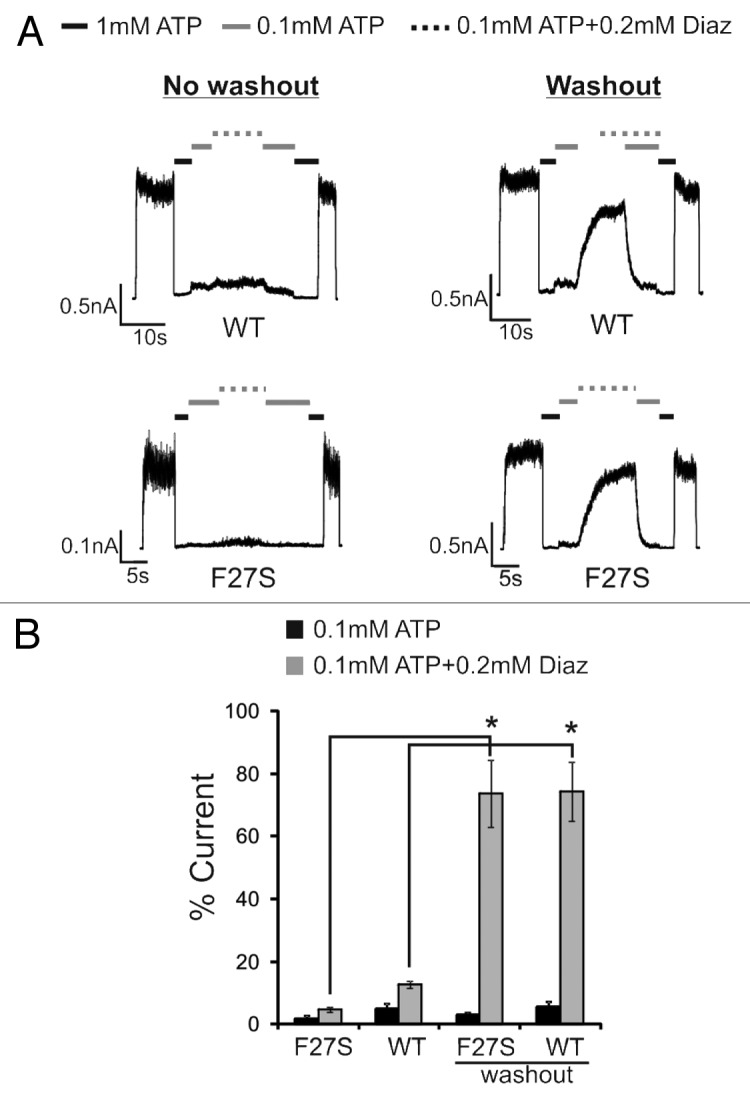
Figure 3. Overnight carbamazepine treatment also impairs channel response to diazoxide, but the effect is reversed upon extensive washout of carbamazepine. (A) Representative inside-out patch-clamp recordings testing diazoxide responses of WT and F27S channels expressed in cells treated overnight with 10 µM carbamazepine with (right) or without (left) 2 h of washout prior to recording. (B) Quantification of diazoxide response. Currents in 0.1 mM ATP or 0.1 mM ATP plus 0.2 mM diazoxide were expressed as percent of those observed in K-INT. Asterisks indicate statistical significance in diazoxide response between no washout and > 2-h of washout of carbamazepine prior to recording (P < 0.05, n = 3) by 2-tailed, unpaired Student t test.
From the above experiments we cannot necessarily conclude that carbamazepine interferes with channel response to MgADP directly, as prolonged drug incubation and washout could change the cellular environment to indirectly affect channel behavior. We therefore tested whether carbamazepine can block the stimulatory effect of MgADP when acutely applied to channels in isolated membrane patches. WT channels from cells not pretreated with carbamazepine were exposed to bath solutions containing various combinations of ATP, MgADP and carbamazepine in inside-out patch-clamp recording. As shown in Figure 4A, addition of 0.25 mM ADP to 0.1 mM ATP (free [Mg2+] ~1 mM) antagonized the inhibitory effect of 0.1 mM ATP and stimulated channel activity (compare time points 1 and 2 in Fig. 4A). Subsequent exposure to a solution containing the same concentrations of ATP and ADP but with an additional 10 µM carbamazepine led to a gradual decline in channel activity (compare time points 2 and 3 in Fig. 4A). Returning channels back to K-INT solution (time point 4) recovered channel activity to ~70–80% of the original currents observed in K-INT immediately after patch excision, which is in the range typically observed with WT KATP channels in solutions containing Mg2+ that causes rundown. Interestingly, exposure of the same patch to 0.1 mM ATP followed by 0.1 mM ATP plus 0.25 mM ADP showed that the channels were now unable to respond to the stimulatory effect of MgADP, in contrast to that seen in the first round of 0.1 mM ATP plus 0.25 mM ADP exposure (compare time points 2 and 5). Further exposure to the solution containing 0.1 mM ATP, 0.25 mM ADP and 10 µM carbamazepine caused the currents to decline even more (compare time points 5 and 6). These results show that carbamazepine exerts an acute inhibitory effect on the channel, rendering it unable to open in the presence of MgADP.

