

Abstract
Humans are colonized by thousands of bacterial species, but it is difficult to assess the metabolic and pathogenic potential of the majority of these because they have yet to be cultured. Here, we characterize an uncultivated vaginal mycoplasma tightly associated with trichomoniasis that was previously known by its 16S rRNA sequence as “Mnola.” In this study, the mycoplasma was found almost exclusively in women infected with the sexually transmitted pathogen Trichomonas vaginalis, but rarely observed in women with no diagnosed disease. The genomes of four strains of this species were reconstructed using metagenome sequencing and assembly of DNA from four discrete mid-vaginal samples, one of which was obtained from a pregnant woman with trichomoniasis who delivered prematurely. These bacteria harbor several putative virulence factors and display unique metabolic strategies. Genes encoding proteins with high similarity to potential virulence factors include two collagenases, a hemolysin, an O-sialoglycoprotein endopeptidase and a feoB-type ferrous iron transport system. We propose the name “Candidatus Mycoplasma girerdii” for this potential new pathogen.
Introduction
Application of next-generation sequencing to the study of the human microbiome is rapidly transforming our understanding of the diversity of the microbial communities that inhabit the human body [1]. However, progress towards the identification of specific microbiome signatures or specific organisms with strong links to disease states has proven elusive. We characterize a new vaginal mycoplasma species “Candidatus Mycoplasma girerdii”, previously identified only by its 16S rRNA sequence [2], that exhibits a strong and unique association with the sexually transmitted pathogen Trichomonas vaginalis.
Organisms of the Mycoplasma and Ureaplasma genera are collectively referred to as mycoplasmas. They lack cell walls, have small genomes and are often dependent on their hosts. Mycoplasmas of the female urogenital tract are associated with bacterial vaginosis (BV), pelvic inflammatory disease, preterm labor and preterm birth [3], [4]. These mycoplasmas are among the most common organisms to invade the amniotic cavity, and their carriage is associated with chorioamnionitis in preterm premature rupture of membranes (PPROM) [5]. Moreover, uncultivated and uncharacterized bacterial species also invade the amniotic cavity and likely impact pregnancy outcome [6]. Mycoplasmas can induce inflammatory cytokines in the host [4], and they are more prevalent in the vaginal flora of HIV-infected women [7]. Ureaplasmas have been associated with complications during pregnancy [4], M. genitalium with pelvic inflammatory disease, cervicitis, endometritis and salpingitis [8], and M. hominis with BV [3] and trichomoniasis [9], [10]. M. genitalium is an emerging sexually-transmitted infection, which causes nongonococcal urethritis in men. Despite these associations with disease, M. hominis and Ureaplasma are also common in apparently healthy women. T. vaginalis causes trichomoniasis, the most common non-viral sexually transmitted infection worldwide [11]. Trichomoniasis often accompanies low levels of lactobacilli [12] and BV, and has been implicated in an array of pregnancy complications [13], [14]. Although the extracellular eukaryotic parasite binds to vaginal epithelial cells and is hemolytic [15], the mechanisms of its pathogenesis remain enigmatic.
Martin et al. [2] recently described a 16S rRNA sequence from an unknown Mycoplasma, which they called “Mnola”, in vaginal secretions and found it to be strongly associated with presence of T. vaginalis in the study. Shortly thereafter, Costello et al. [16] reported a phylotype with a 16S rRNA sequence exhibiting ∼99% identity to that of the new mycoplasma described herein and by Martin et al. [2] as a predominant taxon in the oral sample of a low birth-weight infant (24.5 wks) and speculated it may have been acquired by vertical transmission during delivery. Hyman et al. also subsequently identified a partial 16S rRNA sequence from a vaginal sample of a woman who delivered full term that is 99% similar to “Ca. M. girerdii” [17]. We independently identified this phylotype and its association with T. vaginalis, first reported by Martin et al. In the current work, we confirm and extend the characterization of this new Mycoplasma using metagenomic strategies, present the genomic sequences of four independently identified strains, one of which was isolated from a pregnant woman who subsequently delivered preterm, and we propose “Candidatus Mycoplasma girerdii” for its name.
