

Abstract
The concept of synthetic lethality (the creation of a lethal phenotype from the combined effects of mutations in two or more genes) has recently been exploited in various efforts to develop new genotype-selective anticancer therapeutics. These efforts include screening for novel anticancer agents, identifying novel therapeutic targets, characterizing mechanisms of resistance to targeted therapy, and improving efficacies through the rational design of combination therapy. This review discusses recent developments in synthetic lethality anticancer therapeutics, including poly ADP-ribose polymerase inhibitors for BRCA1- and BRCA2-mutant cancers, checkpoint inhibitors for p53 mutant cancers, and small molecule agents targeting RAS gene mutant cancers. Because cancers are caused by mutations in multiple genes and abnormalities in multiple signaling pathways, synthetic lethality for a specific tumor suppressor gene or oncogene is likely cell context-dependent. Delineation of the mechanisms underlying synthetic lethality and identification of treatment response biomarkers will be critical for the success of synthetic lethality anticancer therapy.
Introduction
Genetic and epigenetic alterations that lead to the functional deregulations of several signaling and metabolic pathways are known to be the major driving forces behind carcinogenesis and cancer progression.1 Those functional deregulations in cancer cells have been exploited for pathway-targeted anticancer therapy. Small molecules and antibodies that directly inhibit critical nodes in oncogenic signaling networks, most notably kinases or enzymes, have been used to treat various cancers in humans,1,2 resulting in substantial improvement in clinical symptoms and outcomes in a subset of cancer patients. However, many critical nodes in oncogenic signaling networks may not be targeted directly by small molecules or antibodies. For example, functional losses in tumor suppressor genes caused by gene mutations or deletions may not be restored through small molecules. Moreover, the functions of some intracellular oncogene products, such as RAS and c-MYC, have been found to be difficult to modulate directly through small molecules.3 Nevertheless, functional alterations in nondruggable targets may lead to changes in signal transduction and metabolism that render the mutant cells more susceptible to functional changes in other genes or to pharmaceutical interventions aimed at other targets, providing an opportunity to selectively eliminate those mutant cells through synthetic lethality. Synthetic lethality (the creation of a lethal phenotype from the combined effects of mutations in two or more genes4) offers the potential to eliminate malignant cells by indirectly targeting cancer-driving molecules that are difficult to target directly with small molecules or antibodies.
The concept of synthetic lethality is illustrated in Figure 1A. The two genes A and B are synthetic lethal if the mutations in any one of them will not change the viability of a cell or an organism, but simultaneous mutations in both A and B genes will result in a lethal phenotype. This concept has has been used in genetic studies to determine functional interactions and compensation among genes for decades5 and has recently been exploited for the development of new genotype-selective anticancer agents,6−8 identification of novel therapeutic targets for cancer treatment,9−11 and characterization of genes associated with treatment response.12−14 For example, if gene A in Figure 1B is mutated, small interfering RNA (siRNA) or small molecules targeting the genes X, Y, or Z would likely induce synthetic lethality in cells with an abberant A but not in the cells with a wild-type A. Therefore, using paired isogenic cell lines with and without abberant A, one can screen for siRNA or compounds that specifically kill the cells with an abberant A.
Figure 1.
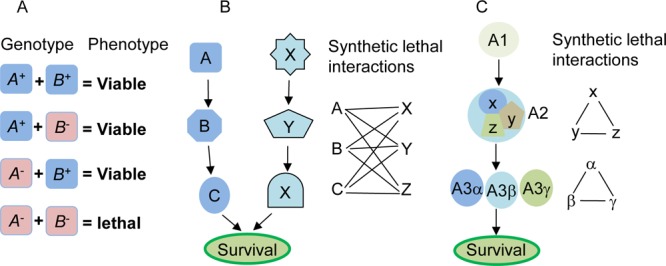
Concept and models of synthetic lethal interactions. (A) Synthetic lethality between genes A and B. A+ and B+ represent wild types, while A– and B– represent mutants. Synthetic lethality refers to a lethal phenotype observed only in the combination group of A– and B–. (B) An essential survival function is regulated by two pathways conducted by A, B, C and X, Y, X, respectively. A functional change in either of these pathways is insufficient to induce viability changes. However, the simultaneous presence of mutations or dysfunctions in both pathways, such as a mutation in A and any mutation in X, Y, or Z, induces lethal phenotype. Thus, A is synthetic lethal with X, Y, and Z, and vice versa. (C) An essential survival function is regulated by pathway A alone, in which A2 is a multiprotein complex composed of X, Y, and Z; and A3 has homologues of α, β and γ. Synthetic lethality may exist among X, Y, and Z and among A3α, β, and γ.
Several models of interactions among genes and/or proteins have been proposed to account for synthetic lethality,15,16 including the components of parallel pathways that together regulate an essential biological function, the presence of homologous genes or protein isomers derived from the same ancestral gene (paralogs), subunits of an essential multiprotein complex, and components of a single linear essential pathway (Figure 1B,C). Studies in yeast revealed that synthetic lethal interactions occurred significantly more frequently among genes with the same mutant phenotype, among genes encoding proteins with the same subcellular localization, and among genes involved in similar biological processes, such as those in parallel or compensating pathways or bridging bioprocesses.17 For a particular tumor suppressor gene or oncogene, synthetic lethality can be identified by using isogenic cell lines to screen an siRNA library for synthetic lethal genes or a chemical library for synthetic lethal compounds. This review discusses recent advances in the development of synthetic lethality based anticancer therapeutics. In particular, it emphasizes the development of anticancer agents that target DNA damage response and oncogene Ras pathways through a synthetic lethality approach.
DNA Damage Response Pathway
The TP53 gene, which encodes tumor suppressor protein p53, a master transcriptional regulator of cellular response to DNA damage, is commonly inactivated in about 50% of human cancers by either gene mutations or degradation through HDM2.18,19 Moreover, pathways involved in DNA damage response are often constitutively activated in a majority of tumors, even in early stages of tumor development and in tumor specimens from untreated patients, presumably because of oncogene-mediated deregulation of DNA replication.20
Different mechanisms are used in cells in response to different types of DNA damage. Single-strand breaks (SSBs) activate poly ADP-ribose polymerase (PARP) and are repaired mainly by PARP-mediated base-excision repair, while double-strand breaks (DSBs) are repaired by the mechanisms of homologous recombination (HR) and nonhomologous end joining (NHEJ).21 PARP can be activated by binding to SSBs,22−24 leading to SSB repair through base excision mechanisms (Figure 2). However, if SSBs are not repaired, they will cause a blockage or collapse of DNA replication forks during DNA synthesis and the formation of DSBs. DSBs can also be incurred by endogenous and exogenous DNA-damaging agents such as ionizing radiation.
Figure 2.
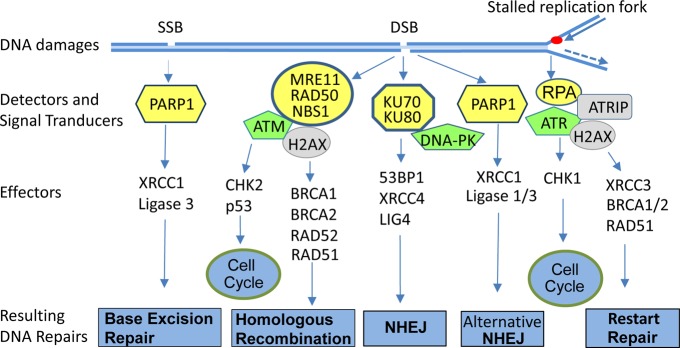
DNA damage repair pathways. Single-strand break (SSB), double-strand break (DSB), and single strand DNA derived from DNA damage or stalled replication fork are recognized by various sensor molecules (marked yellow), leading to activation of signal transducers (marked green), which in turn activate different DNA repair pathways and checkpoint pathways, thereby preventing transmission of the genetic lesion to the daughter cells. Those parallel pathways provide opportunities of eliminating some cancer cells with mutations in those pathways through synthetic lethality.
DSBs are detected by the MRE11/RAD50/NBS1 complex or by Ku70/Ku80 heterodimers. The single-strand DNA present at stalled replication forks or generated by processing of DSBs is recognized by replication protein A (RPA).25 The assembly of those sensor molecules in the damaged DNA sites leads to the recruitment and activation of signal transducers, including three phosphatidylinositol 3-kinase related kinases (PIKKs) (ataxia telangiectasia mutated (ATM), ATM- and Rad3-related (ATR), and DNA-dependent protein kinase (DNA-PK)) that in turn activate downstream effectors, resulting in the activation of checkpoint and DNA repair pathways.25,26 The phosphorylation of H2AX by ATM, ATR, or DNA-PK at S139 (known as γH2AX) triggers the recruitment of DNA repair proteins and leads to the assembly of DNA repair complexes at the damaged DNA sites.25,27 Consequently, the cell cycle progression is arrested to permit the repair of the damaged DNA, thereby preventing transmission of the genetic lesion to the daughter cells (Figure 2).
Both BRCA1 and BRCA2 are required for the assembly of protein complexes for HR, including recruitment of recombinase RAD51 to the DNA DSBs.28,29 DSB repair through HR is mostly error free and can occur only in the late S and G2 phases of the cell cycle because it requires an intact sister chromatid as a template for repair. In contrast, NHEJ involves Ku70/80, DNA-PK, XRCC4, and DNA ligase IV, often results in gene deletion or translocation, and can occur in all phases of the cell cycle.25 Most NHEJ occurs independent of the MRE11/RAD50/NBS1 complex and ATM activation because the Ku70/Ku80 heterodimer can directly bind to the ends of double-strand DNA, recruit DNA-PK to the site, and initiate an NHEJ process.30 In the absence of Ku proteins, an alternative NHEJ initiates with the involvement of PARP1, XRCC1, and ligase I/III31 (Figure 2).
PARP and PARP Inhibitors
PARP is a group of ADP-ribose transferase enzymes that catalyze polyADP-ribosylation of proteins by transferring ADP-ribose groups from the donor substrate nicotinamide adenine dinucleotide (NAD+) to glutamic acid, aspartic acid, and lysine residues in the acceptor proteins, thereby regulating the functions of those proteins. Seventeen PARP family members have been identified in humans.32 DNA-dependent PARP subfamily members PARPs 1–3 have been reported to regulate various DNA damage response processes. The recognition and binding to damaged DNA structures by PARP1 and PARP2, either through their own DNA binding domain or through interaction with damaged DNA-binding protein 2,22−24 lead to the activation of their enzymatic activity and the polymerization of ADP-ribose units of a number of proteins, including PARP1/2, histones, topoisomerase, and DNA-PK.33,34 The polyADP-ribose on those proteins provides a docking site for recruiting cell cycle checkpoint proteins and DNA repair proteins (e.g., p53, XRCC1, DNA-PK, Ku70, and ATM) to the sites of DNA lesions,35−38 thereby regulating various processes of DNA repair, including base-excision repair, HR, and NHEJ. PARP1 regulates base-excision repair by interaction with XRCC1,39 DNA polymerase β,40 and the base-excision repair enzymes apurinic/apyrimidinic endonuclease 1 41 and ALC1.42 PARP1 also interacts with DNA-PK and Ku and is required for an alternative and PARP-dependent NHEJ pathway.43,44 Moreover, PARP1 participates in HR by interacting with MRE11 and ATM.36,45 Although PARP1–/– embryonic stem cells and embryonic fibroblasts exhibit normal repair of DNA DSBs and RAD51 foci formation,46PARP1–/– mice have increased deletion mutations and insertions and/or rearrangements in vivo after treatment with the alkylating agent N-nitrosobis(2-hydroxypropyl)amine.47 Interestingly, PARP1–/– mice are viable and fertile and do not develop spontaneous tumors, possibly because of functional compensation from PARP2, as PARP1 and PARP2 double knockout is embryonically lethal.48 Nevertheless, PARP1–/– mice or cells exhibit defective DNA SSB repair and increased HR, sister chromatid exchange, and chromosome instability.49 PARP1, but not PARP2, is required for the survival of cells with defects in the HR pathway because knockdown of both PARP1 and BRCA2 significantly reduces the survival of human cells, whereas knockdown of both PARP2 and BRCA2 has no effect on cell survival.50 The mechanisms underlying the synthetic lethality of PARP1 and BRCA genes are still not clear, although evidence suggests that it might be caused by the deregulation of NHEJ,51 increased spontaneous DNA breaks that need to be repaired by HR,52 or the suppression of BRCA and RAD51 expression by E2F4/p130-mediated transcriptional repression53 caused by PARP1 inhibition.
