

Abstract
The second messenger pathways linking receptor activation at the membrane to changes in the nucleus are just beginning to be unraveled in neurons. The work presented here attempts to identify in striatal neurons the pathways that mediate cAMP response element–binding protein (CREB) phosphorylation and gene expression in response to NMDA receptor activation. We investigated the phosphorylation of the transcription factor CREB, the expression of the immediate early genec-fos, and the induction of a transfected reporter gene under the transcriptional control of CREB after stimulation of ionotropic glutamate receptors. We found that neither AMPA/kainate receptors nor NMDA receptors were able to stimulate independently a second messenger pathway that led to CREB phosphorylation orc-fos gene expression. Instead, we saw a consecutive pathway from AMPA/kainate receptors to NMDA receptors and from NMDA receptors to L-type Ca2+ channels. AMPA/kainate receptors were involved in relieving the Mg2+ block of NMDA receptors, and NMDA receptors triggered the opening of L-type Ca2+ channels. The second messenger pathway that activates CREB phosphorylation and c-fos gene expression is likely activated by Ca2+ entry through L-type Ca2+ channels. We conclude that in primary striatal neurons glutamate-mediated signal transduction is dependent on functional L-type Ca2+ channels.
Keywords: glutamate, NMDA, AMPA, kainate, L-type Ca2+ channels, CREB, c-fos
The striatum (caudate, putamen, and nucleus accumbens) has been implicated in movement disorders like Parkinson’s disease and Huntington’s disease (Vonsattel et al., 1985;DiFiglia, 1990; Langston, 1996). The striatum also plays a critical role in drug addiction (Pich et al., 1997; Volkow et al., 1997), schizophrenia (Buchsbaum et al., 1992; Siegel et al., 1993; Busatto and Kerwin, 1997; Heckers, 1997), and memory (Knowlton et al., 1996). Neuroplasticity of the striatum enables compensation for the loss of dopamine (Bernheimer et al., 1973; Neve et al., 1982; Calne et al., 1985; Burns, 1991; Hornykiewicz, 1993) and contributes to both the therapeutic potential of neuroleptics (Burt et al., 1977; Eastwood et al., 1994, 1997; Mijnster et al., 1996) and the addictive properties of drugs of abuse (Nestler et al., 1993; Self and Nestler, 1995; Hyman, 1996; Hyman and Nestler, 1996). Synaptic plasticity forms the basis of learning and memory and involves mechanisms such as protein phosphorylation and protein synthesis, leading to the strengthening of preexisting synapses and the establishment of new synaptic connections (Stanton and Sarvey, 1984; Deadwyler et al., 1987; Matthies, 1989;Nairn and Shenolikar, 1992; Schulman, 1995; Bailey et al., 1996). The examination of the mechanisms of gene regulation in the striatum is vital for our understanding of striatal plasticity, striatal function, and malfunction and for the treatment of disorders of the striatum.
The excitatory amino acid l-glutamate (glutamate) participates in processes from neuronal communication to plasticity and neuropathology via its interaction with ionotropic and metabotropic receptors (Greenamyre and Porter, 1994; Michaelis, 1998). Ionotropic glutamate receptors are classified into AMPA, kainate, and NMDA receptors (Hollmann and Heinemann, 1994; Schoepfer et al., 1994; Michaelis, 1998; Ozawa et al., 1998). AMPA/kainate receptors gate ion channels that promote Na+ influx and, to a lesser extent, Ca2+ influx (Hollmann et al., 1991;Egebjerg and Heinemann, 1993; Kohler et al., 1993; Lomeli et al., 1994) and desensitize rapidly (Lomeli et al., 1994; Mosbacher et al., 1994).
NMDA-type glutamate receptor channels bind glycine and glutamate (Ascher and Nowak, 1987; Johnson and Ascher, 1987; Hollmann and Heinemann, 1994) and are permeable to Ca2+(MacDermott et al., 1986; Connor et al., 1988) as well as Na+ and K+ ions (Ascher and Nowak, 1987; Kandel et al., 1991). At resting potential, NMDA receptors are blocked by Mg2+ (Mayer et al., 1984; MacDermott et al., 1986; Ruppersberg et al., 1993; Schoepfer et al., 1994). This Mg2+ block can be relieved by depolarization, which can be achieved by activation of AMPA/kainate receptors.
Another route for Ca2+ entry into striatal neurons is provided by voltage-operated Ca2+ channels (VOCCs) (Sanna et al., 1986; Thayer et al., 1986). The α1 subunit of VOCCs determines the properties of the Ca2+ channel (Hofmann et al., 1994; McCleskey, 1994; Walker and De Waard, 1998) and forms the pore through which Ca2+ ions enter the cell (Catterall, 1991; Walker and De Waard, 1998). Ca2+ channels that contain the α1C or α1D subunit are classified as dihydropyridine-sensitive L-type Ca2+ channels (Hofmann et al., 1994; McCleskey, 1994; Walker and De Waard, 1998). The L-type Ca2+channel is activated by strong depolarization from relatively depolarized holding potentials and shows almost no inactivation by depolarization (Tsien et al., 1988). Therefore, it opens only after a strong stimulus and, after opened, causes extensive Ca2+ influx (Catterall and Striessnig, 1992). These characteristics and the distribution of L-type Ca2+channels on somata and at the base of dendrites (Westenbroek et al., 1990; Schild et al., 1995) provide a favorable setting to mediate gene regulation in the nucleus.
Ca2+-stimulated second messenger pathways can activate the transcription factor cAMP response element–binding protein (CREB) (Sheng et al., 1991; Sun et al., 1994; Thompson et al., 1995; Bito et al., 1996; Deisseroth and Tsien, 1997). CREB constitutively binds with high affinity to the cAMP-responsive element (CRE) and becomes a transcriptional activator after phosphorylation of Ser133 (Montminy and Bilezikjian, 1987; Gonzalez and Montminy, 1989; Montminy et al., 1990). CREB has been linked to memory formation (Bourtchuladze et al., 1994; Yin et al., 1994), neuroplasticity (Murphy and Segal, 1997), and long-term potentiation (Impey et al., 1996). The immediate early genec-fos is activated by CREB (Sheng et al., 1990). The promoter of the c-fos gene contains the cAMP and Ca2+–responsive element (CaRE), which interacts with CREB (Sheng et al., 1990; Ghosh et al., 1994). The CaRE site integrates several second messenger pathways (Bonni et al., 1995; Ahn et al., 1998) and is one of the preeminent regulatory sites of thec-fos promoter (Robertson et al., 1995). Like CREB phosphorylation, c-fos is induced after NMDA receptor stimulation (Cole et al., 1989; Aronin et al., 1991; Lerea and McNamara, 1993; Dave and Tortella, 1994) and after L-type Ca2+ channel activation (Murphy et al., 1991; Misra et al., 1994).
We show here that in primary striatal cultures, glutamate via activation of NMDA receptors mediates CREB phosphorylation and gene expression via L-type Ca2+ channels.
MATERIALS AND METHODS
Drugs. NMDA, (±)AMPA hydrobromide, kainate (kainic acid), dizocilpine maleate [(+)MK 801 hydrogen maleate], (±)2-amino5-phosphonopentanoic acid (APV), DNQX, 2,5-dimethyl-4-[2-(phenylmethyl)benzoyl]-1H-pyrrole-3-carboxylic acid methylester (FPL 64176), 1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-3,4-dihydro-5H-2,3-benzodiazepine (GYKI 52466) hydrochloride, tetrodotoxin citrate (TTX), (±)verapamil hydrochloride, nifedipine, bicuculline, and picrotoxin were purchased from Research Biochemicals (Natick, MA), and l-glutamate was purchased from Sigma (St. Louis, MO). The Ser133CREB antiserum (Ginty et al., 1993), the CREB antiserum, and the Fos antiserum were purchased from Upstate Biotechnology (Lake Placid, NY). The antiserum against the α1C Ca2+ channel was purchased from Alomone Labs (Jerusalem, Israel).
Primary striatal cultures. Primary striatal cultures were prepared as described previously, with minor modifications (Konradi et al., 1996; Rajadhyaksha et al., 1998). Striata were dissected under a stereomicroscope from 18-d-old Sprague Dawley rat fetuses. Tissue was resuspended in 2 ml of defined medium [50% F12/DMEM and 50% DMEM (Life Technologies, Gaithersburg, MD) with the following supplements per liter of medium: 4 gm of dextrose, 1× B27, 10 ml of penicillin–streptomycin liquid (Life Technologies), and 25 mm HEPES]. The tissue was mechanically dissociated with a fire-narrowed Pasteur pipette; the cells were resuspended in defined medium to 106 cells/ml and plated in six-well plates (Costar, Cambridge, MA) at 2 × 106 cells/well. Plates were pretreated with 2 ml of a 1:500-diluted sterile solution of polyethylenimine in water for 24 hr, washed twice with sterile water, coated with 2.5% serum-containing PBS solution for at least 4 hr, and aspirated just before plating. All experiments were performed with cells 6–8 d in culture and repeated at least once in an independent dissection. As determined by HPLC analysis, glutamate levels in the medium on the day of the experiments ranged from 1 to 5 μm. The neuron to astroglia ratio was below 25:1, as established by immunocytochemical staining with the glial fibrillary acid protein (Dako, Carpinteria, CA) and counterstaining with 1% cresyl violet.
Defined salt solutions. To have comparable parameters, none of the defined salt solutions contained sodium bicarbonate. Sodium bicarbonate was replaced by N-methyl-d-glucamine (45 mm). Control salt solutions contained 110 mm NaCl, 2 mm MgSO4, 1.8 mm CaCl2, 400 μm glycine, 45 mmN-methyl-d-glucamine, 0.5% phenol red, 3 gm/l dextrose, and 20 mm HEPES-KOH. In experiments without Na+, NaCl was replaced by 110 mmN-methyl-d-glucamine. In experiments without Ca2+ or without Mg2+, either ion was left out. Salt solutions were adjusted with HCl to pH 7.2. In experiments that used NMDA as an agonist, 10 μm glutamate was added 18 hr before the experiment. All salt solutions were added 18 hr before the experiment to avoid false results because of media change. Neurons were carefully monitored for neuronal death.