Results and Discussion
As part of the Vaginal Human Microbiome Project at Virginia Commonwealth University, we generated 16S rRNA gene-based microbiome profiles for 1,361 mid-vaginal samples collected from women visiting outpatient clinics and an additional 110 samples collected in a labor and delivery unit [18]. Our analyses revealed a novel mycoplasma phylotype that represented the most abundant bacterium observed in 25 mid-vaginal samples (i.e., 25/1,471), including at least one from a woman who experienced preterm labor. Twenty-two of these 25 (88%) women had a clinically diagnosed vaginal infection (Table 1), and all but one of these 22 women for whom vaginal pH was recorded exhibited an elevated pH value greater than 4.5 (median pH value = 5.8), an indicator of vaginal dysbiosis. Although microbiome profiles based on 16S rRNA gene surveys are not always accurate measures of the proportions of bacterial taxa present in a sample for a variety of reasons (e.g., biases inherent in DNA extraction, PCR and related sequencing technologies and variations in the number 16S rRNA genes per genome in different species), it is clear that this mycoplasma represents a very abundant taxon in the vaginal samples collected from these 25 women.
Table 1. Characteristics of 73 “Ca. M. girerdii” positive vaginal samples.
| VCU_ID | % “Ca. M. girerdii” | qRT-PCR T. vaginalis | Current clinical diagnosis | Race/ethnicity | Vaginal pH | T. vaginalis genotype | Dominant taxon |
| VCU_NT02 | 99.90 | + | Trichomoniasis | African American | 4.4 | ND | “Ca. M. girerdii” |
| VCU_CD82 | 99.70 | + | Trichomoniasis | African American | 6 | 2 | “Ca. M. girerdii” |
| VCU_LN42 | 97.50 | + | Trichomoniasis | African American | ND | 1 | “Ca. M. girerdii” |
| VCU_QM60 | 94.90 | + | Trichomoniasis | African American | 5.3 | 1 | “Ca. M. girerdii” |
| VCU_NT41† * | 92.90 | + | Preterm labor and delivery | African American | 6.5 | ND | “Ca. M. girerdii” |
| VCU_FQ09 | 90.60 | + | Trichomoniasis | African American | 5.5 | 2 | “Ca. M. girerdii” |
| VCU_NT49 | 90.40 | + | None | African American | 5.6 | ND | “Ca. M. girerdii” |
| VCU_CT62† | 89.90 | + | Trichomoniasis | Hispanic | 6 | 2 | “Ca. M. girerdii” |
| VCU_KH69 | 88.80 | + | Trichomoniasis | Caucasian | 7 | 1 | “Ca. M. girerdii” |
| VCU_GK81 | 88.00 | + | Trichomoniasis, Bacterial vaginosis | African American | 5.8 | AMB | “Ca. M. girerdii” |
| VCU_NT63 | 87.30 | + | Bacterial vaginosis | African American | 6 | ND | “Ca. M. girerdii” |
| VCU_NT94 | 85.00 | + | Not Available | African American | ND | ND | “Ca. M. girerdii” |
| VCU_AM41 | 84.20 | + | Trichomoniasis | African American | 5.8 | 2 | “Ca. M. girerdii” |
| VCU_NT05 | 82.50 | + | Yeast infection | Other (African) | 7 | ND | “Ca. M. girerdii” |
| VCU_OL06 | 75.60 | + | Trichomoniasis | African American | 7 | 1 | “Ca. M. girerdii” |
| VCU_NT03 | 73.70 | + | Bacterial vaginosis | African American | 7 | ND | “Ca. M. girerdii” |
| VCU_NT54 | 60.10 | + | Trichomoniasis | African American | ND | ND | “Ca. M. girerdii” |
| VCU_MP27 | 59.60 | + | Trichomoniasis | African American | 5 | 1 | “Ca. M. girerdii” |
| VCU_NT95 | 58.80 | + | None | African American | 7 | ND | “Ca. M. girerdii” |
| VCU_BK46 | 56.10 | + | Trichomoniasis | African American | 5.5 | 2 | “Ca. M. girerdii” |
| VCU_NT31 | 51.50 | + | Genital warts | African American | 5 | ND | “Ca. M. girerdii” |
| VCU_NT27 | 49.70 | + | Yeast infection | African American | 5.5 | ND | “Ca. M. girerdii” |
| VCU_NT44† | 48.40 | + | Yeast infection | African American | 5.5 | ND | “Ca. M. girerdii” |
| VCU_NT22 | 46.10 | + | Bacterial vaginosis | African American | 5.2 | ND | “Ca. M. girerdii” |
