

Abstract
Objective:
To review the current evidence and make practice recommendations regarding the diagnosis and treatment of limb-girdle muscular dystrophies (LGMDs).
Methods:
Systematic review and practice recommendation development using the American Academy of Neurology guideline development process.
Results:
Most LGMDs are rare, with estimated prevalences ranging from 0.07 per 100,000 to 0.43 per 100,000. The frequency of some muscular dystrophies varies based on the ethnic background of the population studied. Some LGMD subtypes have distinguishing features, including pattern of muscle involvement, cardiac abnormalities, extramuscular involvement, and muscle biopsy findings. The few published therapeutic trials were not designed to establish clinical efficacy of any treatment.
Principal recommendations:
For patients with suspected muscular dystrophy, clinicians should use a clinical approach to guide genetic diagnosis based on clinical phenotype, inheritance pattern, and associated manifestations (Level B). Clinicians should refer newly diagnosed patients with an LGMD subtype and high risk of cardiac complications for cardiology evaluation even if they are asymptomatic from a cardiac standpoint (Level B). In patients with LGMD with a known high risk of respiratory failure, clinicians should obtain periodic pulmonary function testing (Level B). Clinicians should refer patients with muscular dystrophy to a clinic that has access to multiple specialties designed specifically to care for patients with neuromuscular disorders (Level B). Clinicians should not offer patients with LGMD gene therapy, myoblast transplantation, neutralizing antibody to myostatin, or growth hormone outside of a research study designed to determine efficacy and safety of the treatment (Level R). Detailed results and recommendations are available on the Neurology® Web site at Neurology.org.
Limb-girdle muscular dystrophies (LGMDs) are a group of hereditary myopathies characterized by predominantly proximal muscle weakness (pelvic and shoulder girdles).1 Initially described as a clinical phenotype, they are now recognized as a heterogeneous group of myopathies that vary in severity and may affect persons at all ages from childhood through adulthood. The LGMDs are classified into 2 main groups depending on the inheritance pattern: LGMD1, autosomal dominant; and LGMD2, autosomal recessive. Appended to this numeric division is a letter designating the order of discovery for each chromosomal locus.2,3
This guideline reviews the current evidence and makes practice recommendations regarding the diagnosis and treatment of LGMDs. We also review other hereditary myopathies that may be considered forms of LGMD (e.g., hereditary inclusion body myopathies [hIBMs], Emery-Dreifuss muscular dystrophy [EDMD], Becker muscular dystrophy [BMD], and manifesting carriers of dystrophin mutations). We also discuss non–limb-girdle adult-onset myopathies that are genotypically identical to the LGMDs (e.g., Miyoshi distal myopathy, which is allelic to LGMD2B) and myofibrillar myopathies (MFM).
Duchenne dystrophy, congenital muscular dystrophy, myotonic dystrophy, and facioscapulohumeral dystrophy are not included in this guideline, as they will be discussed in forthcoming guidelines. Table e-1 on the Neurology® Web site at Neurology.org delineates the most recent classification of what is considered a muscular dystrophy in adults who were included in this review. We use the terms LGMD and muscular dystrophy interchangeably to refer to the disorders reviewed in this guideline.
The principal audience for this guideline is clinicians caring for patients with muscular dystrophies.
DESCRIPTION OF THE ANALYTIC PROCESS
The systematic review and practice recommendations were developed according to the processes described in the 2004 and 2011 American Academy of Neurology (AAN) guideline development process manuals.4,5 Appendices e-1 through e-6 provide further background on development of this guideline.
To inform recommendations regarding the diagnosis, management, and treatment of LGMD, the authors performed systematic reviews to answer the following questions:
What is the frequency of genetically confirmed LGMD subtypes?
How often do patients with muscular dystrophy and its specific subtypes have specific clinical features, including ethnic predilection, diagnostic patterns of weakness, respiratory and cardiac complications, laboratory abnormalities (e.g., elevated creatine kinase [CK]), specific patterns on imaging, and muscle biopsy features?
Are there effective therapies for muscular dystrophies?
The conclusions from the systematic reviews informed the practice recommendation development process. The author panel developed actionable practice recommendations following an explicit structured process. Levels of obligation associated with each recommendation were assigned using a modified Delphi process.5
ANALYSIS OF EVIDENCE
What is the frequency of genetically confirmed LGMD subtypes?
Overall, the LGMDs are uncommon disorders.6 The most common adult-onset muscular dystrophy presenting with limb-girdle weakness is BMD, with an estimated prevalence of 2.38–7.29 per 100,000.6–10 Most of the other specific LGMD disorders are rare, with estimated prevalences ranging from 0.07 per 100,000 (LGMD2D and LGMD2E) to 0.43 per 100,000 (LGMD2I).6 The frequency of some muscular dystrophies varies based on the ethnic background of the population (e.g., LGMD2C is more common in Roma and Tunisian populations, with a prevalence of 0.13 per 100,000).6,11–14 We found no studies estimating the frequency of disorders due to genetic defects in DNAJB6, TRIM32, FHL1, MYH7, filamin C, VCP, matrin-3, selenoprotein, cavin, nebulin, nesprin, KLHL9, and Welander distal myopathies.
How often do patients with muscular dystrophy and its specific subtypes have specific clinical features, including important respiratory and cardiac complications, laboratory abnormalities, and muscle biopsy features?
Common features.