Immunoblots. Primary rat striatal cultures were harvested in boiling sample buffer (62.5 mm Tris-HCl, pH 6.8, 20% glycerol, 2% SDS, 5% β-mercaptoethanol, and 0.025% bromophenol blue). Cell lysates were sonicated and centrifuged for 10 min. Equal volumes of the lysates were loaded on 12% polyacrylamide gels for phospho-CREB and CREB immunoblots or on 8% gels for Fos and α1C Ca2+ channel–subtype immunoblots. Protein was transferred to polyvinylidene fluoride membrane (Immobilon-P; 0.45 mm; Millipore, Bedford, MA) and blocked in blocking buffer (5% nonfat dry milk in PBS and 0.1% Tween 20) for 1 hr. The blots were incubated in primary antibody (1:1000 anti-Ser133-phospho-CREB or anti-CREB; 1:10,000 anti-Fos; or 1:500 anti-α1C) for 2 hr followed by three washes for 10 min in blocking buffer. This was followed by a 1 hr incubation in goat anti-rabbit horseradish peroxidase–linked IgG (Vector Laboratories, Burlingame, CA) at a dilution of 1:3000. Blots were washed three times for 10 min in blocking buffer, developed with the Renaissance detection system (Dupont NEN, Wilmington, DE), and exposed to autoradiographic film (Kodak, Rochester, NY). Kaleidoscope-prestained standards (Bio-Rad, Hercules, CA) were used for protein size determination. Phospho-CREB and CREB bands were detected at the 43 kDa standard. The Fos band was detected at 60 kDa.
Calcium phosphate transfections. Transfection of primary striatal neurons was performed following the protocol of Xia et al. (1996). Embryonic day 18 striatal neurons were transfected on 4 d in vitro (DIV). The DNA/calcium phosphate precipitate was prepared by mixing the DNA in 250 mmCaCl2 with an equal volume of 2× HEPES-buffered saline (0.14 mm NaCl, 0.025 mm HEPES, and 0.7 μm Na2HPO4). The precipitate was allowed to form for 1 hr at room temperature. Fifteen minutes before addition of the DNA mixture, the conditioned culture medium was removed from the cells and replaced with 1.5 ml of F12/DMEM (Life Technologies). The conditioned media were kept under 5% CO2. The DNA mixture (100 μl) was added dropwise to each well of a six-well plate and rocked gently. Plates were incubated for 80 min in a 5% CO2 incubator. After 80 min the cells were shocked with 500 μl of 2% DMSO in F12/DMEM for 2 min and washed twice with 1.5 ml of F12/DMEM. The conditioned media were added back to the cells, and the plates were incubated in a 5% CO2incubator at 37°C. For all transfections, 6 μg of total DNA was used per well (35 mm) of a six-well plate. Forty-eight hours after transfection, cells were treated with the respective drugs for 6 hr. Media were aspirated, and plates were quick-frozen on liquid nitrogen and stored at −80°C.
3xCRE-luciferase construct. A DNA sequence containing three CRE sequences (TGACGTCA) was fused to a minimal Rous sarcoma virus promoter (enhancerless) and was inserted into the pA3Pluc vector (Maxwell et al., 1989) 5′ of a luciferase reporter gene.
Luciferase assay. The luciferase assay was performed using the Promega luciferase assay kit (Madison, WI). Cells were lysed in 150 μl of 1× cell culture lysis reagent (25 mmTris-phosphate, pH 7.8, 2 mm DTT, 2 mm1,2-diaminocyclohexane-N,N,N′,N′-tetra-acetic acid, 10% glycerol, and 1% Triton X-100); 100 μl of the lysate was used for the luciferase reporter assay. Luciferase activity was measured for a period of 10 sec using a luminometer (EG & G Berthold, Oak Ridge, TN), and light intensity was expressed as relative light units.
Antisense experiments. Antisense sequences were targeted to the translation initiation sites. Both antisense and scrambled antisense oligonucleotides were synthesized by the Massachusetts General Hospital core DNA synthesis facility. The first and last three nucleotides were phosphorothioate modified to avoid degradation of the oligonucleotides and also to reduce the cytotoxic effect observed with fully phosphorothioate-modified oligonucleotides. Striatal cultures were treated according to the method ofBito et al. (1996) with a mixture of 5 μm α1C (GGCCCGAATCATTGTGACTCCAGT) and 5 μm α1D (TCATCATCATCATCATCATCCACG) antisense oligonucleotides for L-type Ca2+ channel antisense experiments or a mixture of 5 μm NR1 (TGCTCATGAGCTCCGGGCACAGCG), 5 μm NR2A (CAATCTGCCCATGGTCGCCACTTA), and 5 μm NR2B (ACTCTGCGCTGGGCTTCATCTTCA) antisense oligonucleotides for the NMDA receptor experiment. Oligonucleotides were added 30 min after cells were plated and daily thereafter until 4 DIV. Twenty-four hours after the last addition, cells were treated with the respective agonists. For scrambled antisense DNA controls, approximately every third and fourth nucleotide of the antisense constructs were exchanged (α1C, GACCTGAGTTACTGGCATATCCGC; α1D, TAAACCTCTCCATTCTCGTACACA; NR1, TGCCTAGTAGCTCGCGGACCAGGC; NR2A, ACATCGTCATCGCGCTGATCTCAC; and NR2B, TATGCCTCGGCGTTCAGCTCCATT).
c-fos Northern blot analysis. Medium was aspirated, and striatal neurons were lysed in 500 μl of lysis buffer (50 mm Tris, pH 8.0, 100 mm NaCl, 5 mmMgCl2, and 0.5% NP-40). After a 5 min incubation on ice, lysates were transferred into microcentrifuge tubes and centrifuged for 2 min at 14,000 rpm at 4°C; the supernatant was transferred, and SDS was added to a final concentration of 0.2%. Cells were extracted with phenol, followed by a chloroform extraction and ethanol precipitation. RNA was size-separated on a 1.2% denaturing agarose gel (1 m paraformaldehyde) in 3-(N-morpholino)propanesulfonic acid (MOPS) buffer (20 mm MOPS, pH 7.0, 5 mm sodium acetate, and 1 mm EDTA), electroblotted onto a nylon membrane (GeneScreen; DuPont, Billerica, MA), and hybridized with a c-fosriboprobe (Riboprobe system; Promega). Northern blots were analyzed with a PhosphorImager (Molecular Dynamics, Sunnyvale, CA), with the IP lab imaging software. Expression of cyclophilin mRNA was used as a loading control (Danielson et al., 1988).
Statistical analyses. Autoradiographic films were scanned with the Hewlett Packard Scan Jet. Because of the narrow range of film (approximately one order of magnitude), the data obtained for immunoblots are not comparable with the data obtained forc-fos Northern blots, which were analyzed with a PhosphorImager that has a range of five orders of magnitude. Because the data are compiled of many different Northern blots and immunoblots, they had to be normalized to internal, untreated controls. Thus, the data are semiquantitative and are not based on absolute numbers. Data were analyzed with one-way ANOVAs. The Tukey–Kramer honestly significant difference was used to analyze differences between the groups, whereas the Dunnett’s test was used for comparisons of treatment groups with controls. The JMP computer program (SAS Institute, Cary, NC) was used for data analysis.
RESULTS
Glutamate induces CREB phosphorylation and Fos protein expression in primary striatal cultures
Glutamate (50 μm) induced Ser133CREB phosphorylation in rat primary striatal cultures within 5 min of treatment (Fig. 1A,P-CREB). Ser133 CREB phosphorylation peaked at 15 min and returned to basal levels 10 hr after glutamate was added. When immunoblots were reprobed with a CREB antiserum that is indiscriminate to the state of CREB phosphorylation, no treatment-mediated regulation was seen (Fig. 1A,CREB). In most blots, the CREB antiserum and the Ser133 CREB antiserum revealed a double band slightly >40 kDa, but some variability in the resolution of the double band and/or staining of the upper band was observed with either antiserum. Because the two bands had identical patterns of regulation, these variations were not a problem. A temporal analysis of Fos protein levels demonstrated an increase between 2 and 5 hr after the onset of glutamate treatment (Fig. 1A, Fos). In a different time course, cultures were exposed to glutamate for limited times (Fig. 1B), refed with medium lacking glutamate, and harvested 15 min after glutamate treatment had begun. A 3 min exposure to glutamate yielded maximum levels of CREB phosphorylation, comparable with treatment for the entire 15 min (Fig.1B; see also Fig. 6D for a time course similar to that of Fig. 1A). In all subsequent experiments agonists were present for the entire treatment period, which was 15 min in experiments that examined Ser133CREB phosphorylation.
Fig. 1.

Glutamate induces Ser133 CREB phosphorylation and Fos protein in primary striatal cultures.A, Immunoblots of rat primary striatal cultures were treated with glutamate (50 μm) for 5 min to 10 hr (times indicated above theblots). P-CREB, CREB was phosphorylated 5 min after the addition of glutamate and remained phosphorylated for at least 5 hr. CREB, An antiserum that is indifferent to the state of phosphorylation of CREB showed that the protein levels of CREB were not changed. Fos, Fos protein was induced 2–5 hr after the addition of glutamate. CREB and Ser133CREB had a molecular weight of ∼43 kDa, whereas Fos protein was slightly below the 60 kDa protein marker. B, Primary striatal cultures were exposed to glutamate (50 μm) for 1–15 min, at which time glutamate was removed from the cells. All cells were harvested 15 min after the addition of glutamate. Immunoblots were developed with Ser133 CREB antiserum (Ginty et al., 1993). All treatments are shown in duplicates.
Fig. 6.

