

Abstract
Recognition of DNA by proteins relies on direct interactions with specific DNA-functional groups, along with indirect effects that reflect variable energetics in the response of DNA sequences to twisting and bending distortions induced by proteins. Predicting indirect readout requires knowledge of the variations in DNA curvature and flexibility in the affected region, which we have determined for a series of DNA-binding sites for the E2 regulatory protein by using the cyclization kinetics method. We examined 16 sites containing different noncontacted spacer sequences, which vary by more than three orders of magnitude in binding affinity. For 15 of these sites, the variation in affinity was predicted within a factor of 3, by using experimental curvature and flexibility values and a statistical mechanical theory. The sole exception was traced to differential magnesium ion binding.
Because many proteins deform DNA upon binding (1, 2), it is reasonable to expect that protein–DNA association might be facilitated by enhanced ease of DNA deformation and a match between intrinsic DNA shape in solution and the strained DNA conformation in the complex. The lack of a simple correspondence code between amino acids and DNA bases in hydrogen bonding, or direct readout, makes a general prediction of protein–DNA affinity impossible at this time. However, variations in contributions from indirect readout due to sequence-dependent shape and mechanical properties may be predictable, yielding a partial structural code for protein–DNA interaction (3). Many attempts to test this idea have been performed during the last two decades with different proteins, such as nucleosome (4–6), cAMP-binding protein (7), 434 repressor (8, 9), TATA box-binding protein (10), and E2 protein (11).
Among the impediments to realization of this objective is the difficulty of accurately determining the multiple DNA parameters involved, including magnitude and direction of curvature, helical twist, and bending and torsional flexibilities, for a variety of sequences corresponding to the region of indirect readout. The DNA cyclization method (1, 12, 13), in the high-throughput format we recently described (14), coupled with a statistical mechanical theory (15) for extracting the curvature and flexibility parameters from the data, provides a solution to this problem. Although the extent of variation of DNA flexibility with sequence remains controversial, recent results from the cyclization kinetics method show that a DNA sequence with high histone affinity (TATAAACGCC) has a nearly 2-fold smaller bending force constant and 35% less torsional rigidity than generic DNA (6). Also, an AT repeating sequence has 28% lower bending rigidity (14).
Experimental testing of the accuracy of prediction of indirect readout requires that the nucleotides involved not be also engaged in direct interactions. A system that meets this requirement is the noncontacted spacer region in the DNA-binding site for the E2 protein encoded by the human papillomavirus (HPV) type 16 genome (11). E2 proteins bind at multiple sites on the viral genome, regulate all papillomavirus genes, and are involved in viral DNA replication. The E2-binding sites of all viral strains incorporate a highly conserved dodecameric sequence of the general form ACCGNNNNCGGT, where N4 (referred to as the “spacer”) is variable. Their ordered binding by E2 is thought to be crucial for viral activities. The DNA-binding domains (DBDs) of the E2 proteins from different papillomavirus strains are highly conserved. Comparison between crystal structures of the HPV-18 E2 DBD bound by high- and low-affinity DNA sequences shows that protein configurations, DNA trajectories, and direct protein–DNA hydrogen bonds are nearly the same, independent of the spacer sequences (16). The DNA molecules are bent by 45°, with an overall bending direction toward the minor groove of the spacer. There are no direct contacts between the protein and the spacer sequences. However, variations in spacer-dependent DNA-binding affinity for E2 proteins from different papillomavirus strains do not exclusively result from different DNA global structures and mechanical properties. It has been postulated that depending on which E2 proteins were used, the spacer sequence may be discriminated to variable extent by the distribution of positive charges on the DNA-interaction surface of the proteins due to long-range electrostatic interactions (11). The DBD of HPV-16 E2 was adopted for our study because the free protein crystal structure shows that this E2 protein has the minimal positive charges adjacent to the inferred spacer-sequence-binding site, which leads to a stronger dependence of the binding affinity on the spacer sequence compared with its homologues (17), thus fulfilling the requirement of maximal decoupling of indirect from direct readout.