Our systematic review identified features common to most patients with muscular dystrophy. Most patients present with slowly progressive symmetrical weakness. The age at onset is usually adolescence to early adulthood but is highly variable, ranging from early childhood (LGMD2N, LGMD2P, LGMD1E, FHL1, BAG3)15–40,e1,e2 to late adult life (e.g., Welander myopathy, Udd distal myopathy).e3–e11 Although the limb-girdle pattern of weakness affecting proximal muscles of the arms and legs is the most common, other patterns, including scapuloperoneal weakness and distal weakness, are not rare. Indeed, a single genotypic variety can present with different patterns of weakness in different patients. For example, mutations in the titin gene can manifest with limb-girdle weakness, a distal myopathy affecting predominantly anterior leg compartment muscles (Udd distal myopathy), or early ventilatory/respiratory failure.e3,e8–e17 Patients with desmin mutations can present with limb-girdle, scapuloperoneal, or distal pattern of muscle weakness.29,32,34,36–39,e1 Lamin A/C mutations cause both limb-girdle and humeroperoneal phenotypes.e18–e23 Conversely, a single phenotype can result from more than one genotype. Miyoshi myopathy, manifesting with asymmetric leg weakness affecting posterior leg compartment muscles, can be seen in mutations in dysferlin (LGMD2B)e24–e38 and ANO5 (LGMD2L).e39–e42 The humeroperoneal pattern of weakness is seen in EDMD, which can be due to mutations in the emerin,e19,e43,e44 lamin A/C,e18–e23 FHL1,20,22,e45 TMEM43/LUMA,e46 and nesprin 1 and 2 genes (not reviewed in this guideline). Serum CK levels vary widely between patients with the same disorder, ranging from normal to greater than 10 times above normal levels, and can be as much as 100 times normal in some cases (e.g., LGMD2B).e36 EMG shows short-duration, small-amplitude motor units with early recruitment in weak muscles; findings may be subtle in mild cases. Routine muscle biopsy often shows nonspecific myopathic features, but some dystrophies have specific diagnostic features (discussed below).
Distinguishing features.
Although there are few pathognomonic features, many LGMD disorders have distinguishing features. These features for the major LGMD disorders are enumerated in table e-2. In addition to inheritance patterns and overall patterns of weakness, distinguishing clinical characteristics include the early development of foot drop (e.g., MFM27,30,32−34,36,38,39,e47–e65); asymmetry in muscle weakness (e.g., LGMD1A,27,e47–e54 LGMD2L,e39–e42,e66,e67 MFM 27,30,32−34,36,38,39,e47–e65); limb contractures (lamin A/C myopathies,e18–e23 EDMD,e43,e68–e70 BAG325,e59); prominent muscle cramps (LGMD1Ce71–e73); family or personal history of frontotemporal dementia, Paget disease of bone, or motor neuron disease (hIBMPFD)e74–e90; ancestry (e.g., northern European for LGMD2Ie91); scapular winging (e.g., sarcoglycanopathies,12,13,e92–e94 LGMD2Ae95–e100); calf hypertrophy (e.g., BMD,10,11,e101–e152 LGMD2Ie91,e153–e164); cardiomyopathy (e.g., LGMD2I,e91,e153,e154,e156,e160–e172 BMDe101,e103,e104,e107,e108,e111,e113,e114,e116,e120–e122,e124,e126,e129,e132,e133,e137,e139,e151,e152); or cardiac conduction system abnormalities (e.g., laminopathy,e18,e173–e183 desminopathy27,29−40,e1,e2). Rippling muscle phenomenon and percussion-induced muscle contractions are noted in LGMD1C.e184,e185 Epidermolysis bullosa or congenital pyloric atresia suggest plectin mutations.e186–e215 Distinguishing EMG features include myotonic and pseudomyotonic discharges (the latter characterized by runs of decrescendo positive sharp-wave discharges without the typical waxing and waning of amplitudes and frequencies) in MFM. Muscle biopsy features that can distinguish muscular dystrophies include the presence of rimmed vacuoles (e.g., LGMD1D, hIBM,e76,e77,e89,e216–e221 MFM), reducing bodies/cytoplasmic bodies (FHL119,e222–e225), and derangement of myofibrils consistent with MFM (desmin,e226,e227 myotilin,27,e47,e48,e52 αB-crystallin,e49,e55–e57 ZASP,27,e49,e58,e228 BAG3,25,26 DNAJB6,e229 filamin Ce49,e60–e62). Nemaline rods may be seen in distal myopathies due to nebulin mutations.e230,e231 Reductions of specific proteins on immunohistochemistry suggest deficiencies of these proteins, although the diagnosis needs to be confirmed by genetic testing.
Are there effective therapies for muscular dystrophies?
The systematic review identified only 12 studies evaluating treatments for patients with LGMD. These are summarized below.
Gene therapy.
In 2 randomized double-blind trials,e232,e233 adeno-associated virus gene (AAV) transfer to the extensor digitorum brevis muscle in 6 patients with LGMD2D (α-sarcoglycanopathy) demonstrated that AAV gene therapy probably increases the expression of α-sarcoglycan gene. The clinical relevance of this effect is unknown.
A case series of patients with LGMD2C who received escalating doses of AAV-vector-expressed human γ-sarcoglycan genes into the extensor digitorum communis provided insufficient evidence to determine the clinical effect of AAV-vector-expressed γ-sarcoglycan genes.e234
Myoblast transplantation.
A case series evaluating myoblast transplantation into the tibialis anterior in 3 male participants with BMD pretreated with cyclosporine A provided insufficient evidence to determine the efficacy of myoblast transfer in BMD.e235
Neutralizing antibody to myostatin.
A phase 1 randomized controlled study of a neutralizing antibody (MYO-029) to an endogenous inhibitor of muscle growth (myostatin) performed in 116 participants with different types of muscular dystrophies provided evidence that MYO-029 is probably safe and tolerable in patients with BMD, LGMD2A–E, and LGMD2I. The study was not designed to assess efficacy or long-term safety.e236
Corticosteroids for BMD.