L-type Ca2+ channel blockers inhibit ionotropic glutamate receptor–mediated CREB phosphorylation in primary striatal cultures. A, Ser133 CREB phosphorylation after treatment with NMDA (50 μm), AMPA (50 μm), kainate (50 μm), and FPL 64176 (20 μm) is blocked by the L-type Ca2+channel antagonist nifedipine (20 μm). All treatments are shown in duplicates. The experiment was repeated once. (See also Fig.10.) B, c-fos mRNA induced by glutamate (50 μm), NMDA (50 μm), AMPA (50 μm), kainate (50 μm), and FPL 64176 (20 μm) is inhibited by nifedipine (20 μm). The experiment was repeated three times. (See also Fig. 10.)C, Nifedipine (20 μm) inhibited glutamate (50 μm)- and FPL 64176 (20 μm)–mediated induction of the 3xCRE-luciferase construct (n = 6). Asterisks mark statistically significant differences between groups that are linked in the graph. Data are the average ± SEM. D, Nifedipine (20 μm) blocked glutamate (50 μm)-mediated CREB phosphorylation independent of time of exposure to glutamate. Cultures were exposed to glutamate for thetimes indicated above theblots and harvested immediately after exposure. Pretreatment with nifedipine (20 min) blocked glutamate-mediated CREB phosphorylation at all times. The experiment was repeated three times.E, The GABA receptor antagonist bicuculline (100 μm) did not prevent the inhibitory action of nifedipine (20 μm) on glutamate (50 μm)-mediated CREB phosphorylation. Preincubation with bicuculline did not increase glutamate-mediated CREB phosphorylation, nor did it affect the inhibitory action of nifedipine on glutamate-mediated CREB phosphorylation. − indicates control without agonist.
Activation of ionotropic glutamate receptors and L-type Ca2+ channels induces CREB phosphorylation and gene expression
To analyze the contribution of different ionotropic glutamate receptors to Ser133 CREB phosphorylation, we used agonists specific for each class and found that NMDA (50 μm), AMPA (50 μm), and kainate (50 μm) all induced CREB phosphorylation (Fig.2A). Higher concentrations of these agonists did not induce more CREB phosphorylation (data not shown). The medium contained Mg2+ (2 mm) as well as ambient glutamate levels (2–5 μm, as determined by HPLC measurement). The ambient glutamate levels supported the effect of NMDA on CREB phosphorylation in the presence of Mg2+ (see below). Ser133 CREB phosphorylation was also induced by the L-type Ca2+ channel agonist FPL 64176 (20 μm; Fig. 2A). The phosphorylation of Ser133 CREB coincided with c-fos gene expression in all stimulus paradigms used (Fig. 2B). A luciferase construct under the transcriptional control of three CRE enhancer elements, 3xCRE-luciferase (Fig. 2C), was transfected into primary striatal cultures and was activated by all agonists, comparable with c-fos mRNA (Fig.2B) and Ser133 CREB phosphorylation (Fig. 2A).
Fig. 2.

Activation of ionotropic glutamate receptors and of L-type Ca2+ channels induces CREB phosphorylation, c-fos gene expression, and the3xCRE-luciferase construct in primary striatal cultures.A, Cultures were exposed to glutamate (50 μm), NMDA (50 μm), AMPA (50 μm), kainate (50 μm), and FPL 64176 (FPL; 20 μm) and harvested 15 min after the addition of each drug. Immunoblots were developed with the Ser133 CREB antiserum. All drugs induced CREB phosphorylation to varying degrees. Treatments are shown in duplicates from a representative experiment that was repeated four times.B, Cultures were exposed to glutamate (50 μm), NMDA (50 μm), AMPA (50 μm), kainate (50 μm), and FPL 64176 (20 μm) and harvested 40 min after the addition of each drug. Northern blots were developed with a c-fos riboprobe. All drugs induced c-fos mRNA to varying degrees. Duplicate treatments are shown in two separate blots. The experiment was repeated twice. C, Primary striatal cultures were transfected with a 3xCRE-luciferase construct and treated with glutamate (50 μm), NMDA (50 μm), AMPA (50 μm), kainate (50 μm), and FPL 64176 (20 μm) for 6 hr. Cells were harvested, and luciferase activity was measured. The average fold induction of luciferase activity (± SEM) over control levels is shown (n = 10 for each treatment). − indicates control without agonist.
Glutamate-mediated CREB phosphorylation and gene expression require functional NMDA receptors, Ca2+, and Na+
MK 801 (1 μm) and APV (100 μm), noncompetitive and competitive antagonists of the NMDA receptor, respectively, blocked glutamate (see Fig. 10A)-, NMDA-, AMPA-, and kainate (all 50 μm)-induced CREB phosphorylation (Fig. 3A). Expression of the endogenous c-fos gene (see Figs.3B, 10C) and the 3xCRE-luciferaseconstruct (Fig. 3C) by ionotropic glutamate receptor agonists was also blocked by MK 801.
Fig. 10.

Average fold induction of Ser133 CREB phosphorylation and c-fosgene expression mediated by glutamate or FPL 64176 and inhibited by MK 801, GYKI 52466, or nifedipine. Each antagonist treatment was compared with the agonist treatment within the same blot. Different antagonists were run in separate blots. Levels of induction after agonist treatment were not identical in all blots and are presented in individual bars for each antagonist. Data are the average ± SEM.Asterisks mark statistically significant differences between groups that are linked in the graph. A, Glutamate (50 μm)-mediated CREB phosphorylation was significantly reduced after pretreatment with MK 801 (1 μm; n = 6), GYKI 52466 (50 μm; n = 2), or nifedipine (20 μm; n = 4). B, FPL 64176 (20 μm)–mediated CREB phosphorylation was unaffected by MK 801 (1 μm; n = 9) and by GYKI 52466 (50 μm; n = 2) but was blocked by nifedipine (20 μm; n = 10). C, Glutamate (50 μm)-mediatedc-fos gene expression was significantly reduced after pretreatment with MK 801 (1 μm; n = 10) or nifedipine (20 μm; n = 4). The reduction after pretreatment with GYKI 52466 (50 μm;n = 4) was not significant. D, FPL 64176 (20 μm)–mediated c-fos gene expression was unaffected by MK 801 (1 μm;n = 8) and by GYKI 52466 (50 μm;n = 4) but was blocked by nifedipine (20 μm; n = 6).
Fig. 3.
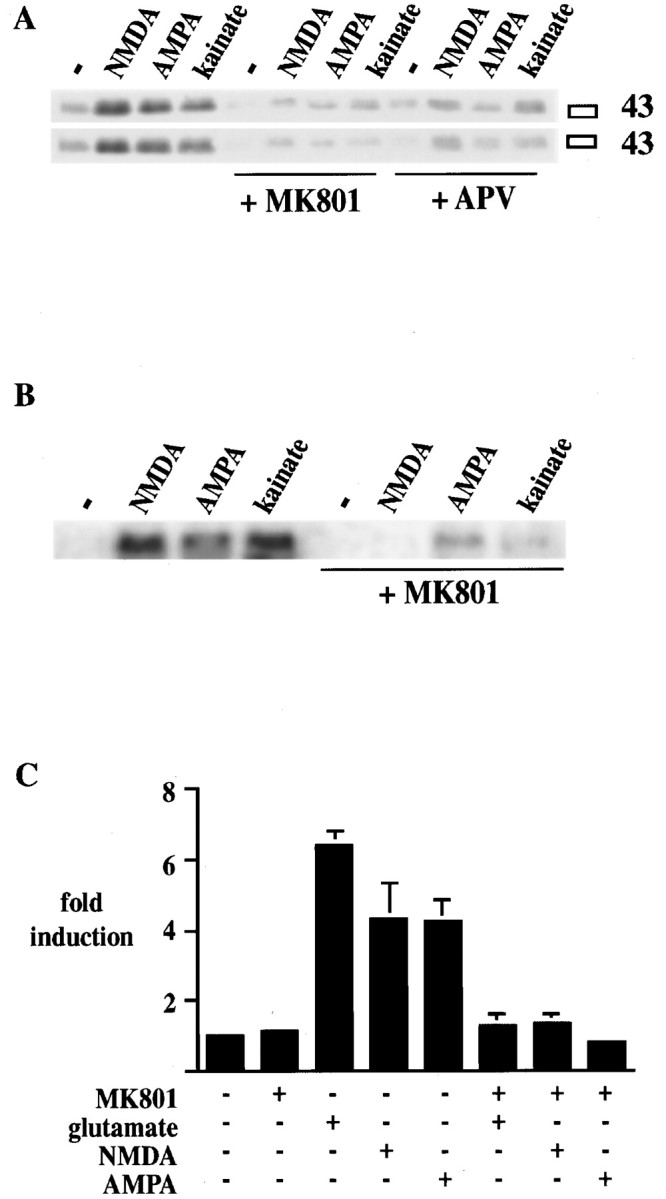
NMDA antagonists block ionotropic glutamate receptor–mediated Ser133 CREB phosphorylation,c-fos gene expression, and the induction of the3xCRE-luciferase construct in primary striatal cultures.A, Ser133 CREB phosphorylation mediated by NMDA (50 μm), AMPA (50 μm), and kainate (50 μm) is blocked by pretreatment for 20 min with the NMDA antagonists MK 801 (1 μm) and APV (100 μm). Duplicate treatments are shown in two separate blots. The experiment was repeated four times. (See also Fig. 10.)B, c-fos gene induction mediated by NMDA (50 μm), AMPA (50 μm), and kainate (50 μm) is blocked by pretreatment for 20 min with the NMDA antagonist MK 801 (1 μm). Duplicate treatments are shown in two separate blots. The experiment was repeated four times. (See also Fig. 10.) C, Induction of the3xCRE-luciferase construct after treatment for 6 hr with glutamate (50 μm), NMDA (50 μm), and AMPA (50 μm) is blocked by pretreatment for 20 min with the NMDA antagonist MK 801 (1 μm;n = 6 for each treatment). The block of induction by MK 801 is significant in all groups. Data are the average ± SEM. − indicates control without agonist.