| VCU_NT71† | 34.90 | + | None | African American | 5 | ND | “Ca. M. girerdii” |
| VCU_RQ481 | 34.77 | + | Trichomoniasis | African American | 5.5 | 2 | Lactobacillus iners |
| VCU_NT75 | 28.15 | + | Bacterial vaginosis | African American | ND | ND | NT |
| VCU_NT96 | 22.56 | + | None | African American | 4.5 | ND | Lactobacillus iners |
| VCU_QQ25 | 17.48 | + | Trichomoniasis | Other (Biracial) | 6.5 | 1 | NT |
| VCU_NT06 | 14.89 | + | Bacterial vaginosis | African American | 5 | ND | Atopobium vaginae |
| VCU_NT64 | 8.63 | + | None | African American | 4.4 | ND | Lactobacillus iners |
| VCU_NT99 | 8.03 | + | None | African American | ND | ND | Lactobacillus iners |
| VCU_VL26 | 7.90 | + | Trichomoniasis | African American | 5.8 | AMB | Prevotella |
| VCU_NT771 | 7.51 | + | Not Available | African American | 6.6 | ND | NT |
| VCU_IV47 | 7.20 | + | Trichomoniasis | African American | 5 | 2 | Gardnerella vaginalis |
| VCU_XN28 | 5.43 | + | Trichomoniasis | African American | 5 | AMB | NT |
| VCU_NT61 | 4.09 | + | None | African American | ND | ND | Lactobacillus iners |
| VCU_NT50 | 3.36 | + | Bacterial vaginosis | African American | 5 | ND | Gardnerella vaginalis |
| VCU_QR65* | 2.98 | + | Trichomoniasis | African American | ND | AMB | Mycoplasma hominis |
| VCU_NT29 | 2.59 | + | Yeast infection | African American | 4.5 | ND | Lactobacillus iners |
| VCU_NT04 | 2.21 | + | Bacterial vaginosis | African American | 5.8 | ND | Atopobium vaginae |
| VCU_NT24 | 2.08 | + | Yeast infection | Other (Biracial) | 5 | ND | Lactobacillus iners |
| VCU_NT55* | 1.90 | − | None | African American | ND | N/A | Lactobacillus crispatus |
| VCU_SY21 | 1.76 | + | Trichomoniasis | Hispanic | 5.5 | 2 | NT |
| VCU_GF833 | 1.64 | + | Trichomoniasis | African American | 5.8 | 1 | Prevotella |
| VCU_NT19 | 1.59 | + | Trichomoniasis | Not available | 6 | ND | Gardnerella vaginalis |
| VCU_LU24 | 1.50 | + | Trichomoniasis, Bacterial vaginosis | African American | 6.1 | 1 | NT |
| VCU_NT52 | 1.40 | − | Trichomoniasis | African American | 5.5 | ND | Gardnerella vaginalis |
| VCU_NT69 | 1.11 | + | None (abnormal discharge) | African American | 4.5 | ND | Lactobacillus iners |
| VCU_BN49 | 1.06 | + | Trichomoniasis, Bacterial vaginosis | African American | 5 | 2 | Gardnerella vaginalis |
| VCU_NT68 | 0.97 | + | None | African American | ND | ND | Lactobacillus iners |
| VCU_NT702 | 0.93 | + | None | African American | 5.8 | ND | Gardnerella vaginalis |
| VCU_NT93 | 0.92 | − | None | African American | 5.3 | N/A | Gardnerella vaginalis |
| VCU_NT21 | 0.86 | + | Yeast infection | African American | 5 | ND | Lactobacillus iners |
| VCU_NT60 | 0.85 | + | Trichomoniasis | African American | 4.4 | ND | BVAB1 |
| VCU_NT67 | 0.61 | + | Yeast infection | Caucasian | 4 | ND | Lactobacillus iners |
| VCU_NT17 | 0.59 | − | None | Caucasian | 4.4 | N/A | Lactobacillus crispatus |
| VCU_NT81 | 0.56 | − | None | African American | 5.5 | N/A | BVAB1 |
| VCU_NT28 | 0.54 | + | None | African American | 4 | ND | Lactobacillus iners |
| VCU_NT582 | 0.44 | + | None | African American | 4.5 | ND | Lactobacillus iners |
| VCU_NT09 | 0.40 | + | None | African American | 4.6 | ND | BVAB1 |
| VCU_QN84 | 0.39 | + | Trichomoniasis | African American | 6.1 | AMB | BVAB1 |
| VCU_NT59 | 0.34 | − | None | Hispanic | ND | N/A | Lactobacillus iners |
| VCU_NT97 | 0.29 | − | None | African American | ND | N/A | Lactobacillus crispatus |
| VCU_NT40 | 0.26 | + | Bacterial vaginosis | African American | 8 | ND | Sneathia amnii |
| VCU_NT733 | 0.20 | + | None | African American | 5.8 | ND | Lactobacillus iners |
