

Abstract

Coordination between the N-terminal gate and the catalytic core of topoisomerase II allows the proper capture, cleavage, and transport of DNA during the catalytic cycle. Because the activities of these domains are tightly linked, it has been difficult to discern their individual contributions to enzyme–DNA interactions and drug mechanism. To further address the roles of these domains, we analyzed the activity of the catalytic core of human topoisomerase IIα. The catalytic core and the wild-type enzyme both maintained higher levels of cleavage with negatively (as compared to positively) supercoiled plasmid, indicating that the ability to distinguish supercoil handedness is embedded within the catalytic core. However, the catalytic core alone displayed little ability to cleave DNA substrates that did not intrinsically provide the enzyme with a transport segment (i.e., substrates that did not contain crossovers). Finally, in contrast to interfacial topoisomerase II poisons, covalent poisons did not enhance DNA cleavage mediated by the catalytic core. This distinction allowed us to further characterize the mechanism of etoposide quinone, a drug metabolite that functions primarily as a covalent poison. Etoposide quinone retained some ability to enhance DNA cleavage mediated by the catalytic core, indicating that it still can function as an interfacial poison. These results further define the distinct contributions of the N-terminal gate and the catalytic core to topoisomerase II function. The catalytic core senses the handedness of DNA supercoils during cleavage, while the N-terminal gate is critical for capturing the transport segment and for the activity of covalent poisons.
Type II topoisomerases are ubiquitous enzymes that regulate DNA supercoiling and remove knots and tangles from the genetic material.1−7 Human cells encode two isoforms, topoisomerase IIα and topoisomerase IIβ. Both enzymes function by passing an intact double helix (the transport segment or T-segment) through a transient double-stranded break that they generate in a separate segment of DNA (the gate segment or G-segment).2,4−7
Because type II topoisomerases generate double-stranded DNA breaks during their catalytic cycle, they can have a profound effect on genomic stability.2,3,5,7−10 To this point, type II topoisomerases are the targets for a number of highly successful anticancer drugs that act by increasing the levels of covalent enzyme-cleaved DNA complexes (cleavage complexes).2,4,7−10 Drugs that act in this manner are called topoisomerase II poisons. The conversion of drug-stabilized cleavage complexes to permanent strand breaks by the movements of replication and transcription complexes can initiate cell death pathways.2,4,7−10
Topoisomerase II poisons can be grouped into two classes: interfacial and covalent.2,4,7,9−11 Interfacial poisons interact with both the protein and the DNA in the active site of the enzyme. They intercalate into the cleaved scissile bond and physically block the ability of topoisomerase II to ligate the cleaved DNA strand.2,9,10,12 In contrast, covalent poisons appear to act distally to the active site of topoisomerase II and form covalent adducts with the enzyme.2,11,13 Although it has not been demonstrated directly, it has been proposed that covalent poisons increase levels of cleavage complexes (at least in part) by closing the N-terminal gate of topoisomerase II.14,15
Topoisomerase II functions as a homodimeric protein. On the basis of homology with DNA gyrase, the enzyme can be divided into three domains: the N-terminal domain, the catalytic core, and the C-terminal domain.1−7 The N-terminal domain contains the site of ATP binding and hydrolysis. ATP binding triggers dimerization of the N-terminal domain, which helps capture the T-segment and closes the N-terminal protein gate.16 This action induces the transport of the T-segment through the open gate in the G-segment.1−7,16 The catalytic core of topoisomerase II contains the active site tyrosine that cleaves and covalently attaches to the DNA. It also forms a second protein gate that allows the T-segment to exit the enzyme following strand passage. The C-terminal domain is the least understood portion of topoisomerase II. It is highly variable and contains nuclear localization sequences and sites of phosphorylation.2,6,7 Although it is not necessary for catalytic activity, the C-terminal domain is involved in the recognition of DNA geometry during strand passage and provides different type II topoisomerases with unique capabilities. In human topoisomerase IIα, the C-terminal domain allows the enzyme to relax positively supercoiled (i.e., overwound) DNA that accumulates ahead of replication forks 10 times faster than it does negatively supercoiled (i.e., underwound) molecules.17,18 In contrast, topoisomerase IIβ relaxes positive and negative DNA supercoils at the same rate.17,18
Although only topoisomerase IIα is able to recognize the handedness of DNA supercoils during relaxation, both isoforms are able to distinguish between positive and negative supercoils during DNA cleavage.19 Topoisomerase IIα and topoisomerase IIβ maintain higher levels of cleavage complexes with underwound as compared to overwound molecules. In spite of the important role played by the C-terminal domain in distinguishing DNA geometry during relaxation, this portion of the enzyme is not involved in recognizing supercoil handedness during DNA cleavage.18 It is not obvious which domain of topoisomerase II is responsible for this recognition. Given the role of the N-terminal gate in capturing the T-segment and that of the catalytic core in cleaving the G-segment, both are likely candidates.
Finally, despite the suggested role of the N-terminal gate in the actions of covalent poisons,14,15 the portion of topoisomerase II that mediates the effects of these compounds has not been established. To further complicate this issue, covalent poisons have been shown to adduct cysteine residues in both the N-terminal gate and the catalytic core of topoisomerase IIα.20,21
To address the issues described above, we characterized the DNA cleavage activity of the catalytic core of human topoisomerase IIα. Results indicate that the catalytic core is sufficient for the enzyme to recognize DNA supercoil handedness during the cleavage reaction. However, the catalytic core alone displayed little ability to cleave DNA substrates that did not intrinsically provide the enzyme with a transport segment (i.e., substrates that did not contain crossovers). Finally, the N-terminal gate is necessary for cleavage enhancement by covalent poisons.
Experimental Procedures
Enzymes and Materials
The truncated hTop2αΔ1175 (containing amino acids 1–1175) was constructed as described previously.22 Human topoisomerase IIα and hTop2αΔ1175 were expressed in Saccharomyces cerevisiae JEL-1Δtop1 and purified as described by Kingma et al.23 The catalytic core of human topoisomerase IIα (containing residues 431–1193)24 was expressed in yeast cells and purified using a Ni2+-nitriloacetic acid agarose column (Qiagen) as described previously.25,26 The enzyme was stored at −80 °C as a 1.5 mg/mL stock in 50 mM Tris-HCl (pH 7.8), 0.1 mM ethylenediaminetetraacetic acid (EDTA), 750 mM KCl, and 5% glycerol. For all of the enzymes examined, the concentration of dithiothreitol carried over from purification protocols was <2 μM in final reaction mixtures.