By using the cyclization method, we have determined the variation in DNA curvature and flexibility for a series of 4-nt spacer sequences in the E2-binding site context. For completeness, we measured the DNA-binding affinity of the variablespacer sequence set under the buffer conditions used for cyclization. Finally, we extended our recent theory of DNA cyclization (15) to replace circle formation constraints by constraints that place each end of the binding sequence at a specific position on the protein, enabling calculation of the relative binding affinity as a function of the structure and mechanical properties of the spacer sequence.
Materials and Methods
DNA Cyclization and Data Analysis. Detailed protocols were published elsewhere (14, 15). The same library of top template strands was used to make constructs by PCR and bottom template strands containing different test sequences were synthesized. The test sequences are three repeats of cgnnnncggt, where nnnn represents the 4-bp spacer sequence.
To measure E2-induced DNA bending and flexibilities, the test sequence contains a single E2-binding site, i.e., AAGTCGCAaccgnnnncggtTGCAAGCTAG, where the binding site is shown in lowercase bold with the spacer either aaac or agct. Recombinant HPV-16 E2 DBD (18) was added to ≈10 nM for AAAC constructs and 20 nM for AGCT constructs, and incubated with DNA in a cuvette for >1 h at 21°C before adding ligase dilution in the same volume (70 μl) to initiate cyclization. A total of 10 nM E2-DBD can only achieve partial occupancy of the AGCT-binding site in its unconstrained state. However, due to the thermodynamic linkage between protein binding and DNA circularization (19, 20), further increase in E2-DBD concentration does not increase the cyclization rate.
Gel-Mobility Shift Assay. Typical experimental data are shown in Fig. 4, which is published as supporting information on the PNAS web site. Consistent with the environment in which DNA global structure and mechanical properties were measured, the same cyclization buffer was used to detect E2-DBD–DNA affinities. As in ref. 17, the E2-binding site is incorporated into the 28-bp duplex region of a DNA hairpin, whose complete nucleotide sequence is GCTTGCAaccgnnnncggtTGCGACTTGCCCCCCAAGTCGCAaccgnnnncggtTGCAAGC, with the E2 site shown in lowercase bold. Chemically synthesized oligos were desalted, 5′-end labeled with [γ-33P]ATP, purified, and scintillation counted. DNA corresponding to 2,000 cpm, with final concentration estimated to be 2–10 pM, was mixed with a series of fresh E2-DBD dilutions and incubated at 21°C for >5 h. The final E2 concentration ranges from 0.01 to 320 nM in a total 40-μl mixture. A 10-μl aliquot was mixed with ≈3 μl of loading buffer, and then immediately loaded onto a 10% gel. DNA bands were quantified by using a PhosphorImager. The ACGT site was used as a control to calculate the relative binding constants for the different binding sites.
Results and Discussion
The Model. Fig. 1 shows the model system that was used to investigate the role of DNA global structure and flexibility. The E2 DBD structure is simplified as a scaffold to provide constraints on DNA only at the ends, where all of the direct protein–DNA interactions occur with an association constant Khalf for each end. With these approximations, the protein–DNA complex can be viewed as a protein-mediated mini-DNA looping system, for which a statistical mechanical model was recently constructed to evaluate the yield of loop formation (Y.Z. and D.M.C., unpublished work). Accordingly, the overall association constant can be expressed as
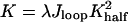 |
[1] |
where λ is a parameter that describes the cooperative binding between the two half-binding sites (ACCG), and Jloop is related to the J factor involved in DNA cyclization, but with an extended definition here. J was originally described by Jacobson and Stockmayer (21) as the effective concentration of one end of polymer chain at another to characterize the size-dependent circularization due to chain entropy effects. Here, we have extended its definition to non-zero end-to-end distance: Given one end of a DNA molecule at the origin, the Jloop factor is the effective concentration of another end at a specified location and orientation of the last base pair. It is proportional to the probability density of the end-constrained DNA configuration within unconstrained configuration space, and completely describes the accompanying DNA elastic energy and entropy changes upon its configurational transition from the free state in solution to the constrained state.