The synthetic lethality of PARP1 and BRCR1/2 genes reported in 2005 by Farmer et al.6 and Bryant et al.50 sparked much interest in the concept of using PARP inhibitors to selectively eliminate BRCA1 or BRCA2 mutant tumor cells. Germ line mutations in BRCA genes predispose carriers to breast, ovarian, and other cancers in an autosomal dominant manner,54,55 with 50–80% penetrance for breast cancer and 30–50% for ovarian cancer.56 Early studies revealed that PARP1 activation facilitates DNA repair and maintenance of genomic integrity and is required for recovery from DNA damage in mice and in cells.49 However, excessive PARP1 activation leads to cell death because of overconsumption and depletion of NAD+ and ATP in the cells, whereas genetic disruption or pharmaceutical inhibition of PARP protects animals from ischemia-induced brain and heart damage.57,58 In addition, PARP1 was found to regulate the transcriptional activity of NFκB and other inflammation-related transcription factors, promoting the expression of inflammation mediators.59 Thus, PARP inhibitors have been investigated for the therapeutic benefits of protecting tissue from ischemia-induced injury, suppressing inflammation, and sensitizing cancer cells to DNA damage-based anticancer therapy.60,61 Several excellent review articles have discussed the development of PARP inhibitors.32,61−63 Briefly, PARP inhibitors used in clinical investigation mostly compete with the ADP-ribose donor substrate NAD+ and inhibit both PARP1 and PARP2. A binding assay with catalytic domains from 13 of the 17 human PARP family members revealed that many of the best known PARP inhibitors, including those used in clinical studies such as olaparib (1), veliparib (2), and rucaparib (3) (Figure 3), bind to several PARP family members, suggesting nonspecific activity of those inhibitors.64
Figure 3.

Structures of PARP inhibitors.
PARP inhibitors are currently under intensive investigations as therapeutic agents for the treatment of cancers with deficiencies in BRCA or other DNA repair proteins. An initial preclinical study by Farmer et al. revealed that the BRCA1/2-defective cells were 57- to 133-fold more sensitive to PARP inhibitors KU0058684 (4) and KU0058948 (5) than wild-type cells.6 At the same time, Bryant et al.50 showed that HR-defective cells with a deficiency in XRCC2, XRCC3, or BRCA2 (XRCC11) were killed by PARP inhibitors such as NU1025 (6) and AG14361 (7) at concentrations that were nontoxic to normal cells.65 Recent studies revealed that defects in other DNA repair proteins, such as ATM,66 MRE11,67 ERCC1,68 and p5369 or PTEN,70 also induce synthetic lethality with the PARP inhibitors. A high-throughput RNA interference screen for 230 known and putative DNA repair proteins revealed additional genes that have synthetic lethality with compound 5, including ATR, PCNA, RAD51, and XRCC1.71 A similar study with a novel potent PARP1/2 inhibitor (BMN 673) (8) and an siRNA library targeting 960 genes, including kinases and kinase-related genes, tumor suppressors, and DNA repair proteins, showed synthetic lethality of 8 with HR and DSB repair genes.72 Moreover, PARP1 inhibitors induced synthetic lethality in cancer cells with positive E26 transformation-specific gene fusions73 and with EGFR inhibitors.74 Because of the critical roles of PARP1 in the DNA repair process, PARP inhibitors have also been intensively tested for sensitizing radiotherapy and chemotherapy that induces DNA damage.
Compounds 1 and 2 are the most extensively investigated inhibitors in clinical trials for the treatment of BRCA1/2-mutated cancers. Other PARP inhibitors under clinical trials are niraparib (MK-4827) (9), 3, and 8. A phase I study of the combination therapy of rucaparib with temzolomide in patients with advanced solid tumors showed that 3 at a dose of 12 mg/m2 inhibited 74–97% of PARP activity in peripheral blood mononuclear cells. A dose of 12 mg/m23 in combination with 200 mg/m2 temozolomide was tolerated, with a dose-limiting toxicity of myelosuppression.75 A phase II study of intravenous administration of 12 mg/m23 with temozolomide in patients with metastatic melanoma showed that 150–200 mg m–2 day–1 temozolomide can be safely given in the combination therapy, with a response rate of about 17% and an increase in progression-free survival over historical controls in metastatic melanoma patients.76 Phase I clinical trials with compound 1 in recurrent/advanced cancer patients with or without BRCA1/2 mutations77,78 revealed that the maximum tolerated dose for 1 was 400 mg twice daily, with dose-limiting toxicities of mood alteration, fatigue, and thrombocytopenia. Clinical benefit was observed in 30–60% of BRCA1/2 mutant breast or ovarian cancer patients.77,78 The pharmacokinetics study revealed a maximum concentration of 2.6–4.8 μg/mL at doses of 400 mg twice daily. The mean maximal PARP inhibitions in peripheral blood mononuclear cells and tumor tissues were 50% and 70%, respectively.79 Mean terminal half-life was about 7–11 h.80 Subsequently, 1 has been evaluated in combination therapy with various conventional anticancer agents, including cisplatin and gemcitabine, topotecan, bevacizumab, dacarbazine, and the VEGFR inhibitor cediranib.81,82 The overall response rates were variable, from 0% to 44%.
A phase II comparison study of 1 versus liposomal doxorubicin treatment for BRCA1/2 mutant ovarian cancer patients who had recurrent tumors after platinum therapy revealed overall response rates of 25–31%, not significantly different from that of doxorubicin alone (18%).83 Phase I trials of a combination of 2 (half-life of about 5 h) with cyclophosphamide or topotecan84 in refractory solid tumors and lymphomas showed promising activity in a subset of patients with BRCA mutations. The maximum tolerated dose was defined as 60 mg of 2 with 50 mg of cyclophosphamide once daily or 0.6 mg m–2 day–1 topotecan administered intravenously on days 1–5 and 10 mg of 2 twice daily on days 1–5 in 21-day cycles. PARP activity was significantly inhibited in peripheral blood mononuclear cells (by 50%) and in tumor biopsies (by 80%). A phase II trial of the combination compared with single-agent cyclophosphamide is ongoing for cases of BRCA mutant ovarian cancer, triple-negative breast cancer, and low-grade lymphoma. The dose-limiting toxicity was myelosuppression. A phase 1 dose-escalation trial of 9 in BRCA mutation carriers and patients with sporadic cancer revealed a maximum tolerated dose of 300 mg/day. Dose-limiting toxicities were fatigue, pneumonitis, and thrombocytopenia. The maximum concentration at 300 mg/day was about 2000–4000 nM. The mean terminal elimination half-life was 36.4 h. Pharmacodynamic analyses confirmed that PARP inhibition exceeded 50% at doses greater than 80 mg/day, and antitumor activity was documented at doses beyond 60 mg/day. Clinical benefit was observed in 40–50% of BRCA1/2 mutation carriers with ovarian or breast cancer. Antitumor activity was also reported in sporadic high-grade serous ovarian cancer, non-small-cell lung cancer, and prostate cancer.85 A phase I study of 8 in solid tumors showed that RECIST and/or CA-125 response occurred at doses of >100 μg/day in 11/17 BRCA carrier ovarian/peritoneal cancer patients.86 In summary, the results from these clinical studies revealed that myelosuppression is the major dose-limiting toxicity of various PARP inhibitors and that clinical benefit is variable in the single-agent and combination therapies, with significant benefit observed in BRCA mutant cancers, consistent with the findings in the preclinical studies.
Checkpoint Inhibitors
The ATM/checkpoint kinase 2 (CHK2) and ATR/CHK1 signaling pathways regulate many common downstream proteins, including p53, CDC25 phosphatases, and Wee1 kinase, thereby regulating G1, S, and G2/M checkpoints (Figure 4). Because activation of oncogenes can cause replication stress and DNA damage, inhibiting checkpoint pathways may trigger synthetic lethality in cancer cells by enhancing the DNA damage-induced apoptosis or senescence, which may be modulated by p53 status. Phosphorylation of p53 on S15 by ATM/ATR and/or on S20 by CHK1/CHK2 stabilizes p53 protein87−89 and up-regulates the expression of p21 (also known as CIP1/WAF1), an inhibitor of cyclin E/cyclin-dependent kinase 2 (CDK2) that controls G1/S progression. In contrast, phosphorylation of CDC25 phosphatases by CHK1/CHK2 promotes degradation of CDC25, which is required for dephosphorylation and activation of cyclin B/CDK1 kinases that control the transition of G2/M phases90 (Figure 4). Evidence has shown that ATR/CHK1 is critical in regulating the activity of CDC25 and S/G2 checkpoints.91,92 Although p53 also regulates the G2 checkpoint,93 G2 arrest is normally induced in p53-deficient cancers, suggesting that some p53-independent mechanisms are sufficient for G2 checkpoint functioning. Recent studies showed that p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38 mitogen-activated protein (MAP) kinase/MAP kinase-activated protein kinase 2 (MK2) pathway for survival after DNA damage.94 Evidence has shown that p53-mediated apoptosis or cell cycle arrest is modulated by ATM and CHK2 activities.95 In cells and tumors that lacked a functional p53 pathway, inactivation of ATM or its downstream molecule CHK2 was sufficient to globally sensitize the cells to genotoxic chemotherapy with cisplatin or doxorubicin.95 In contrast, in p53 wild-type cells, the inhibition of ATM or CHK2 resulted in a substantial survival benefit, suggesting that a combination of cisplatin and doxorubicin with inhibitors of ATM and CHK2 could benefit patients with p53 mutant tumors. Genome-wide small hairpin RNA screening also revealed that inhibiting ATM or MET induced synthetic lethality with a p53 stabilizing/activating compound nutlin-3 (10) (Figure 5) and converted the cellular response from cell cycle arrest to apoptosis in various cancer cell types without affecting the expression of key p53 target genes.96 Although ATM inhibitors such as CP466722 (11) and KU59403 (12) do not have single-agent activity in cancer cell lines, they could sensitize cancer cells to ionizing radiation and/or genotoxic chemotherapeutics.97
Figure 4.
