

Abstract
Neuroplasticity serves an important role for normal striatal function and in disease states. One route to neuroplasticity involves activation of the transcription factor cyclic 3′,5′-adenosine monophosphate (cyclic AMP) response element binding protein (CREB) by phosphorylation of the amino acid 133Ser. Dopamine and glutamate, the two predominant neurotransmitters in the striatum, induce CREB phosphorylation in primary cultures of rat striatum through cyclic AMP and Ca2+ pathways. Here we present the role of N-methyl-D-aspartate receptors and Ca2+ in cyclic AMP-mediated CREB phosphorylation.
Keywords: Striatum, Dopamine, N-Methyl-D-aspartate, DARPP-32, Cyclic AMP response element binding protein, Cyclic AMP, Protein kinase A, Ca2+/calmodulin kinase, Calcineurin, Protein phosphatase 1
Introduction
Neuroplasticity is the term used to define the adaptability of the brain and its cellular components to changing requirements. The central mechanism of neuroplasticity is the modification of efficacy and number of synapses during development and throughout life, thus modeling the brain and adjusting its functions. Neuroplasticity requires intraneuronal processes to both change cellular properties due to arising demands and to store information about the modified properties. Control of protein synthesis accomplishes these requirements [1–4]. Stimulus-dependent protein modification, in particular cyclic 3′,5′-adenosine monophosphate (cyclic AMP)- and Ca2+-mediated phosphorylation, confers neuroplasticity by changing protein function, RNA and protein synthesis, and neuronal protein composition [5–7].
The short review and the data presented here examine the effect of Ca2+- and cyclic AMP-mediated second messenger pathways on the phosphorylation and activation of the transcription factor cyclic AMP response element binding protein (CREB) in the striatum.
Role of Neuroplasticity in the Striatum
The striatum (caudate, putamen and nucleus accumbens) plays a central role in movement. It has been implicated in movement disorders like Parkinson’s disease and Huntington’s disease [8–10]. In the early stages of Parkinson’s disease, neuroplasticity enables the striatum to compensate for the loss of dopamine to the extent that clinical symptoms only become apparent after a significant loss of dopamine has occurred [11–14].
The striatum also plays a critical role in more complex processes, e.g. in behavioral changes due to drug abuse [15, 16] and schizophrenia [17–20]; more recently, the striatum has been implicated in certain forms of memory [21]. Neuroplasticity not only affects normal and abnormal striatal function, but it influences therapeutic drug treatments and must be considered in treatment strategies. It plays a role in the benficial effects of therapeutical drugs, and it is responsible for deleterious side effects. For example, in pharmacologic interventions such as haloperidol treatment of schizophrenia, the beneficial effects take time to develop [22], as do unwanted side effects, e.g. tardive dyskinesia [23]. The late onset of both responses is suggestive of neuroplasticity.
The use of drugs like cocaine or amphetamine causes long-term neuronal modifications [15, 16, 24]. Addiction and tolerance, manifestations of neuroplasticity, reinforce drug-seeking behavior. After cessation of drug use, withdrawal effects like dysphoria, anhedonia, and drug craving are further evidence for the neuronal adaptation to drug abuse. Many of these neuronal modifications hamper successful treatment, and some will prevail for prolonged periods of time [16].
The examination of the mechanisms of striatal neuroplasticity is vital to our understanding of striatal function and malfunction, and for treatments of disorders of the striatum.
Mechanisms of Neuroplasticity in the Striatum
The two major neurotransmitters in the striatum are dopamine, in afferents from the substantia nigra, and glutamate, in cortical afferents [25]. The majority of striatal neurons are medium-sized, spiny [26, 27], and synthesize the inhibitory neurotransmitter γ-aminobutyric acid (GABA) [25]. Most GABA-ergic neurons synapse with nigrostriatal and corticostriatal neurons [25], but differ in their expression of neuromodulators, receptors, and Ca2+-binding proteins [25, 28]. The GABA-ergic neurons of the striatum express receptors of the dopamine receptor family, which consists of five members (D1–D5) [29]. All dopamine receptors described to date are coupled to G proteins [30] and are subdivided into D1-like receptors (D1 and D5 receptors) and D2-like receptors (D2, D3, and D4 receptors) [29, 30]. D1 and D2 receptors dominate in the striatum [31] and are expressed mostly on separate neuronal populations [32–35]. Activation of D1 receptors raises cyclic AMP levels [36]. This increase in cyclic AMP levels leads to activation of protein kinase A (PKA), which affects the phosphorylation states of other proteins, notably dopamine and cyclic AMP-regulated phospho-protein (DARPP-32), which is selectively enriched in GABA-ergic striatal neurons [37, 38].
The GABA-ergic neurons of the striatum also express members of the glutamate receptor family [39–41], including N-methyl-D-aspartate (NMDA) receptors [40, 42]. An important property of NMDA channels is the mediation of Ca2+ influx upon stimulation [43, 44] which contributes to memory-related processes, e.g. long-term potentiation and synaptic plasticity [45–47]. Ca2+ asserts this influence through second messenger pathways. Ca2+ entering through NMDA receptors stimulates kinases and phosphatases which affect protein phosphorylation, e.g. the kinases Ca2+/calmodulin (CaM) kinase [48, 49] and protein kinase C (PKC) [50, 51] and the protein phosphatase calcineurin (protein phosphatase 2B) [52, 53]. These kinases and phosphatases form elements of a cascade capable to induce widespread changes in neurons.
DARPP-32 phosphorylation is a convergence point of NMDA and D1 receptor pathways. Upon phosphorylation by PKA [54], DARPP-32 inhibits protein phosphatases 1 (PP1) and 2A (PP2A) [55]. Calcineurin, a phosphatase activated by NMDA receptors, dephosphorylates DARPP-32 and disables its function as a PP1/PP2A inhibitor (fig. 1) [52, 56].
Fig. 1.

Role of dopamine/cyclic AMP and NMDA/Ca2+ in the activation of protein phosphatases and dephosphorylation of 133Ser-CREB in the striatum. The level of CREB phosphorylation is determined by the relative activity of kinases and phosphatases. The phosphatases responsible for dephosphorylation of 133Ser-CREB are PP1 and PP2A [65, 66]. The DARPP-32 pathway is involved in the regulation of PP1/2A in the striatum [55]. DARPP-32 is a neuronal phosphoprotein which upon phosphorylation on 34Thr [54, 111, 112], becomes a very potent PP1/2A inhibitor [55]. Dopamine-activated PKA phosphorylates DARPP-32 on 34Thr. Therefore, dopamine and cyclic AMP activation promotes CREB phosphorylation via inhibition of phosphatase activity. Stimulation of NMDA receptors induces the calcium-dependent phosphatase calcineurin, the dephosphorylation of DARPP-32 on 34Thr, the disinhibition of PP1/2A [52, 56, 111] and the dephosphorylation of CREB [66]. Insert: CaM kinase IV activated by NMDA receptors [75, 113], and PKA activated by D1 receptors [30, 57, 60, 114] are involved in the phosphorylation of CREB on 133Ser.
Thus, in response to increased synaptic levels of dopamine and glutamate, the GABA-ergic neurons activate specific second messenger pathways that lead to changes in protein phosphorylation. Altered protein phosphorylation can influence gene expression and protein synthesis via transcription factors. The transcription factor CREB is activated through phosphorylation [57] and is an important regulator of gene expression in the striatum.
CREB Phosphorylation as a Model of Striatal Neuroplasticity
CREB is a transcription factor constitutively bound to specific DNA enhancer elements [58]. Phosphorylation of the amino acid 133Ser [59, 60] enables CREB to associate with CREB binding protein and to recruit RNA polymerase II to the promoter [61–64]. PP1 and PP2A deactivate CREB by dephosphorylation of 133Ser (fig. 1) [65–67].