| VCU_NT88 | 0.20 | − | None | African American | 4 | N/A | Lactobacillus iners |
| VCU_XP87 | 0.15 | + | Trichomoniasis, Bacterial vaginosis | African American | ND | 2 | BVAB1 |
| VCU_NT89 | 0.13 | − | Bacterial vaginosis | African American | 4.5 | N/A | Streptococcus agalactiae |
| VCU_NT07 | 0.13 | − | None | Hispanic | ND | N/A | Lactobacillus iners |
| VCU_NT26 | 0.12 | + | Bacterial vaginosis | Other (Multiracial) | 5 | ND | Gardnerella vaginalis |
| VCU_NT62 | 0.11 | ND | Bacterial vaginosis | African American | 5.3 | N/A | Gardnerella vaginalis |
| VCU_NT91 | 0.10 | − | None | Caucasian | 4 | N/A | Lactobacillus iners |
“Ca. M. girerdii” was detected in the mid-vaginal microbiome profile at 0.1% or more of total reads. (ND, not determined; AMB, ambiguous; N/A, not applicable; NT, no type).
* Recruited from Labor & Delivery Unit.
“Ca. M. girerdii” sequenced genomes VCU-M1, VCU-JB1, VCU-PA1 and VCU-G1 in listed order.
Three “Ca. M. girerdii” positive samples were from women who enrolled in the study more than once. VCU_GF83 was collected 424 days after VCU_NT73; VCU_NT58 was collected 246 days after VCU_NT70; VCU_NT77 was collected 117 days after VCU_RQ48.
We examined the association between vaginal carriage of the novel bacterium, even as a minor component of the microbiome, and common clinically diagnosed vaginal infections. The association between “Ca. M. girerdii” and trichomoniasis was highest of several vaginal organisms of the female urogenital tract with a relative risk of 20.12 (Table 2). M. hominis, which has previously been linked with trichomoniasis [9], [10], exhibits a much weaker association with a relative risk of 2.53, likely at least in part due to its strong association with BV. We did not find “Ca. M. girerdii” to be associated with an elevated relative risk for BV as diagnosed by Amsel’s criteria [19]. Amsel’s criterion assessment provides a dichotomous test with a relatively high specificity, but relatively low sensitivity [20]. BV assessed the Nugent’s Gram-stain criteria [21] represents the continuum of alterations in vaginal flora. Both pregnant [22] and non-pregnant [2] women with intermediate Nugent scores have been reported to be more likely to have trichomoniasis. While Nugent scores were not recorded in this study, the 16S rRNA microbiome profiles (Figure S1) are consistent with the hypothesis that women co-infected with “Ca. M. girerdii” and T. vaginalis may also be more likely to have intermediate flora.
Table 2. Associations of vaginal carriage of bacterial taxa with common vaginal infections.
| Relative Risk (95% Confidence Interval) | |||
| Trichomoniasis | Bacterial vaginosis | Yeast infection | |
| “Ca. M girerdii” | 20.12 (7.75–48.34) | 0.88 (0.24–1.53) | 0.86 (0.00–1.98) |
| M. hominis | 2.53 (0.85–6.83) | 2.08 (1.61–2.68) | 0.80 (0.44–1.30) |
| U. parvum/U. urealyticum | 1.36 (0.40–3.49) | 0.62 (0.45–0.84) | 1.18 (0.74–1.75) |
| Gardnerella vaginalis | 4.45 (0.91– Infinity*) | 7.17 (4.05–21.78) | 0.81 (0.54–1.32) |
| Atopobium vaginae | 1.79 (0.70–9.10) | 5.02 (3.50–8.43) | 0.56 (0.35–0.83) |
| BVAB2 | 0.66 (0.17–1.80) | 3.25 (2.54–4.30) | 0.45 (0.22–0.75) |
Bootstrap (n = 1,000) samples were selected from the outpatient clinic population to reflect the outpatient community composition. Median relative risk and 95% bootstrap confidence intervals are shown. A bacterial taxon was considered present in the mid-vaginal sample if at least 0.1% of the metagenomic 16S rRNA gene microbiome profile reads classified to the taxon. Vaginal infection was determined by clinical diagnosis using Amsel’s criteria for BV.
