

Conspectus
Protein structures are not static but sample different conformations over a range of amplitudes and time scales. These fluctuations may involve relatively small changes in bond angles or quite large rearrangements in secondary structure and tertiary fold. The equilibrium between discrete structural substates on the microsecond to millisecond time scale is sometimes termed conformational exchange. Protein dynamics and conformational exchange are believed to provide the basis for many important activities, such as protein–protein and protein–ligand interactions, enzymatic activity and protein allostery; however, for many proteins, the dynamics and conformational exchange that lead to function are poorly defined.
Spectroscopic methods, such as NMR, are among the most important methods to explore protein dynamics and conformational exchange; however, they are difficult to implement in some systems and with some types of exchange events. Site-directed spin labeling (SDSL) is an EPR based approach that is particularly well-suited to high molecular-weight systems such as membrane proteins. Because of the relatively fast time scale for EPR spectroscopy, it is an excellent method to examine exchange. Conformations that are in exchange are captured as distinct populations in the EPR spectrum, and this feature when combined with the use of methods that can shift the free energy of conformational substates allows one to identify regions of proteins that are in dynamic exchange. In addition, modern pulse EPR methods have the ability to examine conformational heterogeneity, resolve discrete protein states, and identify the substates in exchange.
Protein crystallography has provided high-resolution models for a number of membrane proteins; but because of conformational exchange, these models do not always reflect the structures that are present when the protein is in a native bilayer environment. In the case of the Escherichia coli vitamin B12 transporter, BtuB, the energy coupling segment of this protein undergoes a substrate-dependent unfolding, which acts to couple this outer-membrane protein to the inner-membrane protein TonB. EPR spectroscopy demonstrates that the energy coupling segment is in equilibrium between ordered and disordered states, and that substrate binding shifts this equilibrium to favor an unfolded state. However, in crystal structures of BtuB, this segment is resolved and folded within the protein, and neither the presence of this equilibrium nor the substrate-induced change is revealed. This is a result of the solute environment and the crystal lattice, both of which act to stabilize one conformational substate of the transporter.
Using SDSL, it can be shown that conformational exchange is present in other regions of BtuB and in other members of this transporter family. Conformational exchange has also been examined in systems such as the plasma membrane SNARE protein, syntaxin 1A, where dynamics are controlled by regulatory proteins such as munc18. Regulating the microsecond to millisecond time scale dynamics in the neuronal SNAREs is likely to be a key feature that regulates assembly of the SNAREs and neurotransmitter release.
Membrane proteins perform extremely important functions in the cell, but present a significant challenge in the area of structural biology. They are generally more difficult to express and crystallize than globular proteins, and high-resolution NMR is limited in molecular weight and requires that the protein be incorporated into a micelle or other membrane mimetic environment. Despite these difficulties, the number of high-resolution membrane protein structures has dramatically increased in recent years, although they continue to represent just a small fraction of the total number of structures in the Protein Data Bank.
In cases where we have a good high-resolution crystal structure, a molecular description of the function of a membrane protein is usually still lacking. Missing from these structural models are molecular descriptions of alternate protein conformations, conformations that are present in equilibrium, and conformations that represent higher energy or excited states. These conformational states and equilibria are significant, because they represent structural states that may mediate protein function.
In this Account, we will describe a few cases from our own work in which protein conformational exchange has been quantitated and describe how the presence of an equilibrium among protein conformations may bias what is seen in high-resolution crystal structures.
Site-Directed Spin Labeling and Protein Conformational Exchange
Site-directed spin labeling (SDSL) when combined with EPR spectroscopy provides a powerful approach to study protein dynamics and structure, and the interested reader will find a number of excellent reviews on this method that employ both continuous wave and pulse EPR techniques.1−6 Continuous wave EPR spectra from nitroxide labels that are engineered into proteins contain information on fast motions in the 0.1 to 100 ns time scale, and the spectra are known to reflect secondary structure, backbone motion and tertiary contact of the label.7,8 Processes such as conformational exchange occur on a much slower time scale, usually in the microsecond to millisecond time scale. Although these slower motions do not modulate the shape of an EPR spectrum, conformational exchange typically places the label in two or more environments where the label executes different motions on the nanosecond time scale. As a result, two or more components representing different modes of motion of the nitroxide side chain are usually seen in the EPR spectrum when a protein segment is in conformational exchange. Monitoring and quantitating these components in the EPR spectrum provides an excellent approach to characterize the energetics and presence of conformational exchange.
Pulse EPR techniques are now routinely used to measure dipolar interactions between pairs of spin labels in proteins. Methods such as double electron–electron resonance (DEER; also referred to as pulse electron double resonance, PELDOR) can yield distances and distance distributions between nitroxide labels with high resolution out to distances of 60 Å or more.5 This method is being used to characterize membrane protein conformational transitions,9,10 to validate crystal structures in membrane environments,11 and to define the structure of protein complexes.12 Pulse EPR measurements such as DEER are presently carried out in frozen samples and can reveal structural heterogeneity. Although there is no information on dynamics in these data, protein conformations that are in exchange and significantly populated are expected to result in multiple components in the distance distribution.
To characterize conformational exchange, either from the distance distribution observed by DEER or from motional components that are seen in an EPR spectrum, it is important to verify that these components are in equilibrium and that they arise from protein conformers and not from rotameric states of the spin label. In the following presentation, we discuss two examples where EPR spectroscopy has been used to characterize and quantitate protein conformational exchange. In the final section of this Account, we summarize the general approaches that may be used to identify and quantitate conformational exchange with EPR spectroscopy.