CREB activates immediate early genes as well as neuromodulator genes located in striatal neurons, e.g. c-fos, proenkephalin, somatostatin and prodynorphin [57, 68–74]. Gene regulation by CREB phosphorylation provides a mechanism for neuroplasticity [75–77]. Not surprisingly, CREB has been linked to memory formation in mollusks, fruit flies, and mammals [60, 78–81], and has been associated with long-term potentiation [82].
Cyclic AMP and Ca2+ pathways can induce phosphorylation of 133Ser-CREB [59, 60, 76]. Experiments with cell cultures have demonstrated that the catalytic subunit of PKA can separate from the regulatory subunit and translocate to the nucleus to phosphorylate nuclear proteins [83, 84]. Since 133Ser can be phosphorylated by PKA in vitro [60, 83, 85], it is likely that PKA causes CREB phosphorylation in the striatum in response to D1 receptor stimulation. In the NMDA pathway, Ca2+ entering through NMDA receptors binds CaM, which translocates to the nucleus and activates CaM kinase [86]. Thus dopamine released from nigrostriatal terminals and glutamate released from corticostriatal terminals could potentially independently have identical effects on gene expression of GABA-ergic striatal neurons. However, data obtained in vivo [71], in primary striatal culture [73], and in organotypcial culture [87] suggest that dopamine-mediated phosphorylation of CREB in the striatum is dependent on a factor supplied by the NMDA receptor cascade.
Interaction of Dopamine and Glutamate/NMDA Receptors in the Striatum in the Control of CREB Phosphorylation and Gene Expression
CREB phosphorylation presents an ideal mechanism to examine D1 and NMDA receptor-induced striatal synaptic plasticity: it integrates cyclic AMP and Ca2+ stimuli into long-term adaptive changes. The examination of the role of various phosphatases and kinases in dopamine/cyclic AMP- and glutamate/NMDA-mediated CREB phosphorylation is vital to our understanding of striatal neuroplasticity.
In vivo rat experiments in our lab with the indirect D1 receptor agonists amphetamine and cocaine show an induction of c-fos mRNA in the striatum 15 min after drug administration (unpubl. data). This induction is prevented by pretreatment with a noncompetitive NMDA receptor antagonist, MK801, indicating a dependence on NMDA receptors [73, 88, 89]. Since several mechanisms could explain how dopamine and glutamate may interact extrastriatally through neuronal circuits as well as intra-striatally, work in our lab has focused on their intraneuronal interaction. In primary striatal cultures, which lack the circuitry found in vivo, we corroborated the inhibition of dopamine-mediated c-fos expression by NMDA antagonists [73], demonstrating an intraneuronal interaction of the two neurotransmitter systems. In addition, we found dopamine-mediated c-fos expression is dependent on extracellular Ca2+ in the cultures [73]. In the present paper, we use primary cultures of the rat striatum to study the alliance of glutamate/NMDA and dopamine receptors in the mediation of CREB phosphorylation.
Materials and Methods
Materials
Dopamine, the D1 receptor agonist (±) SKF82958 hydrobromide ((±)-chloro-APB hydrobromide [90]), and the NMDA receptor antagonist (+)-MK801 hydrogen maleate (MK801) [91] were obtained from RBI (Natick, Mass.). The adenylyl cyclase-activating drug forskolin [92] was obtained from Sigma (St. Louis, Mo.). NMDAR1 antibody and 133Ser-phospho-CREB antibody [85] were purchased from UBI (Lake Placid, N.Y.); α-phospho-Ser 897 NMDAR1 (phospho-NMDAR1) antibody was a gift from Dr. Richard L. Huganir [93].
Rat Primary Striatal Cultures
Striata were dissected under a stereomicroscope from 18-day-old Sprague-Dawley rat fetuses. Tissue was resuspended in 2 ml of defined medium to prevent glial growth (50% DMEM/F12 and 50% DMEM, Gibco, Gaithersburg, Md.) with the following supplements per liter of medium: 4 g dextrose, 20 ml B27, 10 ml penicillin/streptomycin liquid (Gibco), 25 ml 1 M Hepes. The tissue was mechanically dissociated with a fire-narrowed Pasteur pipette, the cells were resuspended to 106 cells/ml, and plated in 6-well plates (Costar, Cambridge, Mass.) at 2 × 106 cells/well. Plates were pretreated with two ml of 1:500 diluted solution of polyethylenimine in 50 mM sodium borate pH 7.4 for 24 h, washed twice with water, left with serum-containing medium for at least 4 h and aspirated just prior to plating. All experiments were performed with cells 6–8 days in culture in duplicates and repeated at least once in an independent dissection. As determined by HPLC analysis, glutamate levels in the medium on the day of the experiments ranged from 1 to 5 μM. Neuron- to-astroglia ratio was below 25:1, as established by immunocytochemical staining with the glial fibrillary acid protein (Dako, Carpenteria, Calif.), and counterstaining with 1% cresyl violet.
For immunoblots, cultures were treated with forskolin, dopamine, or SKF82958 for 15 min. MK801 was preincubated for 20 min. After treatments, media was aspirated and plates quick-frozen on liquid nitrogen and stored at −80° C.
Immunoblots
Primary rat striatal cultures were harvested in boiling sample buffer (62.5 mM Tris-HCl, pH 6.8, 20% glycerol, 2% SDS, 5% β-mercaptoethanol, 0.025% bromphenol blue). Cell lysates were sonicated and centrifuged for 10 min. Equal volumes of the lysates were loaded on a 12% polyacrylamide gel for phospho CREB immunoblots and an 8% gel for NMDAR1 and phospho-NMDAR1 immunoblots. Protein was transferred to PVDF membrane (Immobilon-P, 0.45 mm; Millipore, Bedford, Mass.) and blocked in blocking buffer (5% nonfat dry milk in phosphate-buffered saline, 0.1% Tween 20) for 1 h. The blots were incubated in primary antibody (1:1,000 anti-133Ser-phospho-CREB; 1 mg/ml anti-phospho-NMDAR1; 1:200 anti-NMDAR1) for 2 h followed by 3 washes for 15 min in blocking buffer. This was followed by a 1-hour incubation in goat antirabbit horseradish peroxidase-linked IgG (Vector Laboratories, Burlin-game, Calif.) at a concentration of 1:3,000 dilution for 133Ser-phos-pho-CREB and 1:10,000 dilution for NMDAR1 and phospho-NMDAR1. Blots were washed 3 times for 10 min in phosphate-buffered saline, 0.1% Tween 20, developed with the Renaissance detection system (NEN, Wilmington, Del.), and exposed to an autoradiographic film. Kaleidoscope-prestained standards (Bio-Rad, Hercules, Calif.) were used for protein size determination. Phospho CREB bands were detected just above the 43-kD standard, and NMDAR1 and phospho-NMDAR1 bands ran just above the 118-kD standard. Size markers and NMDAR1 antibodies (indiscriminate to the state of phosphorylation) were used to control for the authenticity of the NMDAR1 protein band detected with the phospho-NMDAR1 antibody.
Cyclic AMP levels were determined with the cyclic AMP [3H] assay system from Amersham (Arlington Heights, Ill.). Levels of cyclic AMP were determined per well of 6-well tissue culture plates (approximately 2 × 106 cells), for at least 3 wells per treatment condition, all analyzed in duplicate.
Statistical Analyses
Autoradiographic films were scanned with a Hewlett Packard Scan Jet. Due to the narrow range of film (approximately one order of magnitude), the data obtained are semiquantitative. Data from cyclic AMP tests and from autoradiographs were analyzed with one-way ANOVAs. The Tukey-Kramer HSD (honestly significant difference) test was used to analyze differences between the groups, while Dunnett’s test was used for comparisons of treatment groups with controls. The JMP computer program (SAS Institute, Cary, N.C.) was used for data analysis.