*For at least 2.5% of the bootstrap samples, all subjects with a trichomoniasis diagnosis were positive for G. vaginalis.
We detected “Ca. M. girerdii” at threshold of at least 0.1% of the 16S profile in 28 of the 63 (44.4%) women with clinically diagnosed trichomoniasis. We also found the new mollicute at less than 0.1% of the 16S rRNA threshold in eight additional women with trichomoniasis. Thus, we were unable to detect “Ca. M. girerdii” in the 16S rRNA gene profiles of only 27 of the 63 (42.9%) women with clinically defined trichomoniasis. In this study, trichomoniasis was clinically diagnosed by wet prep microscopy rather than culture and microbiome profiles were generated using the V1-V3 hypervariable region of the 16S rRNA gene rather than the V4-V6 region used by others [2]. Despite these methodological differences and differences in the study populations, we confirmed a strong association between the presence of T. vaginalis and “Ca. M. girerdii” previously reported as statistically significant (p = 0.026) by Martin et al. [2].
Association of “Ca. M. girerdii” with T. vaginalis
Up to half of all T. vaginalis infections are asymptomatic and undiagnosed [11]. We performed real-time qRT-PCR on all mid-vaginal samples positive for “Ca. M. girerdii” and found that 49 of the 51 (96%) women who carried the mycoplasma at a 1% threshold by 16S rRNA gene profiling also carry T. vaginalis (Figure 1A; Table 1). Even at a lower 16S rRNA threshold of 0.1%, 61 of 72 (85%) of women who carried “Ca. M. girerdii” were T. vaginalis positive. Thus, “Ca. M. girerdii” exhibits an unusually strong correlation with trichomoniasis. We also found that “Ca. M. girerdii” was associated with both of the previously described genotypes of T. vaginalis [23], [24], type 1 and type 2 (Figure S3), indicating a broad-range association with this infectious disease. Both T. vaginalis genotypes have been reported in the HIV-positive women [25]. Additional studies are needed to determine whether “Ca. M. girerdii” co-infection contributes to the increased risk of HIV acquisition and transmission or to adverse pregnancy outcomes associated with trichomoniasis.
Figure 1. Detection of “Ca. Mycoplasma girerdii” in mid-vaginal samples.

Panel (A) shows relative abundance of major taxonomic groups in “Ca. M. girerdii” positive samples (1% 16S rRNA threshold): “Ca. M. girerdii” is colored red (A). Light colored bars represent other taxa. Dark blue circles represent samples positive for T. vaginalis by qRT-PCR, light gray circles represent negative samples. Panels (B–E) show fluorescence in situ hybridization detection of bacteria in mid-vaginal samples from two participants with clinically diagnosed trichomoniasis (subject 1, panels B, C and D; subject 2, panel E) by confocal laser scanning microscopy. Most bacteria were detected with fluorescein-labeled broad-range bacteria probe Eub338 (turquoise). “Ca. M. girerdii” was also stained with a Cy5-labeled probe targeting 16S rDNA (red). Nuclei were labeled with 4′6′-diamidine-2-phenylindole, dehydrochloride (DAPI, blue). Negative control with reverse complementary probe of Eub338 did not hybridize to any bacteria (data not shown). Scale bar = 10 µm.
Interestingly, of 22 women with no diagnosis who were positive for “Ca. M. girerdii”, 14 were also positive for T. vaginalis (Table 1). Lactobacillus crispatus is associated with decreased rates of T. vaginalis infection [12], and we found that the three “Ca. M. girerdii” positive women with a predominance of L. crispatus were negative for T. vaginalis (Figures 2 and S1). Thus, although our data are supportive of a dependent relationship, it appears that “Ca. M. girerdii” may not absolutely require T. vaginalis to colonize the human vagina. Our data suggest vaginal carriage of the new mycoplasma is associated with elevated vaginal pH and African American race (Table 3), risk factors for preterm birth [26], which are also associated with BV [27] and trichomoniasis [28]. Given the tight association of the mycoplasma with T. vaginalis, it is not possible to determine whether the organism is independently associated with these factors.
Figure 2. Cluster analysis of mid-vaginal samples positive for “Ca. M. girerdii”.