TonB-Dependent Transport
Our interest in protein conformational exchange began with an investigation of a family of highly specific bacterial outer-membrane transport proteins that are believed to derive energy from the inner-membrane electrochemical potential by coupling to the transperiplasmic protein TonB.13 These TonB-dependent transporters (TBDTs) bind and transport scarce solutes, including various forms of iron, vitamin B12, nickel, and carbohydrate.14 TBDTs are binding sites for phage and they also act as highly specific receptors for colicins, which are protein antibiotics produced by bacteria.15 This transport system appears to function in the uptake of a broad range of substrates,14 and in some bacteria as many as 67 putative TBDTs have been identified.16
Over 40 high-resolution structural models have been generated for 12 different TonB-dependent transporters, including the Escherichia coli iron transporters FepA,17 FhuA,18,19 and FecA20,21 and the vitamin B12 transporter BtuB.22 TonB-dependent transporters are structurally homologous (Figure 1a) and consist of two distinct domains: a β-barrel formed from 22 antiparallel strands and a 135–160 residue N-terminal globular region, which fills the barrel. The substrates for these transporters bind to large external loops, which vary in size among the different transporters. On the periplasmic surface, all TonB-dependent transporters possess a highly conserved energy coupling motif termed the Ton box. The Ton box is a six or seven residue segment located near the N-terminus that interacts with TonB through β-strand pairing.23,24 Despite the abundance of high-resolution structures of these TBDTs, the molecular mechanisms that mediate transport remain uncharacterized, as they do for many other transport proteins.
Figure 1.
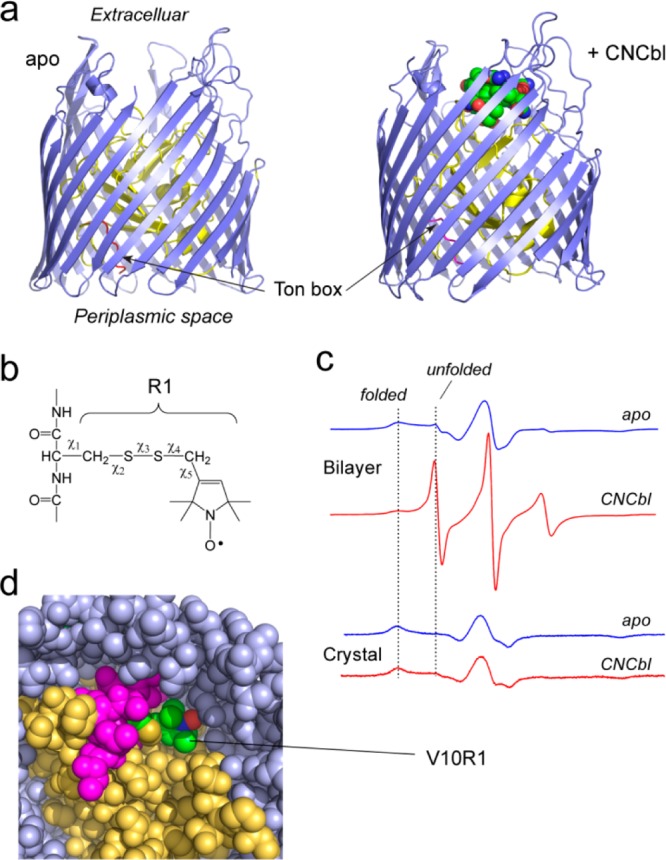
Substrate-dependent conformational changes in BtuB are not seen in the crystal environment. (a) Crystal structures of the Escherichia coli vitamin B12 transporter, BtuB, in the apo and ligand bound forms (PDB IDs 1NQE and 1NQH, respectively). BtuB and all TBDTs consist of a 22-standed β-barrel (blue) and an N-terminal core domain (yellow), which fills the barrel. A conserved motif called the Ton box (red) directly interacts with the inner membrane protein TonB. In both the apo and substrate bound crystal forms, the Ton box is folded within the interior of the protein. (b) The nitroxide side chain, R1, is formed by reaction of a methanethiosulfonate label with a reactive cysteine residue. This label is one of several different labels that have been developed for site-directed spin labeling.6 (c) EPR spectra from BtuB labeled at position 10 in the Ton box (BtuB/V10R1).28 The substrate induced change in the EPR spectrum is observed in bilayers but is not seen in the environment of the protein crystal. (d) Crystal structure of BtuB/V10R1 in the presence of substrate (PDB ID 3M8D).28 The label is in tertiary contact within the protein interior, consistent with the EPR spectra in panel c. All structures were rendered using PyMol (Schrödinger, Portland, OR). Panels c and d reproduced from ref (28). Copyright 2010 Biophysical Society.
EPR Spectroscopy Reveals Structural States That Are Not Observed by Crystallography
In the Escherichia coli vitamin B12 transporter BtuB, SDSL indicates that interactions between BtuB and the inner membrane protein TonB are initiated by a transmembrane signaling event, where substrate binding on the extracellular surface of BtuB unfolds the Ton box25,26 so that it projects up to 30 Å into the periplasmic space.27,28
Spin labels, such as the nitroxide side chain R1 (Figure 1b), yield EPR spectra that report upon the state of the Ton box, and in bilayers, EPR spectra from the Ton box clearly reveal this unfolding transition (Figure 1c). The unfolded Ton box is highly disordered and is likely to interact with TonB through a process resembling a fly casting mechanism,29 where interactions with TonB are enhanced by the dynamic state of the Ton box. Remarkably, no evidence for this transition is found when the apo and ligand bound crystal structures of BtuB are compared (Figure 1a); and in these structures, the Ton box remains folded in the protein interior in the presence of substrate. Consistent with this result, the substrate-dependent transition is not observed by EPR when the spin-labeled protein is removed from the bilayer and crystallized under the same conditions as those used to obtain the structures. Diffraction of the spin labeled protein crystal of BtuB (Figure 1d) also indicates that the Ton box and its associated spin label remain folded within the interior of the protein in the presence of substrate and that the incorporation of the spin label into the Ton box does not significantly perturb the structure of the Ton box or the core of BtuB.28 Thus, the two methods, EPR and crystallography, are in agreement provided the measurements are made under the same experimental conditions.