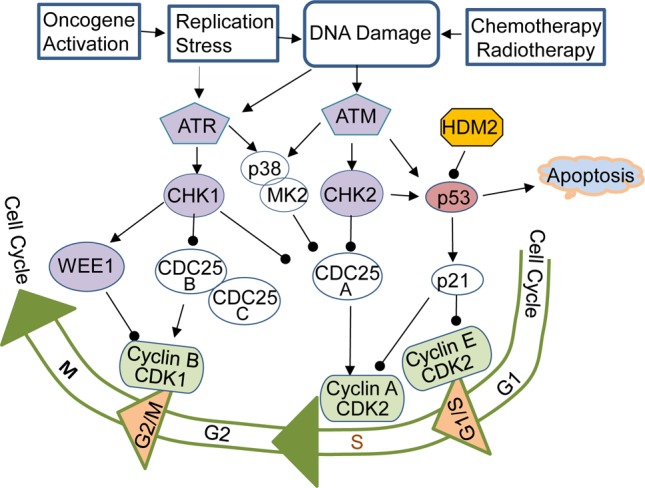
Checkpoint pathways and cell cycle regulations. DNA damage caused by physical, chemical, and biological factors, such as oncogene activation, activates phosphatidylinositol 3-kinase related kinases ATM and ATR, leading to the activation of CHK2, CHK1, and p38/MK2, which further regulate activity and stability of cell cycle regulators, including p53 and CDC25. p53/p21 is the major regulator of G1/S checkpoint. The loss of p53 function in cancer cells makes the cells addicting to the S and/or G2/M checkpoints for DNA repaire and survival. Inhibiting S and/or G2/M checkpoint regulators may induce synthetic lethality in p53 mutant cells when DNA is damaged. Arrow indicates activation, and the line terminating with a black circle indicates suppression.
Figure 5.

Structures of nutlin-3 and inhibitors of ATM/ATR.
Similar results have shown that inhibiting ATR and CHK1 exacerbates levels of oncogene-induced replicative stress, promoting the cell killing of oncogene-overexpressing cells and sensitizing tumor cells to DNA-damaging therapy. Inhibiting ATR by a dominant negative construct selectively sensitizes G1 checkpoint-deficient cells to DNA damage-induced lethal premature chromatin condensation.98 Knockdown of ATR expression selectively enhanced cisplatin sensitivity in human colorectal cancer cells with inactivated p53, whereas the restoration of p53 in ATR-deficient cells increased cell survival after cisplatin treatment.99 Reduced ATR expression prevented the development of Myc-induced lymphomas or pancreatic tumors in mice by enhancing Myc-induced replicative stress and apoptosis, which is more pronounced in p53-deficient cells.100 Transgenic mice with diminished ATR function in the skin have increased apoptosis after ultraviolet exposure and reduced ultraviolet-induced skin carcinogenesis, suggesting that the inhibition of the replication checkpoint function may have therapeutic and/or preventive benefits.101 Inhibiting ATR also induced synthetic lethality in XRCC1-deficient cancer cells102 and sensitized cancer cells to DNA-damaging chemotherapeutic agents.103 Synthetic lethality in XRCC1-deficient cancer cells is also elicited by the ATM inhibitor KU55933 (13) and the DNA-PK inhibitor NU7441 (14).104
A cell-based screening of 623 PI3K inhibitors led researchers to identify an mTOR- and ATR-selective inhibitor, ETP-46464 (15), that induces replicative stress and synthetic lethality in p53-deficient or cyclin E-overexpressing cells.105 The IC50 values of 15 for mTOR and ATR are 0.6 and 14 nM, respectively, but >36 nM for DNAPK, PI3K, and ATM. AZ20 (16), an ATR inhibitor derived from the mTOR inhibitor sulfonylmorpholinopyrimidine, was reported to have single-agent activity when administered orally in mice bearing the xenograft established from colon cancer cell line LoVo.106 Through high-throughput screening and compound optimization, Charrier et al. developed an ATR-selective inhibitor, VE-821 (17), with IC50 values for ATR, ATM, and DNAPK of 12 nM, >8 μM, and >8 μM, respectively.107 In vitro study revealed that treatment with 17 alone induced selective killing of ATM- or p53-deficient cancer cells but only reversibly limited cell cycle progression in normal cells. 17 also increased cisplatin potency 10-fold in HCT116 cells, suggesting that inhibiting ATR could potentiate the efficacies of radiotherapy and genotoxic drugs.108 VE-822 (18), an analogue of 17, was found to have in vivo activity in blocking ATR and sensitizing pancreatic cancer cells to gemcitabine-based chemoradiation therapy.109 Nevertheless, no clinical studies with ATM or ATR inhibitors have yet been reported.
In comparison to the development of ATM/ATR inhibitors, the development of CHK inhibitors, specifically CHK1 inhibitors, is more advanced possibly because it is easier to perform enzymatic analysis on CHK1/CHK2. CHK1 and CHK2 have a highly conserved kinase domain but have distinct overall protein structures.110 However, several small molecular inhibitors inhibit both CHK1 and CHK2 with a similar potency. In an Eμ-Myc-driving lymphoma model, inhibiting CHK1, CHK2, or both with small molecule inhibitors induced cell death, although the Eμ-Myc p53 null lymphoma cells were more sensitive to a dual CHK1/CHK2 inhibitor than to a CHK1-specific inhibitor.111 AZD7762 (19) (Figure 6), a dual CHK1/2 inhibitor, also enhances radiosensitivity- and chemotherapy-induced apoptosis in p53 mutant and/or p21-deficient tumor cells to a greater extent than in p53 wild-type tumor cells.112,113 The potentiation of DNA damaging agent-induced apoptosis by CHK inhibitors is possibly caused by abrogation of the G2 checkpoint and/or inhibition of HR DNA repair.114
Figure 6.

Structures of CHK1/2 inhibitors.
Gene knockout studies revealed that knockout of CHK1 or CHK2 caused defects in the induction and/or maintenance of irradiation-induced G2 arrest.115,116 Intriguingly, knockout of ATM or CHK2 is viable for cells and animals, although phenotypes of ataxia telangiectasia, chromosomal abnormality, and the development of thymic lymphomas are observed in ATM knockout,117 whereas resistance to ionizing radiation-induced apoptosis is observed in CHK2 knockout.115,118 In contrast, knockout of ATR or CHK1 is lethal for cells and embryos with defects in the G2/M DNA damage checkpoint,116,119 indicating that the ATR/CHK1 pathway is essential for cell survival. Therefore, inhibitors of ATR and/or CHK1 could be more potent and toxic than those of ATM and/or CHK2. A synthetic lethality siRNA screening of 572 kinases for improving gemcitabine or cisplatin response in pancreatic or ovarian cancer cells revealed the greatest potentiation by siRNA targeting of ATR and/or CHK1.120 Similar results were obtained by inhibiting CHK1 with small molecule inhibitors such as PD407824 (a dual inhibitor of CHK1 and Wee1) (20).120
A phase I study on 19 single-agent therapy and combination therapy with gemcitabine in patients with advanced solid tumors showed that the dose-limiting toxicities are cardiac and liver function abnormalities and myelosuppression. No objective responses were observed, although disease stabilization was observed in some patients.121 UCN-01 (7-hydroxystaurosporin) (21), a protein kinase C inhibitor that also inhibits many other kinases, including CHK1, and abrogates the G2 checkpoint has been reported to enhance the effectiveness of genotoxic agents in p53-deficient cells.122 A phase I study of 21 in combination with perifosine or irinotecan showed some partial response in p53-defective triple-negative breast cancer,123 whereas a phase II trial in metastatic melanoma showed that 21 as a single agent is not active in refractory melanoma.124 Both 19 and 21 have been found to be not favorable for further development because of pharmacokinetic and toxicity issues. MK-8776 (SCH900776) (22) and LY2603618 (23) are CHK1 selective inhibitors (50- to 500-fold more active than on CHK2)125,126 that are being used in clinical trials for the treatment of leukemias and solid tumors. Cardiac toxicity and myelosuppression were observed in phase I trials of both 22 and 23 in combination therapy with cytarabine and pemetrexed, respectively.127,12822 was used in combination with cytarabine to treat refractory acute leukemias. Complete remissions occurred in 8 (33%) of 24 patients, mostly at a dose of 40 mg/m2 or higher of 22. The maximum tolerated dose for 22 was approximately 56 mg/m2.12723 in combination with pemetrexed (500 mg/m2) for the treatment of solid tumors revealed a maximum tolerated dose for 23 of 150 mg/m2; some partial response or disease stabilization was observed. 23 is currently in phase II trials in combination with cisplatin and pemetrexed.126,129 Some CHK2-selective inhibitors have been identified, including PV1019 (24)130 and CCT241533 (25).131 Both agents were reported to be highly selective against CHK1 and have radioprotective effects in mouse thymocytes.130,131 As single agents, both 24 and 25 had mild antitumor activity but were found to potentiate the cytotoxicity of genotoxic agents and PARP inhibitors, respectively.130 However, clinical evaluation is not available for these agents.
Oncogenic RAS and Anti-RAS Therapeutics
Activating mutations in three oncogenic RAS genes (H-, N-, and K-RAS) is among the first and the most common genetic alterations identified in human cancers, occurring in approximately 30% of human tumors.132 The KRAS gene encodes two splicing isoforms, a major KRAS 4B and a minor KRAS 4A. Therefore, mammals have four small (21 kDa) oncogenic Ras proteins of about 190 amino acids in size, with the first 165 aa conserved in the N-terminal for all the RAS proteins. The KRAS 4B, HRAS, and NRAS isoforms are ubiquitously expressed, whereas KRAS 4A is expressed mainly in kidney, liver, and gastrointestinal tissues.133 As a subfamily of small guanine nucleotide-binding proteins, RAS proteins cycle between an active guanosine triphosphate (GTP) bound form and an inactive guanosine diphosphate (GDP) bound form.134 Binding of RAS with GTP is facilitated by guanine nucleotide exchange factors through catalyzing the release of GDP and is required for the interaction of RAS with target proteins. The intrinsic GTPase activity that is enhanced by GTPase-activating proteins converts GTP to GDP, leading to a GDP-bound, inactive RAS. RAS mutations that diminish the GTPase activity or decrease the GDP binding capacity render RAS in a constitutively active, GTP-bound status. In the absence of a RAS mutation, increased Ras activity is frequently detected in human cancer because of gene amplification,135 overexpression,136 an increase in upstream signals from tyrosine-kinase growth-factor receptors such as HER2 and EGFR,137 and altered expression of micro-RNA such as let-7.138 Increased RAS activity is associated with resistance to chemotherapy and radiotherapy, leading to a poor prognosis.139,140
As a key mediator in the signaling transduction for a variety of growth factors, cytokines, and hormones, RAS proteins are transferred to the inner leaflet of the plasma membrane, where they interact with a diversity of membrane receptors and execute signal transduction in a variety of signaling pathways that govern cell growth, proliferation, differentiation, and death. Several steps of posttranslational modifications are critical for trafficking RAS proteins to the plasma membrane, including farnesylation at the cysteine residue of the carboxy-terminal CAAX motif and methylation of farnesyl cysteine at the C-terminal.141 The enzymes involved in these processes, such as farnesyltransferase,142 geranylgeranyltransferase,143 and isoprenylcysteine carboxyl methyltransferase,144 have been intensively investigated in preclinical and clinical trials for anti-RAS therapy. Small molecules binding irreversibly to the KRAS (G12C) mutant protein145 or interfering with Ras–effector protein interaction146 have recently been reported. Targeting the Ras downstream pathways, particularly the RAF/MEK/ERK and PI3K/AKT/mTOR pathways,147 has also been investigated for inhibiting Ras mutant cancer cells. A clinical trial with biomarker-integrated targeted therapy for lung cancer has revealed that sorafenib (26), a pan-RAF and VEGFR inhibitor, has impressive benefits for KRAS mutant patients.148 However, selective inhibition of BRAF with a dominant negative construct149 in mice or with BRAF-selective inhibitors such as vemurafenib (27) in patients150 promoted the development and/or progression of RAS mutant cancers possibly because of the activation of other RAF isoforms, such as RAF-1. Thus far, effective anti-RAS therapeutics is not clinically available.