Fig. 1.

Illustration of the E2 protein-mediated mini-DNA looping system. (A) The conserved binding sites for all E2 proteins, where the 4-bp spacer sequence (N4) can be any base. (B) Crystal structure of HPV-18 E2 DBD complexed with CAACCGAATTCGGTTG (16). E2 DBD is a homodimer, with its two monomers colored red and blue, respectively. Each monomer inserts an α-helix in the major groove of ACCG, holding the DNA in an arc and leaving the spacer AATT uncontacted by the E2 protein. (C) Calculated equilibrium DNA axes in the free states in solution and in the constrained states in E2–DNA complexes. The scaffold conformation is derived from measured global DNA curvature and twist-induced by HPV-16 E2 DBD binding. All of the DNA-binding sites are aligned by global translation and rotation to overlap their base pairs at one end and then deformed with minimum energy such that the base pairs at the other end coincide with that of the scaffold. The strained state is modeled with six constraints at this end, which is similar to the case in DNA cyclization, with its corresponding J factor calculated. The bending angles shown here are amplified by 1.5-fold for better clarity.
The Jloop factor can be accurately calculated from a statistical mechanical approach we recently developed for constrained DNA molecules when their curvature, helical repeat, and flexibilities are known (15). In this method, the mechanical equilibrium configuration of the end-constrained DNA molecule is first calculated and its Jloop factor is then evaluated by integrating the small configurational fluctuations around it. Although not formulated with a J factor, Eq. 1 was derived by Crothers and Metzger (22) to investigate the thermodynamic linkage between monomeric antibody-binding strengths and the overall association constants of multivalent antibodies, and was recently revisited for study of protein–DNA interactions for proteins with two domains connected by flexible linkers (23). Our approach introduces flexibility by means of the more accurate worm-like chain model. It is worth emphasizing that enhanced DNA flexibility does not always guarantee higher affinity, because flexible chains have a greater entropy loss due to the constraints imposed by complex formation. The theory used here takes into account both bending energy and chain entropy effects.
Our strategy is to detect DNA global structures and mechanical properties in solution for a variety of E2-binding sites with the high-throughput DNA cyclization approach, which measures J factors for cyclized DNA molecules (13). The test sequences contain three repeats of the spacer sequence (for heightened sensitivity), but because the binding site spans 12 bp, maintaining a 10-bp spacing requires overlap of the consensus sequences. Because the repeat used was CGNNNNCGGT, the curvature and other properties determined do not correspond directly to the entire binding site. However, there is good correspondence between the overall bend angles we measured and the available crystallographic data on the free DNA targets (3, 24). The essential point in our measurement is to determine the variation of curvature and mechanical properties with spacer sequence, which is rigorously performed by our constructs.
With sequence-dependent properties in hand, the next step is to calculate the Jloop factors for the constrained state in the complex by using the derived DNA curvature and flexibility parameters, and finally compare their relative values, Jrel, with the experimental relative binding constants Krel. The binding constant for the half-site, Khalf, is estimated to be <10-5·M-1; but its precise value is not available due to significant nonspecific binding at high E2 DBD concentrations, as demonstrated by a competition assay shown in Fig. 5, which is published as supporting information on the PNAS web site, and by other investigators (25). Therefore, the comparison was performed for relative values to test the equation Krel = Jrel. Because only the 4-bp spacer sequences are varied, the binding sites are identified by their spacer sequences below.