Why Is the Structural Transition in the Ton Box Seen in Bilayers but Not Observed in the Protein Crystal?
By use of both EPR and protein crystallography, the source of the differences between bilayer and crystal conditions have been determined.28 There appear to be two environmental factors that play a significant role in the case of the BtuB Ton box, and both act to alter the energy of the folded state relative to the unfolded state. The first factor involves the solutes or precipitants used in crystallography, and the second is the crystal lattice itself.
In protein crystallography, solutes or combinations of solutes are used as precipitants, and they help drive the protein out of solution to produce a protein crystal. In membrane protein crystallography, poly(ethylene glycol)s (PEGs) are frequently used as precipitants usually in combination with salts. Poly(ethylene glycol)s belong to a family of solutes sometimes termed stabilizing osmolytes, and there is significant literature on the effect of these solutes on protein stability and activity.30−32 These solutes are excluded from a region around the protein interface that is accessible to water. This exclusion, which may simply be due to the size of the solute, requires energy, and as a result these osmolytes raise the energy of the protein and reduce its solubility.33 Solutes such as PEGs are known to increase the stability of a protein in its native form relative to its unfolded form and can even be used to refold proteins that are destabilized by mutagenesis. This stabilization occurs because solutes such as PEGs raise the energy of the unfolded (more hydrated) form relative to the folded (native) form. The action of stabilizing osmolytes such as PEGs is in contrast with the effect of solutes such as urea and guanidinium. These destabilizing solutes interact with the protein and help solubilize the protein in solution, thereby stabilizing the unfolded state relative to the native state.33
A careful examination of the EPR spectra from the BtuB Ton box indicates that there are two motional components in the spectra that represent the folded and unfolded states, and it is possible to demonstrate that these states exist in equilibrium as depicted in Figure 2. Shown in Figure 2b is a titration of the EPR spectrum from the BtuB Ton box with increasing concentrations of PEG 400 beginning with the substrate bound state. The narrower component in the EPR spectra results from the unfolded state of the Ton box and decreases with increasing PEG concentrations, while the broader component results from the folded state of the Ton box and increases with PEG addition. From these spectra, the free energy difference between folded and unfolded forms may be determined and plotted as a function of solution osmolality. A linear behavior with a negative slope is expected and observed, where the slope is related to the solvent accessible surface area that is unfolded during the conformational transition. In the case of PEG 400, the free energy of this structural transition is altered by 0.7 kcal/mol per molal of solute, which is in rough agreement with the expected change in hydrated surface area.34
Figure 2.
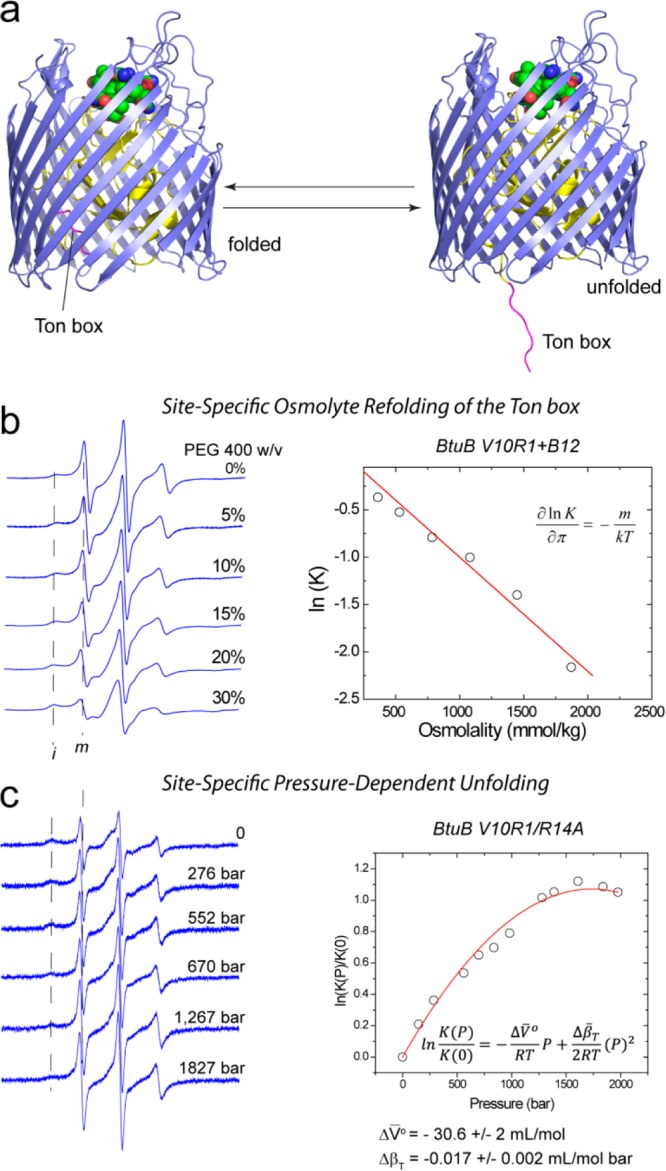
The BtuB Ton box is in conformational exchange. (a) When substrate is bound, the Ton box (in red) is in equilibrium between folded and unfolded states. (b) Titration of BtuB/V10R1 with PEG 400 shifts the equilibrium from the unfolded to the folded state (seen as components m and i in the EPR spectra), and the energy to unfold the Ton box (−kT ln(K)) increases in a linear fashion as the solution osmolality, π, is increased.34 (c) Pressure has opposite effect on the Ton box equilibrium, and it promotes Ton box unfolding (unpublished). In this case, the Ton box in BtuB is partially destabilized by an R14A mutation. Fitting the conformational energy change with pressure yields the volume change, ΔV̅0, and isothermal compressibility change, Δβ̅T, during the Ton box unfolding transition.52
As mentioned above, the crystal lattice also plays a role in the Ton box equilibrium. As seen in Figure 1c, the substrate-dependent unfolding is largely blocked for BtuB in the protein crystal. In fact, careful examination of the EPR spectrum from the crystal indicates that a small fraction of the Ton box unfolds in the presence of substrate (representing less than 0.5% of the total spins). From the change in equilibrium when BtuB is moved from the bilayer to the crystal environment, the free energy of this conformational equilibrium is estimated to shift by about 3 kcal/mol.28 To determine the contributions made by the lattice, the EPR spectrum of the labeled BtuB, V10R1, may be recorded in the crystallization buffer under conditions where the protein concentration is lowered and protein crystals do not form. A comparison of this spectrum with those obtained from bilayer and crystal environments indicates that about half of this 3 kcal/mol energy change is due to the solute conditions and about half is due to the crystal lattice. Since the protein–protein contacts in the unit cell are made with the BtuB β-barrel and do not sterically interfere with the Ton box unfolding, interactions made by BtuB within the protein lattice of the crystal must act to lower the energy of the folded state of Ton box.