Indirect anti-Ras therapy with the synthetic lethality-based approach has recently been investigated by several groups. It is noteworthy that expression of oncogenic RAS in primary normal human or rodent cells often results in apoptosis or senescence, whereas expression of oncogenic RAS in immortal cells or cells with inactivation of p53, p16, or the transcriptional activator interferon regulatory factor 1 leads to transformation and tumorigenesis,151−153 suggesting that RAS transformed cells have additional signaling context for survival, which provides opportunity of synthetic lethality based anti-RAS therapy. In fact, RAS pathways interact with many other cancer related pathways. Several studies on siRNA library screens with isogenic cell lines harboring mutant and wild-type RAS genes have revealed synthetic lethal interactions of oncogenic RAS with some key nodes in cancer signaling network, including Polo-like kinase 1 (PLK1), a serine/threonine protein kinase that regulates cell mitosis;10 the transcription factor Wilms tumor 1 (WT1);154 TANK-binding kinase 1 (TBK1), a noncanonical IκB kinase that regulates the stability of IκB;9 spleen tyrosine kinase (SYK);155 and CDK411 (Figure 7). Some small-molecule inhibitors of PLK1, CDK4, and SYK are already being used in clinical trials for anticancer therapy.156,157 The synthetic lethality interactions of those targets with oncogenic RAS suggest that RAS gene mutation might be used as a marker to identify responders.
Figure 7.
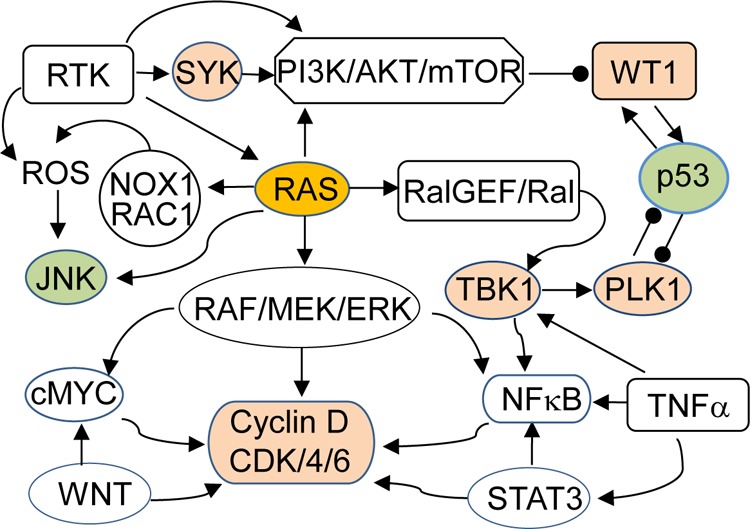
Ras signaling pathways and synthetic lethal interactions. Receptor tyrosine kinases (RTK) activate RAS and PI3K/AKT/mTOR pathways. RAS in turn activates RAF/MEK/ERK, PI3K/AKT/mTOR, and RalGEF/Ral pathways and crosstalks with RTK, WNT, c-Jun N-terminal kinases (JNK), reactive oxidative species (ROS), STAT3, and tumor necrosis factor α (TNFα)/NFκB pathways. Several key nodes in those signaling networks, such as TBK1, PLK1, CDK4, SYK, and WT1 (colored tan), have been reported to have synthetic lethal interactions with oncogenic RAS. In contrast, activation of p53 and JNK may lead to apoptosis. Arrow indicates activation, and the line terminating with a black circle indicates suppression.
Several innovative anticancer agents that selectively induce cytotoxic effects in cancer cells with RAS mutations have been identified by screening chemical libraries, including triphenyltetrazolium (28) and a sulfinyl cytidine derivative (29) (Figure 8) that demonstrated approximately 6-fold selectivity for cell lines containing mutant KRAS;158 erastin (30), which exhibited lethal selectivity in human tumor cells harboring mutations in the HRAS, KRAS, or BRAF oncogenes by acting on mitochondrial voltage-dependent anion channels and inducing oxidative cell death;7 lanperisone (31);159 and oncrasin-1 (32).8 Interestingly, 30,16031,159 and analogues of 32(161,162) all induced cell-killing effects in RAS mutant tumor cells by triggering oxidative stress, although through different underlying mechanisms. While 30 may act on mitochondrial voltage-dependent anion channels and induce oxidative cell death,16032 and its analogues act on RNA polymerase II, protein kinase Cι (PKCι), and STAT3.8,163,164 Most of the molecules involved in oncrasin-induced antitumor activity are directly involved either in signal transductions in RAS, MYC, and STAT3 pathways or in causing synthetic lethality in cells with elevated activity of RAS or MYC oncogenes.
Figure 8.

Structures of oncogenic Ras synthetic lethal agents.
Similar to the agents identified through other approaches, substantial efforts will be required for optimization of compounds identified through synthetic lethality screening. Effort has been made in the Developmental Therapeutics Program at the National Cancer Institute and in our own laboratory for compound optimization of oncrasin 1. One of the most active analogue compounds, NSC-743380 (33), was tested in vitro with the NCI-60 cell panel and 50 non-small-cell lung carcinoma cell lines and was highly active (IC50 between 10 nM and 1 μM) in 30 of 102 cancer cell lines tested.164,165 In 50 non-small-cell lung cancer cell lines tested, 16 (32%) were highly responsive to this compound (IC50 < 0.33 μM), including many KRAS mutant cancer cell lines,8,165 suggesting that a large subset of cancer patients may potentially benefit from treatment with this agent. In vivo studies performed in our laboratory and by the National Cancer Institute’s Developmental Therapeutics Program have shown that intraperitoneal administration of 33 at doses of 67–150 mg/kg caused complete tumor regression or significant growth suppression in some xenograft tumor models,164,165 suggesting that this compound has the potential to move to further development. Nevertheless, most compounds developed by synthetic lethality screening for oncogenic RAS are still at the preclinical evaluation stage.
Future Prospects
The enthusiasm for developing synthetic lethality based anticancer therapeutics has been increasing recently, with increasing numbers of publications on this topic. Most potential therapeutic targets or therapeutic agents developed through the concept of synthetic lethality are identified by cell-based screening with pairs of isogenic cell lines. Although this approach may facilitate development of genotype-specific anticancer therapeutics, several studies have demonstrated that synthetic lethality is likely cell context dependent. For example, KRAS mutant cancer cell lines have been characterized as either KRAS-dependent or KRAS-independent, on the basis of cell-killing effects induced by knockdown of the KRAS gene itself.155 When 45 genes that have synthetic lethality with EGFR inhibitors in the cervical adenocarcinoma cell line A431 were tested for sensitization to erlotinib or cetuximab in seven other cell lines, none of the genes sensitized all tested cell lines, although several of the genes sensitized three to five of the cell lines.12 Similarly, resistance to the synthetic lethality of PARP inhibitors in BRCA1 and BRCA2 mutant cancers has been observed both in experimental tumor models and in clinical trials.166 The differences in genetic and/or epigenetic backgrounds in individual cells may explain the cell-context-dependent synthetic lethality observed in various studies. Because a tumor may harbor an average of 30–70 mutated genes,1 an individual primary tumor or established cancer cell line may carry multiple concomitantly activated oncogenes or inactivated tumor suppressor genes. The activation in other signaling pathways may provide redundant input that drives and maintains downstream survival signaling, resulting in resistance to therapeutic agents that target a particular genetic lesion either directly or indirectly through synthetic lethality. Moreover, even though some passenger mutations in tumor cells may not contribute to tumorigenesis, these mutations may affect drug response because of altered drug metabolism or drug efflux. As a result, querying for the synthetic lethality partners of an oncogenic KRAS gene can identify different candidate genes or lead compounds in different cell lines.9,10,154 Similarly, a synthetic lethality gene–agent combination identified in one cell line may not necessarily be applicable to another cell line.12 Mechanistic delineation of molecules or pathways that mediate responses to synthetic lethality anticancer therapeutics and the identification of predictive biomarkers for treatment responses will be critical for the successful development of synthetic lethality anticancer therapeutics. On the other hand, the concept of synthetic lethality has also been explored for characterizing mechanisms of resistance and for combination therapy or multimodality therapy to enhance the efficacy of anticancer drugs.12−14 Simultaneous targeting the redundant survival pathways is expected to overcome resistance and/or enhance efficacy in anticancer therapy. However, toxicity may increase as well.
Our own experience in developing synthetic-lethality-based anticancer therapeutics suggests that mechanistic characterization163,167 and compound optimization164,168 could be challenging and time-consuming. The biological functions of compounds identified through cell-based synthetic lethality screening are largely unknown. Technological developments in chemical biology, such as conjugating compounds to biotin or resin beads and performing affinity-based target precipitation and subsequent protein identification by mass spectrometry analysis, may facilitate target and mechanistic characterization. However, conjugating and immobilizing a small compound may change its biological function and protein-binding specificity. Alternatively, target proteins may be enriched by protecting the targets from protease-mediated degradation through the binding of unmodified compounds to their targets. The results may depend on the efficiency and specificity of the protection. Robust efforts on in vivo optimization of the lead compound are also essential in drug development, as compounds with similar chemical structures and in vitro activity may have dramatically different in vivo toxicity and efficacy profiles.
Acknowledgments
I thank Shuhong Wu for assistance in preparation of the manuscript and Luanne Jorewicz in the Department of Scientific Publications of The University of Texas MD Anderson Cancer Center, TX, for editorial review of the manuscript. The research was supported by National Institutes of Health/National Cancer Institute Grant R01 CA 124951.