Negatively supercoiled pBR322 plasmid DNA was prepared using a Plasmid Mega Kit (Qiagen) as described by the manufacturer. Positively supercoiled pBR322 DNA was prepared by treating negatively supercoiled molecules with recombinant Archaeoglobus fulgidus reverse gyrase.17,27 The number of positive supercoils was comparable to the number of negative supercoils in the original pBR322 preparations.17 For experiments comparing positively and negatively supercoiled DNA, the negatively supercoiled plasmid was processed in a manner identical to that of the positively supercoiled molecules except that reverse gyrase was omitted from reaction mixtures. Relaxed pBR322 plasmid DNA was generated by treating negatively supercoiled pBR322 with topoisomerase I and purified as described previously.28
Etoposide, benzoquinone, and thymoquinone were purchased from Sigma-Aldrich. Etoposide was stored at room temperature as a 20 mM solution in 100% dimethyl sulfoxide (DMSO). Benzoquinone was stored at −20 °C as a 20 mM solution in water. Thymoquinone was stored at 4 °C as a 40 mM solution in 100% DMSO. The quinolone CP-115,953 was a gift from T. D. Gootz and P. R. McGuirk (Pfizer). It was stored at −20 °C as a 40 mM solution in 0.1 N NaOH and diluted 5-fold with 10 mM Tris-HCl (pH 7.9) immediately prior to use. Etoposide quinone was synthesized as described previously29−31 and stored at 4 °C as a 20 mM solution in 100% DMSO.
Plasmid DNA Cleavage
DNA cleavage reactions were carried out using the procedure of Fortune and Osheroff.32 Reaction mixtures contained 10 nM pBR322 and 150 nM wild-type topoisomerase IIα, 80 nM hTop2αΔ1175, or 430 nM catalytic core in a total of 20 μL of cleavage buffer [10 mM Tris-HCl (pH 7.9), 5 mM MgCl2, 100 mM KCl, 0.1 mM EDTA, and 2.5% (v/v) glycerol]. Reaction mixtures were incubated at 37 °C for 6 min, and enzyme–DNA cleavage complexes were trapped by the addition of 2 μL of 5% sodium dodecyl sulfate (SDS) followed by 2 μL of 250 mM EDTA (pH 8.0). Proteinase K (2 μL of a 0.8 mg/mL solution) was added, and samples were incubated at 45 °C for 30 min to digest the enzyme. Samples were mixed with 2 μL of agarose loading dye [60% sucrose in 10 mM Tris-HCl (pH 7.9), 0.5% bromophenol blue, and 0.5% xylene cyanol FF], heated at 45 °C for 2 min, and subjected to electrophoresis in 1% agarose gels in 40 mM Tris-acetate (pH 8.3) and 2 mM EDTA containing 0.5 μg/mL ethidium bromide. DNA bands were visualized by UV light and quantified using an Alpha Innotech digital imaging system. DNA cleavage was monitored by the conversion of supercoiled plasmid to linear molecules.
Note that lower levels of hTop2αΔ1175 were used in reaction mixtures, because in the presence of Mg2+, the protein displays levels of DNA cleavage ∼2-fold higher than those of wild-type topoisomerase IIα.22 Conversely, higher levels of the catalytic core were used because they display levels of baseline DNA cleavage that are lower than those of the wild-type enzyme in reaction mixtures containing Mg2+ (see Figure 2).
Figure 2.
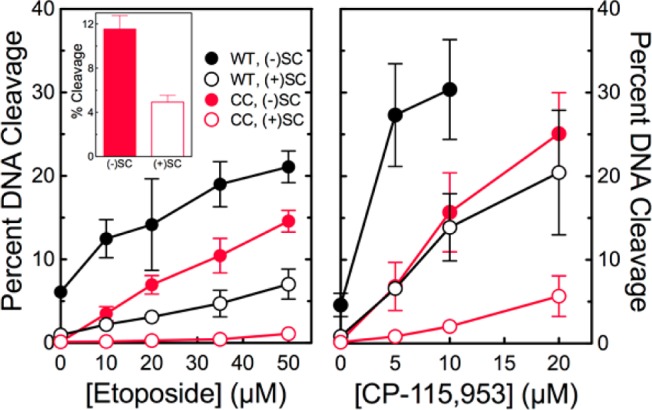
The catalytic core of human topoisomerase IIα preferentially cleaves negatively supercoiled DNA in the presence of etoposide and CP-115,953. The ability of wild-type topoisomerase IIα (WT, black) and the catalytic core (CC, red) to cleave negatively [(−)SC, filled circles] or positively [(+)SC, empty circles] supercoiled plasmid DNA in the presence of etoposide (left) or CP-115,953 (right) is shown. Results for CP-115,953 are not shown at concentrations above 10 μM with the wild-type enzyme because the drug induced multiple cleavage events per plasmid. The inset shows cleavage induced by the catalytic core in the presence of 250 μM etoposide. Error bars represent the standard deviation of at least three independent experiments.
DNA cleavage reactions were carried out in the presence of 0–250 μM etoposide, 0–20 μM CP-115,953, or 0–100 μM benzoquinone, thymoquinone, or etoposide quinone. Alternatively, MgCl2 was omitted from the cleavage buffer, and reaction mixtures contained 0–5 mM CaCl2.
DNA Cleavage Site Utilization
DNA cleavage sites were mapped using a modification33 of the procedure of O’Reilly and Kreuzer.34 A unique derivative of pUC19 (pMP-bcr6) was used as the substrate for DNA cleavage site utilization experiments. The substrate was generated by modifying pUC19 to include a region of PML intron 6 that contains an established breakpoint associated with therapy-related acute promyelocytic leukemia. The genomic DNA of human CEM cells was prepared using standard protocols according to the manufacturer’s instructions for the DNeasy Blood and Tissue Kit (Qiagen). 5′-GGGGGGATCCTTCTGCAAAGGCCACCTACC-3′ and 5′-AGGGGAAGCTTCACTGTCCCCATTCTCAGC-3′ primers were synthesized for amplifying a 319 bp region of the PML gene (44157–44475 of GenBank accession number NG029036) with the CEM genomic DNA as a template. Purified clones containing the insert were transformed into XL1-Blue cells and sequenced with M13 primers by Vantage (Vanderbilt Technologies for Advanced Sequencing). pMP-bcr6 was linearized by treatment with Acc651. Terminal 5′-phosphates were removed by treatment with calf intestinal alkaline phosphatase and replaced with [32P]phosphate using T4 polynucleotide kinase and [γ-32P]ATP. The DNA was treated with EcoRI, and the 2968 bp singly end-labeled fragment was purified from the small EcoRI–Acc651 fragment by passage through a CHROMA SPIN+TE-100 column (Clontech).