Relative abundance of microbial taxa in mid-vaginal bacterial communities of “Ca. M. girerdii” positive women is shown. The dendrogram was generated using Ward’s method with Manhattan distance. This analysis includes only mid-vaginal samples that exhibited at least 0.1% “Ca. M. girerdii” by 16S rDNA profiling. Clinical diagnosis is indicated in the first bar, and presence of T. vaginalis by RT-PCR is indicated in the second bar (orange designates a negative result and pink designates a positive result). The three samples dominated by L. crispatus and the three samples with the highest prevalence of L. iners were negative for T. vaginalis.
Table 3. “Ca. M. girerdii” is associated with African American race and elevated vaginal pH.
| African American race | Caucasian race | Mid-vaginal pH | |
| “Ca. M. girerdii” | 88.0/74.8(0.003*) | 6.9/17.6(1) | 5.5/5.0(0.006*) |
| Mycoplasma hominis | 89.4/70.1(0*) | 6.2/21.2(1) | 5.5/5.0(0*) |
| Ureaplasma spp. | 75.1/75.3(0.555) | 17.0/17.2(0.571) | 5.0/5.0(0.514) |
| Gardnerella vaginalis | 81.5/59.2(0*) | 11.9/30.8(1) | 5.3/4.6(0*) |
| Atopobium vaginae | 85.6/62.2(0*) | 8.8/27.6(1) | 5.3/4.6(0*) |
| BVAB2 | 88.1/69.0(0*) | 6.5/22.3(1) | 5.5/5.0(0*) |
Mean proportions of the most common racial groups and median values for mid-vaginal pH were calculated for carriers and non-carriers (i.e., 0.1% 16S rRNA microbiome threshold) of the genital mycoplasmas and representative BV-associated species of women recruited from 1,361 women recruited from outpatient clinics. Bootstrap analysis was performed so that values represent the underlying population of the clinic.
*Bootstrap probabilities (n = 1,000) less than 0.05 indicate that the mean proportion or median value is significantly higher for carriers of the species.
Fluorescence in situ hybridization (FISH) on vaginal samples with a representation of “Ca. M. girerdii” (Figure 1B–E, Figure S2) showed that the bacterium is prominent in polymicrobial biofilms sometimes associated with “clue cells” (Figure 1B, 1C, 1E), a characteristic of BV. The mycoplasma was also dispersed with other bacteria and only occasionally co-localized with T. vaginalis (Figure 1D). It is not yet clear whether “Ca. M. girerdii” can enter and replicate inside of T. vaginalis like M. hominis [29], penetrate human cells like M. penetrans [30], or whether the mycoplasma is strictly extracellular. Given that eight women carrying the mycoplasma were negative for T. vaginalis (Table 1), our data suggest “Ca. M. girerdii” is not an obligate symbiont of the parasite as suggested by Martin et al. [2]. Symbiotically-associated M. hominis and T. vaginalis have recently been shown to synergistically upregulate the proinflammatory response [31]. Further studies are required to determine whether a symbiotic relationship between “Ca. M. girerdii” and T. vaginalis may similarly synergize to influence host response.
Genomic and Phylogenetic Analyses of “Ca. M. girerdii”
Our attempts to cultivate “Ca. M. girerdii” have not succeeded. Therefore, we employed the strategy of assembling whole metagenome shotgun sequence reads to complete the genome of a reference strain of this organism. The ∼619 kb genome is ∼28.6% GC content and features sequences for ∼572 putative proteins, 34 structural RNAs and one predicted CRISPR locus (Figure 3). Three additional strains from other samples were similarly assembled and aligned to the reference. Gene synteny was very high among the four strains, but limited with other related species (Figure S4). The four strains of “Ca. M. girerdii” exhibited an average of 99.8% nucleotide identity.
Figure 3. Representation of “Ca. M. girerdii” genomes.

A circular representation of the “Ca. M. girerdii” reference genome (strain VCU_M1) assembled from metagenomic sequences from a mid-vaginal sample. Position 1 is set to the start of the dnaA gene. Outermost circles (1–3) show the alignment (97% or greater identity) of contigs of three different strains from metagenomic assemblies from mid-vaginal samples containing high proportions of “Ca. M. girerdii”. Circle 4 (red) represents the reference strain (VCU_M1). Circles 5 (dark red) and 6 (blue) represent the predicted coding sequences in the forward and reverse orientations respectively. Circle 7 (black) shows the GC content, and circle 8 shows GC skew (pink (-), green (+)).