Glutamate- and NMDA-mediated CREB phosphorylation was dependent on extracellular Ca2+ ions. Removal of Ca2+ from the medium prevented CREB phosphorylation (Fig. 4A). Glutamate receptor–mediated CREB phosphorylation was also dependent on Na+ ions (Fig. 4B). In Na+-free medium, glutamate could not induce CREB phosphorylation (Fig. 4B), even in the absence of the NMDA-blocking ion Mg2+. However, influx through voltage-operated Na+ channels was not necessary for glutamate- or FPL 64176–mediated CREB phosphorylation, because the Na+ channel blocker TTX had no effect (Fig.4C).
Fig. 4.

The role of Na+, Mg2+, and Ca2+ in Ser133 CREB phosphorylation mediated by ionotropic glutamate receptors. Primary striatal cultures were switched to defined salt solutions (see Materials and Methods) and treated 24 hr later as indicated. Immunoblots were developed with the Ser133 phospho-CREB antiserum. A, Ca2+ is necessary for NMDA- and glutamate-mediated phosphorylation of Ser133 CREB. In medium without Ca2+ (−Ca2+) neither NMDA (50 μm; top) nor glutamate (50 μm; bottom) can induce Ser133 CREB phosphorylation. This dependency persists in Mg2+-free medium. A representative experiment that was repeated twice is shown. B, Na+ is necessary for glutamate-mediated phosphorylation of Ser133 CREB. In medium without Na+ (−Na+) glutamate (50 μm) cannot induce Ser133 CREB phosphorylation. This dependency persists in Mg2+-free medium. Treatments are shown in duplicates from a representative experiment that was repeated twice.C, Na+ channels are not involved in Ser133 CREB phosphorylation. The voltage-operated Na+ channel blocker TTX (2 μm) does not block glutamate (50 μm)- or FPL 64176 (20 μm)–mediated CREB phosphorylation. Treatments are shown in duplicates from a representative experiment that was repeated three times. − indicates control without agonist.
Glutamate-mediated CREB phosphorylation and gene expression are supported by AMPA/kainate receptors
The role of AMPA/kainate receptors in glutamate-mediated gene expression was investigated with the AMPA/kainate receptor inhibitors GYKI 52466 (50 μm) and DNQX (100 μm; Fig.5). Because DNQX can bind the glycine site of the NMDA receptor (Patel et al., 1990) and thus has inhibitory properties for NMDA and AMPA/kainate receptors, we used the more specific AMPA/kainate inhibitor GYKI 52466 in experiments that differentiated inhibition of the NMDA receptor from that of AMPA/kainate receptors. Both inhibitors partially blocked glutamate (50 μm)- and NMDA (50 μm)-mediated CREB phosphorylation and c-fos gene expression (DNQX not shown) and completely blocked AMPA (50 μm)- and kainate (50 μm)-mediated CREB phosphorylation and c-fosgene expression (Fig. 5A,B). FPL 64176 (20 μm)–mediated CREB phosphorylation orc-fos gene expression was not affected by GYKI 52466 (see Figs. 5A,B,10B,D) or DNQX (data not shown). In transfection assays with the 3xCRE-luciferase construct, glutamate-and AMPA-mediated induction was blocked by DNQX (Fig.5C).
Fig. 5.

The AMPA/kainate antagonists GYKI 52466 and DNQX block ionotropic glutamate receptors to varying degrees, but they do not block L-type Ca2+ channels in primary striatal cultures. A, Ser133 CREB phosphorylation mediated by AMPA (50 μm) or kainate (50 μm) was fully blocked by GYKI 52466 (GYKI; 50 μm). Glutamate (50 μm)- and NMDA (50 μm)-mediated CREB phosphorylation was partially blocked by GYKI 52466, whereas FPL 64176 (20 μm)–mediated CREB phosphorylation was not blocked by GYKI 52466. Treatments are shown in duplicates. (See also Fig. 10.) B, c-fosgene expression induced by AMPA (50 μm) or kainate (50 μm) was fully blocked by GYKI 52466 (50 μm), whereas c-fos gene expression induced by glutamate (50 μm) or NMDA (50 μm) was partially blocked by GYKI 52466. c-fos gene expression induced by FPL 64176 (20 μm) was not blocked by GYKI 52466. The experiment was repeated three times. (See also Fig. 10.)C, DNQX (100 μm) reduced glutamate (50 μm)-mediated induction and blocked AMPA (50 μm)-mediated induction of the3xCRE-luciferase construct (n = 6).Asterisks mark statistically significant differences between groups that are linked in the graph. Data are the average ± SEM. D, Mg2+ prevents phosphorylation of Ser133 CREB by NMDA (50 μm) in the absence of AMPA/kainate activity. In glutamate-free medium, NMDA (50 μm) has little effect on CREB phosphorylation in the presence of Mg2+ (2 mm) but readily induces CREB phosphorylation in the absence of Mg2+ (compare+Mg2+/−glutamate with−Mg2+/−glutamate). Ambient glutamate levels (2–5 μm) are sufficient to prevent Mg2+ (2 mm) from blocking NMDA-mediated CREB phosphorylation (compare+Mg2+/−glutamate with+Mg2+/+glutamate). In the presence of Mg2+ (2 mm) the AMPA/kainate receptor antagonist GYKI 52466 (50 μm) blocks NMDA (50 μm)-mediated CREB phosphorylation but has little effect in the absence of Mg2+ (compare all three conditions). A representative experiment that was repeated twice is shown. Note that +glutamate indicates the exposure to 10 μm glutamate 24 hr before the experiment until the conclusion of the experiment; −glutamate indicates a change to glutamate-free medium 24 hr before the experiment until the conclusion of the experiment. Small amounts of metabolic glutamate (in the nanomolar range) released during the 24 hr may be present in the medium and explain the slight increase of CREB phosphorylation in the+Mg2+/−glutamate condition. − indicates control without agonist.
AMPA/kainate receptors relieve the Mg2+ block of the NMDA receptor
The role of AMPA/kainate receptors in NMDA-mediated CREB phosphorylation was further revealed in experiments that examined the effect of Mg2+ (Fig. 5D). With ambient glutamate (2–5 μm), NMDA induced CREB phosphorylation despite the presence of Mg2+ (2 mm; Fig.5D,+Mg2+/+glutamate). This phosphorylation was blocked by the AMPA/kainate antagonist GYKI 52466 (50 μm). In the absence of glutamate, NMDA (50 μm) did not induce CREB phosphorylation in the presence of Mg2+ (Fig. 5D,+Mg2+/−glutamate). In the absence of Mg2+, NMDA-induced CREB phosphorylation was independent of AMPA/kainate receptors because it was insensitive to GYKI 52466 and did not require ambient glutamate (Fig. 5D,−Mg2+/−glutamate). These data suggest that AMPA/kainate receptors are necessary for removal of the Mg2+ block of NMDA receptors.
L-type Ca2+ channels are necessary for glutamate-mediated CREB phosphorylation and gene expression
The role of L-type Ca2+ channels in CREB phosphorylation and gene expression stimulated by ionotropic glutamate receptors was examined with the L-type Ca2+ channel blocker nifedipine (Fig. 6). Nifedipine (20 μm) blocked CREB phosphorylation induced by glutamate, NMDA, AMPA, or kainate (all 50 μm) or by the L-type Ca2+ channel agonist FPL 64176 (20 μm; see Figs. 6A,D,10A,B). The block of glutamate receptor–mediated CREB phosphorylation was also observed with another L-type Ca2+ channel antagonist, verapamil (data not shown). Likewise, induction of c-fos gene expression by ionotropic glutamate receptor agonists (50 μm) and by FPL 64176 (20 μm) was blocked by nifedipine (20 μm; see Figs. 6B,10C,D). In transfection experiments with the 3xCRE-luciferase construct, nifedipine (20 μm) inhibited glutamate- and FPL 64176–induced increases in luciferase expression (Fig. 6C). Because a transient phosphorylation of CREB in the presence of nifedipine may not have been detected after 15 min, we harvested cultures between 1 and 15 min after glutamate stimulation in the presence or absence of nifedipine (20 μm). Nifedipine blocked glutamate-mediated CREB phosphorylation at all times examined (Fig. 6D; note that this time course is different from the time course in Fig.1B, because the cells were harvested immediately after treatment). Because neurons in culture synapse onto each other, we examined whether the GABA antagonists bicuculline (Fig.6E) or picrotoxin (data not shown) affect the inhibitory potency of nifedipine on glutamate-mediated Ser133 CREB phosphorylation. Neither antagonist stimulated CREB phosphorylation, nor did either affect the potency of nifedipine. In line with the excitatory properties of GABA in early development (Cherubini et al., 1991), both antagonists slightly inhibited CREB phosphorylation after glutamate treatment.
An antisense oligonucleotide approach was used to confirm these results. A knockdown of L-type Ca2+ channels with antisense oligonucleotides against the α1C and α1D subunits of Ca2+ channels diminished glutamate- and FPL 64176–induced CREB phosphorylation significantly (Fig.7A,B), supporting a role for L-type Ca2+ channels in glutamate-mediated CREB phosphorylation. With an antibody against α1C, a decrease in α1C protein was detected in cultures treated with antisense oligonucleotides (Fig. 7A, α1C). No change in total CREB protein was seen (Fig. 7A,CREB).
Fig. 7.