Results
Treatment of cultures with the cyclic AMP-inducing drug, forskolin (20 μM), caused rapid phosphorylation of CREB, as shown in an immunoblot with a 133Ser-phos-pho-CREB antiserum (fig. 2). Phosphorylation of 133Ser-CREB was already apparent at 5 min, peaked at 15 min, and returned to basal levels after 10 h (fig. 2). The maximum level of induction was 8-fold. No significant changes in CREB protein levels were observed. Cells for all subsequent experiments were harvested 15 min after exposure to agonists. Inhibitors were added between 15 and 30 min before the addition of agonists, with the particular times indicated in the figures.
Fig. 2.
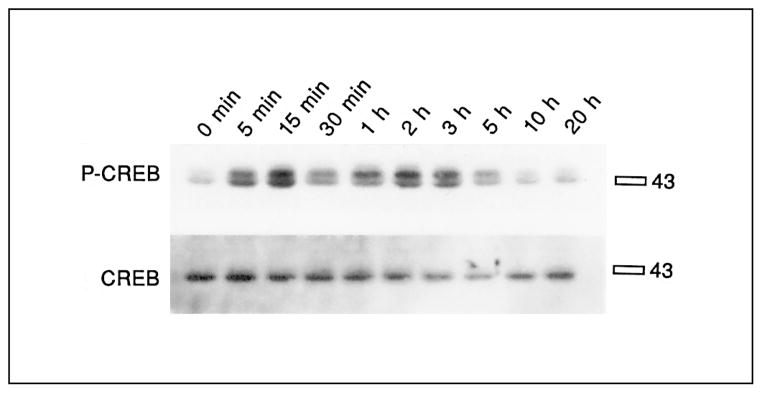
Time course of CREB phosphorylation after stimulation with forskolin in primary striatal cultures. Primary striatal cultures were treated with forskolin (20 μM) for the times indicated, medium was removed and cells were frozen in liquid nitrogen until the day of the experiment. The immunoblot was developed with 133Ser-phos-pho-CREB antibody (upper blot), and reprobed with CREB antiserum (lower blot). The specific bands ran slightly above the 43-kD marker. No significant difference of 133Ser-phospho-CREB induction was observed between 5 min and 3 h. No significant differences of CREB levels were observed at any time points (n = 2).
Dopamine and the full D1 agonist SKF82958 [90] induced significantly 133Ser-CREB phosphorylation in primary striatal cultures (fig. 3, table 1). MK801 pretreatment reduced basal levels of CREB phosphorylation and blocked D1 receptor-mediated CREB phosphorylation (fig. 3, table 1). CREB phosphorylation stimulated by low concentrations of forskolin (2.5 or 5 μM) was at least partly blocked by MK801 pretreatment (fig. 4, table 2). However, CREB phosphorylation induced by high levels of forskolin (20 μM) was not blocked by MK801 pretreatment (fig. 5, table 3). In fact, a significant enhancement of forskolin-mediated CREB phosphorylation was observed when pretreated with MK801. The effectiveness of MK801 as NMDA antagonist was verified in this experiment by the MK801-mediated block of glutamate-induced CREB phosphorylation (fig. 5, table 3).
Fig. 3.
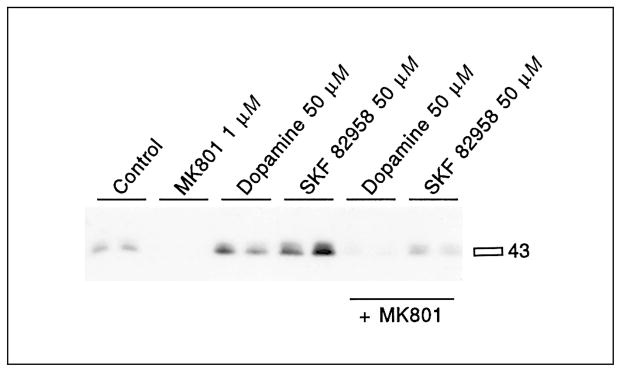
Dopamine and SKF82958-mediated 133Ser-CREB phosphorylation is inhibited by the NMDA antagonist MK801. MK801 (1 μM) was added 30 min before dopamine (50 μM) or SKF82958 (50 μM). Both dopamine and SKF82958 induced 133Ser-CREB phosphorylation. Pretreatment with MK801 for 30 min prevented the induction of CREB phosphorylation. See table 1 for statistical analyses.
Table 1.
Statistical analysis of CREB phosphorylation mediated by dopamine and SKF82958
| Mean ± SD | n | |
|---|---|---|
| Control | 1.0±0.1 | 8 |
| MK801 (1 μM) | 0.2±0.2 | 8 |
| Dopamine (50 μM) | 2.6±0.9a, + | 8 |
| SKF82958 (50 μM) | 3.7±1.0a, * | 8 |
| MK801 (1 μM)/dopamine (50 μM) | 0.6±0.5+ | 8 |
| MK801 (1 μM)/SKF82958 (50 μM) | 0.6±0.5* | 8 |
Mean values and standard deviation (SD) of the fold induction of 133Ser-CREB phosphorylation, as determined by immunoblots, are shown. Dopamine and SKF82958 caused significant increases in CREB phosphorylation. MK801 blocked dopamine- and SKF82958-mediated CREB phosphorylation. Values with the same symbols (+,*) are significantly different (Tukey-Kramer HSD).
Significantly different from control (Dunnett’s method).
Fig. 4.

CREB phosphorylation mediated by low concentrations of forskolin is inhibited by the NMDA antagonist MK801. Cells exposed to 2.5 or 5 μM forskolin show increased CREB phosphorylation. This increase is blocked by pretreatment with MK801 (1 μM) for 30 min (last 4 lanes). See table 2 for statistical analyses.
Table 2.
Statistical analysis of CREB phosphorylation mediated by low concentrations of forskolin
| Mean ± SD | n | |
|---|---|---|
| Control | 1.0±0.3 | 4 |
| MK801 (1 μM) | 0.6±0.1 | 4 |
| Forskolin (2.5 μM) | 3.2±0.4a,b | 4 |
| Forskolin (5 μM) | 3.9±0.8a,+ | 4 |
| MK801 (1 μM)/forskolin (2.5 μM) | 2.4±0.5a,b | 4 |
| MK801 (1 μM)/forskolin (5 μM) | 2.4±0.1a,+ | 4 |
Mean values and standard deviation (SD) of the fold induction of 133Ser-CREB phosphorylation, as determined by immunoblots, are shown. Forskolin (2.5 and 5 μM) caused significant increases in CREB phosphorylation. MK801 blocked partially forskolin-mediated CREB phosphorylation. The block of 2.5 μM forskolin by MK801 did not reach significance with the Tukey-Kramer HSD test. Values with the same symbol (+) are significantly different (Tukey-Kramer HSD).
Significantly different from control (Dunnett’s method).
p < 0.05 (t-test).
Fig. 5.

CREB phosphorylation mediated by high concentrations of forskolin is not inhibited by the NMDA antagonist MK801. Cells exposed to 20 μM forskolin show increased CREB phosphorylation, which is further increased after MK801 (1 μM) treatment. Glutamate-mediated CREB phosphorylation is blocked by MK801. MK801 was added 30 min before forskolin (20 μM). See table 3 for statistical analyses.
Table 3.
Statistical analysis of CREB phosphorylation mediated by high concentrations of forskolin
| Mean ± SD | n | |
|---|---|---|
| Control | 1.0±0.3 | 5 |
| MK801 (1 μM) | 0.4±0.2 | 5 |
| Forskolin (20 μM) | 7.8±0.9a, + | 5 |
| Glutamate (50 μM) | 5.0±1.0a, * | 5 |
| MK801 (1 μM)/forskolin (20 μM) | 10.6±1.5a, + | 5 |
| MK801 (1 μM)/glutamate (50 μM) | 0.6±0.5* | 5 |
Mean values and standard deviation (SD) of the fold induction of 133Ser-CREB phosphorylation, as determined by immunoblots, are shown. Forskolin and glutamate caused significant increases in CREB phosphorylation. MK801 blocked glutamate-mediated CREB phosphorylation, and significantly enhanced forskolin (20 μM)-mediated CREB phosphorylation. Values with the same symbols (+,*) are significantly different (Tukey-Kramer HSD).