Thus, both the solutes used in protein crystallography and the crystal lattice act to alter the energetics of the Ton box equilibrium in BtuB, and both factors act to stabilize the least hydrated or the most compact form of the Ton box. Conformational exchange and crystallization conditions also account for differences observed between the detergent and cubic (meso phase) structures of BtuB.35
Are the Solute Effects Seen for the BtuB Ton Box Unique, or Do They Extend to Other Sites and Membrane Proteins?
In the case of BtuB and related TBDTs, the effects of solutes have generally been observed in regions of the protein that function in molecular recognition.35 In addition to the Ton box in BtuB, the Ton box in FecA is also highly sensitive to solutes, and this segment in FecA appears to be in conformational exchange between a state where it is interacting with or dissociated from an N-terminal transcriptional regulatory motif.36 The loop regions of TBDTs are also highly sensitive to solutes such as PEGs.37 Shown in Figure 3a are EPR spectra recorded for site 188 in the second extracellular loop of BtuB. In the apo state, the EPR spectrum is characteristic of that expected from a loop that is dynamic. When Ca2+ (a coligand of vitamin B12) is bound to BtuB, the spectrum changes indicating that motion of the label is more ordered and motion in the loop is dampened. When PEG3350 is added together with Ca2+, the spectrum broadens and the nitroxide side chain comes into tertiary contact with another region of the protein.
Figure 3.
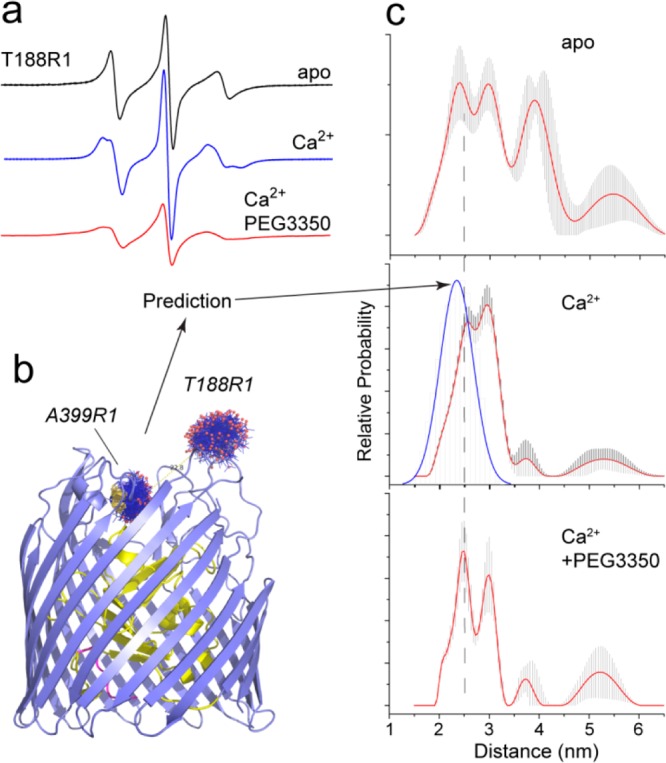
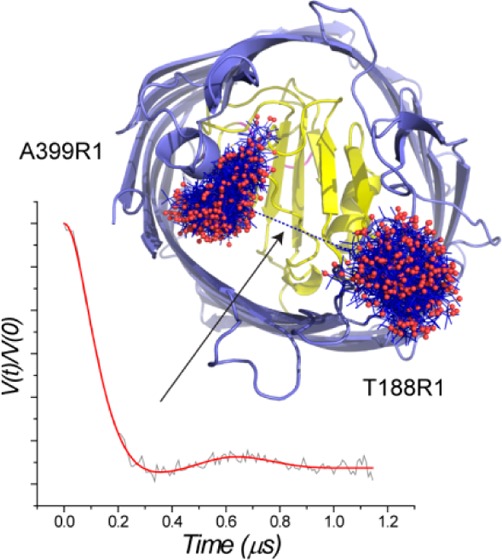
Conformational heterogeneity in the extracellular ligand binding loops of BtuB. (a) X-band EPR spectra from T188R1, which is located in the second extracellular loop of BtuB, in the apo state and in the presence of calcium and calcium with the addition of 30% w/v PEG 3350.37 (b) Positions and allowable conformers of the R1 label at sites 399 and 188 in BtuB. Position 399 is in the barrel and does not exhibit changes in line shape with substrate or PEG addition. The label conformers and the expected distance distribution (blue trace in part c, second panel) were calculated using the PYMOL plug-in MTSSL Wizard,38 which takes into account steric constraints imposed by the structure but does not otherwise bias label conformers based upon torsional potentials or experimentally populated rotamer states.53 (c) Distance distributions obtained by DEER between 399R1 and 188R1 in the apo state, in the presence of Ca2+, and in the presence of Ca2+ with 30% w/v PEG 3350 (top to bottom, respectively).37 The expected distance distribution in the Ca2+-bound state based upon the corresponding crystal structure PDB ID 1NQG is shown (blue trace, center panel). The error bars in the distribution represent fits that have an RMSD within 15% of the best solution. All DEER data were analyzed with the software package DeerAnalysis.54
In BtuB, distance distributions have been measured using DEER between sites on the loops and positions in the barrel. These measurements provide information on conformational heterogeneity in the loops, and Figure 3c shows distributions measured between sites 188 and 399 (Figure 3b). The distribution is broad in the apo state and distances range from 15 to 40 Å. In the presence of Ca2+, the distribution narrows with the most populated distance near 30 Å. The predicted distance distribution based upon the Ca2+-bound crystal structure may be estimated from the sterically accessible label conformers that are compatible with the crystal structure.38 As seen in Figure 3c, the peak distance measured experimentally for Ca2+-bound BtuB is about 7 Å longer than that predicted from the crystal structure, suggesting that the bilayer structure samples a more open state than that seen in the protein crystal. Addition of the precipitant PEG3350 alters the distance distribution so that shorter distances are more highly populated, and the most populated distance observed in the presence of PEG3350 (24 Å) is close to that predicted based upon the crystal structure (23 Å).