Reaction mixtures contained 1 nM labeled pMP-bcr6 and 60 nM wild-type human topoisomerase IIα or 115 nM catalytic core in 50 μL of DNA cleavage buffer (containing 5 mM Ca2+) in the absence or presence of compounds. Reaction mixtures were incubated at 37 °C for 6 min, and enzyme–DNA cleavage complexes were trapped by the addition of 5 μL of 5% SDS followed by 4 μL of 250 mM EDTA (pH 8.0). Proteinase K (5 μL of a 0.8 mg/mL solution) was added, and samples were incubated at 45 °C for 30 min to digest the enzyme. DNA products were precipitated with ethanol and resuspended in 5 μL of polyacrylamide gel loading buffer [10% agarose gel loading buffer, 80% formamide, 100 mM Tris-borate (pH 8.3), and 2 mM EDTA]. Samples were subjected to electrophoresis in denaturing 6% polyacrylamide sequencing gels. Gels were dried in vacuo, and DNA cleavage products were visualized with a Bio-Rad Molecular Imager FX.
Results and Discussion
Recognition of Supercoil Geometry
Human type II topoisomerases can distinguish the handedness of DNA supercoils during scission and maintain levels of cleavage complexes with negatively supercoiled DNA that are ∼2–4-fold higher than those seen with positively supercoiled molecules (Figure 1, left).19Several lines of evidence indicate that the C-terminal domain of topoisomerase II is not involved in this recognition.18,19 Most notably, deletion of the C-terminal domain of human topoisomerase IIα does not affect the ability of the enzyme to preferentially cleave underwound molecules.18
Figure 1.
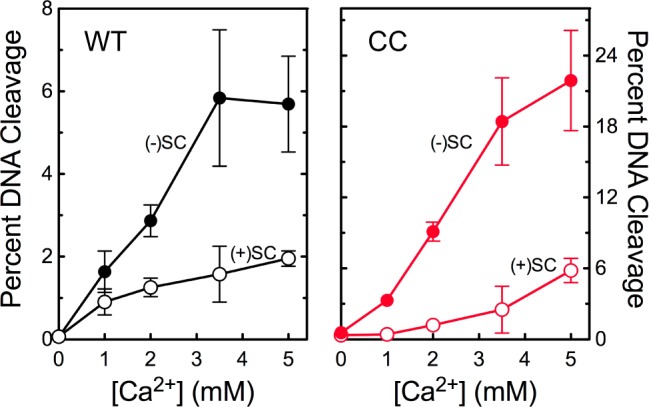
The catalytic core of human topoisomerase IIα preferentially cleaves negatively supercoiled DNA in the presence of Ca2+. The ability of wild-type topoisomerase IIα (WT, left) and the catalytic core (CC, right) to cleave negatively [(−)SC, filled circles] or positively [(+)SC, empty circles] supercoiled plasmid DNA is shown. Error bars represent the standard deviation of at least three independent experiments.
It is not clear which portion of topoisomerase IIα is responsible for the recognition of DNA geometry during cleavage. Although DNA cleavage is mediated by the catalytic core of the enzyme, rates of cleavage can be modulated by changes in the N-terminal gate.35,36 Therefore, the ability of the catalytic core of topoisomerase IIα to cleave negatively and positively supercoiled DNA was assessed. As shown in Figure 1 (right), the catalytic core retained the ability to recognize supercoil handedness and preferentially cleaved negatively supercoiled plasmid.
The experiments described above substituted Ca2+ for Mg2+ as the required divalent metal ion37 to generate levels of DNA cleavage that were high enough to reliably quantify enzyme-mediated DNA scission under conditions that did not include anticancer drugs. Previous work demonstrated that Ca2+ does not affect DNA cleavage site selection by topoisomerase IIα or the ability of type II enzymes to recognize DNA supercoil geometry.19,38 Although the catalytic core displayed a preference for negatively supercoiled DNA that was similar to that of wild-type topoisomerase IIα (Figure 1), it exhibited unexpectedly high levels of DNA cleavage in the presence of Ca2+. The underlying basis for this high level of Ca2+-supported DNA cleavage is not known.
To further confirm the ability of the catalytic core to distinguish supercoil geometry, DNA cleavage was examined in the presence of Mg2+ and topoisomerase II poisons (Figure 2). Etoposide and CP-115,953 are well-characterized interfacial poisons that do not intercalate into DNA (which would change the apparent topology of the plasmid substrate).39,40 Similar to results seen with Ca2+, the catalytic core maintained higher levels of cleavage complexes with negatively as compared to positively supercoiled DNA in the presence of Mg2+ and interfacial topoisomerase II poisons.
These results provide strong evidence that the ability to distinguish the geometry of DNA supercoils during cleavage is embedded in the catalytic core of human topoisomerase IIα.
Role of the N-Terminal Gate of Topoisomerase IIα in Mediating Interactions with the T-Segment of DNA
Topoisomerase II binds negatively supercoiled DNA at sites of helix–helix crossovers, and it has been proposed that this ability to bind DNA crossovers allows topoisomerase II to distinguish between relaxed and supercoiled molecules.38,41 Although it has not been rigorously demonstrated, it is believed that the two DNA helices at the crossover become the G- and T-segments.
Studies with oligonucleotides indicate that binding of the T-segment greatly stimulates topoisomerase II-mediated cleavage of the G-segment.36,42 These findings suggest that the T-segment plays an important role in the ability of topoisomerase II to identify and relax DNA supercoils. Although the N-terminal gate of the protein plays a critical role in capturing the T-segment and passing it through the DNA gate, it is not known whether the initial interaction with the T-segment is mediated by this portion of topoisomerase II or by the catalytic core of the enzyme. Therefore, we examined the ability of the catalytic core of topoisomerase IIα to cleave DNA that does not contain intrinsic crossovers.
In the first experiment, a linearized plasmid was used as the DNA substrate. Etoposide was included in experiments to increase levels of DNA scission. As seen in Figure 3, wild-type human topoisomerase IIα was able to cleave the DNA in the absence or presence of drug. Cleavage was enhanced when ATP was added to reaction mixtures. One interpretation of this finding is that in the presence of ATP, the N-terminal gate is able to capture the T-segment, thereby stimulating DNA scission. In contrast, the catalytic core of the enzyme was unable to cleave the linearized plasmid under any of the conditions described above. Furthermore, no DNA cleavage was seen when the concentration of the catalytic core was increased 5-fold or when Mg2+ was used in place of Ca2+ in reaction mixtures (not shown). These findings suggest that the N-terminal gate plays a critical role in mediating interactions with the T-segment.