A 12-month randomized crossover studye237 in 4 boys with BMD provided evidence that prednisolone 0.35 g/kg/day is probably effective in improving isometric muscle strength in patients with BMD after 3 months of treatment.
Growth hormone for BMD.
A randomized studye238 evaluating the effects of subcutaneous growth hormone (sGH) in 10 patients with BMD provided insufficient evidence to support or refute the use of sGH to improve cardiac and pulmonary function in patients with BMD.
Hand training program in Welander distal myopathy.
A case series of a hand training program in 12 patients with Welander distal myopathy provided insufficient evidence to support or refute the benefit of the exercise program.e239
Endurance training.
Two case series studying the effect of endurance training in 9 ambulatory patients with LGMD2Ie161 and 11 men with BMDe136 provided insufficient evidence to determine the benefit of endurance training to improve maximal oxygen uptake, maximal workload, and other patient-reported outcomes.
Two additional case seriese240,e241 evaluating the effects of exercise on hIBM3 secondary to a defect in the MYH2 gene provided insufficient evidence to assess the effect of endurance training on maximum workload, muscle strength, or change in the expression of myosin isoforms on muscle biopsy after statistical corrections for multiple outcome measures.
MAJOR PRACTICE RECOMMENDATIONS
The recommendations below encompass 3 major areas: diagnosis, evaluation, and management of muscular dystrophies. The full recommendation set is available online.
Diagnosis of muscular dystrophies.
The case for genetic diagnosis.
Accurate diagnosis of the muscular dystrophies is important for patients, their families, and efficient and cost-effective use of medical resources.
Diagnosis assists in defining the long-term prognosis, since some dystrophies are more rapidly progressive, involve the cardiorespiratory systems more frequently, or are associated with other disorders. The identification of these dystrophies through genetic testing will not only inform long-term prognosis but will also assist in directing care more efficiently (e.g., more frequent cardiorespiratory monitoring and prophylactic treatments such as pacer/defibrillator placement for those disorders known to be associated with cardiac involvement). Precise identification of the disorder also eliminates the need for repeated testing for an acquired, treatable disorder such as an inflammatory myopathy, because some dystrophies have inflammation on muscle biopsy, making diagnosis difficult on the basis of routine biopsy findings. In addition, the temptation to try immunosuppressive agents repeatedly, looking for a therapeutic response, is not unusual when there is no diagnosis and the patient is worsening. This exposes patients to potentially serious side effects of immunosuppressive medications. Patients on immunosuppressants need regular monitoring, adding logistical difficulties to a population that may have significantly impaired mobility. Health care costs are increased by repeated investigations, immunosuppressive treatments, and laboratory monitoring. Although establishing a genetic diagnosis is expensive on the front end, the costs of continued investigation for other causes and the risks and expenses associated with empiric trials of immunosuppressants make a strong case for establishing a genetic diagnosis, which often provides patients a sense of closure. Establishing a genetic diagnosis is crucial for genetic counseling to inform decision-making about having children and for screening of offspring. Treatment of cardiomyopathy, arrhythmias, and ventilatory failure prolongs life and improves quality of life in patients with other neuromuscular diseases.e242–e247
The approach to genetic diagnosis.
The wide variation in phenotypic expression of the LGMDs makes establishing a clinical diagnosis a challenge. Our systematic review found that muscular dystrophies have few features that are pathognomonic of a specific disorder. An algorithmic approach emphasizing the pattern of weakness, the inheritance pattern, and the presence of distinguishing clinical and muscle biopsy features can efficiently narrow the differential diagnosis to a few disorders. The overall conceptual approach is outlined in figure 1. See figures 2–5 and figures e-1 and e-2 for more detailed algorithms based on the specific pattern of weakness and suspected inheritance patterns.
Figure 1. Conceptual approach to a patient with a suspected limb-girdle muscular dystrophy.
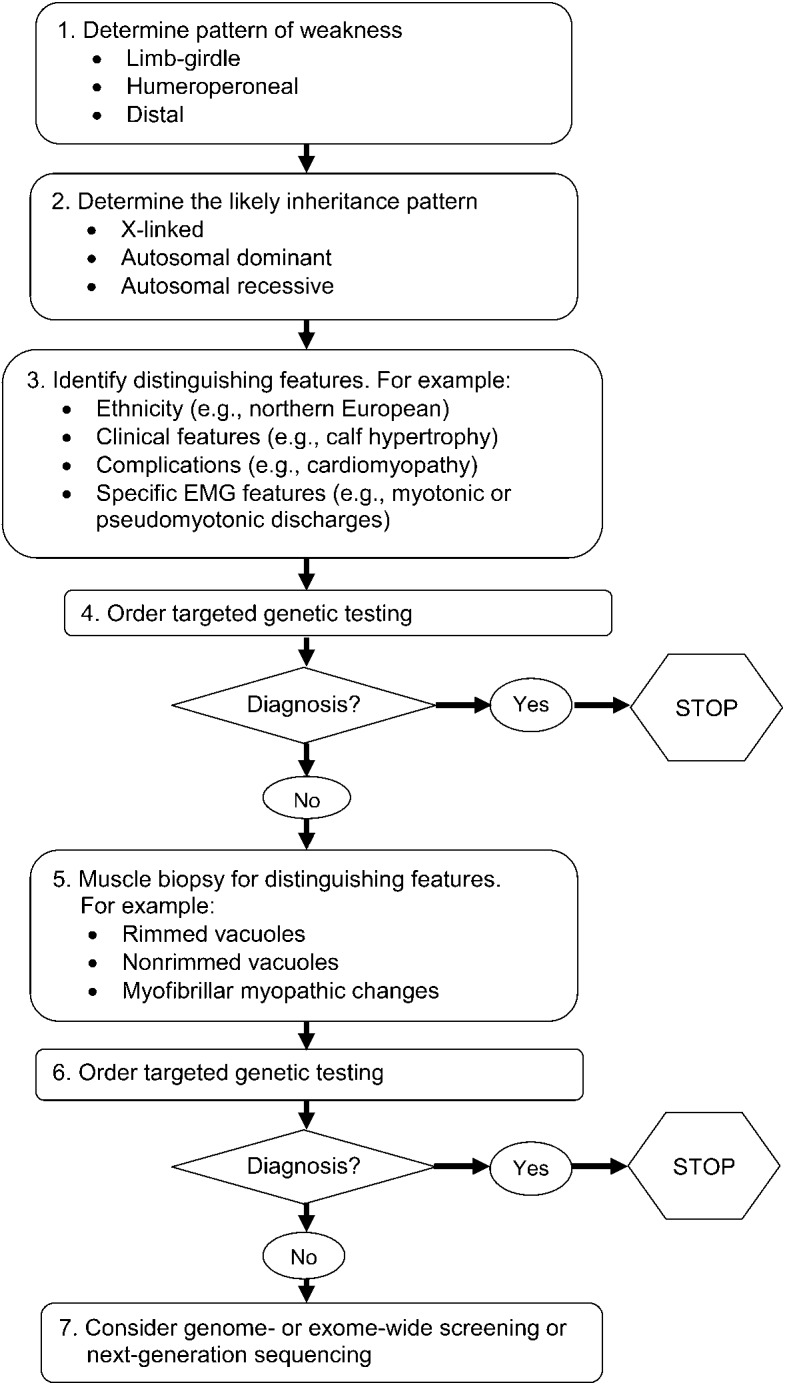
Figure 2. General classification of myopathies.