These EPR measurements indicate that the ligand binding loops of these TBDTs exist in multiple conformations and that these conformations are in equilibrium. Moreover, observations on these transporters indicate that crystal structures of transporters and other dynamic membrane proteins represent one compact substate among an ensemble of conformers that are normally sampled by the protein.
Conformational Exchange in the Membrane Fusion Machinery
Membrane fusion is mediated by SNAREs (soluble N-ethylmaleimide-sensitive factor attachment receptor proteins). In neuronal exocytosis, syntaxin 1A and SNAP25 are plasma membrane associated SNAREs that assemble with the vesicle membrane SNARE synaptobrevin into a four-helical bundle that drives neurotransmitter release.39 The core SNARE complex is extremely stable, but assembly of these SNARE proteins is highly regulated and coordinated, so that membrane fusion is rapid and precisely timed. Conformational fluctuations in syntaxin may control SNARE assembly, which is believed to take place in an N-to C-terminal direction in the SNARE forming motifs. As a result, some of our work on the fusion process has been focused on the structure and dynamics of syntaxin.
Syntaxin appears to be in equilibrium between two forms: an open form, where the segment that participates in SNARE complex formation, the H3 motif, is dissociated from the regulatory Habc domain, and a closed form, where the H3 motif is associated with the Habc domain (see Figure 4a). Distance measurements from the H3 to the Habc domain made by DEER indicate that the central portion of the H3 domain is largely in a closed configuration when the soluble fragment of syntaxin is examined. However, when full-length membrane reconstituted syntaxin is examined, pulse EPR demonstrates that the configuration of the protein is different and predominantly in an open state.40 This difference in behavior with environment likely occurs because of the weak tendency of the SNARE forming heptad repeats (which are laterally amphipathic) to associate with the membrane interface.41
Figure 4.
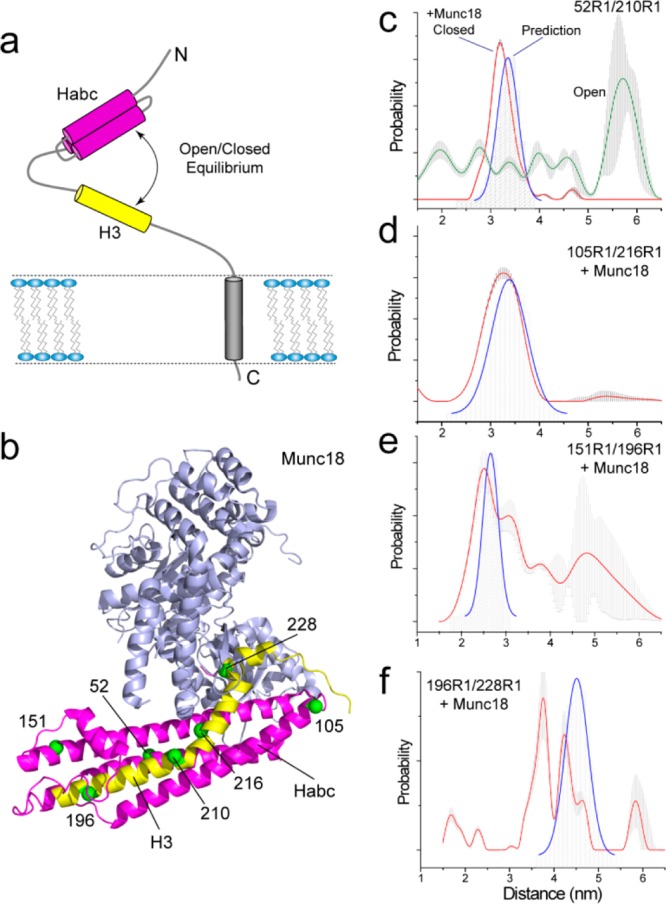
Conformational heterogeneity in the neuronal SNARE protein syntaxin 1A. (a) Syntaxin 1A is believed to exist in an open–closed equilibrium, where the H3 motif (yellow) can be either associated with or dissociated from the regulatory Habc domain (magenta). (b) The crystal structure of the syntaxin 1A/munc18-1 complex (PDB ID 3C98), where syntaxin is in a closed conformation and the H3 motif is resolved and folded along the Habc domain; pairs of labels were placed across syntaxin to make distance measurements between the H3 and Habc regions and along the H3 motif.40 (c) Distance distributions obtained for 52R1/210R1 in full length membrane reconstituted syntaxin in the absence (green trace) and presence of munc18 (red trace); the blue trace shows the prediction based upon the munc18/syntaxin crystal structure. (d, e, f) distance distributions obtained in the presence of munc18 for syntaxin(1–262) for 105R1/216R1, 151R1/196R1, and 196R1/228R1, respectively; the blue trace shows the distance distribution predicted based upon the munc18/syntaxin crystal structure.