Significantly different from control (Dunnett’s method).
Since D1 receptor-mediated CREB phosphorylation is dependent on cyclic AMP induction, we examined the effect of NMDA receptor antagonists on adenylyl cyclase activity. Analysis of cyclic AMP levels showed a dose-dependent increase of cyclic AMP levels after forskolin treatment (fig. 6a,b). MK801 enhanced forskolin-induced cyclic AMP levels at all forskolin concentrations examined (fig. 6a,b). This effect was also observed in the presence of a competitive NMDA antagonist, AP5 (fig. 6c). Glutamate (fig. 6d) and NMDA (500 μM; data not shown) decreased cyclic AMP levels.
Fig. 6.

NMDA antagonists increase forskolin-activated cyclic AMP levels. a Forskolin 2.5 μM and forskolin 5 μM increase cyclic AMP levels in a dose-dependent manner. Pretreatment with MK801 (1 μM) augments forskolin-induced cyclic AMP levels. Analysis done in quadruplicates. b Forskolin 20 μM-induced cyclic AMP levels are augmented by MK801 (1 μM). Analysis done in pentuplicates. c Forskolin (20 μM)-induced cyclic AMP levels are augmented by pre-treatment with AP5 (100 μM). Analysis done in quadruplicates. d Glutamate (25 and 50 μM) decreases cyclic AMP levels induced by forskolin (20 μM). Analysis done in quadruplicates. MK801, AP5 and glutamate were added 30 min before forskolin. All data presented as average ± SD in % of untreated control. Some error bars are below the resolution of the graphs. * = Significantly different from control (Dunnett’s method); #, $ = significant difference between groups (Tukey-Kramer HSD).
In the final set of experiments, we examined how dopamine and elevation of cyclic AMP recruit NMDA receptors. NMDA receptor subunits can be phosphorylated by protein kinases, an event that modulates their function [93, 94]. We therefore examined the possibility of striatal NMDA receptor phosphorylation by forskolin-stimulated PKA.
The pentameric NMDA receptor channel is assembled from receptor subunits of two classes, NMDAR1, and NMDAR2A–D [44]. Each NMDA receptor consists of three NMDAR1 subunits and any two subunits of the NMDAR2 family [95]. The NMDAR1 subtype is a required component of the NMDA receptor [44], and was tested for phosphorylation. An immunoblot with an antibody against phosphorylated NMDAR1 receptor [93] showed that treatment of the cultures with 2.5 or 5 μM forskolin led to potent phosphorylation within 20 min (fig. 7). Phosphorylation of NMDAR1 receptors was independent of the NMDA second messenger pathway, since the NMDA antagonist MK801 did not block the phosphorylation (fig. 7). Maximal phosphorylation of the receptor was achieved at the lowest concentration of forskolin (2.5 μM) and was not augmented by concentrations of up to 50 μM forskolin (table 4).
Fig. 7.

Modulation of NMDA receptor phosphorylation by forskolin. Cells treated with forskolin show phosphorylation of the NMDAR1 protein, as revealed in an immunoblot with a phospho-NMDAR1 receptor antibody. MK801 does not block phosphorylation. All nine samples were run in one gel, transferred, and the membrane was cut before the last three lanes. The left side was developed with an antibody specific for the phosphorylated form of the NMDAR1 receptor, the right side was developed with an antibody specific for the NMDAR1 receptor. The NMDAR1 receptor runs slightly above the 118-kD marker. The phospho-NMDAR1 receptor antibody stains 2 bands around 120 kD, the smaller of which is also detected by the NMDAR1 antiserum and was considered the specific band. See table 4 for statistical analyses.
Table 4.
Statistical analysis of NMDA receptor phosphorylation by forskolin
| Mean ± SD | n | |
|---|---|---|
| Control | 1.0±0.15 | 9 |
| Forskolin (2.5 μM) | 6.5±2.27a | 9 |
| Forskolin (5 μM) | 5.4±2.24a | 5 |
| Forskolin (20 M) | 7.0±2.99a | 6 |
| Forskolin (50 M) | 5.4±1.38a | 6 |
Mean values and standard deviation of the fold induction of the phosphorylation of the NMDAR1 receptor, as determined by immunoblots, are shown. All concentrations of forskolin examined caused significant increases in NMDAR1 phosphorylation. The maximal levels of NMDAR1 phosphorylation were already observed at the lowest concentrations of forskolin used.
Significantly different from control (Dunnett’s method).
Discussion
The two predominant neurotransmitters in the striatum, dopamine and glutamate, induce CREB phosphorylation in rat primary striatal cultures. We found various levels of interaction between dopamine and NMDA pathways which are relevant for CREB phosphorylation: first, an inhibitory effect of NMDA receptor stimulation on forskolin-activated cyclic AMP levels, second, a recruitment of the NMDA pathway for 133Ser-CREB phosphorylation by PKA, and third, we found indirect evidence for a regulation of the CREB phosphatase PP1/2A through the phosphorylation/dephosphorylation of DARPP-32 by Ca2+ and cyclic AMP pathways [52, 55, 56].
Mechanism of CREB Phosphorylation after Treatment with Dopamine and Low Concentrations of Forskolin
Functional NMDA receptors are critical for glutamate, dopamine, and low forskolin-mediated CREB phosphorylation. Though NMDA receptor antagonists enhance the forskolin-induced increase in cyclic AMP levels, they block dopamine/low forskolin-induced 133Ser-CREB phosphorylation. The enhancement of cyclic AMP levels indicates a negative regulation of cyclic AMP synthesis by the NMDA/Ca2+ pathway, which is supported by a reduction of stimulated cyclic AMP levels by glutamate and NMDA. The data are in accordance with studies showing that the predominant adenylyl cyclases in the striatum, type V and X, are inhibited by Ca2+ [96, 97]. Moreover, phosphodiesterase, the enzyme which metabolizes cyclic AMP, is stimulated by Ca2+ pathways [98]. Since NMDA receptors have opposite effects on cyclic AMP synthesis and CREB phosphorylation, facilitation of CREB phosphorylation by NMDA receptors must occur downstream of cyclic AMP induction.
PKA is activated by cyclic AMP [99], and NMDA antagonists increase low forskolin-stimulated cyclic AMP levels. Why then does PKA not phosphorylate CREB in the presence of NMDA antagonists? This question may be answered by a study of the spatiotemporal distribution of cyclic AMP in neurons, which concluded that cyclic AMP is produced primarily in fine neurites [100]. PKA activation after dopamine or low forskolin stimulation may be limited to the synaptic neuropil and may not reach the cell soma and the nucleus (fig. 8a) [100]. The target for PKA phosphorylation must therefore be close to the postsynaptic membrane. Our data on NMDAR1 phosphorylation by PKA elucidate a possible mechanism: PKA phosphorylates NMDAR1 receptors and alters NMDA activity [101], possibly by removing the Mg2+ block [102]. Phospho-NMDA receptors activate Ca2+ influx, CaM, and a ‘CREB kinase’. The CREB kinase, most likely CaM kinase IV [75, 86], phosphorylates CREB (fig. 8b). Inhibition of NMDA receptors interrupts this pathway and prevents CREB phosphorylation by dopamine or low forskolin. Though further experiments are needed to prove this hypothesis conclusively, it should be noted that a similar mechanism has been described for NMDAR1 phosphorylation by PKC [102, 103].
Fig. 8.