Global DNA Structure in the Complex. Because the crystal structure of the HPV16 E2–DNA complex is not available, we measured the protein-induced global curvature and twist by DNA cyclization (19) for single copies of high- and low-affinity full-binding sites, represented by AAAC and AGCT, respectively. Results from the total length assay are shown in Fig. 2. Matches in the measured J factors for the two binding sites indicate that their global DNA structures and flexibilities are nearly identical and independent of affinity (Table 1), which is consistent with DNA bending of high- and low-affinity sites measured by circular permutation assays using full-length HPV-16 E2 protein (26) and the crystal structures of HPV-18 E2–DBD-DNA complexes (16), but in contrast with cAMP-binding protein–DNA complexes in which the magnitude of DNA curvature correlates with affinity (7). DNA twist barely changes upon E2 binding. Protein binding dramatically reduces the apparent local DNA bending and torsional flexibilities, which is consistent with the rigid scaffold simplification of E2 DBD with regard to the constraints on DNA configuration. Compared with DNA cyclization in the presence of HPV-16 E2 DBD, the J factors for the free-binding sites in the absence of E2 protein differ dramatically, showing their distinct global structures and flexibilities in the free state.
Fig. 2.

J factors for DNA constructs of variable overall length containing E2-binding site AAAC or AGCT in the presence or absence of E2 DBD. The best-fit parameters for E2 bound DNA are given in Table 1.
Table 1. Global structural parameters of E2 protein–DNA complexes.
| Spacer sequence | Bend angle,* ° | Bending flexibility,† rms, ° | Twist/bp, ° | Twist flexibility,† rms, ° |
|---|---|---|---|---|
| AAAC | —41.6 ± 0.8 | 3.41 ± 0.06 | 34.39 ± 0.12 | 3.7 ± 0.3 |
| AGCT | —42.3 ± 1.2 | 3.38 ± 0.08 | 34.36 ± 0.15 | 3.7 ± 0.5 |
The bend is by roll at the central dinucleotide step
The averages of the bending magnitudes and twists are used to construct the scaffold configuration: DNA curvature is modeled with a single roll angle of -42° at the middle dinucleotide step, zero tilt and roll for other steps, and 34.38° twist for all steps. All of the 12-bp-binding sites with different spacer sequences, each with characteristic global structure and mechanical properties, are aligned with this scaffold configuration such that the positions and orientations of their first and last base pairs are fixed, and the corresponding Jloop factors are calculated. The constructed scaffold state using parameters from DNA cyclization does not represent the actual bending profile of DNA in the complex, but resembles its global features of interest, filtering out some local irregularities that cannot be detected by cyclization.
Derived DNA Properties and Binding Affinities. Table 2 exhibits the experimental global structures and flexibilities for 16 binding sites in their free state, along with their relative binding constants, determined by the electrophoretic mobility shift assay. Here again, as for the bound state, DNA curvature is modeled by a single roll angle that approximates the global bending of the double helix. Hence, such values may be either larger or smaller than individual roll angles observed in the crystal structures, where DNA curvature is rather smooth and partitioned along several base pair steps. The solution-derived roll angles of the E2-DNA targets, in a coordinate frame at the center of the spacer (except for AAAA; see Table 2), vary from -10° for AAAA to + 2.5° for GGCC; the negative sign indicates bending into the minor groove, as in the protein-bound state. The bending flexibilities, expressed as rms fluctuations in roll and tilt angles, vary from 4.45° for GGCC to 6.40° for ATAT. Note that the bending force constant varies with the inverse square of the rms fluctuation angle, so the force constant for GGCC is ≈2-fold larger than for ATAT. The twisting flexibilities have a similar range, but as discussed below, twist flexibility has only a minor effect on the binding constant when DNA twist is changed little upon binding.