Figure 4. Carbamazepine acutely inhibits channel response to MgADP and MgGTP in isolated membrane patches. (Ai) Representative inside-out patch-clamp recording from COSm6 cells expressing WT channels without overnight carbamazepine treatment. Channels were exposed sequentially to various bath solutions as indicated by the bars above the recording. (Aii) Average currents at the various time points marked in the current trace shown in (Ai) are expressed as percent of those observed immediately after patch excision into K-INT solution. Asterisks indicate statistical significance (P < 0.05, n = 3) between the 2 time points using 2-tailed, paired Student t test. (Bi) Representative recording showing the stimulatory effect of 1 mM GTP and inhibition of this effect by carbamazepine. Note the patch was excised into K-INT solution and allowed to run down to a steady level before being exposed to 1mM GTP (free [Mg2+]~1mM). (Bii) Quantification of channel activity in 1 mM GTP or 1 mM GTP plus 10 µM carbamazepine as % of currents observed in K-INT. * P < 0.05 by 2-tailed, unpaired Student t test, n = 6 and 3 for 1 mM GTP and 1 mM GTP plus 10 µM carbamazepine, respectively.
The effects of adenine nucleotides ATP and ADP on KATP channel activity in the presence of Mg2+, such as the scheme used in the above experiment, are complex due to both inhibition via Kir6.2 and stimulation via SUR1. We therefore further examined how carbamazepine affects channel response to another Mg-nucleotide, MgGTP, which has been reported to undergo hydrolysis at SUR1 to stimulate channel activity but is much less potent than MgATP in inhibiting the channel via Kir6.2. We found that 1mM MgGTP, a concentration previously reported to increase channel activity without significant inhibition on Kir6.2,22 stimulated WT channel activity by 23.5 ± 1.8%, and 10µM carbamazepine abolished the stimulatory effect of MgGTP and caused further decline in channel activity below that observed in control K-INT solution (41.9 ± 2.1 of control) (Fig. 4B). This result further supports the notion that carbamazepine disrupts the ability of SUR1 to stimulate channel activity by Mg-nucleotides.
Discussion
Carbamazepine is a clinically used anticonvulsant and has been reported to modulate voltage-gated sodium channels,23 Ca2+ channels,24 and GABAA receptors.25 Our recent study showing that carbamazepine, within the concentration range currently used to treat epilepsy and trigeminal neuralgia, corrects a subset of trafficking-impaired KATP channels to the cell surface expands the pharmacological targets of this drug. The study presented here demonstrates that carbamazepine also exerts a potent effect on KATP channel gating by abolishing channel response to the physiological activator MgADP. Since the inhibitory effect was observed in WT channels in isolated membrane patches, we propose that carbamazepine interacts directly with the channel complex or another closely associated protein/lipid to modulate channel gating. The finding that carbamazepine abolishes channel response to MgADP explains why mutant channels rescued by overnight carbamazepine treatment fail to open when cells are subjected to metabolic inhibition to mimic glucose deprivation, since the ability of KATP channels to open in response to glucose starvation relies on their ability to be activated by MgADP.
KATP channel stimulation by MgADP is conferred by SUR1. A prevailing model asserts that this regulation of Kir6.2 activity is triggered by a conformational change in SUR1 consequent to MgATP hydrolysis to MgADP at the NBD2. Our results show that carbamazepine inhibited channel response to MgATP/ADP as well as response to diazoxide and MgGTP. MgGTP has been proposed to undergo hydrolysis by SUR1 to stimulate channel activity22; and diazoxide as well as other potassium channel openers have also been proposed to stimulate channel activity by increasing ATPase activity of SUR.26 Thus, it is possible that carbamazepine may affect SUR1 ATPase activity to block channel response to Mg-nucleotides and diazoxide. However, it is worth noting that the hydrolysis model remains controversial.2 Recent studies by Ortiz et al. argue that Mg-nucelotide hydrolysis may not be required for conformational change in SUR1.27,28 Also, neonatal diabetes-causing mutations in the NBDs of SUR1 have been reported to either increase or decrease SUR1 ATPase activity,29,30 suggesting that the ATPase activity may not necessarily predict channel response to Mg-nucleotides. Moreover, it is equally possible that carbamazepine inhibits response to Mg-nucleotides and diazoxide by disrupting their binding or transduction steps required for coupling with Kir6.2. Clearly, additional studies are needed to sort out these possibilities.