Glossary
Abbreviations Used
- AKT
protein kinase B
- ALC1
amplified in liver cancer protein 1
- ATM
ataxia telangiectasia-mutated
- ATR
ataxia telangiectasia-mutated and Rad3 related
- BRCA
breast cancer
- CA-125
cancer antigen 125
- CDC
cell division cycle
- CDK
cyclin-dependent kinase
- CHK
checkpoint kinase
- DNA-PK
DNA-dependent protein kinase
- DSB
DNA double-strand break
- EGFR
epidermal growth factor receptor
- ERCC
excision repair cross-complementing rodent repair deficiency
- ERK
extracellular signal-regulated kinase
- H2AX
histone H2AX
- HDM2
human homologue of double minute 2
- HER2
human epidermal growth factor receptor 2
- HR
homologous recombination
- JNK
c-Jun N-terminal kinase
- MAP
mitogen activated protein
- MEK
mitogen-activated protein kinase kinase
- MET
mesenchymal epithelial transition factor
- MK2
mitogen-activated protein kinase-activated protein kinase 2
- MRE11
meiotic recombination 11 homologue
- mTOR
mammalian target of rapamycin
- MYC
v-myc avian myelocytomatosis viral oncogene homologue
- NBS1
Nijmegen breakage syndrome 1
- NFκB
nuclear factor κ-light-chain-enhancer of activated B cells
- NHEJ
nonhomologous end joining
- NOX1
NADPH oxidase 1
- PARP
poly ADP-ribose polymerase
- PCNA
proliferating cell nuclear protein
- PI3K
phosphoinositide 3-kinase
- PIKK
phosphatidylinositol 3-kinase related kinase
- PKC
protein kinase C
- PLK1
Polo-like kinase 1
- PTEN
phosphatase and tensin homologue
- RAC1
ras-related C3 botulinum toxin substrate 1
- RAF
rapidly accelerated fibrosarcoma
- RalGEF
Ral guanine nucleotide exchange factor
- RAS
rat sarcoma
- RECIST
response evaluation criteria in solid tumors
- ROS
reactive oxidative species
- RPA
replication protein A
- RTK
receptor tyrosine kinase
- siRNA
small interfering RNA
- SSB
DNA single-strand break
- STAT3
signal transducer and activator of transcription 3
- SYK
spleen tyrosine kinase
- TBK1
TANK-binding kinase 1
- TNFα
tumor necrosis factor α
- TP53
tumor protein p53
- VEGFR
vascular endothelial growth factor receptor
- WNT
wingless-type MMTV integration site family
- WT1
Wilms tumor 1
- XRCC
X-ray repair complementing defective repair in Chinese hamster cells
Biography
Bingliang Fang received his M.D. degree in human genetics at Hamburg University (Germany) in 1989. He has been a faculty member in the University of Texas MD Anderson Cancer since 1995 and is currently a Professor in the Department of Thoracic and Cardiovascular Surgery at MD Anderson Cancer Center, TX. His primary research interest is innovative anticancer therapy, the mechanisms of action of and/or resistance to anticancer agents, and molecular signatures or biomarkers for predicting treatment response or early diagnosis of cancers. One of his current research projects is to develop synthetic lethality anticancer therapeutics.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Vogelstein B.; Papadopoulos N.; Velculescu V. E.; Zhou S.; Diaz L. A. Jr.; Kinzler K. W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M. N.; Halling-Brown M. D.; Tym J. E.; Workman P.; Al-Lazikani B. Objective assessment of cancer genes for drug discovery. Nat. Rev. Drug Discovery 2013, 12, 35–50. [DOI] [PubMed] [Google Scholar]
- Hartwell L. H.; Szankasi P.; Roberts C. J.; Murray A. W.; Friend S. H. Integrating genetic approaches into the discovery of anticancer drugs. Science 1997, 278, 1064–1068. [DOI] [PubMed] [Google Scholar]
- Dobzhansky T. H. Genetics of natural populations. XIII. Recombination and variability and populations of drosophila pseudoobscura. Genetics 1946, 31, 269–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi J. C. Synthetic lethality and semi-lethality among functionally related mutants of Drosophila melanfgaster. Genetics 1968, 59, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H.; McCabe N.; Lord C. J.; Tutt A. N.; Johnson D. A.; Richardson T. B.; Santarosa M.; Dillon K. J.; Hickson I.; Knights C.; Martin N. M.; Jackson S. P.; Smith G. C.; Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Dolma S.; Lessnick S. L.; Hahn W. C.; Stockwell B. R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [DOI] [PubMed] [Google Scholar]
- Guo W.; Wu S.; Liu J.; Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res. 2008, 68, 7403–7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie D. A.; Tamayo P.; Boehm J. S.; Kim S. Y.; Moody S. E.; Dunn I. F.; Schinzel A. C.; Sandy P.; Meylan E.; Scholl C.; Frohling S.; Chan E. M.; Sos M. L.; Michel K.; Mermel C.; Silver S. J.; Weir B. A.; Reiling J. H.; Sheng Q.; Gupta P. B.; Wadlow R. C.; Le H.; Hoersch S.; Wittner B. S.; Ramaswamy S.; Livingston D. M.; Sabatini D. M.; Meyerson M.; Thomas R. K.; Lander E. S.; Mesirov J. P.; Root D. E.; Gilliland D. G.; Jacks T.; Hahn W. C. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J.; Emanuele M. J.; Li D.; Creighton C. J.; Schlabach M. R.; Westbrook T. F.; Wong K. K.; Elledge S. J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137, 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puyol M.; Martin A.; Dubus P.; Mulero F.; Pizcueta P.; Khan G.; Guerra C.; Santamaria D.; Barbacid M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010, 18, 63–73. [DOI] [PubMed] [Google Scholar]
- Astsaturov I.; Ratushny V.; Sukhanova A.; Einarson M. B.; Bagnyukova T.; Zhou Y.; Devarajan K.; Silverman J. S.; Tikhmyanova N.; Skobeleva N.; Pecherskaya A.; Nasto R. E.; Sharma C.; Jablonski S. A.; Serebriiskii I. G.; Weiner L. M.; Golemis E. A. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci. Signaling 2010, 3, ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehurst A. W.; Bodemann B. O.; Cardenas J.; Ferguson D.; Girard L.; Peyton M.; Minna J. D.; Michnoff C.; Hao W.; Roth M. G.; Xie X. J.; White M. A. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 2007, 446, 815–819. [DOI] [PubMed] [Google Scholar]
- Dai B.; Yoo S. Y.; Bartholomeusz G.; Graham R. A.; Majidi M.; Yan S.; Meng J.; Ji L.; Coombes K.; Minna J. D.; Fang B.; Roth J. A. KEAP1-dependent synthetic lethality induced by AKT and TXNRD1 inhibitors in lung cancer. Cancer Res. 2013, 73, 5532–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin W. G. Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [DOI] [PubMed] [Google Scholar]
- Le Meur N.; Gentleman R. Modeling synthetic lethality. Genome Biol. 2008, 9, R135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong A. H.; Lesage G.; Bader G. D.; Ding H.; Xu H.; Xin X.; Young J.; Berriz G. F.; Brost R. L.; Chang M.; Chen Y.; Cheng X.; Chua G.; Friesen H.; Goldberg D. S.; Haynes J.; Humphries C.; He G.; Hussein S.; Ke L.; Krogan N.; Li Z.; Levinson J. N.; Lu H.; Menard P.; Munyana C.; Parsons A. B.; Ryan O.; Tonikian R.; Roberts T.; Sdicu A. M.; Shapiro J.; Sheikh B.; Suter B.; Wong S. L.; Zhang L. V.; Zhu H.; Burd C. G.; Munro S.; Sander C.; Rine J.; Greenblatt J.; Peter M.; Bretscher A.; Bell G.; Roth F. P.; Brown G. W.; Andrews B.; Bussey H.; Boone C. Global mapping of the yeast genetic interaction network. Science 2004, 303, 808–813. [DOI] [PubMed] [Google Scholar]
- Hollstein M.; Sidransky D.; Vogelstein B.; Harris C. C. p53 mutations in human cancers. Science 1991, 253, 49–53. [DOI] [PubMed] [Google Scholar]
- Olivier M.; Hollstein M.; Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor Perspect. Biol. 2010, 2, a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J.; Horejsi Z.; Koed K.; Kraemer A.; Tort F.; Zieger K.; Guldberg P.; Sehested M.; Nesland J. M.; Lukas C.; Orntoft T.; Lukas J.; Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers J. H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [DOI] [PubMed] [Google Scholar]
- Langelier M. F.; Planck J. L.; Roy S.; Pascal J. M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A. A.; Timinszky G.; Arribas-Bosacoma R.; Kozlowski M.; Hassa P. O.; Hassler M.; Ladurner A. G.; Pearl L. H.; Oliver A. W. The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat. Struct. Mol. Biol. 2012, 19, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg M. S.; Lindh M.; Acs K.; Vrouwe M. G.; Pines A.; van A. H.; Mullenders L. H.; Dantuma N. P. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J. Cell Biol. 2012, 197, 267–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi A. A.; Jeggo P. A. The repair and signaling responses to DNA double-strand breaks. Adv. Genet. 2013, 82, 1–45. [DOI] [PubMed] [Google Scholar]
- Sperka T.; Wang J.; Rudolph K. L. DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol. 2012, 13, 579–590. [DOI] [PubMed] [Google Scholar]
- Matsuoka S.; Ballif B. A.; Smogorzewska A.; McDonald E. R. III; Hurov K. E.; Luo J.; Bakalarski C. E.; Zhao Z.; Solimini N.; Lerenthal Y.; Shiloh Y.; Gygi S. P.; Elledge S. J. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [DOI] [PubMed] [Google Scholar]
- Chen J.; Silver D. P.; Walpita D.; Cantor S. B.; Gazdar A. F.; Tomlinson G.; Couch F. J.; Weber B. L.; Ashley T.; Livingston D. M.; Scully R. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol. Cell 1998, 2, 317–328. [DOI] [PubMed] [Google Scholar]
- Pellegrini L.; Yu D. S.; Lo T.; Anand S.; Lee M.; Blundell T. L.; Venkitaraman A. R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 2002, 420, 287–293. [DOI] [PubMed] [Google Scholar]
- Ramsden D. A.; Gellert M. Ku protein stimulates DNA end joining by mammalian DNA ligases: a direct role for Ku in repair of DNA double-strand breaks. EMBO J. 1998, 17, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D.; Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat. Struct. Mol. Biol. 2010, 17, 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson B. A.; Kraus W. L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [DOI] [PubMed] [Google Scholar]
- Kim M. Y.; Zhang T.; Kraus W. L. Poly(ADP-ribosyl)ation by PARP-1: “PAR-laying” NAD+ into a nuclear signal. Genes Dev. 2005, 19, 1951–1967. [DOI] [PubMed] [Google Scholar]
- Ruscetti T.; Lehnert B. E.; Halbrook J.; Trong H. L.; Hoekstra M. F.; Chen D. J.; Peterson S. R. Stimulation of the DNA-dependent protein kinase by poly(ADP-ribose) polymerase. J. Biol. Chem. 1998, 273, 14461–14467. [DOI] [PubMed] [Google Scholar]
- Ahel I.; Ahel D.; Matsusaka T.; Clark A. J.; Pines J.; Boulton S. J.; West S. C. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 2008, 451, 81–85. [DOI] [PubMed] [Google Scholar]
- Haince J. F.; Kozlov S.; Dawson V. L.; Dawson T. M.; Hendzel M. J.; Lavin M. F.; Poirier G. G. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 2007, 282, 16441–16453. [DOI] [PubMed] [Google Scholar]
- Chou D. M.; Adamson B.; Dephoure N. E.; Tan X.; Nottke A. C.; Hurov K. E.; Gygi S. P.; Colaiacovo M. P.; Elledge S. J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 18475–18480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleschke J. M.; Kleczkowska H. E.; Strohm M.; Althaus F. R. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem. 2000, 275, 40974–40980. [DOI] [PubMed] [Google Scholar]