Table 2. Comparisons between the predictions of the relative binding constants and experimental measurements.
| Spacer | Roll*, ° | Twist, ° | Bending flex, ° | Twisting flex, ° | Jrel | Krel | Jrel/Krel |
|---|---|---|---|---|---|---|---|
| TAAT | —6.5 (0.4) | 34.26 (0.16) | 5.73 (0.06) | 4.8 (0.4) | 281 (41) | 340 (60) | 0.8 |
| ATAT | —4.6 (0.6) | 34.28 (0.18) | 6.40 (0.04) | 5.1 (0.4) | 292 (42) | 320 (34) | 0.9 |
| AATT | —7.5 (0.5) | 34.32 (0.14) | 5.41 (0.09) | 4.8 (0.3) | 262 (49) | 300 (52) | 0.9 |
| AATG | —6.2 (0.6) | 34.47 (0.17) | 5.61 (0.05) | 4.9 (0.4) | 223 (43) | 296 (65) | 0.8 |
| AAAC | —8.4 (0.6) | 34.52 (0.16) | 4.94 (0.04) | 4.4 (0.4) | 198 (43) | 260 (22) | 0.8 |
| AAGT | —5.6 (0.6) | 34.61 (0.18) | 5.45 (0.06) | 4.8 (0.4) | 156 (29) | 214 (47) | 0.7 |
| AAAA | —10 (0.5) | 34.85 (0.15) | 4.86 (0.05) | 4.5 (0.5) | 121 (23) | 160 (25) | 0.8 |
| TTAA | —0.2 (0.3) | 34.51 (0.13) | 5.31 (0.06) | 5.2 (0.4) | 19 (4) | 9.4 (5.3) | 2.0 |
| TATA | —3.0 (0.5) | 34.60 (0.13) | 6.02 (0.05) | 5.4 (0.5) | 132 (20) | 3.0 (1.1) | 44† |
| GATC | 0.9 (0.5) | 34.21 (0.14) | 4.71 (0.04) | 4.6 (0.5) | 2.7 (0.7) | 1.5 (0.1) | 1.8 |
| ACGT‡ | 1.7 (0.4) | 34.08 (0.12) | 4.53 (0.05) | 4.1 (0.3) | 1 | 1 | 1 |
| GGCC | 2.5 (0.5) | 34.04 (0.19) | 4.45 (0.03) | 4.2 (0.5) | 0.45 (0.12) | 0.71 (0.04) | 0.6 |
| GACC | 1.9 (0.5) | 34.42 (0.11) | 4.52 (0.05) | 3.9 (0.3) | 0.92 (0.29) | 0.7 (0.2) | 1.3 |
| AGCT | 2.2 (0.7) | 34.40 (0.21) | 4.52 (0.07) | 4.3 (0.5) | 0.72 (0.33) | 0.6 (0.3) | 1.2 |
| GTAC | 2.4 (0.5) | 34.33 (0.15) | 4.60 (0.04) | 4.7 (0.4) | 0.85 (0.26) | 0.5 (0.2) | 1.7 |
| CGCG | 2.3 (0.6) | 34.29 (0.17) | 4.51 (0.04) | 5.1 (0.5) | 0.55 (0.18) | 0.18 (0.03) | 3.1 |
| Average | —2.4 (4.5) | 34.39 (0.21) | 5.10 (0.62) | 4.7 (0.4) | 106 (115) | 119 (143) | 1.2† (0.7) |
The roll is for the middle dinucleotide step of the spacer sequence for all the binding sites, except for AAAA, for which the roll is at the step one base pair to the right. Values in parentheses signify SD of the measurement
The outlying value of 44 for Jrel/Krel for TATA differs by ≫ 10 sd from the mean and has been excluded from calculation of the average
This sequence is used as a reference for the relative J factor and binding constant. Its absolute dissociation constant is 12 nM
Binding affinities expressed relative to the reference sequence ACGT vary by more than three orders of magnitude, from 0.18 to 340 (Table 2). There is a clear division between sequences with relative affinity above 100 (AAAA and above) and those with relative affinity below 10 (TTAA and below). Taking into account the two-fold symmetry of the E2 protein (which allows the spacer sequence to be read in either direction), nearly any base can appear in each of four positions of the spacer to achieve both high and low affinities, in agreement with the lack of direct base recognition in the spacer. Change of the spacer base pairs causes significant variation in the global DNA structure and flexibilities. In general, AT-rich sequences bend toward the minor groove and/or have higher bending and twisting flexibilities. However, there is no obvious correlation between bending magnitude and flexibility: the ATAT sequence has highest bending flexibility (6.4°) and moderate curvature (-4.6°), whereas TTAA is straight (-0.2°), but very flexible (5.3°). The TA dinucleotide step has been generally found to be more flexible than other steps (27, 28). However, it is found here