Figure 3.
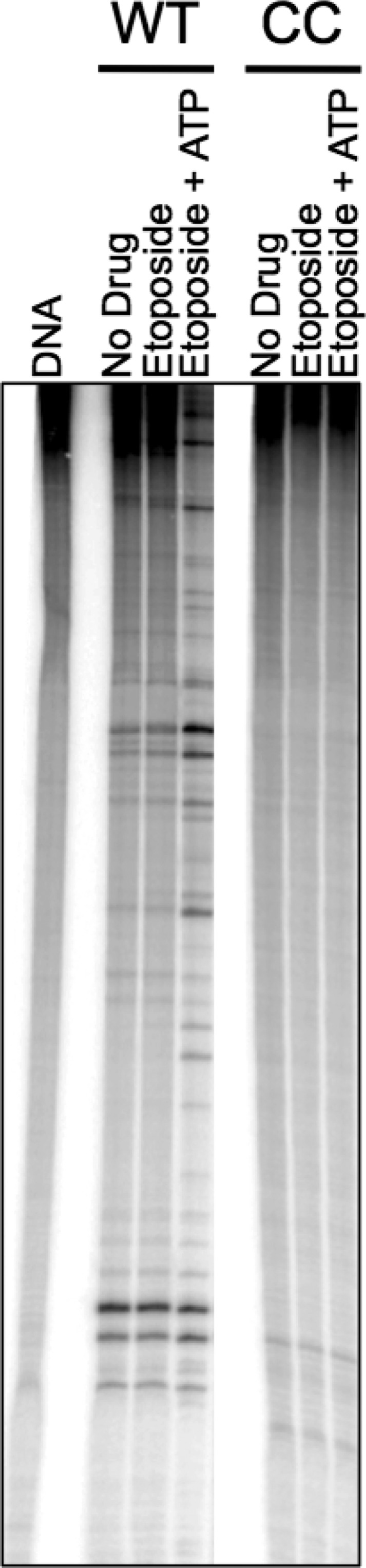
Effects of etoposide on sites of DNA cleavage mediated by wild-type human topoisomerase IIα and the catalytic core. An autoradiogram of a polyacrylamide gel depicting DNA sites cleaved by wild-type topoisomerase IIα (WT) and the catalytic core (CC) is shown. Reaction mixtures contained no enzyme (DNA), enzyme in the absence of drug (No Drug), or enzyme in the presence of 20 μM etoposide with or without 1 mM ATP. Lanes shown were taken from different portions of the same gel. The autoradiogram is representative of three independent experiments.
To further explore this conclusion, the ability of wild-type topoisomerase IIα and the catalytic core to cleave relaxed DNA was examined (Figure 4). In contrast to negatively supercoiled plasmid, which contains numerous inherent DNA crossovers, relaxed molecules contain few if any intrinsic sites of helix–helix justaposition. For this reason, topoisomerase II preferentially cleaves negatively supercoiled over relaxed molecules (compare scission in Figures 2 and 4).38
Figure 4.
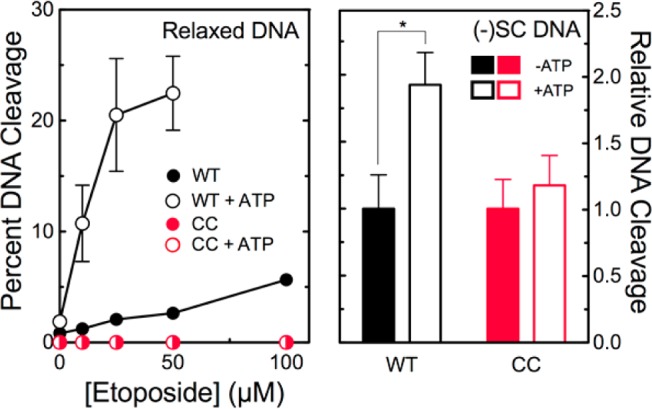
Effects of ATP on cleavage of relaxed and negatively supercoiled DNA by wild-type human topoisomerase IIα and the catalytic core. The ability of wild-type topoisomerase IIα (WT, black) and the catalytic core (CC, red) to cleave relaxed DNA (left) or negatively supercoiled DNA (right) in the presence of etoposide is shown. Experiments were conducted in the absence (filled symbols) or presence (empty symbols) of 500 μM ATP. Experiments with negatively supercoiled DNA included 50 μM etoposide. Error bars represent the standard deviation of at least three independent experiments. The statistically significant difference is denoted with an asterisk (*p < 0.01).
Wild-type topoisomerase IIα was able to cleave relaxed plasmid (Figure 4, left), suggesting that the full-length enzyme can capture a transport helix even when there are few intrinsic crossovers in the DNA substrate. When ATP was added to reaction mixtures, levels of cleavage increased ∼9-fold (left), consistent with the conclusion that ATP induces closing of the N-terminal gate, stabilizing the capture of the T-segment. Although ATP also stimulates the ability of the intact enzyme to cleave negatively supercoiled plasmid (Figure 4, right), this enhancement is much smaller (∼2-fold). The presence of high levels of helix–helix crossovers in the negatively supercoiled plasmid facilitates interactions between the intact enzyme and helix–helix crossovers, partially obviating the need for T-segment capture by the addition of ATP.
In contrast to topoisomerase IIα that contained its N-terminal gate, the catalytic core was unable to cleave relaxed plasmid in the absence or presence of ATP (Figure 4, left). Moreover, the addition of ATP did not enhance cleavage of negatively supercoiled plasmid (right).
These experiments lead to the conclusion that the catalytic core of human topoisomerase IIα cannot efficiently capture the T-segment. More importantly, they also lead to the conclusion that the N-terminal gate of the enzyme plays a critical role in mediating the initial interaction with the T-segment. In the absence of this protein domain, the catalytic core is able to cleave supercoiled plasmid primarily because the substrate carries intrinsic DNA crossovers. Thus, the substrate is able to present the T-segment to the enzyme, even in the absence of the protein domain that normally mediates the interaction with the second DNA double helix.