- Wei L.; Nakajima S.; Hsieh C. L.; Kanno S.; Masutani M.; Levine A. S.; Yasui A.; Lan L. Damage response of XRCC1 at sites of DNA single strand breaks is regulated by phosphorylation and ubiquitylation after degradation of poly(ADP-ribose). J. Cell Sci. 2013, 126, 4414–4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R.; Lavrik O. I.; Kim S. J.; Kedar P.; Yang X. P.; Vande Berg B. J.; Wilson S. H. DNA polymerase beta-mediated long patch base excision repair. Poly(ADP-ribose)polymerase-1 stimulates strand displacement DNA synthesis. J. Biol. Chem. 2001, 276, 32411–32414. [DOI] [PubMed] [Google Scholar]
- Sukhanova M. V.; Khodyreva S. N.; Lebedeva N. A.; Prasad R.; Wilson S. H.; Lavrik O. I. Human base excision repair enzymes apurinic/apyrimidinic endonuclease1 (APE1), DNA polymerase beta and poly(ADP-ribose) polymerase 1: interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucleic Acids Res. 2005, 33, 1222–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahel D.; Horejsi Z.; Wiechens N.; Polo S. E.; Garcia-Wilson E.; Ahel I.; Flynn H.; Skehel M.; West S. C.; Jackson S. P.; Owen-Hughes T.; Boulton S. J. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009, 325, 1240–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couto C. A.; Wang H. Y.; Green J. C.; Kiely R.; Siddaway R.; Borer C.; Pears C. J.; Lakin N. D. PARP regulates nonhomologous end joining through retention of Ku at double-strand breaks. J. Cell Biol. 2011, 194, 367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour W. Y.; Borgmann K.; Petersen C.; Dikomey E.; Dahm-Daphi J. The absence of Ku but not defects in classical non-homologous end-joining is required to trigger PARP1-dependent end-joining. DNA Repair 2013, 12, 1134–1142. [DOI] [PubMed] [Google Scholar]
- Aguilar-Quesada R.; Munoz-Gamez J. A.; Martin-Oliva D.; Peralta A.; Valenzuela M. T.; Matinez-Romero R.; Quiles-Perez R.; Menissier-de Murcia J.; de Murcia G.; Ruiz de Almodovar M.; Oliver F. J. Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol. Biol. 2007, 8, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. G.; Cortes U.; Patnaik S.; Jasin M.; Wang Z. Q. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 2004, 23, 3872–3882. [DOI] [PubMed] [Google Scholar]
- Shibata A.; Kamada N.; Masumura K.; Nohmi T.; Kobayashi S.; Teraoka H.; Nakagama H.; Sugimura T.; Suzuki H.; Masutani M. PARP-1 deficiency causes an increase of deletion mutations and insertions/rearrangements in vivo after treatment with an alkylating agent. Oncogene 2005, 24, 1328–1337. [DOI] [PubMed] [Google Scholar]
- Menissier de Murcia J.; Ricoul M.; Tartier L.; Niedergang C.; Huber A.; Dantzer F.; Schreiber V.; Ame J. C.; Dierich A.; LeMeur M.; Sabatier L.; Chambon P.; de Murcia G. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003, 22, 2255–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Murcia J. M.; Niedergang C.; Trucco C.; Ricoul M.; Dutrillaux B.; Mark M.; Oliver F. J.; Masson M.; Dierich A.; LeMeur M.; Walztinger C.; Chambon P.; de Murcia G. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 7303–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant H. E.; Schultz N.; Thomas H. D.; Parker K. M.; Flower D.; Lopez E.; Kyle S.; Meuth M.; Curtin N. J.; Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917 (Erratum appears in Nature 2007447, 346). [DOI] [PubMed] [Google Scholar]
- Patel A. G.; Sarkaria J. N.; Kaufmann S. H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 3406–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant H. E.; Helleday T. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006, 34, 1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegan D. C.; Lu Y.; Stachelek G. C.; Crosby M. E.; Bindra R. S.; Glazer P. M. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 2201–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou A.; Pharoah P. D.; Narod S.; Risch H. A.; Eyfjord J. E.; Hopper J. L.; Loman N.; Olsson H.; Johannsson O.; Borg A.; Pasini B.; Radice P.; Manoukian S.; Eccles D. M.; Tang N.; Olah E.; Anton-Culver H.; Warner E.; Lubinski J.; Gronwald J.; Gorski B.; Tulinius H.; Thorlacius S.; Eerola H.; Nevanlinna H.; Syrjakoski K.; Kallioniemi O. P.; Thompson D.; Evans C.; Peto J.; Lalloo F.; Evans D. G.; Easton D. F. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130 (Erratum appears in Am. J. Hum. Genet. 200373, 709). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson D.; Easton D. F.; Cancer incidence in BRCA1 mutation carriers. J. Natl. Cancer Inst. 2002, 94, 1358–1365. [DOI] [PubMed] [Google Scholar]
- Roy R.; Chun J.; Powell S. N. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiemermann C.; Bowes J.; Myint F. P.; Vane J. R. Inhibition of the activity of poly(ADP ribose) synthetase reduces ischemia–reperfusion injury in the heart and skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson M. J.; Sampei K.; Mandir A. S.; Hurn P. D.; Traystman R. J.; Bao J.; Pieper A.; Wang Z. Q.; Dawson T. M.; Snyder S. H.; Dawson V. L. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 1997, 3, 1089–1095. [DOI] [PubMed] [Google Scholar]
- Ha H. C.; Hester L. D.; Snyder S. H. Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 3270–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese C. R.; Almassy R.; Barton S.; Batey M. A.; Calvert A. H.; Canan-Koch S.; Durkacz B. W.; Hostomsky Z.; Kumpf R. A.; Kyle S.; Li J.; Maegley K.; Newell D. R.; Notarianni E.; Stratford I. J.; Skalitzky D.; Thomas H. D.; Wang L. Z.; Webber S. E.; Williams K. J.; Curtin N. J. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J. Natl. Cancer Inst. 2004, 96, 56–67. [DOI] [PubMed] [Google Scholar]
- Jagtap P.; Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discovery 2005, 4, 421–440. [DOI] [PubMed] [Google Scholar]
- Rouleau M.; Patel A.; Hendzel M. J.; Kaufmann S. H.; Poirier G. G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraris D. V. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J. Med. Chem. 2010, 53, 4561–4584. [DOI] [PubMed] [Google Scholar]
- Wahlberg E.; Karlberg T.; Kouznetsova E.; Markova N.; Macchiarulo A.; Thorsell A. G.; Pol E.; Frostell A.; Ekblad T.; Oncu D.; Kull B.; Robertson G. M.; Pellicciari R.; Schuler H.; Weigelt J. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat. Biotechnol. 2012, 30, 283–288. [DOI] [PubMed] [Google Scholar]
- Skalitzky D. J.; Marakovits J. T.; Maegley K. A.; Ekker A.; Yu X. H.; Hostomsky Z.; Webber S. E.; Eastman B. W.; Almassy R.; Li J.; Curtin N. J.; Newell D. R.; Calvert A. H.; Griffin R. J.; Golding B. T. Tricyclic benzimidazoles as potent poly(ADP-ribose) polymerase-1 inhibitors. J. Med. Chem. 2003, 46, 210–213. [DOI] [PubMed] [Google Scholar]
- Weston V. J.; Oldreive C. E.; Skowronska A.; Oscier D. G.; Pratt G.; Dyer M. J.; Smith G.; Powell J. E.; Rudzki Z.; Kearns P.; Moss P. A.; Taylor A. M.; Stankovic T. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 2010, 116, 4578–4587. [DOI] [PubMed] [Google Scholar]
- Vilar E.; Bartnik C. M.; Stenzel S. L.; Raskin L.; Ahn J.; Moreno V.; Mukherjee B.; Iniesta M. D.; Morgan M. A.; Rennert G.; Gruber S. B. MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res. 2011, 71, 2632–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postel-Vinay S.; Bajrami I.; Friboulet L.; Elliott R.; Fontebasso Y.; Dorvault N.; Olaussen K. A.; Andre F.; Soria J. C.; Lord C. J.; Ashworth A. A high-throughput screen identifies PARP1/2 inhibitors as a potential therapy for ERCC1-deficient non-small cell lung cancer. Oncogene 2013, 32, 5377–5387. [DOI] [PubMed] [Google Scholar]
- Williamson C. T.; Kubota E.; Hamill J. D.; Klimowicz A.; Ye R.; Muzik H.; Dean M.; Tu L.; Gilley D.; Magliocco A. M.; McKay B. C.; Bebb D. G.; Lees-Miller S. P. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol. Med. 2012, 4, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes-Pereira A. M.; Martin S. A.; Brough R.; McCarthy A.; Taylor J. R.; Kim J. S.; Waldman T.; Lord C. J.; Ashworth A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C. J.; McDonald S.; Swift S.; Turner N. C.; Ashworth A. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair 2008, 7, 2010–2019. [DOI] [PubMed] [Google Scholar]
- Cardnell R. J.; Feng Y.; Diao L.; Fan Y. H.; Masrorpour F.; Wang J.; Shen Y.; Mills G. B.; Minna J. D.; Heymach J. V.; Byers L. A. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin. Cancer Res. 2013, 19, 6322–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner J. C.; Ateeq B.; Li Y.; Yocum A. K.; Cao Q.; Asangani I. A.; Patel S.; Wang X.; Liang H.; Yu J.; Palanisamy N.; Siddiqui J.; Yan W.; Cao X.; Mehra R.; Sabolch A.; Basrur V.; Lonigro R. J.; Yang J.; Tomlins S. A.; Maher C. A.; Elenitoba-Johnson K. S.; Hussain M.; Navone N. M.; Pienta K. J.; Varambally S.; Feng F. Y.; Chinnaiyan A. M. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell 2011, 19, 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowsheen S.; Cooper T.; Stanley J. A.; Yang E. S. Synthetic lethal interactions between EGFR and PARP inhibition in human triple negative breast cancer cells. PLoS One 2012, 7, e46614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer R.; Jones C.; Middleton M.; Wilson R.; Evans J.; Olsen A.; Curtin N.; Boddy A.; McHugh P.; Newell D.; Harris A.; Johnson P.; Steinfeldt H.; Dewji R.; Wang D.; Robson L.; Calvert H. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 7917–7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer R.; Lorigan P.; Steven N.; Scott L.; Middleton M. R.; Wilson R. H.; Mulligan E.; Curtin N.; Wang D.; Dewji R.; Abbattista A.; Gallo J.; Calvert H. A phase II study of the potent PARP inhibitor, rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemother. Pharmacol. 2013, 71, 1191–1199. [DOI] [PubMed] [Google Scholar]
- Fong P. C.; Yap T. A.; Boss D. S.; Carden C. P.; Mergui-Roelvink M.; Gourley C.; De Greve J.; Lubinski J.; Shanley S.; Messiou C.; A’Hern R.; Tutt A.; Ashworth A.; Stone J.; Carmichael J.; Schellens J. H.; de Bono J. S.; Kaye S. B. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [DOI] [PubMed] [Google Scholar]
- Tutt A.; Robson M.; Garber J. E.; Domchek S. M.; Audeh M. W.; Weitzel J. N.; Friedlander M.; Arun B.; Loman N.; Schmutzler R. K.; Wardley A.; Mitchell G.; Earl H.; Wickens M.; Carmichael J. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 2010, 376, 235–244. [DOI] [PubMed] [Google Scholar]
- Bundred N.; Gardovskis J.; Jaskiewicz J.; Eglitis J.; Paramonov V.; McCormack P.; Swaisland H.; Cavallin M.; Parry T.; Carmichael J.; Dixon J. M. Evaluation of the pharmacodynamics and pharmacokinetics of the PARP inhibitor olaparib: a phase I multicentre trial in patients scheduled for elective breast cancer surgery. Invest. New Drugs 2013, 31, 949–958. [DOI] [PubMed] [Google Scholar]