that these steps can appear in both high- and low-affinity E2-binding sites (ATAT vs. GTAC), suggesting a context-dependent effect. These observations are in accord with the high conformational variability of alternating ATAT sequences observed in crystal structures (29, 30) on one hand, and the structural features of GTAC tetranucleotides (e.g., local major-groove bending) on the other (3). AAAA is relatively rigid and exhibits the maximum bend angle of 10°, which is consistent with previous results (31). Its lowest affinity among the high-affinity class of binding sites may be related to the nonsymmetrical conformation of this target, where the bending direction is one step away from the center, unlike all of the other binding sites. Such an arrangement may not be optimal for a symmetric protein dimer, and the DNA duplex would have to writhe to bind E2, leading to free-energy loss relative to the symmetrically bent DNA targets. In contrast to AT-rich sequences, GC-rich sequences curve slightly into the major groove, and are more rigid. Similar bending characteristics were observed in crystal structures of free E2 DNA targets with spacers ACGT (3), GTAC (3) AATT (24), and AAAA (H. Rozenberg, M. Kitayner, and Z.S., unpublished work).
Comparison Between Experiment and Theory. The apparently complex dependence of binding affinity on DNA properties is accurately predicted by the theory. Table 2 shows Jrel and Krel values, which give the Jloop and K values in terms of their ratio to the value for the reference ACGT sequence. Because K is proportional to Jloop, the ratio Jrel/Krel should be unity if the prediction is accurate. Even though the binding affinities span three orders of magnitude, the calculated Jrel/Krel ratio is indeed close to unity; the average (excluding the outlying TATA sequence) is 1.2 ± 0.7. As is evident from Table 2, the one striking exception is the TATA sequence, whose measured affinity is 44-fold lower than predicted. We found that this disparity is much reduced at Mg2+ concentrations <10 mM, and nearly eliminated in Mg2+-free buffers. Preliminary experiments show that the TATA sequence releases approximately twice as many Mg2+ ions as the average of the other sequences upon E2 binding (Y.Z., Z.X., and D.M.C., unpublished work). Also, the anomaly of this sequence may be linked to its propensity to adopt A-DNA-like conformations (i.e., bending into the major groove) as a result of the two T-A hinges (32).
The contrasting sensitivity of the affinity to the multiple parameters is further confirmed by the calculated dependence of the J factor on these parameters, shown in Fig. 3. The J factor dramatically changes with DNA curvature and bending flexibility. In contrast, the J factor changes are <2-fold when DNA twist and torsional flexibility vary in their accessible regions. In a case such as the E2–DNA complex, in which there is virtually no twist change upon binding, there is nothing to be gained by an increase in the torsional flexibility, but there is entropy to be lost because the increased twisting fluctuations are quenched in the complex. For example, an increase of the torsional flexibility of ATAT from 4.0° to 5.1° decreases its calculated relative J factor from 372 to 292. On the other hand, in a case such as 434 repressor, in which the protein twists the DNA upon binding, the J factor and binding constant should show high sensitivity to DNA torsional flexibility (9).
Fig. 3.