Role of the N-Terminal Gate of Human Topoisomerase IIα in Mediating the Actions of Covalent Poisons
A number of environmental, dietary, and medicinal compounds act as covalent topoisomerase II poisons.13−15,21,43−48 Compared to interfacial poisons, the mechanistic basis for the actions of covalent poisons is less well understood. These compounds form adducts with the enzyme.11,13,20,21 At present, only cysteine adducts have been characterized. It has been proposed that the ability of covalent poisons to close the N-terminal gate plays an important role in mediating their ability to increase levels of topoisomerase II–DNA cleavage complexes.14,15 However, modified residues have been identified in both the N-terminal gate and the catalytic core.20,21
To explore the role of the N-terminal gate in the actions of covalent poisons, the effects of benzoquinone43 and thymoquinone48 on DNA cleavage mediated by wild-type topoisomerase IIα and the catalytic core were examined. Effects on cleavage mediated by a truncated human enzyme lacking the C-terminal domain (hTop2αΔ1175) also were determined as a control. Benzoquinone and thymoquinone displayed similar abilities to increase the level of cleavage complexes formed with full-length topoisomerase IIα or hTop2αΔ1175 and negatively supercoiled plasmid (Figure 5). This finding demonstrates that the C-terminal domain plays no significant role in mediating the actions of covalent topoisomerase II poisons. In marked contrast, neither compound displayed any ability to enhance DNA cleavage mediated by the catalytic core. This result indicates that the N-terminal gate of topoisomerase IIα is critical for the actions of covalent poisons. Furthermore, it provides yet another distinction between interfacial poisons [which do not require the N-terminal gate to stimulate topoisomerase II-mediated DNA cleavage (see Figure 2)] and covalent poisons.
Figure 5.
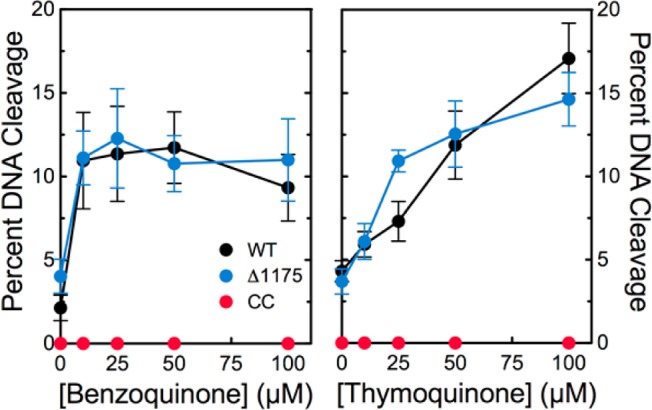
Covalent poisons do not enhance DNA cleavage mediated by the catalytic core of human topoisomerase IIα. Effects of benzoquinone (left) and thymoquinone (right) on DNA cleavage mediated by wild-type human topoisomerase IIα (black), the catalytic core (red), and hTop2αΔ1175 (blue) are shown. Error bars represent the standard deviation of at least three independent experiments.
In further contrast to interfacial poisons, covalent topoisomerase II poisons display the hallmark characteristic of inactivating the enzyme when the two are incubated prior to the addition of DNA.13,43 Even though the inactivation can be explained by the ability of covalent poisons to close the N-terminal protein gate (thus preventing DNA from entering the active site of topoisomerase II),15,20 this proposed mechanism is controversial. Indeed, treatment of human topoisomerase IIα with benzoquinone or PCB quinones blocks the ability of the enzyme to cleave oligonucleotides that are able to bind to the protein and diffuse into the active site without entering through the protein gate.20 This finding implies that mechanisms besides the proposed closing of the N-terminal gate may contribute to enzyme inactivation by covalent poisons.
To address this controversy, benzoquinone and thymoquinone were incubated with wild-type topoisomerase IIα, hTop2αΔ1175, or the catalytic core prior to the addition of negatively supercoiled plasmid, and the effects on DNA cleavage were assessed. Assays with the catalytic core were carried out in the presence of Ca2+ to raise baseline levels of DNA cleavage (see Figure 1). As seen in Figure 6, benzoquinone and thymoquinone inactivated all three enzymes. Thus, while covalent poisons require the N-terminal gate to stimulate DNA cleavage mediated by topoisomerase II, they do not require this portion of the protein to inactivate the enzyme. Although the closing of the N-terminal gate may contribute to topoisomerase II inactivation, clearly other mechanisms can produce a similar effect.
Figure 6.
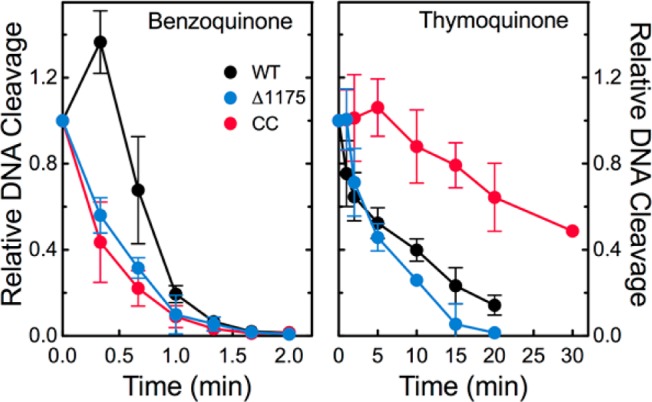
Covalent poisons inactivate human topoisomerase IIα enzymes when incubated with the protein prior to the addition of DNA. The DNA cleavage activities of wild-type human topoisomerase IIα (black), the catalytic core (red), and hTop2αΔ1175 (blue) were monitored in the presence of 50 μM benzoquinone (left) or 50 μM thymoquinone (right). DNA cleavage levels were calculated relative to cleavage levels induced when the drug and the enzyme were not incubated prior to DNA addition. Error bars represent the standard deviation of at least three independent experiments.
Mechanism of Action of Etoposide Quinone
Etoposide has been linked to the generation of treatment-related acute myeloid leukemias,49−52 and etoposide quinone, a metabolite of etoposide,30,53 has been implicated in this process.54 These leukemias feature rearrangements of the MLL gene at chromosomal band 11q23 and are believed to be triggered by drug-induced DNA cleavage events mediated by human type II topoisomerases.49−52 Although etoposide is an interfacial topoisomerase II poison, several studies indicate that etoposide quinone acts primarily as a covalent poison.31,55,56 However, it is not known whether the covalent interaction of the quinone with topoisomerase II masks the fact that the metabolite also can act as an interfacial poison.
Previous studies indicate that the pendant E-ring of etoposide is critical to its actions as an interfacial poison (Figure 7).57−59 Substitution of either the 3′- or 5′-methoxy groups with a hydroxyl moiety has little effect on drug activity.55,60 Thus, the catechol metabolite of etoposide displays an activity (and mechanism) similar to that of the parent drug. Removal of the 4′-hydroxyl moiety or substitution by a methoxy group greatly compromises the activity of etoposide.57,59 However, it is not known whether substitution by a carbonyl group affects the ability of etoposide to function as an interfacial poison.