- Yamamoto N.; Nokihara H.; Yamada Y.; Goto Y.; Tanioka M.; Shibata T.; Yamada K.; Asahina H.; Kawata T.; Shi X.; Tamura T. A phase I, dose-finding and pharmacokinetic study of olaparib (AZD2281) in Japanese patients with advanced solid tumors. Cancer Sci. 2012, 103, 504–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan A.; Carter C. A.; Kelly R. J.; Gutierrez M.; Kummar S.; Szabo E.; Yancey M. A.; Ji J.; Mannargudi B.; Woo S.; Spencer S.; Figg W. D.; Giaccone G. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin. Cancer Res. 2012, 18, 2344–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. F.; Tolaney S. M.; Birrer M.; Fleming G. F.; Buss M. K.; Dahlberg S. E.; Lee H.; Whalen C.; Tyburski K.; Winer E.; Ivy P.; Matulonis U. A. A phase 1 trial of the poly(ADP-ribose) polymerase inhibitor olaparib (AZD2281) in combination with the anti-angiogenic cediranib (AZD2171) in recurrent epithelial ovarian or triple-negative breast cancer. Eur. J. Cancer 2013, 49, 2972–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye S. B.; Lubinski J.; Matulonis U.; Ang J. E.; Gourley C.; Karlan B. Y.; Amnon A.; Bell-McGuinn K. M.; Chen L. M.; Friedlander M.; Safra T.; Vergote I.; Wickens M.; Lowe E. S.; Carmichael J.; Kaufman B. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J. Clin.Oncol. 2012, 30, 372–379. [DOI] [PubMed] [Google Scholar]
- Kummar S.; Chen A.; Ji J.; Zhang Y.; Reid J. M.; Ames M.; Jia L.; Weil M.; Speranza G.; Murgo A. J.; Kinders R.; Wang L.; Parchment R. E.; Carter J.; Stotler H.; Rubinstein L.; Hollingshead M.; Melillo G.; Pommier Y.; Bonner W.; Tomaszewski J. E.; Doroshow J. H. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011, 71, 5626–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu S. K.; Schelman W. R.; Wilding G.; Moreno V.; Baird R. D.; Miranda S.; Hylands L.; Riisnaes R.; Forster M.; Omlin A.; Kreischer N.; Thway K.; Gevensleben H.; Sun L.; Loughney J.; Chatterjee M.; Toniatti C.; Carpenter C. L.; Iannone R.; Kaye S. B.; de Bono J. S.; Wenham R. M. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013, 14, 882–892. [DOI] [PubMed] [Google Scholar]
- de Bono J. S.; Mina L. A.; Gonzalez M.; Curtin N. J.; Wang E.; Henshaw J. W.; Chadha M.; Sachdev J. C.; Matei D.; Jameson G. S.; Ong M.; Basu B.; Wainberg Z. A.; Byers L. A.; Chugh R.; Dorr A.; Kaye S. B.; Ramanathan R. K. First-in-human trial of novel oral PARP inhibitor BMN 673 in patients with solid tumor. J. Clin. Oncol. 2013, 31, 2580.23733753 [Google Scholar]
- Banin S.; Moyal L.; Shieh S.; Taya Y.; Anderson C. W.; Chessa L.; Smorodinsky N. I.; Prives C.; Reiss Y.; Shiloh Y.; Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [DOI] [PubMed] [Google Scholar]
- Chehab N. H.; Malikzay A.; Appel M.; Halazonetis T. D. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev. 2000, 14, 278–288. [PMC free article] [PubMed] [Google Scholar]
- Tibbetts R. S.; Brumbaugh K. M.; Williams J. M.; Sarkaria J. N.; Cliby W. A.; Shieh S. Y.; Taya Y.; Prives C.; Abraham R. T. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999, 13, 152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzelli M.; Draetta G. F. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003, 4, 671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliby W. A.; Roberts C. J.; Cimprich K. A.; Stringer C. M.; Lamb J. R.; Schreiber S. L.; Friend S. H. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998, 17, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uto K.; Inoue D.; Shimuta K.; Nakajo N.; Sagata N. Chk1, but not Chk2, inhibits Cdc25 phosphatases by a novel common mechanism. EMBO J. 2004, 23, 3386–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocente S. A.; Abrahamson J. L.; Cogswell J. P.; Lee J. M. p53 regulates a G2 checkpoint through cyclin B1. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 2147–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt H. C.; Aslanian A. S.; Lees J. A.; Yaffe M. B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H.; Reinhardt H. C.; Bartkova J.; Tommiska J.; Blomqvist C.; Nevanlinna H.; Bartek J.; Yaffe M. B.; Hemann M. T. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009, 23, 1895–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan K. D.; Padilla-Just N.; Henry R. E.; Porter C. C.; Kim J.; Tentler J. J.; Eckhardt S. G.; Tan A. C.; DeGregori J.; Espinosa J. M. ATM and MET kinases are synthetic lethal with nongenotoxic activation of p53. Nat. Chem. Biol. 2012, 8, 646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey M. A.; Zhao Y.; Kyle S.; Richardson C.; Slade A.; Martin N. M.; Lau A.; Newell D. R.; Curtin N. J. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol. Cancer Ther. 2013, 12, 959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nghiem P.; Park P. K.; Kim Y.; Vaziri C.; Schreiber S. L. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 9092–9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangster-Guity N.; Conrad B. H.; Papadopoulos N.; Bunz F. ATR mediates cisplatin resistance in a p53 genotype-specific manner. Oncogene 2011, 30, 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga M.; Campaner S.; Lopez-Contreras A. J.; Toledo L. I.; Soria R.; Montana M. F.; D’Artista L.; Schleker T.; Guerra C.; Garcia E.; Barbacid M.; Hidalgo M.; Amati B.; Fernandez-Capetillo O. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasumi M.; Lemos B.; Bradner J. E.; Thibodeau R.; Kim Y. S.; Schmidt M.; Higgins E.; Koo S. W.; Angle-Zahn A.; Chen A.; Levine D.; Nguyen L.; Heffernan T. P.; Longo I.; Mandinova A.; Lu Y. P.; Conney A. H.; Nghiem P. Protection from UV-induced skin carcinogenesis by genetic inhibition of the ataxia telangiectasia and Rad3-related (ATR) kinase. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 13716–13721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R.; Abdel-Fatah T.; Perry C.; Moseley P.; Albarakti N.; Mohan V.; Seedhouse C.; Chan S.; Madhusudan S. Ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase inhibition is synthetically lethal in XRCC1 deficient ovarian cancer cells. PLoS One 2013, 8, e57098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntoon C. J.; Flatten K. S.; Wahner Hendrickson A. E.; Huehls A. M.; Sutor S. L.; Kaufmann S. H.; Karnitz L. M. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013, 73, 3683–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R.; bdel-Fatah T.; Abbotts R.; Hawkes C.; Albarakati N.; Seedhouse C.; Ball G.; Chan S.; Rakha E. A.; Ellis I. O.; Madhusudan S. Targeting XRCC1 deficiency in breast cancer for personalized therapy. Cancer Res. 2013, 73, 1621–1634. [DOI] [PubMed] [Google Scholar]
- Toledo L. I.; Murga M.; Zur R.; Soria R.; Rodriguez A.; Martinez S.; Oyarzabal J.; Pastor J.; Bischoff J. R.; Fernandez-Capetillo O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote K. M.; Blades K.; Cronin A.; Fillery S.; Guichard S. S.; Hassall L.; Hickson I.; Jacq X.; Jewsbury P. J.; McGuire T. M.; Nissink J. W.; Odedra R.; Page K.; Perkins P.; Suleman A.; Tam K.; Thommes P.; Broadhurst R.; Wood C. Discovery of 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1H-indole (AZ20): a potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 2013, 56, 2125–2138. [DOI] [PubMed] [Google Scholar]
- Charrier J. D.; Durrant S. J.; Golec J. M.; Kay D. P.; Knegtel R. M.; MacCormick S.; Mortimore M.; O’Donnell M. E.; Pinder J. L.; Reaper P. M.; Rutherford A. P.; Wang P. S.; Young S. C.; Pollard J. R. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 2011, 54, 2320–2330. [DOI] [PubMed] [Google Scholar]
- Reaper P. M.; Griffiths M. R.; Long J. M.; Charrier J. D.; MacCormick S.; Charlton P. A.; Golec J. M.; Pollard J. R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [DOI] [PubMed] [Google Scholar]
- Fokas E.; Prevo R.; Pollard J. R.; Reaper P. M.; Charlton P. A.; Cornelissen B.; Vallis K. A.; Hammond E. M.; Olcina M. M.; Gillies M. W.; Muschel R. J.; Brunner T. B. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker T. H.; Usui T.; Petrini J. H. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair 2009, 8, 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrao P. T.; Bukczynska E. P.; Johnstone R. W.; McArthur G. A. Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene 2012, 31, 1661–1672. [DOI] [PubMed] [Google Scholar]
- Landau H. J.; McNeely S. C.; Nair J. S.; Comenzo R. L.; Asai T.; Friedman H.; Jhanwar S. C.; Nimer S. D.; Schwartz G. K. The checkpoint kinase inhibitor AZD7762 potentiates chemotherapy-induced apoptosis of p53-mutated multiple myeloma cells. Mol. Cancer Ther. 2012, 11, 1781–1788. [DOI] [PubMed] [Google Scholar]
- Origanti S.; Cai S. R.; Munir A. Z.; White L. S.; Piwnica-Worms H. Synthetic lethality of Chk1 inhibition combined with p53 and/or p21 loss during a DNA damage response in normal and tumor cells. Oncogene 2013, 32, 577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan M. A.; Parsels L. A.; Zhao L.; Parsels J. D.; Davis M. A.; Hassan M. C.; Arumugarajah S.; Hylander-Gans L.; Morosini D.; Simeone D. M.; Canman C. E.; Normolle D. P.; Zabludoff S. D.; Maybaum J.; Lawrence T. S. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010, 70, 4972–4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao A.; Kong Y. Y.; Matsuoka S.; Wakeham A.; Ruland J.; Yoshida H.; Liu D.; Elledge S. J.; Mak T. W. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 2000, 287, 1824–1827. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Guntuku S.; Cui X. S.; Matsuoka S.; Cortez D.; Tamai K.; Luo G.; Carattini-Rivera S.; DeMayo F.; Bradley A.; Donehower L. A.; Elledge S. J. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Ashley T.; Brainerd E. E.; Bronson R. T.; Meyn M. S.; Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996, 10, 2411–2422. [DOI] [PubMed] [Google Scholar]
- Takai H.; Naka K.; Okada Y.; Watanabe M.; Harada N.; Saito S.; Anderson C. W.; Appella E.; Nakanishi M.; Suzuki H.; Nagashima K.; Sawa H.; Ikeda K.; Motoyama N. Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J. 2002, 21, 5195–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E. J.; Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14, 397–402. [PMC free article] [PubMed] [Google Scholar]
- Azorsa D. O.; Gonzales I. M.; Basu G. D.; Choudhary A.; Arora S.; Bisanz K. M.; Kiefer J. A.; Henderson M. C.; Trent J. M.; Von Hoff D. D.; Mousses S. Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. J. Transl. Med. 2009, 7, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto T.; Esaki T.; Hirai F.; Arita S.; Nosaki K.; Makiyama A.; Kometani T.; Fujimoto C.; Hamatake M.; Takeoka H.; Agbo F.; Shi X. Phase I, dose-escalation study of AZD7762 alone and in combination with gemcitabine in Japanese patients with advanced solid tumours. Cancer Chemother. Pharmacol. 2013, 72, 619–627. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Fan S.; Eastman A.; Worland P. J.; Sausville E. A.; O’Connor P. M. UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J. Natl. Cancer Inst. 1996, 88, 956–965. [DOI] [PubMed] [Google Scholar]