Cyclic AMP mediates CREB phosohorylation by two different mechanism. a The distribution of activated PKA after stimulation with low levels of forskolin or dopamine, and after high levels of forskolin. b Low levels of forskolin increase local PKA activity which causes phosphorylation of the NMDAR1 receptor. Kinases activated intraneuronally by the NMDA receptor pathway phosphorylate CREB. NMDA receptors also stimulate PP1 activity through the DARPP-32 pathway, but the equilibrium favors kinase activity, as demonstrated by increased 133Ser-CREB phosphorylation. Therefore, inhibition of NMDA receptors prevents CREB phosphorylation. The bold arrow points out the predominant action of the NMDA receptor pathway in the presence of low concentrations of forskolin. c High levels of forskolin stimulate sufficient amounts of PKA to directly phosphorylate 133Ser-CREB. Kinases activated by NMDA receptors cannot further contribute to CREB phosphorylation, while the activation of calcineurin and the opposition to DARPP-32 phosphorylation attenuates CREB phosphorylation. The net effect of the NMDA receptor pathway is a decrease of high forskolin-stimulated 133Ser-phospho-CREB levels. Therefore, inhibition of NMDA receptors increases CREB phosphorylation. The bold arrow points out the predominant action of the NMDA receptor pathway in the presence of high concentrations of forskolin.
Activation of PKA in the synaptic neuropil not only promotes phosphorylation of NMDA receptor proteins, but also of DARPP-32 (fig. 8b). Calcineurin, activated by NMDA receptors, dephosphorylates DARPP-32, which releases PP1/2A (fig. 1) [52, 56]. Thus, dopamine/low forskolin, through the PKA and the NMDA second messenger pathway, have a contradictory influence on DARPP-32 phosphorylation, the inhibition of PP1/2A, and the dephosphorylation of CREB (fig. 1). Since the net effect of dopamine/low forskolin administration is an increase in CREB phosphorylation, the dominant component contributed by the NMDA pathway is the kinase activity (fig. 8b).
Mechanism of CREB Phosphorylation after Treatment with High Concentration of Forskolin
Experiments with the adenylyl cyclase-activating drug forskolin demonstrate the complexity of the interaction of the cyclic AMP and the NMDA pathway. With a different extent of cyclic AMP stimulation, the dominance of different kinases and phosphatases in the pathway changes. Moderate induction of cyclic AMP, as mediated by dopamine or low concentrations of forskolin, requires functional NMDA receptors to produce CREB phosphorylation. High levels of forskolin induce 133Ser-CREB phosphorylation independently of NMDA receptors that is not blocked by NMDA antagonists. This again can be explained by the spatiotemporal distribution of stimulated PKA [100]. High concentrations of forskolin cause widespread PKA activation, which directly mediates phosphorylation of CREB, independent of NMDA-activated kinases (fig. 8a,c). Stimulation with high amounts of forskolin also causes NMDAR1 receptor phosphorylation and, in addition to the cyclic AMP pathway, supports the NMDA-mediated kinase and phosphatase pathway (fig. 8c). Because high doses of forskolin directly phosphorylate 133Ser-CREB, the discernible factor contributed by the NMDA pathway is the phosphatase calcineurin, which supports PP1/2A activity via the DARPP-32 pathway [52, 56]. While CaM kinase IV does not further enhance PKA-mediated CREB phosphorylation, calcineurin, counteracting PKA-mediated DARPP-32 phosphorylation, supports the CREB dephosphorylation pathway (fig. 8c). Inhibition of NMDA receptors decreases calcineurin and PP1/2A activity [104]. In addition, NMDA antagonists significantly enhance forskolin-stimulated cyclic AMP levels, which, via increased PKA activity, could augment CREB phosphorylation. Thus, inhibition of the NMDA receptor not only fails to block high forskolin-mediated CREB phosphorylation, but actually increases total amount of 133Ser-phospho-CREB.
Role of NMDA Receptors in Dopamine-Mediated CREB Phosphorylation
The mechanism of dopamine-mediated CREB phosphorylation is comparable to the mechanism used by low forskolin, indicating that under conditions of physiologic stimulation, dopamine modulates CREB phosphorylation predominantly via NMDA receptors. The role of NMDA receptors in dopamine and low forskolin-mediated CREB phosphorylation confirms the prominent position of NMDA receptors in synaptic plasticity [48, 51, 105–107]. However, the potency and stimulus duration of cyclic AMP activation determines the extent of the dependence on NMDA receptors, e.g. increasing cyclic AMP stimulation is decreasingly dependent on NMDA receptors. Higher amounts of cyclic AMP can diffuse retrogradely to the nucleus [100], stimulate PKA and phosphorylate 133Ser-CREB. Thus, while there is ample cross talk between Ca2+ and PKA pathways, PKA can mediate synaptic plasticity independently of NMDA receptors [4, 99, 108–110].
Acknowledgments
The phospho-NMDAR1 antibody was a gift of Dr. Richard Huganir. The authors would like to acknowledge the input of the members of the writing seminar under the guidance of Ellie Hackett and Theodor Stern, at Massachusetts General Hospital, Department of Psychiatry. This work was supported by the National Alliance for Research on Schizophrenia and Depression (C.K.), and a National Institute of Drug Abuse grant DA07134 (C.K.).
References
- 1.Matthies H. In search of cellular mechanisms of memory. Prog Neurobiol. 1989;32:277–349. doi: 10.1016/0301-0082(89)90024-5. [DOI] [PubMed] [Google Scholar]
- 2.Stanton PK, Sarvey JM. Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J Neurosci. 1984;4:3080–3088. doi: 10.1523/JNEUROSCI.04-12-03080.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deadwyler SA, Dunwiddie T, Lynch G. A critical level of protein synthesis is required for long-term potentiation. Synapse. 1987;1:90–95. doi: 10.1002/syn.890010112. [DOI] [PubMed] [Google Scholar]
- 4.Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA. 1996;93:13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulman H. Protein phosphorylation in neuronal plasticity and gene expression. Curr Opin Neurobiol. 1995;5:375–381. doi: 10.1016/0959-4388(95)80051-4. [DOI] [PubMed] [Google Scholar]
- 6.Roche KW, Tingley WG, Huganir RL. Glutamate receptor phosphorylation and synaptic plasticity. Curr Opin Neurobiol. 1994;4:383–388. doi: 10.1016/0959-4388(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 7.Nairn AC, Shenolikar S. The role of protein phosphatases in synaptic transmission, plasticity and neuronal development. Curr Opin Neurobiol. 1992;2:296–301. doi: 10.1016/0959-4388(92)90118-5. [DOI] [PubMed] [Google Scholar]
- 8.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Langston JW. The etiology of Parkinson’s disease with emphasis on the MPTP story. Neurology. 1996;47:S153–160. doi: 10.1212/wnl.47.6_suppl_3.153s. [DOI] [PubMed] [Google Scholar]
- 10.DiFiglia M. Excitotoxic injury of the neostriatum: A model for Huntington’s disease. Trends Neurosci. 1990;13:286–289. doi: 10.1016/0166-2236(90)90111-m. [DOI] [PubMed] [Google Scholar]
- 11.Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20:415–455. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