Variation of the J factor with each parameter for global DNA structure and mechanical properties. Unless indicated as variable, the default parameters used for calculations shown here are 0° kink, 34.45° twist, 4.678° bending flexibility, and 4.388° twisting flexibility, respectively. The kink is a roll at the middle of the binding site. Note the difference in scale for the J factor between the two images.
Summary and Discussion. In general, the relationships between DNA sequence, global DNA structure and mechanical properties, and DNA-protein affinity studied here, indicate that AT-rich sequences tend to bind protein better than GC-rich sequences when bending toward the minor groove is required, as seen for nucleosomes (4–6), cAMP-binding protein (7), and 434 repressor (8). The high-affinity sequences all bend toward the minor groove and simultaneously have high bendability compared with the low-affinity sites, which are straight, bent into the major groove, or rigid. Hence, DNA curvature and bending flexibility are the principle components determining, and equally contributing to, the affinity changes. Both the magnitude and direction of curvature are important. In contrast to DNA bending and bendability, DNA twist and torsional flexibility only slightly modulate the affinities.
The spacer sequences of the natural DNA-binding sites of HPV-16 (AATT, TTTT, AAAT, and AAAC) are AT-rich of the kind showing considerable intrinsic bending into the minor groove (Table 2). The AATT sequence exhibits the highest affinity in this series (26) in agreement with the current study, most likely due to its symmetric configuration and enhanced bendability (Table 2). The dramatically lower affinity (by three orders of magnitude) shown by the wild-type TTTT target (ACCGTTTTGGGT) is mainly a consequence of substitution of a conserved cytosine by a guanine base at the 3′ end of the dodecameric target (26). The biological consequence (transcriptional activation/repression or viral DNA replication) of E2 binding to each of the four binding sites on the genome is different. Thus, both high- and low-affinity binding sites are critical to the life cycle of the pathogenic virus.
We show that differential binding affinity in the HPV-16 E2 system is determined by indirect readout and that these effects can be accurately predicted by theory. Such studies provide insights into structure–function relationships in gene regulation.
Acknowledgments
Y.Z. thanks Jing Wang for technical assistance. This work was supported by National Institutes of Health Grants GM 21966 (to D.M.C.) and CA66964 and GM63582 (to R.S.H.); the National Foundation for Cancer Research in support of the National Foundation for Cancer Research–Yale Center for Protein and Nucleic Acid Chemistry; and the Israel Science Foundation (to Z.S.).
Abbreviations: HPV, human papillomavirus; DBD, DNA-binding domain.
References
- 1.Bloomfield, V. A., Crothers, D. M. & Tinoco, I. (1999) Nucleic Acids: Structures, Properties, and Functions (University Science Books, Sausalito, CA).
- 2.Dickerson, R. E. (1998) Nucleic Acids Res. 26, 1906-1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rozenberg, H., Rabinovich, D., Frolow, F., Hegde, R. S. & Shakked, Z. (1998) Proc. Natl. Acad. Sci. USA 95, 15194-15199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drew, H. R. & Travers, A. A. (1985) J. Mol. Biol. 186, 773-790. [DOI] [PubMed] [Google Scholar]