The reference genome exhibits irregular GC skews with no distinctive inversion (Figure 3), which is indicative of the high genome plasticity that is typical for mycoplasmas and consistent with the overall lack of synteny with related species. In the reference strain, one putative CRISPR locus containing four 34-nucleotide repeats and three 35-nucleotide spacers was identified in the genome with a consensus direct repeat sequence of 5′-AAGTATTAATATTCCAAGTAGTGTAACTAGTATT. Genes in the rRNA operon were organized 5′-16S-23S-5S, and no tRNAs were identified in the intergenic transcribed spacer regions as with most Mycoplasma and Ureaplasma species [32]. Like other mycoplasmas, “Ca. M. girerdii” possesses a minimal number of tRNAs and utilizes UGA as a tryptophan codon. We identified 31 putative tRNAs: 11 amino acids represented by a single anticodon; seven amino acids (Gly, Lys, Ser, Thr, Trp, Met) represented by two anticodons; and two amino acids (Leu, Arg) represented by three anticodons. Some, but not all, mycoplasmas have lost the tRNA-Trp gene that utilizes the TGG codon[33], but “Ca. M. girerdii” appears to have both. We identified two tRNA-Trp genes, one that utilizes the UGA codon with an observed codon frequency of ∼0.87% and another that utilizes the TGG codon with an observed codon frequency of ∼0.13%. The dnaA and dnaN genes are co-localized, but recF appears to be absent and gyrB is only distantly linked. As with M. penetrans and U. urealyticum [34], no clusters of DnaA boxes were identified upstream of the dnaA gene, as only one 9-mer with two base differences from the DnaA box consensus (5′-TTATCCACA) was identified in that region.
Homologs to putative virulence factors, including collagenases, a hemolysin, an O-sialoglycoprotein endopeptidase, and a feoB-like iron transport system, were identified in all four strains. Intriguingly, tensile strength of fetal membranes is imparted by collagens, and thus bacterial collagenase activity could facilitate fetal membrane rupture. “Ca. M. girerdii” appears to lack the superoxide dismutase gene, but encodes a complete desulfoferrodoxin-type superoxide reductase system that likely functions to protect against oxidative stress. A ∼16 kb plasmid is apparently present at approximately two copies per “Ca. M. girerdii” cell in the sample containing the reference strain, but was not observed in the samples containing the other strains. It carries a plasmid replication initiator protein, two genes resembling components of a type IV secretion system and ∼9 hypothetical genes. Because of its prevalence in the former sample and its similarity to plasmids from related organisms, it is possible that this may represent the first plasmid associated with a mycoplasma in this phylogenetic group (see below).
Phylogenetic analysis of 16S rRNA genes shows that “Ca. M. girerdii” is most closely related to other uncultivated organisms identified by 16S rRNA sequence: the organism reported by Martin et al. [2], an organism identified by Costello et al. in oral samples of a low birth weight neonate [16], and other organisms from bovine rumen [35], the gut of termites [36]–[38] and Asiatic elephant and Somali wild ass feces [39] (Figure 4). Interestingly, the environments of the gut of lower termites, the rumen of cattle and other foregut fermenters and the cecum of hindgut fermenters (e.g., Asiatic elephant and Somali wild ass) are all models of symbiosis where diverse groups of organisms including bacteria and protozoa contribute to carbohydrate fermentation and benefit the host by assisting with plant digestion.
Figure 4. Phylogenetic tree of 16SrRNA shows uncultured “Ca. M. girerdii” clusters most closely with other uncultivated organisms in the Pneumoniae Group.

The maximum likelihood tree was inferred by RAxML 7.2.7 using the gamma-distributed heterogeneity rate categories with 1,000 bootstraps. The 16S rRNA gene alignments were manually inspected. The Hominis Group is shaded in blue, the Pneumoniae Group in green, the Hemoplasma Group in gray and the Spiroplasma Group in purple. The 16S rRNA sequence of “Ca. M. girerdii” VCU_M1, “Ca. M. girerdii” VCU_PA1, “Ca. M. girerdii” VCU JB1 and “Ca. M. girerdii” VCU_G1 were identical. “Ca. M. girerdii” groups most closely with “Mnola”, which shows 100% identity in 16S rRNA sequence, uncultivated organisms from the oral sample of a low birth weight infant (HG764209, HG764210, and HG764212) and uncultivated species from rumen and termite gut in the Pneumoniae Group. A partial 16S rRNA sequence from the vaginal sample of a woman who delivered full term (JX871253) also exhibits 99% identity with “Ca. M. girerdii”, but was not included in the analysis due to its length.