- 12.Hornykiewicz O. Parkinson’s disease and the adaptive capacity of the nigrostriatal dopamine system: Possible neurochemical mechanisms. Adv Neurol. 1993;60:140–147. [PubMed] [Google Scholar]
- 13.Burns RS. Subclinical damage to the nigrostriatal dopamine system by MPTP as a model of preclinical Parkinson’s disease: A review. Acta Neurol Scand Suppl. 1991;136:29–36. doi: 10.1111/j.1600-0404.1991.tb05017.x. [DOI] [PubMed] [Google Scholar]
- 14.Calne DB, Langston JW, Martin WR, Stoessl AJ, Ruth TJ, Adam MJ, Pate BD, Schulzer M. Positron emission tomography after MPTP: Observations relating to the cause of Parkinson’s disease. Nature. 1985;317:246–248. doi: 10.1038/317246a0. [DOI] [PubMed] [Google Scholar]
- 15.Self DW, Nestler EJ. Molecular mechanisms of drug reinforcement and addiction. Annu Rev Neurosci. 1995;18:463–495. doi: 10.1146/annurev.ne.18.030195.002335. [DOI] [PubMed] [Google Scholar]
- 16.Hyman SE. Addiction to cocaine and amphetamine. Neuron. 1996;16:901–904. doi: 10.1016/s0896-6273(00)80111-7. [DOI] [PubMed] [Google Scholar]
- 17.Buchsbaum MS, Haier RJ, Potkin SG, Nuechterlein K, Bracha HS, Katz M, Lohr J, Wu J, Lottenberg S, Jerabek PA. Frontostriatal disorder of cerebral metabolism in never-medicated schizophrenics. Arch Gen Psyhiatry. 1992;49:935–942. doi: 10.1001/archpsyc.1992.01820120023005. [DOI] [PubMed] [Google Scholar]
- 18.Siegel BV, Jr, Buchsbaum MS, Bunney WE, Jr, Gottschalk LA, Haier RJ, Lohr JB, Lottenberg S, Najafi A, Nuechterlein KH, Potkin SG, et al. Cortical-striatal-thalamic circuits and brain glucose metabolic activity in 70 unmedicated male schizophrenic patients. Am J Psychiatry. 1993;150:1325–1336. doi: 10.1176/ajp.150.9.1325. [DOI] [PubMed] [Google Scholar]
- 19.Heckers S. Neuropathology of schizophrenia: Cortex, thalamus, basal ganglia, and neurotransmitter-specific projection systems. Schizophr Bull. 1997;23:403–421. doi: 10.1093/schbul/23.3.403. [DOI] [PubMed] [Google Scholar]
- 20.Busatto GF, Kerwin RW. Schizophrenia, psychosis, and the basal ganglia. Psychiatr Clin North Am. 1997;20:897–910. doi: 10.1016/s0193-953x(05)70351-8. [DOI] [PubMed] [Google Scholar]
- 21.Knowlton BJ, Mangels JA, Squire LR. A neostriatal habit learning system in humans. Science. 1996;273:1399–1402. doi: 10.1126/science.273.5280.1399. [DOI] [PubMed] [Google Scholar]
- 22.Hyman SE, Nestler EJ. Initiation and adaptation: A paradigm for understanding psychotropic drug action. Am J Psychiatry. 1996;153:151–162. doi: 10.1176/ajp.153.2.151. [DOI] [PubMed] [Google Scholar]
- 23.Kane JM. Tardive dyskinesia: Epidemiological and clinical presentation. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: The Fourth Generation of Progress. New York: Raven Press; 1995. pp. 1485–1495. [Google Scholar]
- 24.Nestler EJ, Hope BT, Widnell KL. Drug addiction: A model for the molecular basis of neural plasticity. Neuron. 1993;11:995–1006. doi: 10.1016/0896-6273(93)90213-b. [DOI] [PubMed] [Google Scholar]
- 25.Smith AD, Bolam JP. The neural network of the basal ganglia as revealed by the study of synaptic connections of identified neurones. Trends Neurosci. 1990;13:259–265. doi: 10.1016/0166-2236(90)90106-k. [DOI] [PubMed] [Google Scholar]
- 26.Somogyi P, Hodgson AJ, Smith AD. An approach to tracing neuron networks in the cerebral cortex and basal ganglia. Combination of Golgi staining, retrograde transport of horse-radish peroxidase and anterograde degeneration of synaptic boutons in the same material. Neuroscience. 1979;4:1805–1852. doi: 10.1016/0306-4522(79)90059-9. [DOI] [PubMed] [Google Scholar]
- 27.Kemp JM, Powell TP. The structure of the caudate nucleus of the cat: Light and electron microscopy. Philos Trans R Soc Lond B Biol Sci. 1971;262:383–401. doi: 10.1098/rstb.1971.0102. [DOI] [PubMed] [Google Scholar]
- 28.Graybiel AM. Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci. 1990;13:244–254. doi: 10.1016/0166-2236(90)90104-i. [DOI] [PubMed] [Google Scholar]
- 29.Sibley DR, Monsma FJ, Jr, McVittie LD, Gerfen CR, Burch RM, Mahan LC. Molecular neurobiology of dopamine receptor subtypes. Neurochem Int. 1992;20:17S–22S. doi: 10.1016/0197-0186(92)90205-6. [DOI] [PubMed] [Google Scholar]
- 30.Seeman P, Van Tol HHM. Dopamine receptor pharmacology. Curr Opin Neurol Neurosurg. 1993;6:602–608. [PubMed] [Google Scholar]
- 31.Levey AI, Hersch SM, Rye DB, Sunahara RK, Niznik HB, Kitt CA, Price DL, Maggio R, Brann MR, Ciliax BJ, et al. Localization of D1 and D2 dopamine receptors in brain with subtype-specific antibodies. Proc Natl Acad Sci USA. 1993;90:8861–8865. doi: 10.1073/pnas.90.19.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 33.Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- 34.LeMoine C, Normand E, Guitteny AF, Fouque B, Teoule R, Bloch B. Dopamine receptor gene expression by enkephalin neurons in rat fore-brain. Proc Natl Acad Sci USA. 1990;87:230–234. doi: 10.1073/pnas.87.1.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Moine C, Normand E, Bloch B. Phenotypical characterization of the rat striatal neurons expressing the D1 dopamine receptor gene. Proc Natl Acad Sci USA. 1991;88:4205–4209. doi: 10.1073/pnas.88.10.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monsma FJ, Jr, Mahan LC, McVittie LD, Gerfen CR, Sibley DR. Molecular cloning and expression of a D1 dopamine receptor linked to adenylyl cyclase activation. Proc Natl Acad Sci USA. 1990;87:6723–6727. doi: 10.1073/pnas.87.17.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walaas SI, Greengard P. DARPP-32, a dopamine- and adenosine 3′:5′-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. I. Regional and cellular distribution in the rat brain. J Neurosci. 1984;4:84–98. doi: 10.1523/JNEUROSCI.04-01-00084.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ouimet CC, Miller PE, Hemmings HCJ, Walaas SI, Greengard P. DARPP-32, a dopamine-and adenosine 3′:5′-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. III. Immunocytochemical localization. J Neurosci. 1984;4:111–124. doi: 10.1523/JNEUROSCI.04-01-00111.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bischoff S, Barhanin J, Bettler B, Mulle C, Heinemann S. Spatial distributions of kainate receptor subunit mRNA in the mouse basal ganglia and ventral mesencephalon. J Comp Neurol. 1997;379:541–562. doi: 10.1002/(sici)1096-9861(19970324)379:4<541::aid-cne6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 40.Chen Q, Veenman CL, Reiner A. Cellular expression of ionotropic glutamate receptor subunits on specific striatal neuron types and its implication for striatal vulnerability in glutamate receptor-mediated excitotoxicity. Neuroscience. 1996;73:715–731. doi: 10.1016/0306-4522(96)00011-5. [DOI] [PubMed] [Google Scholar]
- 41.Bernard V, Gardiol A, Faucheux B, Bloch B, Agid Y, Hirsch EC. Expression of glutamate receptors in the human and rat basal ganglia: Effect of the dopaminergic denervation on AMPA receptor gene expression in the striatopallidal complex in Parkinson’s disease and rat with 6-OHDA lesion. J Comp Neurol. 1996;368:553–568. doi: 10.1002/(SICI)1096-9861(19960513)368:4<553::AID-CNE7>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 42.Chen Q, Reiner A. Cellular distribution of the NMDA receptor NR2A/2B subunits in the rat striatum. Brain Res. 1996;743:346–352. doi: 10.1016/s0006-8993(96)01098-0. [DOI] [PubMed] [Google Scholar]