- Gojo I.; Perl A.; Luger S.; Baer M. R.; Norsworthy K. J.; Bauer K. S.; Tidwell M.; Fleckinger S.; Carroll M.; Sausville E. A. Phase I study of UCN-01 and perifosine in patients with relapsed and refractory acute leukemias and high-risk myelodysplastic syndrome. Invest. New Drugs 2013, 31, 1217–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.; Christensen S. D.; Frankel P. H.; Margolin K. A.; Agarwala S. S.; Luu T.; Mack P. C.; Lara P. N. Jr.; Gandara D. R. A phase II study of cell cycle inhibitor UCN-01 in patients with metastatic melanoma: a California Cancer Consortium trial. Invest. New Drugs 2012, 30, 741–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzi T. J.; Paruch K.; Dwyer M. P.; Labroli M.; Shanahan F.; Davis N.; Taricani L.; Wiswell D.; Seghezzi W.; Penaflor E.; Bhagwat B.; Wang W.; Gu D.; Hsieh Y.; Lee S.; Liu M.; Parry D. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol. Cancer Ther. 2011, 10, 591–602. [DOI] [PubMed] [Google Scholar]
- Arora S.; Bisanz K. M.; Peralta L. A.; Basu G. D.; Choudhary A.; Tibes R.; Azorsa D. O. RNAi screening of the kinome identifies modulators of cisplatin response in ovarian cancer cells. Gynecol. Oncol. 2010, 118, 220–227. [DOI] [PubMed] [Google Scholar]
- Karp J. E.; Thomas B. M.; Greer J. M.; Sorge C.; Gore S. D.; Pratz K. W.; Smith B. D.; Flatten K. S.; Peterson K.; Schneider P.; Mackey K.; Freshwater T.; Levis M. J.; McDevitt M. A.; Carraway H. E.; Gladstone D. E.; Showel M. M.; Loechner S.; Parry D. A.; Horowitz J. A.; Isaacs R.; Kaufmann S. H. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor Sch 900776 in refractory acute leukemias. Clin. Cancer Res. 2012, 18, 6723–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss G. J.; Donehower R. C.; Iyengar T.; Ramanathan R. K.; Lewandowski K.; Westin E.; Hurt K.; Hynes S. M.; Anthony S. P.; McKane S. Phase I dose-escalation study to examine the safety and tolerability of LY2603618, a checkpoint 1 kinase inhibitor, administered 1 day after pemetrexed 500mg/m(2) every 21 days in patients with cancer. Invest. New Drugs 2013, 31, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maugeri-Sacca M.; Bartucci M.; De Maria R. Checkpoint kinase 1 inhibitors for potentiating systemic anticancer therapy. Cancer Treat. Rev. 2013, 39, 525–533. [DOI] [PubMed] [Google Scholar]
- Jobson A. G.; Lountos G. T.; Lorenzi P. L.; Llamas J.; Connelly J.; Cerna D.; Tropea J. E.; Onda A.; Zoppoli G.; Kondapaka S.; Zhang G.; Caplen N. J.; Cardellina J. H.; Yoo S. S.; Monks A.; Self C.; Waugh D. S.; Shoemaker R. H.; Pommier Y. Cellular inhibition of checkpoint kinase 2 (Chk2) and potentiation of camptothecins and radiation by the novel Chk2 inhibitor PV1019 [7-nitro-1H-indole-2-carboxylic acid {4-[1-(guanidinohydrazone)-ethyl]-phenyl}-amide]. J. Pharmacol. Exp. Ther. 2009, 331, 816–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell J. J.; Welsh E. J.; Matijssen C.; Anderson V. E.; Antoni L.; Boxall K.; Urban F.; Hayes A.; Raynaud F. I.; Rigoreau L. J.; Raynham T.; Aherne G. W.; Pearl L. H.; Oliver A. W.; Garrett M. D.; Collins I. Structure-based design of potent and selective 2-(quinazolin-2-yl)phenol inhibitors of checkpoint kinase 2. J. Med. Chem. 2011, 54, 580–590. [DOI] [PubMed] [Google Scholar]
- Fernandez-Medarde A.; Santos E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pells S.; Divjak M.; Romanowski P.; Impey H.; Hawkins N. J.; Clarke A. R.; Hooper M. L.; Williamson D. J. Developmentally-regulated expression of murine K-ras isoforms. Oncogene 1997, 15, 1781–1786. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi D.; Hall A. Ras and Rho GTPases: a family reunion. Cell 2000, 103, 227–238. [DOI] [PubMed] [Google Scholar]
- Hoa M.; Davis S. L.; Ames S. J.; Spanjaard R. A. Amplification of wild-type K-ras promotes growth of head and neck squamous cell carcinoma. Cancer Res. 2002, 62, 7154–7156. [PubMed] [Google Scholar]
- Coleman W. B.; Throneburg D. B.; Grisham J. W.; Smith G. J. Overexpression of c-K-ras, c-N-ras and transforming growth factor beta co-segregate with tumorigenicity in morphologically transformed C3H 10T1/2 cell lines. Carcinogenesis 1994, 15, 1005–1012. [DOI] [PubMed] [Google Scholar]
- Buday L.; Downward J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 1993, 73, 611–620. [DOI] [PubMed] [Google Scholar]
- Johnson S. M.; Grosshans H.; Shingara J.; Byrom M.; Jarvis R.; Cheng A.; Labourier E.; Reinert K. L.; Brown D.; Slack F. J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [DOI] [PubMed] [Google Scholar]
- Bernhard E. J.; Stanbridge E. J.; Gupta S.; Gupta A. K.; Soto D.; Bakanauskas V. J.; Cerniglia G. J.; Muschel R. J.; McKenna W. G. Direct evidence for the contribution of activated N-ras and K-ras oncogenes to increased intrinsic radiation resistance in human tumor cell lines. Cancer Res. 2000, 60, 6597–6600. [PubMed] [Google Scholar]
- Diaz L. A. Jr.; Williams R. T.; Wu J.; Kinde I.; Hecht J. R.; Berlin J.; Allen B.; Bozic I.; Reiter J. G.; Nowak M. A.; Kinzler K. W.; Oliner K. S.; Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock J. F. Ras proteins: different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–384. [DOI] [PubMed] [Google Scholar]
- Sebti S. M.; Adjei A. A. Farnesyltransferase inhibitors. Semin. Oncol. 2004, 31, 28–39. [DOI] [PubMed] [Google Scholar]
- Berndt N.; Hamilton A. D.; Sebti S. M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter-Vann A. M.; Baron R. A.; Wong W.; dela Cruz J.; York J. D.; Gooden D. M.; Bergo M. O.; Young S. G.; Toone E. J.; Casey P. J. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 4336–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem J. M.; Peters U.; Sos M. L.; Wells J. A.; Shokat K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima F.; Yoshikawa Y.; Ye M.; Araki M.; Matsumoto S.; Liao J.; Hu L.; Sugimoto T.; Ijiri Y.; Takeda A.; Nishiyama Y.; Sato C.; Muraoka S.; Tamura A.; Osoda T.; Tsuda K.; Miyakawa T.; Fukunishi H.; Shimada J.; Kumasaka T.; Yamamoto M.; Kataoka T. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 8182–8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusconi P.; Caiola E.; Broggini M. RAS/RAF/MEK inhibitors in oncology. Curr. Med. Chem. 2012, 19, 1164–1176. [DOI] [PubMed] [Google Scholar]
- Kim E. S.; Herbst R. S.; Wistuba I. I.; Lee J. J.; Blumenschein G. R. Jr.; Tsao A.; Stewart D. J.; Hicks M. E.; Erasmus J. Jr.; Gupta S.; Alden C. M.; Liu S.; Tang X.; Khuri F. R.; Tran H. T.; Johnson B. E.; Heymach J. V.; Mao L.; Fossella F.; Kies M. S.; Papadimitrakopoulou V.; Davis S. E.; Lippman S. M.; Hong W. K. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discovery 2011, 1, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidorn S. J.; Milagre C.; Whittaker S.; Nourry A.; Niculescu-Duvas I.; Dhomen N.; Hussain J.; Reis-Filho J. S.; Springer C. J.; Pritchard C.; Marais R. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010, 140, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan M. K.; Rampal R.; Harding J. J.; Klimek V. M.; Chung Y. R.; Merghoub T.; Wolchok J. D.; Solit D. B.; Rosen N.; Abdel-Wahab O.; Levine R. L.; Chapman P. B. Progression of RAS-mutant leukemia during RAF inhibitor treatment. N. Engl. J. Med. 2012, 367, 2316–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N.; Ishihara M.; Kitagawa M.; Harada H.; Kimura T.; Matsuyama T.; Lamphier M. S.; Aizawa S.; Mak T. W.; Taniguchi T. Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell 1994, 77, 829–839. [DOI] [PubMed] [Google Scholar]
- Serrano M.; Lin A. W.; McCurrach M. E.; Beach D.; Lowe S. W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Cox A. D.; Der C. J. The dark side of Ras: regulation of apoptosis. Oncogene 2003, 22, 8999–9006. [DOI] [PubMed] [Google Scholar]
- Vicent S.; Chen R.; Sayles L. C.; Lin C.; Walker R. G.; Gillespie A. K.; Subramanian A.; Hinkle G.; Yang X.; Saif S.; Root D. E.; Huff V.; Hahn W. C.; Sweet-Cordero E. A. Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J. Clin. Invest. 2010, 120, 3940–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.; Greninger P.; Rhodes D.; Koopman L.; Violette S.; Bardeesy N.; Settleman J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt Y.; Lampkin T. Targeting Polo-like kinase in cancer therapy. Clin. Cancer Res. 2010, 16, 384–389. [DOI] [PubMed] [Google Scholar]
- Dickson M. A.; Tap W. D.; Keohan M. L.; D’Angelo S. P.; Gounder M. M.; Antonescu C. R.; Landa J.; Qin L. X.; Rathbone D. D.; Condy M. M.; Ustoyev Y.; Crago A. M.; Singer S.; Schwartz G. K. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J. Clin. Oncol. 2013, 31, 2024–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrance C. J.; Agrawal V.; Vogelstein B.; Kinzler K. W. Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat. Biotechnol. 2001, 19, 940–945. [DOI] [PubMed] [Google Scholar]
- Shaw A. T.; Winslow M. M.; Magendantz M.; Ouyang C.; Dowdle J.; Subramanian A.; Lewis T. A.; Maglathin R. L.; Tolliday N.; Jacks T. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 8773–8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagoda N.; von Rechenberg M.; Zaganjor E.; Bauer A. J.; Yang W. S.; Fridman D. J.; Wolpaw A. J.; Smukste I.; Peltier J. M.; Boniface J. J.; Smith R.; Lessnick S. L.; Sahasrabudhe S.; Stockwell B. R. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W.; Wei X.; Wu S.; Wang L.; Peng H.; Wang J.; Fang B. Antagonistic effect of flavonoids on NSC-741909-mediated antitumor activity via scavenging of reactive oxygen species. Eur. J. Pharmacol. 2010, 649, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X.; Guo W.; Wu S.; Wang L.; Huang P.; Liu J.; Fang B. Oxidative stress in NSC-741909-induced apoptosis of cancer cells. J. Transl. Med. 2010, 8, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W.; Wu S.; Wang L.; Wang R.; Wei L.; Liu J.; Fang B. Interruption of RNA processing machinery by a small compound 1-[(4-chlorophenyl)methyl]-1H-indole-3-carboxaldehyde (oncrasin-1). Mol. Cancer Ther. 2009, 8, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W.; Wu S.; Wang L.; Wei X.; Liu X.; Wang J.; Lu Z.; Hollingshead M.; Fang B. Antitumor activity of a novel oncrasin analogue is mediated by JNK activation and STAT3 inhibition. PLoS One 2011, 6, e28487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Guo W.; Wu S.; Wang L.; Wang J.; Dai B.; Kim E. S.; Heymach J. V.; Wang M.; Girard L.; Minna J.; Roth J. A.; Swisher S. G.; Fang B. Antitumor activity of a novel STAT3 inhibitor and redox modulator in non-small cell lung cancer cells. Biochem. Pharmacol. 2012, 83, 1456–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers J. E.; Kersbergen A.; Boon U.; Sol W.; van D. L.; Zander S. A.; Drost R.; Wientjens E.; Ji J.; Aly A.; Doroshow J. H.; Cranston A.; Martin N. M.; Lau A.; O’Connor M. J.; Ganesan S.; Borst P.; Jonkers J.; Rottenberg S. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discovery 2013, 3, 68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X.; Guo W.; Wu S.; Wang L.; Lu Y.; Xu B.; Liu J.; Fang B. Inhibiting JNK dephosphorylation and induction of apoptosis by novel anticancer agent NSC-741909 in cancer cells. J. Biol. Chem. 2009, 284, 16948–16955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Wang L.; Guo W.; Liu X.; Liu J.; Wei X.; Fang B. Analogues and derivatives of oncrasin-1, a novel inhibitor of the C-terminal domain of RNA polymerase II and their antitumor activities. J. Med. Chem. 2011, 54, 2668–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]