The differential diagnosis of myopathy includes several diverse conditions, both inherited and acquired. The neuromuscular examination, ancillary laboratory tests, and EMG assist in the differential diagnosis of these disorders. The term other muscular dystrophies is used here to indicate hereditary disorders of muscle that have 3 major phenotypes of weakness: limb-girdle, humeroperoneal, and distal. MD = muscular dystrophy. *Other muscular dystrophy phenotypes. Limb-girdle pattern of weakness: symmetric weakness predominantly affecting the proximal legs and arms. Distal muscles may be involved but to a much lesser extent. Neck flexors and extensors may be involved. Humeroperoneal: humeral muscles (biceps and triceps) and the anterior compartment of the distal leg muscles. May be asymmetric. Distal: weakness involving the anterior or posterior compartments of the distal legs or the distal arm/forearm muscles. Other patterns, such as distal arm/proximal leg (inclusion body myositis), ptosis/ophthalmoplegia (myasthenia gravis, myotonic dystrophy, some congenital myopathies, oculopharyngeal muscular dystrophy, mitochondrial myopathy), and neck extensor weakness (dropped head syndrome) (amyotrophic lateral sclerosis, myasthenia gravis, inflammatory myopathies, isolated neck extensor myopathy), may be noted and suggest specific diagnoses other than limb-girdle muscular dystrophy and variants, as noted in parentheses. **Autosomal dominant, autosomal recessive, or X-linked inheritance may be responsible in sporadic cases. Figures 3–5, e-1, and e-2 discuss the clinical approach to diagnosis using the inheritance pattern and the pattern of weakness (as outlined in figure 2) as a starting point.
Figure 3. Diagnostic approach to patients with a limb-girdle pattern of weakness and suspected muscular dystrophy with an autosomal dominant inheritance pattern.

*Autosomal dominant, autosomal recessive, or X-linked inheritance may be responsible in sporadic cases. EDMD = Emery-Dreifuss muscular dystrophy; hIBMPFD = hereditary inclusion body myopathy with Paget disease and frontotemporal dementia; LGMD = limb-girdle muscular dystrophy; VCP = valosin-containing protein.
Figure 4. Diagnostic approach to patients with a limb-girdle pattern of weakness and suspected muscular dystrophy with an autosomal recessive inheritance pattern.

*Autosomal dominant, autosomal recessive, or X-linked inheritance may be responsible in sporadic cases. LGMD = limb-girdle muscular dystrophy.
Figure 5. Diagnostic approach to patients with a limb-girdle pattern of weakness and suspected muscular dystrophy with an X-linked recessive inheritance pattern.

In females, a manifest X-linked disorder may be considered if there is a familial presentation with males more affected than females. *Autosomal dominant, autosomal recessive, or X-linked inheritance may be responsible in sporadic cases. MD = muscular dystrophy.
Recommendations.
For patients with suspected muscular dystrophy, clinicians should use a clinical approach to guide genetic diagnosis based on the clinical phenotype, including the pattern of muscle involvement, inheritance pattern, age at onset, and associated manifestations (e.g., early contractures, cardiac or respiratory involvement) (Level B).
In patients with suspected muscular dystrophy in whom initial clinically directed genetic testing does not provide a diagnosis, clinicians may obtain genetic consultation or perform parallel sequencing of targeted exomes, whole-exome sequencing, whole-genome screening, or next-generation sequencing to identify the genetic abnormality (Level C).
Evaluation and medical management of muscular dystrophies.
Cardiac involvement.
Our systematic review reveals that many (e.g., LGMD1A, LGMD1B, LGMD1D, LGMD1E, LGMD2C–K, LGMD2M–P, BMD, EDMD, and MFM), though not all (e.g., LGMD1C, LGMD2A, LGMD2B, and LGMD2L), muscular dystrophy subtypes have associated cardiac involvement. Muscular dystrophy patients with cardiac involvement often do not have symptoms such as chest pain, pedal edema, or palpitations that precede cardiac morbidity or sudden cardiac death. Serious cardiac manifestations in patients with muscular dystrophy are often identified only with cardiology testing. The detection and appropriate management of cardiac dysfunction are important to reduce morbidity and mortality. Patients with muscular dystrophy often have improved quality of life following appropriate pharmacologic treatment, device placement, or surgical intervention for their cardiac involvement.e248
Recommendations.