- 43.MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 44.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 45.Eccles JC. Calcium in long-term potentiation as a model for memory. Neuroscience. 1983;10:1071–1081. doi: 10.1016/0306-4522(83)90100-8. [DOI] [PubMed] [Google Scholar]
- 46.Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719–721. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- 47.Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in α-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- 48.Fukunaga K, Stoppini L, Miyamoto E, Muller D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1993;268:7863–7867. [PubMed] [Google Scholar]
- 49.Stevens CF, Tonegawa S, Wang Y. The role of calcium-calmodulin kinase II in three forms of synaptic plasticity. Curr Biol. 1994;4:687–693. doi: 10.1016/s0960-9822(00)00153-6. [DOI] [PubMed] [Google Scholar]
- 50.Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S. Modified hippocampal long-term potentiation in PKC gamma-mutant mice. Cell. 1993;75:1253–1262. doi: 10.1016/0092-8674(93)90613-u. [DOI] [PubMed] [Google Scholar]
- 51.Muller D, Buchs PA, Stoppini L, Boddeke H. Long-term potentiation, protein kinase C, and glutamate receptors. Mol Neurobiol. 1991;5:277–288. doi: 10.1007/BF02935551. [DOI] [PubMed] [Google Scholar]
- 52.King MM, Huang CY, Chock PB, Nairn AC, Hemmings HCJ, Chan KF, Greengard P. Mammalian brain phosphoproteins as substrates for calcineurin. J Biol Chem. 1984;259:8080–8083. [PubMed] [Google Scholar]
- 53.Tong G, Shepherd D, Jahr CE. Synaptic desensitization of NMDA receptors by calcineurin. Science. 1995;267:1510–1512. doi: 10.1126/science.7878472. [DOI] [PubMed] [Google Scholar]
- 54.Hemmings HCJ, Williams KR, Konigsberg WH, Greengard P. DARPP-32, a dopamine-and adenosine 3′:5′-monophosphate-regulated neuronal phosphoprotein. I. Amino acid sequence around the phosphorylated threonine. J Biol Chem. 1984;259:14486–14490. [PubMed] [Google Scholar]
- 55.Hemmings HCJ, Greengard P, Tung HY, Cohen P. DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature. 1984;310:503–505. doi: 10.1038/310503a0. [DOI] [PubMed] [Google Scholar]
- 56.Halpain S, Girault JA, Greengard P. Activation of NMDA receptors induces dephosphorylation of DARPP-32 in rat striatal slices. Nature. 1990;343:369–372. doi: 10.1038/343369a0. [DOI] [PubMed] [Google Scholar]
- 57.Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 58.Montminy MR, Bilezikjian LM. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature. 1987;328:175–178. doi: 10.1038/328175a0. [DOI] [PubMed] [Google Scholar]
- 59.Sheng M, Thompson MA, Greenberg ME. CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 60.Dash PK, Karl KA, Colicos MA, Prywes R, Kandel ER. cAMP response element-binding protein is activated by Ca2+/calmodulin- as well as cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1991;88:5061–5065. doi: 10.1073/pnas.88.11.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arias J, Alberts AS, Brindle P, Claret FX, Smeal T, Karin M, Feramisco J, Montminy M. Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature. 1994;370:226–229. doi: 10.1038/370226a0. [DOI] [PubMed] [Google Scholar]
- 62.Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 63.Kwok RP, Lundblad JR, Chrivia JC, Richards JP, Bachinger HP, Brennan RG, Roberts SG, Green MR, Goodman RH. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature. 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- 64.Kee BL, Arias J, Montminy MR, Kee BI. Adaptor-mediated recruitment of RNA polymerase II to a signal-dependent activator. J Biol Chem. 1996;271:2373–2375. doi: 10.1074/jbc.271.5.2373. [DOI] [PubMed] [Google Scholar]
- 65.Hagiwara M, Alberts A, Brindle P, Meinkoth J, Feramisco J, Deng T, Karin M, Shenolikar S, Montminy M. Transcriptional attenuation following cAMP induction requires PP-1 mediated dephosphorylation of CREB. Cell. 1992;70:105–113. doi: 10.1016/0092-8674(92)90537-m. [DOI] [PubMed] [Google Scholar]
- 66.Alberts AS, Montminy M, Shenolikar S, Feramisco JR. Expression of a peptide inhibitor of protein phosphatase 1 increases phosphorylation and activity of CREB in NIH 3T3 fibroblasts. Mol Cell Biol. 1994;14:4398–4407. doi: 10.1128/mcb.14.7.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wadzinski BE, Wheat WH, Jaspers S, Peruski LFJ, Lickteig RL, Johnson GL, Klemm DJ. Nuclear protein phosphatase 2A dephosphorylates protein kinase A-phosphorylated CREB and regulates CREB transcriptional stimulation. Mol Cell Biol. 1993;13:2822–2834. doi: 10.1128/mcb.13.5.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sheng M, McFadden G, Greenberg ME. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- 69.Cole RL, Konradi C, Douglass J, Hyman SE. Neuronal adaptation to amphetamine and dopamine: Molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron. 1995;14:813–823. doi: 10.1016/0896-6273(95)90225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Konradi C, Kobierski LA, Nguyen TV, Heckers S, Hyman SE. The cAMP-response-element-binding protein interacts, but Fos protein does not interact, with the proenkephalin enhancer in rat striatum. Proc Natl Acad Sci USA. 1993;90:7005–7009. doi: 10.1073/pnas.90.15.7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Konradi C, Cole RL, Heckers S, Hyman SE. Amphetamine regulates gene expression in rat striatum via transcription factor CREB. J Neurosci. 1994;14:5623–5634. doi: 10.1523/JNEUROSCI.14-09-05623.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Konradi C, Cole RL, Green D, Senatus P, Leveque JC, Pollack A, Grossbard SJ, Hyman SE. Analysis of the proenkephalin second messenger-inducible enhancer in rat striatal cultures. J Neurochem. 1995;65:1007–1015. doi: 10.1046/j.1471-4159.1995.65031007.x. [DOI] [PubMed] [Google Scholar]
- 73.Konradi C, Leveque JC, Hyman SE. Amphetamine and dopamine-induced immediate early gene expression in striatal neurons depends upon postsynaptic NMDA receptors and calcium. J Neurosci. 1996;16:4231–4239. doi: 10.1523/JNEUROSCI.16-13-04231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Douglass J, McKinzie AA, Pollock KM. Identification of multiple DNA elements regulating basal and protein kinase A-induced transcriptional expression of the rat prodynorphin gene. Mol Endocrinol. 1994;8:333–344. doi: 10.1210/mend.8.3.8015551. [DOI] [PubMed] [Google Scholar]
- 75.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: A Ca2+-and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 76.Bito H, Deisseroth K, Tsien RW. Ca2+ dependent regulation in neuronal gene expression. Curr Opin Neurobiol. 1997;7:419–429. doi: 10.1016/s0959-4388(97)80072-4. [DOI] [PubMed] [Google Scholar]
- 77.Deisseroth K, Bito H, Tsien RW. Signalling from synapse to nucleus: Postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 78.Bourtchuladze R, Frenguelli B, Blendie J, Cioffi D, Schütz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 79.Yin JCP, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T. Induction of a dominant negative CREB transgene specifically blocks long-term memory in drosophila. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- 80.Stevens CF. CREB and memory consolidation. Neuron. 1994;13:769–770. doi: 10.1016/0896-6273(94)90244-5. [DOI] [PubMed] [Google Scholar]