Knockdown of the L-type Ca2+ channel reduces CREB phosphorylation after treatment of cultures with glutamate or with FPL 64176. Knockdown of the NMDA receptor reduces CREB phosphorylation only after treatment with NMDA (50 μm). Striatal cultures were exposed to antisense oligonucleotides (AS) against the α1 subunits of the L-type Ca2+ channel (α1C and α1D) (A, B) or against the NMDA receptor (NR1, NR2A, and NR2B) (C, D) for 5 d and treated with glutamate (50 μm), NMDA (50 μm), or FPL 64176 (20 μm). In control experiments, sister cultures were treated with scrambled antisense oligonucleotides (SAS).A, Immunoblots developed with the Ser133 CREB antiserum (P-CREB), an antiserum against the α1C subtype of the L-type Ca2+ channel (α1C), or an antiserum against CREB (CREB). The antisense treatment reduced the α1C subtype of the L-type Ca2+ channel but not the levels of total CREB protein. Glutamate (50 μm)- and FPL 64176 (20 μm)–mediated CREB phosphorylation was partially blocked by the antisense treatment. B, Average induction of CREB phosphorylation (± SEM) after treatment with glutamate (50 μm) or FPL 64176 (20 μm) in cultures exposed to SAS (control) or AS(n = 4). The reduction in CREB phosphorylation in the presence of antisense oligonucleotides was statistically significant for glutamate-and FPL 64176–treated cultures (marked byasterisks). C, Immunoblots developed with the Ser133 CREB antiserum (P-CREB), an antiserum against the NR1 subtype of the NMDA receptor (NR1), or an antiserum against CREB protein (CREB). Levels of NR1 protein were reduced, whereas levels of total CREB protein were unaffected. The antisense treatment partially blocked NMDA (50 μm)-mediated CREB phosphorylation but did not affect FPL 64176 (20 μm)–mediated CREB phosphorylation. D, Average induction of CREB phosphorylation (± SEM) after treatment with NMDA (50 μm) or FPL 64176 (20 μm) in cultures exposed to SAS (control) or AS(n = 4). The reduction in CREB phosphorylation in the presence of antisense oligonucleotides was statistically significant for NMDA-treated cultures (marked byasterisks).
FPL 64176–mediated CREB phosphorylation and gene expression are independent of functional ionotropic glutamate receptors
The NMDA antagonist MK 801 (1 μm) did not block FPL 64176 (20 μm)–mediated CREB phosphorylation (see Figs.8A, 10B), even at concentrations submaximal for the stimulation of CREB phosphorylation (Fig.8B). Likewise, the AMPA/kainate antagonists GYKI 52466 (50 μm; see Figs.5A, 10B) or DNQX (100 μm; data not shown) had no effect on FPL 64176–mediated CREB phosphorylation, nor did a combination of NMDA and AMPA/kainate receptor antagonists [MK 801 (1 μm) and DNQX (100 μm); Fig. 8C]. Knockdown of NMDA receptors with antisense oligonucleotides had no effect on FPL 64176 (20 μm)–mediated CREB phosphorylation but reduced CREB phosphorylation after NMDA (50 μm) treatment (Fig.7C,D). A combination of antisense oligonucleotides against NR1, NR2A, and NR2B was used in these experiments, and NR1 protein was reduced in antisense-treated cultures (Fig. 7C, NR1). NR1 is a necessary component of all NMDA receptors (Hollmann and Heinemann, 1994), and NR2A and NR2B are the most prevalent NR2 subtypes in the striatum (Standaert et al., 1994; Landwehrmeyer et al., 1995; Mutel et al., 1998). c-fosgene expression activated by L-type Ca2+ channel agonists was also unaffected by the AMPA/kainate antagonist GYKI 52466 (50 μm; see Figs. 5B, 10D) and the NMDA antagonist MK 801 (1 μm; Fig.8D). Combinations of FPL 64176 and AMPA or of FPL 64176 and NMDA were unaffected by MK 801 or by GYKI 52466 (Fig.9), although these combinations were blocked by the L-type Ca2+ channel antagonist nifedipine. Moreover, Ser133 CREB phosphorylation induced by a combination of the glutamate receptor agonists AMPA and NMDA was significantly blocked by nifedipine (20 μm), MK 801 (1 μm), and GYKI 52466 (50 μm; Fig. 9). A comparison of the inhibitory properties of MK 801, GYKI 52466, and nifedipine on glutamate- and FPL 64176–activated CREB phosphorylation and c-fos gene expression is shown in Figure10.
Fig. 8.

Ionotropic glutamate receptor antagonists cannot block FPL 64176–mediated Ser133 CREB phosphorylation in primary striatal cultures. A, The NMDA antagonist MK 801 (1 μm) does not block FPL 64176 (20 μm)–mediated CREB phosphorylation. All treatments are shown in duplicates. The experiment was repeated four times in duplicates. (See also Fig. 9.) B, Lower concentrations of FPL 64176 (0.5 and 1 μm) are also not blocked by MK 801. All treatments are shown in duplicates. C, A mixture of DNQX (100 μm) and MK 801 (1 μm) does not block FPL 64176 (20 μm)–mediated CREB phosphorylation. All treatments are shown in duplicates. The experiment was repeated twice. D, c-fos gene expression induced by FPL 64176 (20 μm) is not blocked by MK 801 (1 μm). Duplicate treatments are shown in two separate blots. (See also Fig. 9.) − indicates control without agonist.
Fig. 9.

The induction of Ser133 CREB phosphorylation by ionotropic glutamate receptor agonists together with the L-type Ca2+channel agonist FPL 64176 is inhibited by nifedipine but not by ionotropic glutamate receptor antagonists. Primary striatal cultures were treated with AMPA (50 μm) and NMDA (50 μm), FPL 64176 (20 μm) and AMPA (50 μm), or FPL 64176 (20 μm) and NMDA (50 μm). A, B,Left, Immunoblots with the Ser133phospho-CREB antiserum are shown. Right, The bar graphs show the average fold induction of Ser133 CREB phosphorylation (± SEM) of six experiments. Asterisks mark statistically significant differences from the group treated with the agonist mixture.A, The induction of CREB phosphorylation was significantly blocked by the L-type Ca2+channel antagonist nifedipine (20 μm) in all treatments but was not blocked by the NMDA antagonist MK 801 (1 μm) whenever FPL 64176 was part of the mixture. B, The induction of CREB phosphorylation was not blocked by the AMPA/kainate antagonist GYKI 52466 (50 μm) in the presence of FPL 64176. − indicates control without agonist.
DISCUSSION
Glutamate mediates CREB phosphorylation and gene expression in primary striatal cultures in a Ca2+- and Na+-dependent manner
Glutamate was a potent inducing agent of Ser133CREB phosphorylation in primary striatal cultures. Phosphorylation of Ser133 CREB was dependent on Ca2+and Na+ and reached maximum levels after a 3 min exposure to glutamate. The subsequent expression of Fos protein was observed between 1 and 5 hr. The timing of induction of CREB phosphorylation and Fos protein expression is in agreement with regulation of the c-fos gene by Ser133phospho-CREB. Glutamate treatment had no effect on the levels of CREB protein, consistent with the notion that CREB activates gene expression after it is phosphorylated on Ser133 and not via an increase in its protein levels. Because the c-fos promoter can also be regulated by regulatory elements other than CREB, we examined the regulation of a transfected reporter gene, luciferase, under the exclusive control of three copies of a consensus CREB-binding site (CRE). The regulation of the 3xCRE-luciferase reporter construct by ionotropic glutamate receptor agonists was comparable with the regulation of the endogenous c-fos gene.
AMPA/kainate receptors activate the NMDA receptor by relieving the Mg2+ block
AMPA/kainate receptor agonists induced CREB phosphorylation andc-fos gene expression by an NMDA receptor–dependent mechanism. In glutamate-containing medium, NMDA antagonists blocked AMPA- and kainate-mediated Ser133 CREB phosphorylation and c-fos gene expression, demonstrating the need for functional NMDA receptors. In the presence of Mg2+, AMPA/kainate receptors provided the depolarization that enabled the NMDA receptor to open in response to glutamate and glycine. In fact, the ability of the NMDA receptor to mediate CREB phosphorylation in Mg2+-containing medium was significantly hampered when AMPA/kainate receptors were blocked. Our data suggest that a preexisting interaction of the NMDA receptor with ambient glutamate and glycine levels caused channel opening after the relief of the Mg2+ block by AMPA/kainate receptors. In glutamate-free medium, AMPA/kainate receptor agonists could not induce Ser133 CREB phosphorylation (data not shown). NMDA receptors induced CREB phosphorylation in glutamate-free medium only in the absence of Mg2+ but depended on glutamate (and AMPA/kainate receptors) in the presence of Mg2+.
Two findings support the hypothesis that AMPA/kainate receptors mediate gene expression by removing the Mg2+ block of NMDA receptors; in the absence of extracellular NMDA or glutamate or when NMDA receptors were blocked, AMPA/kainate receptor stimulation could not induce CREB phosphorylation. Furthermore, in the absence of Mg2+, AMPA/kainate receptors were irrelevant for NMDA receptor–mediated phosphorylation of Ser133 CREB.
L-type Ca2+ channels are essential for glutamate-mediated CREB phosphorylation and gene expression in primary striatal cultures
Ionotropic glutamate receptors induced CREB phosphorylation andc-fos gene expression with the assistance of L-type Ca2+ channels. Antagonists of L-type Ca2+ channels were able to reduce significantly the induction of CREB phosphorylation and gene expression by all ionotropic glutamate receptor agonists, alone or in combination. The effect of L-type Ca2+ channel blockers was independent of GABA activity in the cultures, because the potency of the block was not changed by GABA antagonists. Antisense oligonucleotides against the two α1 subtypes of the L-type Ca2+ channel in the brain, α1C and α1D, significantly reduced CREB phosphorylation mediated by glutamate or FPL 64176, whereas antisense oligonucleotides against the NMDA receptor had no effect on FPL 64176–mediated CREB phosphorylation. Glutamate-mediated CREB phosphorylation was dependent on Na+ ions yet independent of TTX-sensitive Na+ channels. This finding suggests that Na+ influx specifically through ionotropic glutamate receptors may be a necessary step toward glutamate-mediated CREB phosphorylation. We hypothesize that Na+ influx through NMDA receptors is important to depolarize the neuron and to trigger the opening of L-type Ca2+ channels. Even though there was Ca2+ in the Na+-free medium, it was not enough to stimulate an intraneuronal second messenger pathway and CREB phosphorylation. This suggests that Na+ and Ca2+ ions have different roles in the signal transduction pathway, Na+ via membrane depolarization viaNMDA receptors and Ca2+ via membrane depolarization via L-type Ca2+ channels and via stimulation of the intracellular second messenger pathway.