The inhibitory effect of carbamazepine on channel activity represents a challenge in the quest to harness the chaperone effect of the drug to treat congenital hyperinsulinism caused by KATP channel trafficking defects. A potential solution to this problem lies in the reversibility of this inhibitory effect. Our results show that although inhibition by carbamazepine is not reversed quickly in the time course (seconds) of the patch-clamp recording experiments (see Fig. 3), it is reversible after the drug has been removed from the culture medium for an extended period of time (30 min–2 h) as we have shown previously by 86Rb+ efflux and here by electrophysiology, much like what we have observed before for the reversible sulfonylurea, tolbutamide. The same reversibility is observed for diazoxide response. Interestingly, we previously observed that diazoxide facilitated functional recovery of carbamazepine-rescued mutant channels in response to metabolic inhibition such that some channel activity was recovered without washout prior to the efflux assay.21 Likely diazoxide and metabolic inhibition act additively or synergistically to reverse the inhibitory effect of carbamazepine. Thus, it is worth exploring the possibility of corrector and potentiator combination therapy in recovering channel function loss due to trafficking mutations.
The gating effect of carbamazepine on KATP channels could also potentially be exploited to treat diseases caused by overactive channels seen in neonatal diabetes, DEND syndrome and Cantu syndrome, much like the way sulfonylureas have been utilized successfully to treat some cases of neonatal diabetes/DEND syndrome caused by gain-of-function KATP channel mutations. One question regarding the use of sulfonylureas to treat DEND syndrome is how efficiently the drugs can pass the blood-brain-barrier.31 Having another channel inhibitor that has been used to treat disease of the central nervous system such as carbamazepine may provide an alternative.
In conclusion, our study shows that in addition to correcting KATP channel trafficking defects, carbamazepine inhibits KATP channel activity by disrupting channel response to MgADP. The remarkable similarities between carbamazepine and sulfonylureas with respect to their effects on KATP channel biogenesis/trafficking and activity suggest they may modulate channel behavior via a common structural mechanism.
Methods
Molecular Biology
Rat Kir6.2 cDNA and hamster SUR1 cDNA constructs were in pCDNAI/Amp and pECE plasmids, respectively. Site-directed mutagenesis was performed using the QuikChange kit from Stratagene and mutations were confirmed by direct sequencing as described previously.21
Patch-clamp recording
COSm6 cells were transfected with SUR1 and Kir6.2 cDNAs along with cDNA encoding the green fluorescent protein to identify transfected cells. Cells were re-plated onto coverslips 24 h post-transfection and treated with carbamazepine or the vehicle control DMSO (0.1% final concentration) overnight. Cells were subjected to inside-out patch voltage-clamp recording with or without 2-h washout of the drugs prior to recording. Micropipettes were pulled from non-heparinized Kimble glass (Fisher) on a horizontal puller (Sutter Instrument) with resistance typically ~1–2 megaohms. For MgADP or diazoxide response, the bath (intracellular) and pipette (extracellular) solutions were K-INT: 140 mM KCl, 10 mM K-HEPES, 1 mM K-EGTA, pH 7.3. ATP and ADP were added as the potassium salt. In solutions containing 0.1mM ATP, 0.1mM ATP and 0.25mM ADP or 0.1mM ATP and 0.2mM diazoxide, MgCl2 were added to yield free [Mg2+] of ~1mM. For GTP stimulation, GTP was added as the sodium salt, with the corresponding pipette solution containing the same concentration of NaCl. Free [Mg2+] was ~1mM. Carbamazepine was made in DMSO and diluted in recording solution to a final DMSO concentration of 0.1%. Recording was performed at room temperature and currents were measured at a membrane potential of -50mV. Inward currents were shown as upward deflections.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the NIH grants DK057699 and DK066485 (to S.L.S.). G.M. is supported by an NIH Ruth L. Kirschstein T32 PMCB Training Grant.