Figure 7.
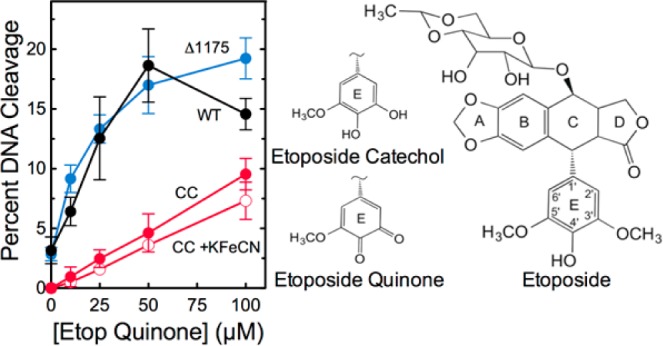
Etoposide quinone enhances DNA cleavage mediated by the catalytic core of human topoisomerase IIα. The effects of etoposide quinone on DNA cleavage mediated by wild-type human topoisomerase IIα (black), the catalytic core (red), and hTop2αΔ1175 (blue) are shown. Cleavage reactions with etoposide quinone and the catalytic core that included 10 μM K3[Fe(CN)6] (empty circles) also are shown. The structure of etoposide and the E-rings of etoposide catechol and etoposide quinone are depicted at right. Error bars represent the standard deviation of at least three independent experiments.
The experiments shown in Figure 5 provide a method for determining whether etoposide quinone can function as an interfacial poison in addition to acting as a covalent poison. If etoposide quinone functions purely as a covalent poison, it should have no effect on DNA cleavage mediated by the catalytic core. However, if it retains the ability to act as an interfacial poison (despite the fact that it can also act as a covalent poison), it should display at least some activity against the catalytic core. As seen in Figure 7, etoposide quinone retains partial activity against the catalytic core of human topoisomerase IIα. It is possible that this activity reflects the fact that a portion of the etoposide quinone preparation has been reduced over time to the catechol, which is an interfacial topoisomerase II poison. To address this possibility, the effect of 10 μM K3[Fe(CN)6] on the ability of etoposide quinone to enhance DNA cleavage mediated by the catalytic core was assessed. The oxidant, which converts the catechol to the quinone,55 had little effect on the actions of etoposide quinone against the catalytic core. Although etoposide quinone functions primarily as a covalent topoisomerase II poison, these findings indicate that it still retains a modest ability to act as an interfacial poison of human topoisomerase IIα.
Conclusions
The N-terminal gate and the catalytic core of type II topoisomerases work coordinately to capture, cleave, and transport DNA during the DNA strand passage reaction. Although this coordination is essential for proper enzyme function, it has obscured the individual contributions of these two domains to important aspects of enzyme–DNA interactions and drug mechanism. Previous studies have shown that the C-terminal domain of human topoisomerase IIα is responsible for DNA geometry recognition during relaxation; however, the present results indicate that the catalytic core is the portion of the enzyme that senses the handedness of DNA supercoils during the cleavage reaction (Figure 8). Conversely, the N-terminal gate plays critical roles in the capture of the T-segment.
Figure 8.

Domains of human topoisomerase IIα and their involvement in DNA geometry recognition and drug activity. The enzyme is divided into three domains: the N-terminal gate (blue, amino acid residues 1–430), which contains the ATPase active site; the catalytic core (red, residues 431–1193), which contains the TOPRIM domain (the portion that binds the catalytic divalent metal ions) and the DNA cleavage/ligation active site tyrosine residue (Y805); and the C-terminal domain (green, residues 1193–1531).2−7 Functions associated with each domain are indicated. Three cysteine residues (C) are indicated in the N-terminal gate (C300, C392, and C405 from left to right). Cys300 has been identified as a site of attachment of isothiocyanate-based covalent topoisomerase II poisons.21 However, it has not been established whether attachment at this residue plays a role in the actions of these agents.21 Cys392 and Cys405 have been identified as sites of attachment of quinone-based covalent poisons,20 and substitution of alanine residues at these positions results in an ∼2-fold resistance to a variety of covalent poisons.20,46,48
The use of different topoisomerase IIα constructs also provided considerable insight into the actions of covalent topoisomerase II poisons (Figure 8). Whereas the N-terminal gate is necessary for the enhancement of DNA cleavage by these compounds, residues within the catalytic core may be responsible for the inhibition of catalytic function that follows the incubation of covalent poisons with topoisomerase II prior to the addition of DNA. Finally, the ability of interfacial poisons, but not covalent poisons, to enhance DNA cleavage mediated by the catalytic core allowed us to further characterize the mechanism of action of etoposide quinone. Although this important drug metabolite functions primarily as a covalent poison, it still retains the ability to act in an interfacial manner.
Acknowledgments
Human topoisomerase IIα was generously provided by Jo Ann Byl. We are grateful to Kendra R. Vann and Lorena Infante Lara for critical reading of the manuscript. R. Hunter Lindsey, Jr., performed the majority of the experiments reported in this paper and was instrumental in formulating the overall concept underlying this project. Unfortunately, he lost his valiant battle against cancer before the manuscript could be completed.
This research was supported by Grant GM033944 (N.O.) from the National Institutes of Health. M.P. was a trainee under Grant T32 CA09582 from the National Institutes of Health. R.E.A. was supported by Graduate Research Fellowship DGE-0909667 from the National Science Foundation. J.E.D. and S.L.M. were supported by funds from the Lipscomb University College of Pharmacy and Health Sciences.
The authors declare no competing financial interest.
Author Status
∇ Deceased.