- 81.Frank DA, Greenberg ME. CREB: A mediator of long-term memory from mollusks to mammals. Cell. 1994;79:5–8. doi: 10.1016/0092-8674(94)90394-8. [DOI] [PubMed] [Google Scholar]
- 82.Impey S, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- 83.Riabowol KT, Fink S, Gilman MZ, Walsh DA, Goodman RH, Feramisco JR. The catalytic subunit of cAMP-dependent protein kinase induces expression of genes containing cAMP-responsive enhancer elements. Nature. 1988;336:83–86. doi: 10.1038/336083a0. [DOI] [PubMed] [Google Scholar]
- 84.Nigg EA, Hilz H, Eppenberger HM, Dutly F. Rapid and reversible translocation of the catalytic subunit of cAMP-dependent protein kinase type II from the Golgi complex to the nucleus. EMBO J. 1985;4:2801–2806. doi: 10.1002/j.1460-2075.1985.tb04006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ginty DD, Kornhauser JM, Thompson MA, Bading H, Mayo KE, Takahashi JS, Greenberg ME. Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and circadian clock. Science. 1993;260:238–241. doi: 10.1126/science.8097062. [DOI] [PubMed] [Google Scholar]
- 86.Deisseroth K, Tsien RW. Calmodulin translocation to the nucleus mediates rapid synaptic control of CREB phosphorylation in hippocampal neurons. Soc Neurosci Abstr. 1997;23:311. [Google Scholar]
- 87.Liu FC, Graybiel AM. Spatiotemporal dynamics of CREB phosphorylation: Transient versus sustained phosphorylation in the developing striatum. Neuron. 1996;17:1133–1144. doi: 10.1016/s0896-6273(00)80245-7. [DOI] [PubMed] [Google Scholar]
- 88.Paul ML, Graybiel AM, David JC, Robertson HA. D1-like and D2-like dopamine receptors synergistically activate rotation and c-fos expression in the dopamine-depleted striatum in a rat model of Parkinson’s disease. J Neurosci. 1992;12:3729–3742. doi: 10.1523/JNEUROSCI.12-10-03729.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Snyder-Keller AM. Striatal c-fos induction by drugs and stress in neonatally dopamine-depleted rats given nigral transplants: Importance of NMDA activation and relevance to sensitization phenomena. Exp Neurol. 1991;113:155–165. doi: 10.1016/0014-4886(91)90171-8. [DOI] [PubMed] [Google Scholar]
- 90.O’Boyle KM, Gaitanopoulos DE, Brenner M, Waddington JL. Agonist and antagonist properties of benzazepine and thienopyridine derivatives at the D1 dopamine receptor. Neuropharmacology. 1989;28:401–405. doi: 10.1016/0028-3908(89)90036-1. [DOI] [PubMed] [Google Scholar]
- 91.Wong EH, Kemp JA, Priestley T, Knight AR, Woodruff GN, Iversen LL. The anticonvulsant MK-801 is a potent N-methyl-D-aspartate antagonist. Proc Natl Acad Sci USA. 1986;83:7104–7108. doi: 10.1073/pnas.83.18.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Metzger H, Lindner E. The positive inotropic-acting forskolin, a potent adenylate cyclase activator. Arzneimittelforschung. 1981;31:1248–1250. [PubMed] [Google Scholar]
- 93.Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem. 1997;272:5157–5166. doi: 10.1074/jbc.272.8.5157. [DOI] [PubMed] [Google Scholar]
- 94.Tingley WG, Roche KW, Thompson AK, Huganir RL. Regulation of NMDA receptor phosphorylation by alternative splicing of the C-terminal domain. Nature. 1993;364:70–73. doi: 10.1038/364070a0. [DOI] [PubMed] [Google Scholar]
- 95.Premkumar LS, Auerbach A. Stoichiometry of recombinant N-methyl-D-aspartate receptor channels inferred from single-channel current patterns. J Gen Physiol. 1997;110:485–502. doi: 10.1085/jgp.110.5.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Paterson JM, Smith SM, Harmar AJ, Antoni FA. Control of a novel adenylyl cyclase by calcineurin. Biochem Biophys Res Commun. 1995;214:1000–1008. doi: 10.1006/bbrc.1995.2385. [DOI] [PubMed] [Google Scholar]
- 97.Katsushika S, Chen L, Kawabe J-I, Nilakantan R, Halnon NJ, Homcy CJ, Ishikawa Y. Cloning and characterization of a sixth adenylyl cyclase isoform: Types V and VI constitute a subgroup within the mammalian adenylyl cyclase family. Proc Natl Acad Sci USA. 1992;89:8774–8778. doi: 10.1073/pnas.89.18.8774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Polli JW, Kincaid RL. Expression of a calmodulin-dependent phosphodiesterase isoform (PDE1B1) correlates with brain regions having extensive dopaminergic innervation. J Neurosci. 1994;14:1251–1261. doi: 10.1523/JNEUROSCI.14-03-01251.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brandon EP, Idzerda RL, McKnight GS. PKA isoforms, neural pathways, and behaviour: Making the connection. Curr Opin Neurobiol. 1997;7:397–403. doi: 10.1016/s0959-4388(97)80069-4. [DOI] [PubMed] [Google Scholar]
- 100.Hempel CM, Vincent P, Adams SR, Tsien RY, Selverston AI. Spatio-temporal dynamics of cyclic AMP signals in an intact neural circuit. Nature. 1996;384:166–169. doi: 10.1038/384166a0. [DOI] [PubMed] [Google Scholar]
- 101.Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci USA. 1993;90:9576–9580. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen L, Huang LYM. Protein kinase C reduces Mg2+ block of NMDA receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- 103.Ben-Ari Y, Aniksztejn L, Bregestovski P. Protein kinase C modulation of NMDA currents: An important link for LTP induction. Trends Neurosci. 1992;15:333–339. doi: 10.1016/0166-2236(92)90049-e. [DOI] [PubMed] [Google Scholar]
- 104.Nishi H, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J Neurosci. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bear MF. A synaptic basis for memory storage in the cerebral cortex. Proc Natl Acad Sci USA. 1996;93:13453–13459. doi: 10.1073/pnas.93.24.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brown TH, Chapman PF, Kairiss EW, Keenan CL. Long-term synaptic potentiation. Science. 1988;242:724–728. doi: 10.1126/science.2903551. [DOI] [PubMed] [Google Scholar]
- 107.Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 108.Harada K, Nagatsugu Y, Ito H, Shingai R. Intracellular cAMP regulates the response to NMDA in hippocampal neurons. Neuroreport. 1991;2:673–676. doi: 10.1097/00001756-199111000-00010. [DOI] [PubMed] [Google Scholar]
- 109.Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- 110.Brandon EP, Zhuo M, Huang YY, Qi M, Gerhold KA, Burton KA, Kandel ER, McKnight GS, Idzerda RL. Hippocampal long-term depression and depotentiation are defective in mice carrying a targeted disruption of the gene encoding the RI beta subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1995;92:8851–8855. doi: 10.1073/pnas.92.19.8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Desdouits F, Siciliano JC, Greengard P, Girault JA. Dopamine- and cAMP-regulated phosphoprotein DARPP-32: Phosphorylation of Ser-137 by casein kinase I inhibits dephosphorylation of Thr-34 by calcineurin. Proc Natl Acad Sci USA. 1995;92:2682–2685. doi: 10.1073/pnas.92.7.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Walaas SI, Aswad DW, Greengard P. A dopamine- and cyclic AMP-regulated phosphoprotein enriched in dopamine-innervated brain regions. Nature. 1983;301:69–71. doi: 10.1038/301069a0. [DOI] [PubMed] [Google Scholar]
- 113.Sun P, Enslen H, Myung PS, Maurer RA. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8:2527–2539. doi: 10.1101/gad.8.21.2527. [DOI] [PubMed] [Google Scholar]
- 114.Montminy MR, Gonzalez GA, Yamamoto KK. Regulation of cAMP-inducible genes by CREB. Trends Neurosci. 1990;13:184–188. doi: 10.1016/0166-2236(90)90045-c. [DOI] [PubMed] [Google Scholar]