The formation of the core SNARE complex is inhibited by munc18-1, which stabilizes syntaxin 1A in its closed conformation.42 Munc18 is an important regulatory protein that is required for membrane fusion. It appears to function in part as a chaperone, preventing syntaxin from aggregating or assembling until the SNARE complex is ready to be assembled. Shown in Figure 4b is the crystal structure of syntaxin in association with munc18, and Figure 4c shows the distance distributions measured by DEER between sites 52 and 210, which are located on the Habc and H3 domains, respectively. In the absence of munc18, the configuration is open (green trace), but the distribution changes dramatically when munc18 binds syntaxin (red trace). The expected distribution that is based upon the syntaxin/munc18 crystal structure (blue trace) closely matches the experimental distribution, where the slight overestimate in distance is within the uncertainty of the prediction.
On the C-terminal end of syntaxin, the measured distances also match the prediction, as is seen for measurements between sites 105 and 216 (Figure 4d). However, at the N-terminal end of syntaxin the EPR measurements yield different and more heterogeneous distance distributions than those expected. Measurements between sites 151 and 196 yield much longer distances than those expected (Figure 4e) and measurements along the length of the H3 segment between 196 and 228 yield both shorter and longer distances than expected (Figure 4f). These differences suggest that the N-terminal end of syntaxin (but not the central part and C-terminal end) is present in conformations much different than those in the crystal structure and may be sampling multiple conformational states. This conclusion is supported by the continuous wave EPR spectra, which show multiple motional components that can be interconverted by titration with a stabilizing osmolyte such as sucrose.40
An interesting possibility now being explored is whether conditions or other regulatory proteins that are thought trigger fusion, such as munc13, alter conformational heterogeneity or exchange in the N-terminal region of syntaxin. To trigger fusion, it may not be necessary to dissociate the H3 motif completely, it may only be necessary to enhance fluctuations at the N-terminus or bring the other SNARE partners into close proximity to the more dynamic syntaxin N-terminus.
General EPR-Based Approaches To Investigate Conformational Exchange in Proteins
At least three approaches based upon EPR may be used to determine the presence of an equilibrium between protein conformations. These include the use of SDSL and stabilizing osmolytes, electron relaxation rate measurements using saturation recovery EPR, and the application of high hydrostatic pressure. As indicated above, the EPR spectra from spin-labeled proteins are frequently composed of two or more motional components that result from the label being present in two or more environments. These components may result from different rotameric states of the label or they may reflect protein conformational exchange. These three approaches also provide a means to distinguish rotomeric from conformational exchange.
Site-directed spin labeling in combination with stabilizing osmolytes is a general approach that may be used to investigate protein conformational exchange. Components in the EPR spectrum that are due to different label rotameric states are not sensitive to osmolytes such as sucrose, whereas components due to protein conformational exchange are.43 A careful study in myoglobin has used this approach to map out dynamics in this protein and demonstrated that the results are consistent with measurements made by NMR.44 Saturation recovery EPR is a method that allows one to make a direct measurement of the nitroxide spin-lattice relation time (T1e), and it provides a different approach to characterize conformational exchange.45 If protein conformational exchange is present, multicomponent EPR spectra will yield multiexponential saturation recovery curves, and the approach will also yield an estimate for exchange lifetimes that take place within the 1 to 70 μs time scale. Rotameric exchange will often yield a single exponential in the recovery curve as observed for labels on the surface of the BtuB barrel.46 Protein conformational exchange is also typically slower than rotameric exchange, having a lifetime that is on order of tens of microseconds or longer.47
High hydrostatic pressure in combination with EPR spectroscopy is being explored as a method to characterize protein conformational substates. High hydrostatic pressure is well-known to denature proteins and to place proteins in excited conformation states, because both of these forms occupy less volume than does the native state.48 Conformers in equilibrium will also be sensitive to pressure, and pressure should populate the more disordered state. As shown in Figure 2c pressure has an effect that is opposite that of stabilizing osmolytes on the BtuB Ton box, and it acts to shift the equilibrium in this segment toward the more disordered unfolded state. Pressure may also be used to distinguish rotameric equilibria from protein conformational exchange. As seen in Figure 2c, ln K(P) is not linear in pressure, which is a result of a significant isothermal compressibility change (Δβ̅T) for this exchange process; however for rotameric exchange ln K(P) is linear with pressure because Δβ̅T is close to zero in these cases.49 An exciting possibility is the use of high pressure to trap protein conformations in excited states that may reveal the details of structural transitions that mediate protein function. More details on how these experiments are set up and how experiments such as DEER are made at high pressure using pulse EPR have been described.6,50,51
Biography
David S. Cafiso was born in San Francisco and did both his undergraduate and graduate studies at the University of California, Berkeley. He received his Ph.D. in 1979 under the direction of Wayne L. Hubbell, followed by postdoctoral work at Stanford University with Harden M. McConnell. In 1981, he joined the faculty in Chemistry at the University of Virginia where he is currently the Alfred Burger Professor of Biological Chemistry. His interests include membranes and membrane proteins, specifically the molecular mechanisms of membrane transport and the molecular mechanisms of membrane fusion.