- 5.Shrader, T. E. & Crothers, D. M. (1989) Proc. Natl. Acad. Sci. USA 86, 7418-7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roychoudhury, M., Sitlani, A., Lapham, J. & Crothers, D. M. (2000) Proc. Natl. Acad. Sci. USA 97, 13608-13613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gartenberg, M. R. & Crothers, D. M. (1988) Nature 333, 824-829. [DOI] [PubMed] [Google Scholar]
- 8.Koudelka, G. B., Harrison, S. C. & Ptashne, M. (1987) Nature 326, 886-888. [DOI] [PubMed] [Google Scholar]
- 9.Koudelka, G. B. & Carlson, P. (1992) Nature 355, 89-91. [DOI] [PubMed] [Google Scholar]
- 10.Bareket-Samish, A., Cohen, I. & Haran, T. E. (2000) J. Mol. Biol. 299, 965-977. [DOI] [PubMed] [Google Scholar]
- 11.Hegde, R. S. (2002) Annu. Rev. Biophys. Biomol. Struct. 31, 343-360. [DOI] [PubMed] [Google Scholar]
- 12.Shore, D., Langowski, J. & Baldwin, R. L. (1981) Proc. Natl. Acad. Sci. USA 78, 4833-4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crothers, D. M., Drak, J., Kahn, J. D. & Levene, S. D. (1992) Methods Enzymol. 212, 3-29. [DOI] [PubMed] [Google Scholar]
- 14.Zhang, Y. L. & Crothers, D. M. (2003) Proc. Natl. Acad. Sci. USA 100, 3161-3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang, Y. L. & Crothers, D. M. (2003) Biophys. J. 84, 136-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim, S. S., Tam, J. K., Wang, A. F. & Hegde, R. S. (2000) J. Biol. Chem. 275, 31245-31254. [DOI] [PubMed] [Google Scholar]
- 17.Hines, C. S., Meghoo, C., Shetty, S., Biburger, M., Brenowitz, M. & Hegde, R. S. (1998) J. Mol. Biol. 276, 809-818. [DOI] [PubMed] [Google Scholar]
- 18.Hegde, R. S. & Androphy, J. (1998) J. Mol. Biol. 284, 1479-1489. [DOI] [PubMed] [Google Scholar]
- 19.Kahn, J. D. & Crothers, D. M. (1992) Proc. Natl. Acad. Sci. USA 89, 6343-6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parvin, J. D., McCormick, R. J., Sharp, P. A. & Fisher, D. E. (1995) Nature 373, 724-727. [DOI] [PubMed] [Google Scholar]
- 21.Jacobson, H. & Stockmayer, W. H. (1950) J. Chem. Phys. 18, 1600-1606. [Google Scholar]
- 22.Crothers, D. M. & Metzger, H. (1972) Immunochemistry 9, 341-357. [DOI] [PubMed] [Google Scholar]
- 23.Zhou, H. X. (2001) Biochemistry 40, 15069-15073. [DOI] [PubMed] [Google Scholar]
- 24.Hizver, J., Rozenberg, H., Frolow, F., Rabinovich, D. & Shakked, Z. (2001) Proc. Natl. Acad. Sci. USA 98, 8490-8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferreiro, D. U. & de Prat-Gay, G. (2003) J. Mol. Biol. 331, 89-99. [DOI] [PubMed] [Google Scholar]
- 26.Thain, A., Webster, K., Emery, D., Clarke, A. R. & Gaston, K. (1997) J. Biol. Chem. 272, 8236-8242. [DOI] [PubMed] [Google Scholar]
- 27.Olson, W. K., Gorin, A. A., Lu, X. J., Hock, L. M. & Zhurkin, V. B. (1998) Proc. Natl. Acad. Sci. USA 95, 11163-11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mack, D. R., Chiu, T. K. & Dickerson, R. E. (2001) J. Mol. Biol. 312, 1037-1049. [DOI] [PubMed] [Google Scholar]
- 29.Yuan, H., Quintana, J. & Dickerson, R. E. (1992) Biochemistry 31, 8009-8021. [PubMed] [Google Scholar]
- 30.Shatzky-Schwartz, M., Arbuckle, N. D., Eisenstein, M., Rabinovich, D., Bareket-Samish, A., Haran, T. E., Luisi, B. F. & Shakked, Z. (1997) J. Mol. Biol. 267, 595-623. [DOI] [PubMed] [Google Scholar]
- 31.Barbic, A., Zimmer, D. P. & Crothers, D. M. (2003) Proc. Natl. Acad. Sci. USA 100, 2369-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guzikevich-Guerstein G. & Shakked Z. (1996) Nat. Struct. Biol. 3, 32-37. [DOI] [PubMed] [Google Scholar]