AMPA/kainate receptors, NMDA receptors, and L-type Ca2+ channels contribute consecutively to the same signal transduction pathway
One important question was whether signals from ionotropic glutamate receptors and L-type Ca2+ channels mediatec-fos gene expression by distinct calcium-signaling pathways, as has been suggested for other culture systems (Bading et al., 1993; Hardingham et al., 1997). It has been found that the two primary cis-acting regulatory elements of thec-fos promoter, the CRE and the serum response element (SRE), are regulated differentially depending on the mode of calcium entry. The CRE is regulated by nuclear calcium from the L-type calcium channels, and the SRE is regulated by cytoplasmic calcium from the NMDA receptor (Bading et al., 1993; Hardingham et al., 1997). However, in our cultures, NMDA receptor–induced c-fos gene expression was dependent on L-type Ca2+ channels. Comparable regulation of CREB phosphorylation and c-fos gene expression was observed. Furthermore, direct activation of the L-type Ca2+ channels with FPL 64176 inducedc-fos gene expression. This suggests that in our striatal cultures, either the SRE is not active or the L-type Ca2+ channel is able to provide all the factors necessary for c-fos transcriptional activation, including the SRE. The pharmacological profile of the ionotropic glutamate receptor antagonists and of the L-type Ca2+ channel blockers allowed us to determine further that AMPA/kainate receptors, NMDA receptors, and L-type Ca2+ channels contribute consecutively to Ser133 CREB phosphorylation (see below).
The sequence of events leading from glutamate receptor activation to gene expression
AMPA/kainate receptor channels open after interaction with glutamate and permit Na+ entry at the synapse (Kandel et al., 1991). The resulting local depolarization removes the Mg2+ block of the NMDA receptor, which permits the NMDA receptor to respond to extracellular glutamate and glycine. Opening of the NMDA receptor channel causes Na+ and Ca2+ influx. Unlike the AMPA/kainate receptor channel that desensitizes rapidly, NMDA receptor channels have long opening times (Ascher and Nowak, 1987; Gasic and Hollmann, 1992). Therefore, NMDA receptors can trigger the opening of L-type Ca2+ channels that open during strong depolarization (Tsien et al., 1988). The activation of L-type Ca2+channels promotes Ca2+ entry along the dendrites and at the cell body (Westenbroek et al., 1990; Schild et al., 1995). Second messengers activated by Ca2+ translocate to the nucleus and phosphorylate CREB (Deisseroth et al., 1998) (Fig.11).
Fig. 11.

Model of the interaction of AMPA/kainate receptors, NMDA receptors, and L-type Ca2+ channels in striatal Ser133 CREB phosphorylation and gene expression. 1, The activation of AMPA/kainate receptors causes Na+ influx and a local depolarization that relieves the Mg2+ block of the NMDA receptor.2a, Activation of the NMDA receptor via ligand binding and depolarization leads to Na+ influx as well as Ca2+ influx. Unlike the AMPA/kainate receptors, NMDA receptors do not rapidly desensitize and allow for a depolarization that is strong enough to trigger the opening of L-type Ca2+ channels. 3, L-type Ca2+ channels allow for Ca2+influx and activation of a kinase pathway that translocates to the nucleus to phosphorylate Ser133 CREB.2b, A signal transduction cascade originating at NMDA receptors that is independent of L-type Ca2+channels is negligible for CREB phosphorylation or c-fosgene expression in primary striatal cultures.
Our results suggest an important role for L-type Ca2+ channels in neuroplasticity of the striatum and confirm previous reports about the involvement of L-type Ca2+ channels in NMDA-mediated plasticity and toxicity (Weiss et al., 1990; Westenbroek et al., 1990; Chetkovich et al., 1991; Sucher et al., 1991; Aroniadou et al., 1993). Under our experimental conditions, NMDA receptors initiated a signal transduction pathway but did not initiate a significant intraneuronal second messenger pathway, either alone or together with AMPA/kainate receptors. Depolarization of L-type Ca2+ channels played a crucial role in the activation of an intraneuronal second messenger pathway.
Although the supportive role of AMPA/kainate receptors for NMDA receptors is in agreement with previous findings in hippocampal culture (Bading et al., 1995), other findings differ. In hippocampal cultures NMDA receptors and L-type Ca2+ channels seem to contribute to independent, parallel pathways rather than to the same pathway (Bading et al., 1993, 1995). Like in hippocampal cultures, L-type Ca2+ channels in the striatum activate the CRE and function independently of NMDA receptors. But although we do not exclude a direct pathway from NMDA receptors to the SRE in the striatum, this pathway in itself is not enough to mediatec-fos gene expression. This difference may be attributed to intrinsic differences between both types of neurons or to the different neurotransmitters released in either culture. Hippocampal neurons are mostly glutamatergic and express very high levels of glutamate receptors (Jarvis et al., 1987; Miyoshi et al., 1991; Sakurai et al., 1991). Striatal cultures are primarily GABAergic and express much lower levels of glutamate receptors (Jarvis et al., 1987; Miyoshi et al., 1991; Sakurai et al., 1991). Because neurons in culture synapse onto each other, hippocampal neurons excite each other after activation, whereas GABA in striatal neurons, dependent on the level of maturity (Cherubini et al., 1991), may be excitatory or inhibitory. To avoid trans-synaptic effects in hippocampal cultures, Na+ channels are often blocked with TTX. We repeated some of our experiments in the presence of TTX but observed results comparable with those of experiments without TTX (data not shown). Thus, there are fundamental differences in glutamate-mediated gene expression in neurons of both brain areas.
Footnotes
This work was supported by the National Alliance for Research on Schizophrenia and Depression (C.K.) and the National Institute of Drug Abuse Grant DA07134 (C.K.).
Correspondence should be addressed to Dr. Christine Konradi, Laboratory of Molecular and Developmental Neuroscience, Massachusetts General Hospital, CNY 2510, Building 149, 13th Street, Charlestown, MA 02129.
REFERENCES
- 1.Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol. 1998;18:967–977. doi: 10.1128/mcb.18.2.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aroniadou VA, Maillis A, Stefanis CC. Dihydropyridine-sensitive calcium channels are involved in the induction of N-methyl-d-aspartate receptor-independent long-term potentiation in visual cortex of adult rats. Neurosci Lett. 1993;151:77–80. doi: 10.1016/0304-3940(93)90050-u. [DOI] [PubMed] [Google Scholar]
- 3.Aronin N, Chase K, Sagar SM, Sharp FR, DiFiglia M. N-Methyl-d-aspartate receptor activation in the neostriatum increases c-fos and fos-related antigens selectively in medium-sized neurons. Neuroscience. 1991;44:409–420. doi: 10.1016/0306-4522(91)90065-v. [DOI] [PubMed] [Google Scholar]
- 4.Ascher P, Nowak L. Electrophysiological studies of NMDA receptors. Trends Neurosci. 1987;10:284–287. [Google Scholar]
- 5.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 6.Bading H, Segal MM, Sucher NJ, Dudek H, Lipton SA, Greenberg ME. N-Methyl-d-aspartate receptors are critical for mediating the effects of glutamate on intracellular calcium concentration and immediate early gene expression in cultured hippocampal neurons. Neuroscience. 1995;64:653–664. doi: 10.1016/0306-4522(94)00462-e. [DOI] [PubMed] [Google Scholar]
- 7.Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA. 1996;93:13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20:415–455. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
- 9.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 10.Bonni A, Ginty DD, Dudek H, Greenberg ME. Serine 133-phosphorylated CREB induces transcription via a cooperative mechanism that may confer specificity to neurotrophin signals. Mol Cell Neurosci. 1995;6:168–183. doi: 10.1006/mcne.1995.1015. [DOI] [PubMed] [Google Scholar]
- 11.Bourtchuladze R, Frenguelli B, Blendie J, Cioffi D, Schütz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 12.Buchsbaum MS, Haier RJ, Potkin SG, Nuechterlein K, Bracha HS, Katz M, Lohr J, Wu J, Lottenberg S, Jerabek PA. Frontostriatal disorder of cerebral metabolism in never-medicated schizophrenics. Arch Gen Psychiatry. 1992;49:935–942. doi: 10.1001/archpsyc.1992.01820120023005. [DOI] [PubMed] [Google Scholar]
- 13.Burns RS. Subclinical damage to the nigrostriatal dopamine system by MPTP as a model of preclinical Parkinson’s disease: a review. Acta Neurol Scand Suppl. 1991;136:29–36. doi: 10.1111/j.1600-0404.1991.tb05017.x. [DOI] [PubMed] [Google Scholar]