Funding Statement
National Institutes of Health, United States
References
- Liu Z.; Deibler R. W.; Chan H. S.; Zechiedrich L. (2009) The why and how of DNA unlinking. Nucleic Acids Res. 37, 661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deweese J. E.; Osheroff N. (2009) The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 37, 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss J. L. (2009) DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 9, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y.; Leo E.; Zhang H.; Marchand C. (2010) DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 17, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos S. M.; Tretter E. M.; Schmidt B. H.; Berger J. M. (2011) All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 12, 827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry A. C., and Osheroff N. (2013) DNA topoisomerases: Type II. In Encyclopedia of Biological Chemistry, pp 163–168, Elsevier Inc., Amsterdam. [Google Scholar]
- McClendon A. K.; Osheroff N. (2007) DNA topoisomerase II, genotoxicity, and cancer. Mutat. Res. 623, 83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss J. L. (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 9, 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y.; Marchand C. (2012) Interfacial inhibitors: Targeting macromolecular complexes. Nat. Rev. Drug Discovery 11, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y. (2013) Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 8, 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketron A. C.; Osheroff N. (2014) Phytochemicals as anticancer and chemopreventive topoisomerase II poisons. Phytochem. Rev. 13, 19–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. C.; Li T. K.; Farh L.; Lin L. Y.; Lin T. S.; Yu Y. J.; Yen T. J.; Chiang C. W.; Chan N. L. (2011) Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 333, 459–462. [DOI] [PubMed] [Google Scholar]
- Wang H.; Mao Y.; Chen A. Y.; Zhou N.; LaVoie E. J.; Liu L. F. (2001) Stimulation of topoisomerase II-mediated DNA damage via a mechanism involving protein thiolation. Biochemistry 40, 3316–3323. [DOI] [PubMed] [Google Scholar]
- Bender R. P.; Lehmler H. J.; Robertson L. W.; Ludewig G.; Osheroff N. (2006) Polychlorinated biphenyl quinone metabolites poison human topoisomerase IIα: Altering enzyme function by blocking the N-terminal protein gate. Biochemistry 45, 10140–10152. [DOI] [PubMed] [Google Scholar]
- Mondrala S.; Eastmond D. A. (2010) Topoisomerase II inhibition by the bioactivated benzene metabolite hydroquinone involves multiple mechanisms. Chem.-Biol. Interact. 184, 259–268. [DOI] [PubMed] [Google Scholar]
- Schmidt B. H.; Osheroff N.; Berger J. M. (2012) Structure of a topoisomerase II-DNA-nucleotide complex reveals a new control mechanism for ATPase activity. Nat. Struct. Mol. Biol. 19, 1147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClendon A. K.; Rodriguez A. C.; Osheroff N. (2005) Human topoisomerase IIα rapidly relaxes positively supercoiled DNA: Implications for enzyme action ahead of replication forks. J. Biol. Chem. 280, 39337–39345. [DOI] [PubMed] [Google Scholar]
- McClendon A. K.; Gentry A. C.; Dickey J. S.; Brinch M.; Bendsen S.; Andersen A. H.; Osheroff N. (2008) Bimodal recognition of DNA geometry by human topoisomerase IIα: Preferential relaxation of positively supercoiled DNA requires elements in the C-terminal domain. Biochemistry 47, 13169–13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClendon A. K.; Osheroff N. (2006) The geometry of DNA supercoils modulates topoisomerase-mediated DNA cleavage and enzyme response to anticancer drugs. Biochemistry 45, 3040–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender R. P.; Ham A. J.; Osheroff N. (2007) Quinone-induced enhancement of DNA cleavage by human topoisomerase IIα: Adduction of cysteine residues 392 and 405. Biochemistry 46, 2856–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R. K.; Zhou N.; Lyu Y. L.; Tsai Y. C.; Lu C. H.; Kerrigan J.; Chen Y. T.; Guan Z.; Hsieh T. S.; Liu L. F. (2011) Dietary isothiocyanate-induced apoptosis via thiol modification of DNA topoisomerase IIα. J. Biol. Chem. 286, 33591–33600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey J. S.; Osheroff N. (2005) Impact of the C-terminal domain of topoisomerase IIα on the DNA cleavage activity of the human enzyme. Biochemistry 44, 11546–11554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma P. S.; Greider C. A.; Osheroff N. (1997) Spontaneous DNA lesions poison human topoisomerase IIα and stimulate cleavage proximal to leukemic 11q23 chromosomal breakpoints. Biochemistry 36, 5934–5939. [DOI] [PubMed] [Google Scholar]
- Wendorff T. J.; Schmidt B. H.; Heslop P.; Austin C. A.; Berger J. M. (2012) The structure of DNA-bound human topoisomerase IIα: Conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 424, 109–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biersack H.; Jensen S.; Gromova I.; Nielsen I.; Westergaard O.; Andersen A. (1996) Active heterodimers are formed from human DNA topoisomerase IIα and IIβ isoforms. Proc. Natl. Acad. Sci. U.S.A. 93, 8288–8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oestergaard V. H.; Bjergbaek L.; Skouboe C.; Giangiacomo L.; Knudsen B. R.; Andersen A. H. (2004) The transducer domain is important for clamp operation in human DNA topoisomerase IIα. J. Biol. Chem. 279, 1684–1691. [DOI] [PubMed] [Google Scholar]
- Rodriguez A. C. (2002) Studies of a positive supercoiling machine: Nucleotide hydrolysis and a multifunctional “latch” in the mechanism of reverse gyrase. J. Biol. Chem. 277, 29865–29873. [DOI] [PubMed] [Google Scholar]
- Aldred K. J.; Breland E. J.; Vlčková V.; Strub M.-P.; Neuman K. C.; Kerns R. J.; Osheroff N. (2014) Role of the water–metal ion bridge in mediating interactions between quinolones and Escherichia coli topoisomerase IV. Biochemistry 53, 5558–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemec J. (1986) Epipodophyllotoxin quinone glucoside derivatives, method of production and use. Patent 4,609,644.