Clinicians should refer newly diagnosed patients with (1) LGMD1A, LGMD1B, LGMD1D, LGMD1E, LGMD2C–K, LGMD2M–P, BMD, EDMD, and MFM or (2) muscular dystrophy without a specific genetic diagnosis for cardiology evaluation, including ECG and structural evaluation (echocardiography or cardiac MRI), even if they are asymptomatic from a cardiac standpoint, to guide appropriate management (Level B).
If ECG or structural cardiac evaluation (e.g., echocardiography) has abnormal results, or if the patient has episodes of syncope, near-syncope, or palpitations, clinicians should order rhythm evaluation (e.g., Holter monitor or event monitor) to guide appropriate management (Level B).
Clinicians should refer muscular dystrophy patients with palpitations, symptomatic or asymptomatic tachycardia or arrhythmias, or signs and symptoms of cardiac failure for cardiology evaluation (Level B).
It is not obligatory for clinicians to refer patients with LGMD2A, LGMD2B, and LGMD2L for cardiac evaluation unless they develop overt cardiac signs or symptoms (Level B).
Dysphagia and nutrition.
Patients with muscular dystrophy may have difficulty receiving adequate oral intake due to dysphagia or inability to feed themselves due to arm weakness. Maintaining adequate nutrition and body weight is important for optimizing strength, function, and quality of life. When oral intake is inadequate, other means of maintaining intake (e.g., gastrostomy or jejunostomy feeding tubes) may be needed to maintain optimal nutrition. There is evidence from related conditions (amyotrophic lateral sclerosis [ALS]) that maintenance of nutrition and body weight prolongs survival.e243
Recommendation.
Clinicians should refer muscular dystrophy patients with dysphagia, frequent aspiration, or weight loss for swallowing evaluation or gastroenterology evaluation to assess and manage swallowing function and aspiration risk, to teach patients techniques for safe and effective swallowing (e.g., chin tuck maneuver, altered food consistencies), and to consider placement of a gastrostomy/jejunostomy tube for nutritional support (Level B).
Pulmonary complications.
Our systematic review demonstrates that some forms of muscular dystrophy (e.g., LGMD2I or MFM) are associated with oropharyngeal or ventilatory muscle weakness and that patients with these forms are at high risk for developing respiratory failure during the course of their disease. Patients with other forms of muscular dystrophy (e.g., LGMD2B and LGMD2L) rarely, if ever, have symptomatic respiratory involvement from their disease. Patients with respiratory failure from neuromuscular-related weakness often do not have symptoms, such as dyspnea, that precede the onset of respiratory failure. Impending respiratory failure in these patients is often identified only with pulmonary function tests. Respiratory failure constitutes a major source of morbidity, interfering with daytime cognitive function and negatively affecting quality of life. In addition, ventilatory and oropharyngeal weakness can threaten survival through the risk of upper airway obstruction or bellows failure (or both).e243 Patients with respiratory failure secondary to muscle weakness often have improved quality of life with noninvasive pulmonary ventilation.e243
Recommendations.
Clinicians should order pulmonary function testing (spirometry and maximal inspiratory/expiratory force in the upright and, if normal, supine positions) or refer for pulmonary evaluation (to identify and treat respiratory insufficiency) in muscular dystrophy patients at the time of diagnosis, or if they develop pulmonary symptoms later in their course (Level B).
In patients with a known high risk of respiratory failure (e.g., those with LGMD2I or MFM), clinicians should obtain periodic pulmonary function testing (spirometry and maximal inspiratory/expiratory force in the upright position and, if normal, in the supine position) or evaluation by a pulmonologist to identify and treat respiratory insufficiency (Level B).
It is not obligatory for clinicians to refer patients with LGMD2B and LGMD2L for pulmonary evaluation unless they are symptomatic (Level C).
Clinicians should refer muscular dystrophy patients with excessive daytime somnolence, nonrestorative sleep (e.g., frequent nocturnal arousals, morning headaches, excessive daytime fatigue), or respiratory insufficiency based on pulmonary function tests for pulmonary or sleep medicine consultation for consideration of noninvasive ventilation to improve quality of life (Level B).
Spinal deformities.
Our systematic review has revealed the risk of evolving musculoskeletal spine deformities, such as scoliosis, kyphosis, or rigid spine syndrome, in various muscular dystrophies (EDMD, rigid spine syndrome). These deformities can result in discomfort and functional impairment, interfering with gait, activities of daily living, and pulmonary function. Their management is important in order to reduce discomfort, preserve mobility or ability to sit in a wheelchair, and reduce pulmonary complications.e248
Recommendations.
Clinicians should monitor patients with muscular dystrophy for the development of spinal deformities to prevent resultant complications and preserve function (Level B).
Clinicians should refer muscular dystrophy patients with musculoskeletal spine deformities to an orthopedic spine surgeon for monitoring and surgical intervention if it is deemed necessary in order to maintain normal posture, assist mobility, maintain cardiopulmonary function, and optimize quality of life (Level B).
Rehabilitative management and treatment of muscular dystrophies.
Clinical rehabilitative management.
Our evidence-based review of the literature on rehabilitative management of muscular dystrophies consisted primarily of single Class III studies. Thus, the currently available data are not adequate to assess the effect of any rehabilitation modality (endurance and strength training, bracing, assistive devices, new computer-based technology). However, the principles of long-term management emphasize maintaining mobility and functional independence for as long as possible, with a focus on maximizing quality of life. The prevention and management of comorbidities, both expected and acquired, is a major part of such management. Another important aspect of management includes proactively preparing patients and their families for the long-term consequences of muscular dystrophies and engaging in discussions regarding end-of-life care. There is evidence from studies in other neuromuscular diseases, including ALS, that a multidisciplinary approach is the most effective way to deliver care.e249
Recommendations.