- 14.Burt DR, Creese I, Snyder SH. Antischizophrenic drugs: chronic treatment elevates dopamine receptor binding in brain. Science. 1977;196:326–328. doi: 10.1126/science.847477. [DOI] [PubMed] [Google Scholar]
- 15.Busatto GF, Kerwin RW. Schizophrenia, psychosis, and the basal ganglia. Psychiatr Clin North Am. 1997;20:897–910. doi: 10.1016/s0193-953x(05)70351-8. [DOI] [PubMed] [Google Scholar]
- 16.Calne DB, Langston JW, Martin WR, Stoessl AJ, Ruth TJ, Adam MJ, Pate BD, Schulzer M. Positron emission tomography after MPTP: observations relating to the cause of Parkinson’s disease. Nature. 1985;317:246–248. doi: 10.1038/317246a0. [DOI] [PubMed] [Google Scholar]
- 17.Catterall WA. Functional subunit structure of voltage-gated calcium channels. Science. 1991;253:1499–1500. doi: 10.1126/science.1654596. [DOI] [PubMed] [Google Scholar]
- 18.Catterall WA, Striessnig J. Receptor sites for Ca2+ channel antagonists. Trends Pharmacol Sci. 1992;13:256–262. doi: 10.1016/0165-6147(92)90079-l. [DOI] [PubMed] [Google Scholar]
- 19.Cherubini E, Gaiarsa JL, Ben-Ari Y. GABA: an excitatory transmitter in early postnatal life. Trends Neurosci. 1991;14:515–519. doi: 10.1016/0166-2236(91)90003-d. [DOI] [PubMed] [Google Scholar]
- 20.Chetkovich DM, Gray R, Johnston D, Sweatt JD. N-Methyl-d-aspartate receptor activation increases cAMP levels and voltage-gated Ca2+ channel activity in area CA1 of hippocampus. Proc Natl Acad Sci USA. 1991;88:6467–6471. doi: 10.1073/pnas.88.15.6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cole AJ, Saffen DW, Baraban JM, Worley PF. Rapid increase of an immediate early gene messenger RNA in hippocampal neurons by synaptic NMDA receptor activation. Nature. 1989;340:474–476. doi: 10.1038/340474a0. [DOI] [PubMed] [Google Scholar]
- 22.Connor JA, Wadman WJ, Hockberger PE, Wong RK. Sustained dendritic gradients of Ca2+ induced by excitatory amino acids in CA1 hippocampal neurons. Science. 1988;240:649–653. doi: 10.1126/science.2452481. [DOI] [PubMed] [Google Scholar]
- 23.Danielson PE, Forss-Petter S, Brow MA, Calavetta L, Douglass J, Milner RJ, Sutcliffe JG. p1B15: a cDNA clone of the rat mRNA encoding cyclophilin. DNA. 1988;7:261–267. doi: 10.1089/dna.1988.7.261. [DOI] [PubMed] [Google Scholar]
- 24.Dave JR, Tortella FC. Regional changes in c-fos mRNA in rat brain after i.v. or i.c.v. NMDA injections. NeuroReport. 1994;5:1645–1648. doi: 10.1097/00001756-199408150-00026. [DOI] [PubMed] [Google Scholar]
- 25.Deadwyler SA, Dunwiddie T, Lynch G. A critical level of protein synthesis is required for long-term potentiation. Synapse. 1987;1:90–95. doi: 10.1002/syn.890010112. [DOI] [PubMed] [Google Scholar]
- 26.Deisseroth K, Tsien RW. Calmodulin translocation to the nucleus mediates rapid synaptic control of CREB phosphorylation in hippocampal neurons. Soc Neurosci Abstr. 1997;23:311. [Google Scholar]
- 27.Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- 28.DiFiglia M. Excitotoxic injury of the neostriatum: a model for Huntington’s disease. Trends Neurosci. 1990;13:286–289. doi: 10.1016/0166-2236(90)90111-m. [DOI] [PubMed] [Google Scholar]
- 29.Eastwood SL, Burnet PW, Harrison PJ. Striatal synaptophysin expression and haloperidol-induced synaptic plasticity. NeuroReport. 1994;5:677–680. doi: 10.1097/00001756-199402000-00004. [DOI] [PubMed] [Google Scholar]
- 30.Eastwood SL, Heffernan J, Harrison PJ. Chronic haloperidol treatment differentially affects the expression of synaptic and neuronal plasticity-associated genes. Mol Psychiatry. 1997;2:322–329. doi: 10.1038/sj.mp.4000238. [DOI] [PubMed] [Google Scholar]
- 31.Egebjerg J, Heinemann SF. Ca2+ permeability of unedited and edited versions of the kainate selective glutamate receptor GluR6. Proc Natl Acad Sci USA. 1993;90:755–759. doi: 10.1073/pnas.90.2.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gasic G, Hollmann M. Molecular neurobiology of glutamate receptors. Annu Rev Physiol. 1992;54:507–536. doi: 10.1146/annurev.ph.54.030192.002451. [DOI] [PubMed] [Google Scholar]
- 33.Ghosh A, Ginty DD, Bading H, Greenberg ME. Calcium regulation of gene expression in neuronal cells. J Neurobiol. 1994;25:294–303. doi: 10.1002/neu.480250309. [DOI] [PubMed] [Google Scholar]
- 34.Ginty DD, Kornhauser JM, Thompson MA, Bading H, Mayo KE, Takahashi JS, Greenberg ME. Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and circadian clock. Science. 1993;260:238–241. doi: 10.1126/science.8097062. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 36.Greenamyre JT, Porter RH. Anatomy and physiology of glutamate in the CNS. Neurology. 1994;44:S7–S13. [PubMed] [Google Scholar]
- 37.Hardingham GE, Chawla S, Johnson CM, Bading H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature. 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- 38.Heckers S. Neuropathology of schizophrenia: cortex, thalamus, basal ganglia, and neurotransmitter-specific projection systems. Schizophr Bull. 1997;23:403–421. doi: 10.1093/schbul/23.3.403. [DOI] [PubMed] [Google Scholar]
- 39.Hofmann F, Biel M, Flockerzi V. Molecular basis for Ca2+ channel diversity. Annu Rev Neurosci. 1994;17:399–418. doi: 10.1146/annurev.ne.17.030194.002151. [DOI] [PubMed] [Google Scholar]
- 40.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 41.Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 42.Hornykiewicz O. Parkinson’s disease and the adaptive capacity of the nigrostriatal dopamine system: possible neurochemical mechanisms. Adv Neurol. 1993;60:140–147. [PubMed] [Google Scholar]
- 43.Hyman SE. Addiction to cocaine and amphetamine. Neuron. 1996;16:901–904. doi: 10.1016/s0896-6273(00)80111-7. [DOI] [PubMed] [Google Scholar]
- 44.Hyman SE, Nestler EJ. Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am J Psychiatry. 1996;153:151–162. doi: 10.1176/ajp.153.2.151. [DOI] [PubMed] [Google Scholar]
- 45.Impey S, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- 46.Jarvis MF, Murphy DE, Williams M. Quantitative autoradiographic localization of NMDA receptors in rat brain using [3H]CPP: comparison with [3H]TCP binding sites. Eur J Pharmacol. 1987;141:149–152. doi: 10.1016/0014-2999(87)90423-7. [DOI] [PubMed] [Google Scholar]
- 47.Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- 48.Kandel ER, Schwartz JH, Jessell TM. Principles of neural science. Elsevier; New York: 1991. [Google Scholar]
- 49.Knowlton BJ, Mangels JA, Squire LR. A neostriatal habit learning system in humans. Science. 1996;273:1399–1402. doi: 10.1126/science.273.5280.1399. [DOI] [PubMed] [Google Scholar]
- 50.Kohler M, Burnashev N, Sakmann B, Seeburg PH. Determinants of Ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: diversity by RNA editing. Neuron. 1993;10:491–500. doi: 10.1016/0896-6273(93)90336-p. [DOI] [PubMed] [Google Scholar]
- 51.Konradi C, Leveque JC, Hyman SE. Amphetamine and dopamine-induced immediate early gene expression in striatal neurons depends upon postsynaptic NMDA receptors and calcium. J Neurosci. 1996;16:4231–4239. doi: 10.1523/JNEUROSCI.16-13-04231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Landwehrmeyer GB, Standaert DG, Testa CM, Penney JB, Jr, Young AB. NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J Neurosci. 1995;15:5297–5307. doi: 10.1523/JNEUROSCI.15-07-05297.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langston JW. The etiology of Parkinson’s disease with emphasis on the MPTP story. Neurology. 1996;47:S153–S160. doi: 10.1212/wnl.47.6_suppl_3.153s. [DOI] [PubMed] [Google Scholar]
- 54.Lerea LS, McNamara JO. Ionotropic glutamate receptor subtypes activate c-fos transcription by distinct calcium-requiring intracellular signaling pathways. Neuron. 1993;10:31–41. doi: 10.1016/0896-6273(93)90239-n. [DOI] [PubMed] [Google Scholar]
- 55.Lomeli H, Mosbacher J, Melcher T, Hoger T, Geiger JR, Kuner T, Monyer H, Higuchi M, Bach A, Seeburg PH. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science. 1994;266:1709–1713. doi: 10.1126/science.7992055. [DOI] [PubMed] [Google Scholar]
- 56.MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 57.Matthies H. In search of cellular mechanisms of memory. Prog Neurobiol. 1989;32:277–349. doi: 10.1016/0301-0082(89)90024-5. [DOI] [PubMed] [Google Scholar]
- 58.Maxwell IH, Harrison GS, Wood WM, Maxwell F. A DNA cassette containing a trimerized SV40 polyadenylation signal which efficiently blocks spurious plasmid-initiated transcription. Biotechniques. 1989;7:276–280. [PubMed] [Google Scholar]
- 59.Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 60.McCleskey EW. Calcium channels: cellular roles and molecular mechanisms. Curr Opin Neurobiol. 1994;4:304–312. doi: 10.1016/0959-4388(94)90090-6. [DOI] [PubMed] [Google Scholar]
- 61.Michaelis EK. Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Prog Neurobiol. 1998;54:369–415. doi: 10.1016/s0301-0082(97)00055-5. [DOI] [PubMed] [Google Scholar]