Research from the author’s laboratory was supported by grants from the NIH (NIGMS GM072694 and NIGMS GM035215).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Jeschke G. Distance measurements in the nanometer range by pulse EPR. ChemPhysChem 2002, 3, 927–932. [DOI] [PubMed] [Google Scholar]
- Fanucci G. E.; Cafiso D. S. Recent advances and applications of site-directed spin labeling. Curr. Opin. Struct. Biol. 2006, 16, 644–653. [DOI] [PubMed] [Google Scholar]
- Jeschke G.; Polyhach Y. Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. PCCP Phys. Chem. Chem. Phys. 2007, 9, 1895–1910. [DOI] [PubMed] [Google Scholar]
- Mchaourab H. S.; Steed P. R.; Kazmier K. Toward the fourth dimension of membrane protein structure: insight into dynamics from spin-labeling EPR spectroscopy. Structure 2011, 19, 1549–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke G. DEER distance measurements on proteins. Annu. Rev. Phys. Chem. 2012, 63, 419–446. [DOI] [PubMed] [Google Scholar]
- Hubbell W. L.; Lopez C. J.; Altenbach C.; Yang Z. Technological advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 2013, 23, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mchaourab H.; Lietzow M.; Hideg K.; Hubbell W. Motion of spin-labeled side-chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry 1996, 35, 7692–7704. [DOI] [PubMed] [Google Scholar]
- Columbus L.; Hubbell W. L. Mapping backbone dynamics in solution with site-directed spin labeling: GCN4–58 bZip free and bound to DNA. Biochemistry 2004, 43, 7273–7287. [DOI] [PubMed] [Google Scholar]
- Altenbach C.; Kusnetzow A. K.; Ernst O. P.; Hofmann K. P.; Hubbell W. L. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 7439–7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masureel M.; Martens C.; Stein R. A.; Mishra S.; Ruysschaert J. M.; Mchaourab H. S.; Govaerts C. Protonation drives the conformational switch in the multidrug transporter LmrP. Nat. Chem. Biol. 2014, 10, 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan G.; Remaut H.; Wang T.; Allen W. J.; Pirker K. F.; Lebedev A.; Henderson N. S.; Geibel S.; Volkan E.; Yan J.; Kunze M. B.; Pinkner J. S.; Ford B.; Kay C. W.; Li H.; Hultgren S. J.; Thanassi D. G.; Waksman G. Crystal structure of the FimD usher bound to its cognate FimC-FimH substrate. Nature 2011, 474, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boura E.; Rozycki B.; Herrick D. Z.; Chung H. S.; Vecer J.; Eaton W. A.; Cafiso D. S.; Hummer G.; Hurley J. H. Solution structure of the ESCRT-I complex by small-angle X-ray scattering, EPR, and FRET spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 9437–9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N.; Guillier M.; Barnard T. J.; Buchanan S. K. TonB-dependent transporters: regulation, structure, and function. Annu. Rev. Microbiol. 2010, 64, 43–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer K.; Rodionov D. A.; de Reuse H. New substrates for TonB-dependent transport: Do we only see the ‘tip of the iceberg’?. Trends Biochem. Sci. 2008, 33, 330–338. [DOI] [PubMed] [Google Scholar]
- Kleanthous C. Swimming against the tide: Progress and challenges in our understanding of colicin translocation. Nat. Rev. Microbiol. 2010, 8, 843–848. [DOI] [PubMed] [Google Scholar]
- Braun V.; Endriss F. Energy-coupled outer membrane transport proteins and regulatory proteins. Biometals 2007, 20, 219–231. [DOI] [PubMed] [Google Scholar]
- Buchanan S. K.; Smith B. S.; Venkatramanil L.; Xia D.; Esser L.; Palnitkar M.; Chakraborty R.; van der Helm D.; Deisenhofer J. Crystal structure of the outer membrane active transporter FepA from Escherichia coli. Nat. Struct. Biol. 1999, 6, 56–63. [DOI] [PubMed] [Google Scholar]
- Locher K. P.; Rees B.; Koebnik R.; Mitschler A.; Moulinier L.; Rosenbusch J. P.; Moras D. Transmembrane signaling across the ligand-gate FhuA receptor: Crystal structures of the free and ferrichrome-bound states reveal allosteric changes. Cell 1998, 95, 771–778. [DOI] [PubMed] [Google Scholar]
- Ferguson A. D.; Hofmann E.; Coulton J. W.; Diederichs K.; Welte W. Siderophore-mediated iron transport: crystal structure of FhuA with bound lipopolysaccharide. Science 1998, 282, 2215–2220. [DOI] [PubMed] [Google Scholar]
- Ferguson A. D.; Chakraborty R.; Smith B. S.; Esser L.; Van der Helm D.; Deisenhofer J. Structural basis of gating by the outer membrane transporter FecA. Science 2002, 295, 1715–1719. [DOI] [PubMed] [Google Scholar]
- Yue W. W.; Grizot S.; Buchanan S. K. Structural evidence for iron-free citrate and ferric citrate binding to the TonB-dependent outer membrane transporter FecA. J. Mol. Biol. 2003, 332, 353–368. [DOI] [PubMed] [Google Scholar]
- Chimento D. P.; Mohanty A. K.; Kadner R. J.; Wiener M. C. Substrate-induced transmembrane signaling in the cobalamin transporter BtuB. Nat. Struct. Biol. 2003, 10, 394–401. [DOI] [PubMed] [Google Scholar]
- Shultis D. D.; Purdy M. D.; Banchs C. N.; Wiener M. C. Outer membrane active transport: Structure of the BtuB:TonB complex. Science 2006, 312, 1396–1399. [DOI] [PubMed] [Google Scholar]
- Pawelek P. D.; Croteau N.; Ng-Thow-Hing C.; Khursigara C. M.; Moiseeva N.; Allaire M.; Coulton J. W. Structure of TonB in complex with FhuA, E. coli outer membrane receptor. Science 2006, 312, 1399–1402. [DOI] [PubMed] [Google Scholar]
- Merianos H. J.; Cadieux N.; Lin C. H.; Kadner R.; Cafiso D. S. Substrate-induced exposure of an energy-coupling motif of a membrane transporter. Nat. Struct. Biol. 2000, 7, 205–209. [DOI] [PubMed] [Google Scholar]
- Fanucci G. E.; Coggshall K. A.; Cadieux N.; Kim M.; Kadner R. J.; Cafiso D. S. Substrate-Induced conformational changes of the perplasmic N-terminus of an outer-membrane transporter by site-directed spin labeling. Biochemistry 2003, 42, 1391–1400. [DOI] [PubMed] [Google Scholar]