Clinicians should refer patients with muscular dystrophy to a clinic that has access to multiple specialties (e.g., physical therapy, occupational therapy, respiratory therapy, speech and swallowing therapy, cardiology, pulmonology, orthopedics, and genetics) designed specifically to care for patients with muscular dystrophy and other neuromuscular disorders in order to provide efficient and effective long-term care (Level B).
Clinicians should recommend that patients with muscular dystrophy have periodic assessments by a physical and occupational therapist for symptomatic and preventive screening (Level B).
While respecting and protecting patient autonomy, clinicians should proactively anticipate and facilitate patient and family decision-making as the disease progresses, including decisions regarding loss of mobility, need for assistance with activities of daily living, medical complications, and end-of-life care (Level B).
For patients with muscular dystrophy, clinicians should prescribe physical and occupational therapy, as well as bracing and assistive devices that are adapted specifically to the patient's deficiencies and contractures, in order to preserve mobility and function and prevent contractures (Level B).
Strength training and aerobic exercise training.
The evidence base regarding the effectiveness of rehabilitation management of muscular dystrophies is limited. However, the available evidence suggests that this population would benefit from strengthening and aerobic fitness training programs. Due to the muscle degeneration in muscular dystrophy, there may be some risk of exercise-induced muscle damage, myoglobinuria, and subsequent overwork weakness following supramaximal, high-intensity exercise. There have been several randomized or quasirandomized controlled trials comparing strength training programs, aerobic exercise programs, or both to nontraining controls in patients with a variety of neuromuscular disorders.e250–e252 On the basis of this literature, both strength training and aerobic exercise programs appear to be safe, without any notable deleterious effects.
Recommendations.
Clinicians may advise patients with muscular dystrophy that aerobic exercise combined with a supervised submaximal strength training program is probably safe (Level C).
Clinicians may advise patients with muscular dystrophy that gentle, low-impact aerobic exercise (swimming, stationary bicycling) improves cardiovascular performance, increases muscle efficiency, and lessens fatigue (Level C).
Clinicians may counsel patients with muscular dystrophy to hydrate adequately, not to exercise to exhaustion, and to avoid supramaximal, high-intensity exercise (Level C).
Clinicians should educate patients with muscular dystrophy who are participating in an exercise program about the warning signs of overwork weakness and myoglobinuria, which include feeling weaker rather than stronger within 30 minutes after exercise, excessive muscle soreness 24–48 hours following exercise, severe muscle cramping, heaviness in the extremities, and prolonged shortness of breath (Level B).
Medical treatments.
The systematic review demonstrated that effects on the clinical course and the long-term safety of gene transfer,e232–e234 myoblast transplantation,e235 neutralizing antibody to myostatin,e236 or growth hormonee238 are yet to be determined.
Recommendation.
Clinicians should not currently offer patients with muscular dystrophy gene therapy, myoblast transplantation, neutralizing antibody to myostatin, or growth hormone outside of a research study designed to determine the efficacy and safety of the treatment (Level R).
Supplementary Material
GLOSSARY
- AAN
American Academy of Neurology
- AAV
adeno-associated virus gene
- ALS
amyotrophic lateral sclerosis
- BMD
Becker muscular dystrophy
- CK
creatine kinase
- EDMD
Emery-Dreifuss muscular dystrophy
- hIBM
hereditary inclusion body myopathy
- LGMD
limb-girdle muscular dystrophy
- MFM
myofibrillar myopathies
- sGH
subcutaneous growth hormone
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Pushpa Narayanaswami: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content, study supervision. Michael Weiss: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Duygu Selcen: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. William David: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Elizabeth Raynor: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Gregory Carter: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Matthew Wicklund: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Richard J. Barohn: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Erik Ensrud: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Robert C. Griggs: study concept and design. Gary Gronseth: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Anthony A. Amato: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content, study supervision.
STUDY FUNDING
Funding for this publication was made possible (in part) by grant DD10-1012 from the Centers for Disease Control and Prevention. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. The remaining funding was provided by the American Academy of Neurology.
DISCLOSURE
P. Narayanaswami has received honoraria from the American Academy of Neurology (AAN) and the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM). M. Weiss has served as a speaker for the AAN, AANEM, American Academy of Physical Medicine & Rehabilitation (AAPM&R), Athena Diagnostics, Nufactor, Walgreens, and Grifols Inc.; serves on speakers' bureaus for Athena Diagnostics and Walgreens; has consulted for Genzyme Corporation, CSL Behring, Questcor Pharmaceuticals, and Washington State Labor and Industries; and has received research funding support from the ALS Therapy Alliance and Northeast ALS Consortium. D. Selcen has served as an editorial board member for Neuromuscular Disorders and has received funding for research from the National Institutes of Health (NIH). W. David and E. Raynor report no disclosures relevant to the manuscript. G. Carter has served as the senior associate editor for Muscle & Nerve, has received honoraria from the AANEM and the Canadian Association of Physical Medicine and Rehabilitation, has received funding for research from the National Institutes on Aging and the National Institute on Disability and Rehabilitation Research, and has testified on a case regarding the use of marijuana in pain. M. Wicklund has served on a scientific advisory board for Sarepta Therapeutics, has served on a speakers' bureau for Genzyme, has received grant funding from Eli Lilly, and has collaborated on research without compensation with Athena Diagnostics. R. Barohn has served as a consultant or on a scientific advisory board for Genzyme, Grifols, MedImmune, and Novartis; has received honoraria from Alexion, Isis, Baxter, Sarepta, and CSL Behring; and has received funding for research from the US Food and Drug Administration (FDA) and the NIH. E. Ensrud reports no relevant disclosures. R. Griggs consults for PTC Therapeutics (Chair of DSMB), Novartis (DSMB), Marathon Pharmaceuticals, Taro Pharmaceuticals, and Viromed (DSMB); receives funding from the NIH, the Italian Telethon (DSMB Chair), the Muscular Dystrophy Association, the Parent Project for Muscular Dystrophy, and the AAN; and receives royalties from Elsevier (for Cecil Essentials and Cecil Textbook of Medicine). G. Gronseth serves as an editorial advisory board member of Neurology Now, is an associate editor of Neurology, and receives honoraria from the AAN. A. Amato has served as a consultant or on scientific advisory boards for MedImmune, Amgen, Biogen, DART, and Baxter; serves as an associate editor for Neurology and Muscle & Nerve; has received royalties from publishing from Neuromuscular Disorders; has received honoraria from the AAN and AANEM; and has received funding for research from Amgen, MedImmune, Novartis, the FDA, and the NIH. Go to Neurology.org for full disclosures.