- 62.Mijnster MJ, Ingham CA, Meredith GE, Docter GJ, Arbuthnott GW. Morphological changes in met(5)-enkephalin-immunoreactive synaptic boutons in the rat neostriatum after haloperidol decanoate treatment. Eur J Neurosci. 1996;8:716–726. doi: 10.1111/j.1460-9568.1996.tb01257.x. [DOI] [PubMed] [Google Scholar]
- 63.Misra RP, Bonni A, Miranti CK, Rivera VM, Sheng M, Greenberg ME. L-type voltage-sensitive calcium channel activation stimulates gene expression by a serum response factor-dependent pathway. J Biol Chem. 1994;269:25483–25493. [PubMed] [Google Scholar]
- 64.Miyoshi R, Kito S, Doudou N, Nomoto T. Influence of age on N-methyl-d-aspartate antagonist binding sites in the rat brain studied by in vitro autoradiography. Synapse. 1991;8:212–217. doi: 10.1002/syn.890080307. [DOI] [PubMed] [Google Scholar]
- 65.Montminy MR, Bilezikjian LM. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature. 1987;328:175–178. doi: 10.1038/328175a0. [DOI] [PubMed] [Google Scholar]
- 66.Montminy MR, Gonzalez GA, Yamamoto KK. Regulation of cAMP-inducible genes by CREB. Trends Neurosci. 1990;13:184–188. doi: 10.1016/0166-2236(90)90045-c. [DOI] [PubMed] [Google Scholar]
- 67.Mosbacher J, Schoepfer R, Monyer H, Burnashev N, Seeburg PH, Ruppersberg JP. A molecular determinant for submillisecond desensitization in glutamate receptors. Science. 1994;266:1059–1062. doi: 10.1126/science.7973663. [DOI] [PubMed] [Google Scholar]
- 68.Murphy D, Segal M. Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc Natl Acad Sci USA. 1997;94:1482–1487. doi: 10.1073/pnas.94.4.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murphy TH, Worley PF, Baraban JM. L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron. 1991;7:625–635. doi: 10.1016/0896-6273(91)90375-a. [DOI] [PubMed] [Google Scholar]
- 70.Mutel V, Buchy D, Klingelschmidt A, Messer J, Bleuel Z, Kemp JA, Richards JG. In vitro binding properties in rat brain of [3H]Ro 25-6981, a potent and selective antagonist of NMDA receptors con-taining NR2B subunits. J Neurochem. 1998;70:2147–2155. doi: 10.1046/j.1471-4159.1998.70052147.x. [DOI] [PubMed] [Google Scholar]
- 71.Nairn AC, Shenolikar S. The role of protein phosphatases in synaptic transmission, plasticity and neuronal development. Curr Opin Neurobiol. 1992;2:296–301. doi: 10.1016/0959-4388(92)90118-5. [DOI] [PubMed] [Google Scholar]
- 72.Nestler EJ, Hope BT, Widnell KL. Drug addiction: a model for the molecular basis of neural plasticity. Neuron. 1993;11:995–1006. doi: 10.1016/0896-6273(93)90213-b. [DOI] [PubMed] [Google Scholar]
- 73.Neve KA, Kozlowski MR, Marshall JF. Plasticity of neostriatal dopamine receptors after nigrostriatal injury: relationship to recovery of sensorimotor functions and behavioral supersensitivity. Brain Res. 1982;244:33–44. doi: 10.1016/0006-8993(82)90901-5. [DOI] [PubMed] [Google Scholar]
- 74.Ozawa S, Kamiya H, Tsuzuki K. Glutamate receptors in the mammalian central nervous system. Prog Neurobiol. 1998;54:581–618. doi: 10.1016/s0301-0082(97)00085-3. [DOI] [PubMed] [Google Scholar]
- 75.Patel J, Zinkand WC, Klika AB, Mangano TJ, Keith RA, Salama AI. 6,7-Dinitroquinoxaline-2,3-dione blocks the cytotoxicity of N-methyl-d-aspartate and kainate, but not quisqualate, in cortical cultures. J Neurochem. 1990;55:114–121. doi: 10.1111/j.1471-4159.1990.tb08828.x. [DOI] [PubMed] [Google Scholar]
- 76.Pich EM, Pagliusi SR, Tessari M, Talabot-Ayer D, Hooft van Huijsduijnen R, Chiamulera C. Common neural substrates for the addictive properties of nicotine and cocaine. Science. 1997;275:83–86. doi: 10.1126/science.275.5296.83. [DOI] [PubMed] [Google Scholar]
- 77.Rajadhyaksha A, Leveque J, Macias W, Barczak A, Konradi C. Molecular components of striatal plasticity: the various routes of cyclic AMP pathways. Dev Neurosci. 1998;20:204–215. doi: 10.1159/000017314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Robertson LM, Kerppola TK, Vendrell M, Luk D, Smeyne RJ, Bocchiaro C, Morgan JI, Curran T. Regulation of c-fos expression in transgenic mice requires multiple interdependent transcription control elements. Neuron. 1995;14:241–252. doi: 10.1016/0896-6273(95)90282-1. [DOI] [PubMed] [Google Scholar]
- 79.Ruppersberg JP, Mosbacher J, Gunther W, Schoepfer R, Fakler B. Studying block in cloned N-methyl-d-aspartate (NMDA) receptors. Biochem Pharmacol. 1993;46:1877–1885. doi: 10.1016/0006-2952(93)90627-9. [DOI] [PubMed] [Google Scholar]
- 80.Sakurai SY, Cha JH, Penney JB, Young AB. Regional distribution and properties of [3H]MK-801 binding sites determined by quantitative autoradiography in rat brain. Neuroscience. 1991;40:533–543. doi: 10.1016/0306-4522(91)90139-f. [DOI] [PubMed] [Google Scholar]
- 81.Sanna E, Head GA, Hanbauer I. Evidence for a selective localization of voltage-sensitive Ca2+ channels in nerve cell bodies of corpus striatum. J Neurochem. 1986;47:1552–1557. doi: 10.1111/j.1471-4159.1986.tb00794.x. [DOI] [PubMed] [Google Scholar]
- 82.Schild D, Geiling H, Bischofberger J. Imaging of L-type Ca2+ channels in olfactory bulb neurones using fluorescent dihydropyridine and a styryl dye. J Neurosci Methods. 1995;59:183–190. doi: 10.1016/0165-0270(94)00181-f. [DOI] [PubMed] [Google Scholar]
- 83.Schoepfer R, Monyer H, Sommer B, Wisden W, Sprengel R, Kuner T, Lomeli H, Herb A, Kohler M, Burnashev N, Günther W, Ruppersberg P, Seeburg P. Molecular biology of glutamate receptors. Prog Neurobiol. 1994;42:353–357. doi: 10.1016/0301-0082(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 84.Schulman H. Protein phosphorylation in neuronal plasticity and gene expression. Curr Opin Neurobiol. 1995;5:375–381. doi: 10.1016/0959-4388(95)80051-4. [DOI] [PubMed] [Google Scholar]
- 85.Self DW, Nestler EJ. Molecular mechanisms of drug reinforcement and addiction. Annu Rev Neurosci. 1995;18:463–495. doi: 10.1146/annurev.ne.18.030195.002335. [DOI] [PubMed] [Google Scholar]
- 86.Sheng M, McFadden G, Greenberg ME. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- 87.Sheng M, Thompson MA, Greenberg ME. CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 88.Siegel BV, Jr, Buchsbaum MS, Bunney WE, Jr, Gottschalk LA, Haier RJ, Lohr JB, Lottenberg S, Najafi A, Nuechterlein KH, Potkin SG, Wu JC. Cortical-striatal-thalamic circuits and brain glucose metabolic activity in 70 unmedicated male schizophrenic patients. Am J Psychiatry. 1993;150:1325–1336. doi: 10.1176/ajp.150.9.1325. [DOI] [PubMed] [Google Scholar]
- 89.Standaert DG, Testa CM, Young AB, Penney JB., Jr Organization of N-methyl-d-aspartate glutamate receptor gene expression in the basal ganglia of the rat. J Comp Neurol. 1994;343:1–16. doi: 10.1002/cne.903430102. [DOI] [PubMed] [Google Scholar]
- 90.Stanton PK, Sarvey JM. Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J Neurosci. 1984;4:3080–3088. doi: 10.1523/JNEUROSCI.04-12-03080.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sucher NJ, Lei SZ, Lipton SA. Calcium channel antagonists attenuate NMDA receptor-mediated neurotoxicity of retinal ganglion cells in culture. Brain Res. 1991;551:297–302. doi: 10.1016/0006-8993(91)90944-q. [DOI] [PubMed] [Google Scholar]
- 92.Sun P, Enslen H, Myung PS, Maurer RA. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8:2527–2539. doi: 10.1101/gad.8.21.2527. [DOI] [PubMed] [Google Scholar]
- 93.Thayer SA, Murphy SN, Miller RJ. Widespread distribution of dihydropyridine-sensitive calcium channels in the central nervous system. Mol Pharmacol. 1986;30:505–509. [PubMed] [Google Scholar]
- 94.Thompson MA, Ginty DD, Bonni A, Greenberg ME. L-type voltage-sensitive Ca2+ channel activation regulates c-fos transcription at multiple levels. J Biol Chem. 1995;270:4224–4235. doi: 10.1074/jbc.270.9.4224. [DOI] [PubMed] [Google Scholar]
- 95.Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- 96.Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, Hitzemann R, Chen AD, Dewey SL, Pappas N. Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature. 1997;386:830–833. doi: 10.1038/386830a0. [DOI] [PubMed] [Google Scholar]
- 97.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 98.Walker D, De Waard M. Subunit interaction sites in voltage-dependent Ca2+ channels: role in channel function. Trends Neurosci. 1998;21:148–154. doi: 10.1016/s0166-2236(97)01200-9. [DOI] [PubMed] [Google Scholar]
- 99.Weiss JH, Hartley DM, Koh J, Choi DW. The calcium channel blocker nifedipine attenuates slow excitatory amino acid neurotoxicity. Science. 1990;247:1474–1477. doi: 10.1126/science.247.4949.1474. [DOI] [PubMed] [Google Scholar]
- 100.Westenbroek RE, Ahlijanian MK, Catterall WA. Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature. 1990;347:281–284. doi: 10.1038/347281a0. [DOI] [PubMed] [Google Scholar]
- 101.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yin JCP, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T. Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]