Glossary
Abbreviations:
- KATP
ATP-sensitive potassium channel
- CBZ
carbamazepine
- Kir6.2
inward rectifier potassium channel 6.2
- SUR1
sulfonylurea receptor 1
- TMD0
transmembrane domain zero
- WT
wild type
References
- 1.Aguilar-Bryan L, Clement JP, 4th, Gonzalez G, Kunjilwar K, Babenko A, Bryan J. Toward understanding the assembly and structure of KATP channels. Physiol Rev. 1998;78:227–45. doi: 10.1152/physrev.1998.78.1.227. [DOI] [PubMed] [Google Scholar]
- 2.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–6. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 3.Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest. 2005;115:2047–58. doi: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O’Cochlain F, Gao F, Karger AB, Ballew JD, Hodgson DM, Zingman LV, et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004;36:382–7. doi: 10.1038/ng1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350:1838–49. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- 6.Harakalova M, van Harssel JJ, Terhal PA, van Lieshout S, Duran K, Renkens I, Amor DJ, Wilson LC, Kirk EP, Turner CL, et al. Dominant missense mutations in ABCC9 cause Cantú syndrome. Nat Genet. 2012;44:793–6. doi: 10.1038/ng.2324. [DOI] [PubMed] [Google Scholar]
- 7.Nichols CG, Singh GK, Grange DK. KATP channels and cardiovascular disease: suddenly a syndrome. Circ Res. 2013;112:1059–72. doi: 10.1161/CIRCRESAHA.112.300514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, Aguilar-Bryan L, Gagel RF, Bryan J. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science. 1995;268:426–9. doi: 10.1126/science.7716548. [DOI] [PubMed] [Google Scholar]
- 9.Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev. 1999;20:101–35. doi: 10.1210/edrv.20.2.0361. [DOI] [PubMed] [Google Scholar]
- 10.Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–83. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- 11.Zingman LV, Alekseev AE, Bienengraeber M, Hodgson D, Karger AB, Dzeja PP, Terzic A. Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron. 2001;31:233–45. doi: 10.1016/S0896-6273(01)00356-7. [DOI] [PubMed] [Google Scholar]
- 12.Babenko AP, Polak M, Cavé H, Busiah K, Czernichow P, Scharfmann R, Bryan J, Aguilar-Bryan L, Vaxillaire M, Froguel P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355:456–66. doi: 10.1056/NEJMoa055068. [DOI] [PubMed] [Google Scholar]
- 13.Koster JC, Remedi MS, Dao C, Nichols CG. ATP and sulfonylurea sensitivity of mutant ATP-sensitive K+ channels in neonatal diabetes: implications for pharmacogenomic therapy. Diabetes. 2005;54:2645–54. doi: 10.2337/diabetes.54.9.2645. [DOI] [PubMed] [Google Scholar]
- 14.Zhou Q, Garin I, Castaño L, Argente J, Muñoz-Calvo MT, Perez de Nanclares G, Shyng SL. Neonatal diabetes caused by mutations in sulfonylurea receptor 1: interplay between expression and Mg-nucleotide gating defects of ATP-sensitive potassium channels. J Clin Endocrinol Metab. 2010;95:E473–8. doi: 10.1210/jc.2010-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP, 4th, Gonzalez G, Aguilar-Bryan L, Permutt MA, Bryan J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–7. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- 16.Shyng SL, Ferrigni T, Shepard JB, Nestorowicz A, Glaser B, Permutt MA, Nichols CG. Functional analyses of novel mutations in the sulfonylurea receptor 1 associated with persistent hyperinsulinemic hypoglycemia of infancy. Diabetes. 1998;47:1145–51. doi: 10.2337/diabetes.47.7.1145. [DOI] [PubMed] [Google Scholar]
- 17.Martin GM, Chen PC, Devaraneni P, Shyng SL. Pharmacological rescue of trafficking-impaired ATP-sensitive potassium channels. Frontiers in physiology. 2013;4:386. doi: 10.3389/fphys.2013.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gribble FM, Tucker SJ, Ashcroft FM. The interaction of nucleotides with the tolbutamide block of cloned ATP-sensitive K+ channel currents expressed in Xenopus oocytes: a reinterpretation. J Physiol. 1997;504:35–45. doi: 10.1111/j.1469-7793.1997.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan F, Lin CW, Weisiger E, Cartier EA, Taschenberger G, Shyng SL. Sulfonylureas correct trafficking defects of ATP-sensitive potassium channels caused by mutations in the sulfonylurea receptor. J Biol Chem. 2004;279:11096–105. doi: 10.1074/jbc.M312810200. [DOI] [PubMed] [Google Scholar]