DISCLAIMER
This statement is provided as an educational service of the American Academy of Neurology and American Association of Neuromuscular & Electrodiagnostic Medicine. It is based on an assessment of current scientific and clinical information. It is not intended to include all possible proper methods of care for a particular neurologic problem or all legitimate criteria for choosing to use a specific procedure. Neither is it intended to exclude any reasonable alternative methodologies. The AAN and AANEM recognize that specific patient care decisions are the prerogative of the patient and the physician caring for the patient, based on all of the circumstances involved. The clinical context section is made available in order to place the evidence-based guidelines into perspective with current practice habits and challenges. Formal practice recommendations are not intended to replace clinical judgment.
CONFLICT OF INTEREST
The American Academy of Neurology and American Association of Neuromuscular & Electrodiagnostic Medicine are committed to producing independent, critical, and truthful clinical practice guidelines (CPGs). Significant efforts are made to minimize the potential for conflicts of interest to influence the recommendations of this CPG. To the extent possible, the AAN and AANEM keep separate those who have a financial stake in the success or failure of the products appraised in the CPGs and the developers of the guidelines. Conflict of interest forms were obtained from all authors and reviewed by an oversight committee prior to project initiation. AAN and AANEM limit the participation of authors with substantial conflicts of interest. The AAN and AANEM forbid commercial participation in, or funding of, guideline projects. Drafts of the guideline have been reviewed by at least 3 AAN committees, at least 1 AANEM committee, a network of neurologists, Neurology peer reviewers, and representatives from related fields. The AAN Guideline Author Conflict of Interest Policy can be viewed at www.aan.com. For complete information on this process, access the 2004 AAN process manual.4
REFERENCES
- 1.Walton JN, Nattrass FJ. On the classification, natural history and treatment of the myopathies. Brain 1954;77:169–231 [DOI] [PubMed] [Google Scholar]
- 2.Bushby KM. Diagnostic criteria for the limb-girdle muscular dystrophies: report of the ENMC Consortium on Limb-Girdle Dystrophies. Neuromuscul Disord 1995;5:71–74 [DOI] [PubMed] [Google Scholar]
- 3.Bushby KM, Beckmann JS. The limb-girdle muscular dystrophies: proposal for a new nomenclature. Neuromuscul Disord 1995;5:337–343 [DOI] [PubMed] [Google Scholar]
- 4.American Academy of Neurology. Guideline Development Process Manual, 2004 ed. St. Paul, MN: American Academy of Neurology; 2004 [Google Scholar]
- 5.American Academy of Neurology. Clinical Practice Guideline Process Manual, 2011 ed. St. Paul, MN: American Academy of Neurology; 2011 [Google Scholar]
- 6.Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain 2009;132:3175–3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bushby KM, Thambyayah M, Gardner-Medwin D. Prevalence and incidence of Becker muscular dystrophy. Lancet 1991;337:1022–1024 [DOI] [PubMed] [Google Scholar]
- 8.Siciliano G, Tessa A, Renna M, Manca ML, Mancuso M, Murri L. Epidemiology of dystrophinopathies in North-West Tuscany: a molecular genetics-based revisitation. Clin Genet 1999;56:51–58 [DOI] [PubMed] [Google Scholar]
- 9.Nakagawa M, Nakahara K, Yoshidome H, et al. Epidemiology of progressive muscular dystrophy in Okinawa, Japan: classification with molecular biological techniques. Neuroepidemiology 1991;10:185–191 [DOI] [PubMed] [Google Scholar]
- 10.Arikawa E, Hoffman EP, Kaido M, Nonaka I, Sugita H, Arahata K. The frequency of patients with dystrophin abnormalities in a limb-girdle patient population. Neurology 1991;41:1491–1496 [DOI] [PubMed] [Google Scholar]
- 11.Kefi M, Amouri R, Driss A, et al. Phenotype and sarcoglycan expression in Tunisian LGMD 2C patients sharing the same del521-T mutation. Neuromuscul Disord 2003;13:779–787 [DOI] [PubMed] [Google Scholar]
- 12.Todorova A, Tournev I, Ninova N, Georgieva V, Kremensky I. Screening for C283Y gamma-sarcoglycan mutation in a high-risk group of Bulgarian Gypsies: evidence for a geographical localization and a non-random distribution among Gypsy subgroups. Community Genet 2002;5:217–221 [DOI] [PubMed] [Google Scholar]
- 13.Merlini L, Kaplan JC, Navarro C, et al. The limb-girdle muscular dystrophy 2C in Gypsies. Acta Myol 2001;20:188–191 [Google Scholar]
- 14.Spengos K, Walter MC, Dekomien G, Papadopoulos K, Lochmüller H, Manta P. C283Y mutation in the gamma-sarcoglycan gene in Greek Gypsies with severe limb girdle muscular dystrophy. Eur J Neurol 2010;17:e41–e42 [DOI] [PubMed] [Google Scholar]
- 15.Godfrey C, Clement E, Mein R, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007;130:2725–2735 [DOI] [PubMed] [Google Scholar]