- Lovett B. D.; Strumberg D.; Blair I. A.; Pang S.; Burden D. A.; Megonigal M. D.; Rappaport E. F.; Rebbeck T. R.; Osheroff N.; Pommier Y. G.; Felix C. A. (2001) Etoposide metabolites enhance DNA topoisomerase II cleavage near leukemia-associated MLL translocation breakpoints. Biochemistry 40, 1159–1170. [DOI] [PubMed] [Google Scholar]
- Jacob D. A.; Mercer S. L.; Osheroff N.; Deweese J. E. (2011) Etoposide quinone is a redox-dependent topoisomerase II poison. Biochemistry 50, 5660–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune J. M.; Osheroff N. (1998) Merbarone inhibits the catalytic activity of human topoisomerase IIα by blocking DNA cleavage. J. Biol. Chem. 273, 17643–17650. [DOI] [PubMed] [Google Scholar]
- Baldwin E. L.; Byl J. A.; Osheroff N. (2004) Cobalt enhances DNA cleavage mediated by human topoisomerase IIα in vitro and in cultured cells. Biochemistry 43, 728–735. [DOI] [PubMed] [Google Scholar]
- O’Reilly E. K.; Kreuzer K. N. (2002) A unique type II topoisomerase mutant that is hypersensitive to a broad range of cleavage-inducing antitumor agents. Biochemistry 41, 7989–7997. [DOI] [PubMed] [Google Scholar]
- Baird C. L.; Harkins T. T.; Morris S. K.; Lindsley J. E. (1999) Topoisomerase II drives DNA transport by hydrolyzing one ATP. Proc. Natl. Acad. Sci. U.S.A. 96, 13685–13690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Jung S. R.; Heo K.; Byl J. A.; Deweese J. E.; Osheroff N.; Hohng S. (2012) DNA cleavage and opening reactions of human topoisomerase IIα are regulated via Mg2+-mediated dynamic bending of gate-DNA. Proc. Natl. Acad. Sci. U.S.A. 109, 2925–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deweese J. E.; Osheroff N. (2010) The use of divalent metal ions by type II topoisomerases. Metallomics 2, 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechiedrich E. L.; Osheroff N. (1990) Eukaryotic topoisomerases recognize nucleic acid topology by preferentially interacting with DNA crossovers. EMBO J. 9, 4555–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W.; Rowe T.; Glisson B.; Yalowich J.; Liu L. (1984) Role of topoisomerase II in mediating epipodophyllotoxin-induced DNA cleavage. Cancer Res. 44, 5857–5860. [PubMed] [Google Scholar]
- Robinson M. J.; Martin B. A.; Gootz T. D.; McGuirk P. R.; Moynihan M.; Sutcliffe J. A.; Osheroff N. (1991) Effects of quinolone derivatives on eukaryotic topoisomerase II: A novel mechanism for enhancement of enzyme-mediated DNA cleavage. J. Biol. Chem. 266, 14585–14592. [PubMed] [Google Scholar]
- Roca J.; Berger J. M.; Wang J. C. (1993) On the simultaneous binding of eukaryotic DNA topoisomerase II to a pair of double-stranded DNA helices. J. Biol. Chem. 268, 14250–14255. [PubMed] [Google Scholar]
- Corbett A. H.; Zechiedrich E. L.; Osheroff N. (1992) A role for the passage helix in the DNA cleavage reaction of eukaryotic topoisomerase II: A two-site model for enzyme-mediated DNA cleavage. J. Biol. Chem. 267, 683–686. [PubMed] [Google Scholar]
- Lindsey R. H. Jr.; Bromberg K. D.; Felix C. A.; Osheroff N. (2004) 1,4-Benzoquinone is a topoisomerase II poison. Biochemistry 43, 7563–7574. [DOI] [PubMed] [Google Scholar]
- Bandele O. J.; Osheroff N. (2008) (−)-Epigallocatechin gallate, a major constituent of green tea, poisons human type II topoisomerases. Chem. Res. Toxicol. 21, 936–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandele O. J.; Clawson S. J.; Osheroff N. (2008) Dietary polyphenols as topoisomerase II poisons: B ring and C ring substituents determine the mechanism of enzyme-mediated DNA cleavage enhancement. Chem. Res. Toxicol. 21, 1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketron A. C.; Gordon O. N.; Schneider C.; Osheroff N. (2013) Oxidative metabolites of curcumin poison human type II topoisomerases. Biochemistry 52, 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmel M. A.; Byl J. A.; Osheroff N. (2013) Epimerization of green tea catechins during brewing does not affect the ability to poison human type II topoisomerases. Chem. Res. Toxicol. 26, 622–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley R. E.; Osheroff N. (2014) Natural products as topoisomerase II poisons: Effects of thymoquinone on DNA cleavage mediated by human topoisomerase IIα. Chem. Res. Toxicol. 27, 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix C. A.; Kolaris C. P.; Osheroff N. (2006) Topoisomerase II and the etiology of chromosomal translocations. DNA Repair 5, 1093–1108. [DOI] [PubMed] [Google Scholar]
- Cowell I. G.; Sondka Z.; Smith K.; Lee K. C.; Manville C. M.; Sidorczuk-Lesthuruge M.; Rance H. A.; Padget K.; Jackson G. H.; Adachi N.; Austin C. A. (2012) Model for MLL translocations in therapy-related leukemia involving topoisomerase IIβ-mediated DNA strand breaks and gene proximity. Proc. Natl. Acad. Sci. U.S.A. 109, 8989–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezoe S. (2012) Secondary leukemia associated with the anti-cancer agent, etoposide, a topoisomerase II inhibitor. Int. J. Environ. Res. Public Health 9, 2444–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton M.; Lindsey R. H. Jr.; Felix C. A.; Grimwade D.; Osheroff N. (2014) Topoisomerase II and leukemia. Ann. N.Y. Acad. Sci. 1310, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Maanen J. M. S.; Retl J.; de Vries J.; Pinedo H. M. (1988) Mechanism of action of antitumor drug etoposide: A review. J. Natl. Cancer Inst. 80, 1526–1533. [DOI] [PubMed] [Google Scholar]
- Felix C. A.; Walker A. H.; Lange B. J.; Williams T. M.; Winick N. J.; Cheung N. K.; Lovett B. D.; Nowell P. C.; Blair I. A.; Rebbeck T. R. (1998) Association of CYP3A4 genotype with treatment-related leukemia. Proc. Natl. Acad. Sci. U.S.A. 95, 13176–13181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob D. A.; Gibson E. G.; Mercer S. L.; Deweese J. E. (2013) Etoposide catechol is an oxidizable topoisomerase II poison. Chem. Res. Toxicol. 26, 1156–1158. [DOI] [PubMed] [Google Scholar]
- Smith N. A.; Byl J. A.; Mercer S. L.; Deweese J. E.; Osheroff N. (2014) Etoposide quinone is a covalent poison of human topoisomerase IIβ. Biochemistry 53, 3229–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilstermann A. M.; Bender R. P.; Godfrey M.; Choi S.; Anklin C.; Berkowitz D. B.; Osheroff N.; Graves D. E. (2007) Topoisomerase II-drug interaction domains: Identification of substituents on etoposide that interact with the enzyme. Biochemistry 46, 8217–8225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long B. H. (1987) Structure-activity relationships of podophyllin congeners that inhibit topoisomerase II. Natl. Cancer Inst. Monogr. 123–127. [PubMed] [Google Scholar]
- Bender R. P.; Jablonksy M. J.; Shadid M.; Romaine I.; Dunlap N.; Anklin C.; Graves D. E.; Osheroff N. (2008) Substituents on etoposide that interact with human topoisomerase IIα in the binary enzyme-drug complex: Contributions to etoposide binding and activity. Biochemistry 47, 4501–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha B. K.; Politi P. M.; Eliot H. M.; Kerrigan D.; Pommier Y. (1990) Structure-activity relations, cytotoxicity and topoisomerase II dependent cleavage induced by pendulum ring analogues of etoposide. Eur. J. Cancer 26, 590–593. [DOI] [PubMed] [Google Scholar]