- 20.Yan FF, Lin YW, MacMullen C, Ganguly A, Stanley CA, Shyng SL. Congenital hyperinsulinism associated ABCC8 mutations that cause defective trafficking of ATP-sensitive K+ channels: identification and rescue. Diabetes. 2007;56:2339–48. doi: 10.2337/db07-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen PC, Olson EM, Zhou Q, Kryukova Y, Sampson HM, Thomas DY, Shyng SL. Carbamazepine as a novel small molecule corrector of trafficking-impaired ATP-sensitive potassium channels identified in congenital hyperinsulinism. J Biol Chem. 2013;288:20942–54. doi: 10.1074/jbc.M113.470948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapp S, Tucker SJ, Ashcroft FM. Activation and inhibition of K-ATP currents by guanine nucleotides is mediated by different channel subunits. Proc Natl Acad Sci U S A. 1997;94:8872–7. doi: 10.1073/pnas.94.16.8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ambrósio AF, Soares-Da-Silva P, Carvalho CM, Carvalho AP. Mechanisms of action of carbamazepine and its derivatives, oxcarbazepine, BIA 2-093, and BIA 2-024. Neurochem Res. 2002;27:121–30. doi: 10.1023/A:1014814924965. [DOI] [PubMed] [Google Scholar]
- 24.Ambrósio AF, Silva AP, Malva JO, Soares-da-Silva P, Carvalho AP, Carvalho CM. Carbamazepine inhibits L-type Ca2+ channels in cultured rat hippocampal neurons stimulated with glutamate receptor agonists. Neuropharmacology. 1999;38:1349–59. doi: 10.1016/S0028-3908(99)00058-1. [DOI] [PubMed] [Google Scholar]
- 25.Granger P, Biton B, Faure C, Vige X, Depoortere H, Graham D, Langer SZ, Scatton B, Avenet P. Modulation of the gamma-aminobutyric acid type A receptor by the antiepileptic drugs carbamazepine and phenytoin. Mol Pharmacol. 1995;47:1189–96. [PubMed] [Google Scholar]
- 26.Bienengraeber M, Alekseev AE, Abraham MR, Carrasco AJ, Moreau C, Vivaudou M, Dzeja PP, Terzic A. ATPase activity of the sulfonylurea receptor: a catalytic function for the KATP channel complex. FASEB J. 2000;14:1943–52. doi: 10.1096/fj.00-0027com. [DOI] [PubMed] [Google Scholar]
- 27.Ortiz D, Gossack L, Quast U, Bryan J. Reinterpreting the action of ATP analogs on K(ATP) channels. J Biol Chem. 2013;288:18894–902. doi: 10.1074/jbc.M113.476887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ortiz D, Voyvodic P, Gossack L, Quast U, Bryan J. Two neonatal diabetes mutations on transmembrane helix 15 of SUR1 increase affinity for ATP and ADP at nucleotide binding domain 2. J Biol Chem. 2012;287:17985–95. doi: 10.1074/jbc.M112.349019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Wet H, Proks P, Lafond M, Aittoniemi J, Sansom MS, Flanagan SE, Pearson ER, Hattersley AT, Ashcroft FM. A mutation (R826W) in nucleotide-binding domain 1 of ABCC8 reduces ATPase activity and causes transient neonatal diabetes. EMBO Rep. 2008;9:648–54. doi: 10.1038/embor.2008.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Wet H, Rees MG, Shimomura K, Aittoniemi J, Patch AM, Flanagan SE, Ellard S, Hattersley AT, Sansom MS, Ashcroft FM. Increased ATPase activity produced by mutations at arginine-1380 in nucleotide-binding domain 2 of ABCC8 causes neonatal diabetes. Proc Natl Acad Sci U S A. 2007;104:18988–92. doi: 10.1073/pnas.0707428104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mlynarski W, Tarasov AI, Gach A, Girard CA, Pietrzak I, Zubcevic L, Kusmierek J, Klupa T, Malecki MT, Ashcroft FM. Sulfonylurea improves CNS function in a case of intermediate DEND syndrome caused by a mutation in KCNJ11. Nat Clin Pract Neurol. 2007;3:640–5. doi: 10.1038/ncpneuro0640. [DOI] [PubMed] [Google Scholar]