- 16.Biancheri R, Falace A, Tessa A, et al. POMT2 gene mutation in limb-girdle muscular dystrophy with inflammatory changes. Biochem Biophys Res Commun 2007;363:1033–1037 [DOI] [PubMed] [Google Scholar]
- 17.Hara Y, Balci-Hayta B, Yoshida-Moriguchi T, et al. A dystroglycan mutation associated with limb-girdle muscular dystrophy. N Engl J Med 2011;364:939–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dinçer P, Balci B, Yuva Y, et al. A novel form of recessive limb girdle muscular dystrophy with mental retardation and abnormal expression of α-dystroglycan. Neuromuscul Disord 2003;13:771–778 [DOI] [PubMed] [Google Scholar]
- 19.Schessl J, Taratuto AL, Sewry C, et al. Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain 2009;132:452–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schoser B, Goebel HH, Janisch I, et al. Consequences of mutations within the C terminus of the FHL1 gene. Neurology 2009;73:543–551 [DOI] [PubMed] [Google Scholar]
- 21.Shalaby S, Hayashi YK, Nonaka I, Noguchi S, Nishino I. Novel FHL1 mutations in fatal and benign reducing body myopathy. Neurology 2009;72:375–376 [DOI] [PubMed] [Google Scholar]
- 22.Chen DH, Raskind WH, Parson WW, et al. A novel mutation in FHL1 in a family with X-linked scapuloperoneal myopathy: phenotypic spectrum and structural study of FHL1 mutations. J Neurol Sci 2010;296:22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schessl J, Zou Y, McGrath MJ, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest 2008;118:904–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarkozy A, Windpassinger C, Hudson J, et al. Phenotypic heterogeneity in British patients with a founder mutation in the FHL1 gene. Eur J Hum Genet 2011;19:1038–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selcen D, Muntoni F, Burton BK, et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol 2009;65:83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Odgerel Z, Sarkozy A, Lee HS, et al. Inheritance patterns and phenotypic features of myofibrillar myopathy associated with a BAG3 mutation. Neuromuscul Disord 2010;20:438–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olivé M, Odgerel Z, Martínez A, et al. Clinical and myopathological evaluation of early- and late-onset subtypes of myofibrillar myopathy. Neuromuscul Disord 2011;21:533–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilchrist JM, Pericak-Vance M, Silverman L, Roses AD. Clinical and genetic investigation in autosomal dominant limb-girdle muscular dystrophy. Neurology 1988;38:5–9 [DOI] [PubMed] [Google Scholar]
- 29.Goldfarb LG, Park KY, Cervenáková L, et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet 1998;19:402–403 [DOI] [PubMed] [Google Scholar]
- 30.Arias M, Pardo J, Blanco-Arias P, et al. Distinct phenotypic features and gender-specific disease manifestations in a Spanish family with desmin L370P mutation. Neuromuscul Disord 2006;16:498–503 [DOI] [PubMed] [Google Scholar]
- 31.Bergman JE, Veenstra-Knol HE, van Essen AJ, et al. Two related Dutch families with a clinically variable presentation of cardioskeletal myopathy caused by a novel S13F mutation in the desmin gene. Eur J Med Genet 2007;50:355–366 [DOI] [PubMed] [Google Scholar]
- 32.Dalakas MC, Dagvadorj A, Goudeau B, et al. Progressive skeletal myopathy, a phenotypic variant of desmin myopathy associated with desmin mutations. Neuromuscul Disord 2003;13:252–258 [DOI] [PubMed] [Google Scholar]
- 33.Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med 2000;342:770–780 [DOI] [PubMed] [Google Scholar]
- 34.Walter MC, Reilich P, Huebner A, et al. Scapuloperoneal syndrome type Kaeser and a wide phenotypic spectrum of adult-onset, dominant myopathies are associated with the desmin mutation R350P. Brain 2007;130:1485–1496 [DOI] [PubMed] [Google Scholar]
- 35.Goudeau B, Rodrigues-Lima F, Fischer D, et al. Variable pathogenic potentials of mutations located in the desmin alpha-helical domain. Hum Mutat 2006;27:906–913 [DOI] [PubMed] [Google Scholar]
- 36.Bär H, Goudeau B, Wälde S, et al. Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum Mutat 2007;28:374–376 [DOI] [PubMed] [Google Scholar]
- 37.Dagvadorj A, Olivé M, Urtizberea JA, et al. A series of West European patients with severe cardiac and skeletal myopathy associated with a de novo R406W mutation in desmin. J Neurol 2004;251:143–149 [DOI] [PubMed] [Google Scholar]
- 38.Olivé M, Armstrong J, Miralles F, et al. Phenotypic patterns of desminopathy associated with three novel mutations in the desmin gene. Neuromuscul Disord 2007;17:443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong D, Wang Z, Zhang W, et al. A series of Chinese patients with desminopathy associated with six novel and one reported mutations in the desmin gene. Neuropathol Appl Neurobiol 2011;37:257–270 [DOI] [PubMed] [Google Scholar]
- 40.Greenberg SA, Salajegheh M, Judge DP, et al. Etiology of limb girdle muscular dystrophy 1D/1E determined by laser capture microdissection proteomics. Ann Neurol 2012;71:141–145 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.