- Xu Q.; Ellena J. F.; Kim M.; Cafiso D. S. Substrate-dependent unfolding of the energy coupling motif of a membrane transport protein determined by double electron-electron resonance. Biochemistry 2006, 45, 10847–10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed D. M.; Horanyi P. S.; Wiener M. C.; Cafiso D. S. Conformational exchange in a membrane transport protein is altered in protein crystals. Biophys. J. 2010, 99, 1604–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker B. A.; Portman J. J.; Wolynes P. G. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 8868–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timasheff S. N. Protein hydration, thermodynamic binding, and preferential hydration. Biochemistry 2002, 41, 13473–13482. [DOI] [PubMed] [Google Scholar]
- Auton M.; Bolen D. W. Application of the transfer model to understand how naturally occurring osmolytes affect protein stability. Methods Enzymol. 2007, 428, 397–418. [DOI] [PubMed] [Google Scholar]
- Rosgen J.; Pettitt B. M.; Bolen D. W. An analysis of the molecular origin of osmolyte-dependent protein stability. Protein Sci. 2007, 16, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellman J. A. Destabilization and stabilization of proteins. Q. Rev. Biophys. 2005, 38, 351–361. [DOI] [PubMed] [Google Scholar]
- Kim M.; Xu Q.; Fanucci G. E.; Cafiso D. S. Solutes modify a conformational transition in a membrane transport protein. Biophys. J. 2006, 90, 2922–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores Jimenez R. H.; Do Cao M. A.; Kim M.; Cafiso D. S. Osmolytes modulate conformational exchange in solvent-exposed regions of membrane proteins. Protein Sci. 2010, 19, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokdad A.; Herrick D. Z.; Kahn A. K.; Andrews E.; Kim M.; Cafiso D. S. Ligand-induced structural changes in the Escherichia coli ferric citrate transporter reveal modes for regulating protein-protein interactions. J. Mol. Biol. 2012, 423, 818–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.; Xu Q.; Murray D.; Cafiso D. S. Solutes alter the conformation of the ligand binding loops in outer membrane transporters. Biochemistry 2008, 47, 670–679. [DOI] [PubMed] [Google Scholar]
- Hagelueken G.; Ward R.; Naismith J. H.; Schiemann O. MtsslWizard: In silico spin-labeling and generation of distance distributions in PyMOL. Appl. Magn. Reson. 2012, 42, 377–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R.; Scheller R. H. SNAREs--engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006, 7, 631–643. [DOI] [PubMed] [Google Scholar]
- Dawidowski D.; Cafiso D. S. Allosteric control of syntaxin 1a by Munc18-1: characterization of the open and closed conformations of syntaxin. Biophys. J. 2013, 104, 1585–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang B.; Dawidowski D.; Ellena J. F.; Tamm L. K.; Cafiso D. S. The SNARE motif of synaptobrevin exhibits an aqueous-interfacial partitioning that is modulated by membrane curvature. Biochemistry 2014, 53, 1485–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt P.; Hattendorf D. A.; Weis W. I.; Fasshauer D. Munc18a controls SNARE assembly through its interaction with the syntaxin N-peptide. EMBO J. 2008, 27, 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez C. J.; Fleissner M. R.; Guo Z.; Kusnetzow A. K.; Hubbell W. L. Osmolyte perturbation reveals conformational equilibria in spin-labeled proteins. Protein Sci. 2009, 18, 1637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez C. J.; Oga S.; Hubbell W. L. Mapping molecular flexibility of proteins with site-directed spin labeling: a case study of myoglobin. Biochemistry 2012, 51, 6568–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges M. D.; Hideg K.; Hubbell W. L. Resolving conformational and rotameric exchange in spin-labeled proteins using saturation recovery EPR. Appl. Magn. Reson. 2010, 37, 363–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed D. M.; Khan A. K.; Horanyi P. S.; Cafiso D. S. Molecular origin of electron paramagnetic resonance line shapes on beta-barrel membrane proteins: the local solvation environment modulates spin-label configuration. Biochemistry 2011, 50, 8792–8803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman K.; Kern D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [DOI] [PubMed] [Google Scholar]
- Akasaka K. Probing conformational fluctuation of proteins by pressure perturbation. Chem. Rev. 2006, 106, 1814–1835. [DOI] [PubMed] [Google Scholar]
- McCoy J.; Hubbell W. L. High-pressure EPR reveals conformational equilibria and volumetric properties of spin-labeled proteins. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 1331–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch M. T.; Horwitz J.; McCoy J.; Hubbell W. L. Circular dichroism and site-directed spin labeling reveal structural and dynamical features of high-pressure states of myoglobin. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E4714–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch M. T.; Yang Z.; Brooks E. K.; Hubbell W. L. Mapping protein conformational heterogeneity under pressure with site-directed spin labeling and double electron-electron resonance. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E1201–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A. Protein fluctuations and the thermodynamic uncertainty principle. Prog. Biophys. Mol. Biol. 1984, 44, 181–214. [DOI] [PubMed] [Google Scholar]
- Warshaviak D. T.; Serbulea L.; Houk K. N.; Hubbell W. L. Conformational analysis of a nitroxide side chain in an α-helix with density functional theory. J. Phys. Chem. B 2011, 115, 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke G.; Chechik V.; Ionita P.; Godt A.; Zimmermann H.; Banham J.; Timmel C. R.; Hilger D.; Jung H. DeerAnalysis2006 - a comprehensive software package for analysing pulsed ELDOR data. Appl. Magn. Reson. 2006, 30, 473–498. [Google Scholar]