A phylogenetic analysis using 57 inferred orthologous proteins placed “Ca. M. girerdii” in the Pneumoniae group with M. penetrans and Mycoplasma iowae, relatively distant from M. hominis (Figure 5). This phylogeny is supported by analysis. Analysis of COGs distributed 415 of the “Ca. M. girerdii” genes among functional categories in a pattern similar to that exhibited by other mycoplasma species (Table S1). The full-length “Ca. M. girerdii” 16S rRNA gene from all four genomes and “Mnola” exhibited 100% identity.
Figure 5. Phylogenetic Tree based on inferred amino acid sequences confirms placement of “Ca. M. girerdii” in the Pneumoniae group.

“Ca. M. girerdii” is located within the Pneumoniae group, denoted in green, in a subclade along with the Ureaplasma species, M. iowae and M. penetrans. The tree was inferred using amino acid sequences of 57 orthologs (Tables S4, S5, and S6). Numbers at nodes correspond to the support values from 1,000 bootstrap replicates.
Metabolic strategies of “Ca. M. girerdii”
Genital mycoplasmas have evolved to utilize various energy sources; M. genitalium, M. hominis, and ureaplasmas use glucose, arginine and urea respectively. Our metabolic reconstructions suggest that “Ca. M. girerdii” is glycolytic like M. genitalium and encodes all enzymes for utilization of glucose as an energy source (Figure 6). Arginine dihydrolase pathway and urease genes are absent, thus “Ca. M. girerdii” is not predicted to utilize arginine and urea. Catabolism of galactose, mannose, sucrose, maltose, glycogen, starch or glycerol is not predicted, and the roles of genes in the lactose/galatose pathways are unclear. “Ca. M. girerdii” possesses genes for a putative IIA component of the lactose-specific phosphotransferase system (MGM1_4770), ribose/galactose-ABC-type transporter system (MGM1_3070, MGM13080) and a galactose-6-phosphate isomerase (MGM1_4760/MGM1_4750). However, other genes required for lactose and galactose catabolism, including 6-phospho-beta-galactosidase, tagatose-6-phosphate kinase and tagatose-bisphosphate aldolase, were not identified. Moreover, the L-lactate dehydrogenase gene (MGM1_4130) has an apparent frameshift. Other components of the phosphotransferase system (PTS) system were also identified, including the HPr phosphocarrier protein (MGM1_1420) and an HPr phosophatase/kinase (MGM1_4210), which likely functions in the regulation of carbon metabolism. While all eight subunits for the F1F0 ATPase complex were identified (MGM1_4310 through MGM1_4380), these genes are thought to be involved in maintenance of the proton gradient rather than ATP generation in mycoplasma species as the cytochrome components are absent.
Figure 6. Unique strategies of “Ca. M. girerdii”.

Putative transporters, enzymes involved in carbohydrate metabolism and virulence factors are represented in red for “Ca. M. girerdii”. Comparisons with other genital mycoplasmas are indicated with color-coded boxes: M. genitalium (MG, blue) U. parvum (UP, green) and M. hominis (MH, purple). Arrows indicate direction of transport. Light gray arrows represent metabolic strategies unique to other genital mycoplasma. Metabolic reconstruction was performed using ASGARD and careful inspection of manual annotations.
Unique among the genital mycoplasmas, the “Ca. M. girerdii” genome encodes serine dehydratase (MGM1_2560, MGM1_0390), alanine dehydrogenase (MGM1_5480, MGM1_1820), and 2′,3′-cyclic-nucleotide 2′-phosphodiesterase (MGM1_1930) that may permit use of alternate energy sources in the absence of glucose: L-alanine, L-serine, and 2′,3′ cyclic AMP. No serine dehydratases have been previously described for the mollicutes, and while alanine dehydrogenase has been described for Acholeplasma laidlawii [40] and annotated for M. mycoides (ADH22225.1), M. mobile (AAT27586.1), M. leachii (ADR24467.1), and M. putrefaciens (YP_004790384.1), neither the gene nor the enzyme has been previously identified in any of the genital mycoplasmas or organisms classified in the Pneumoniae group. Moreover, 2′,3′-Cyclic phosphodiesters may be available in the environment as intermediate products in the hydrolysis of RNA by ribonuclease I. This strategy has been proposed for Yersinia enterocolitica [41], which has been shown to grow on 2′,3′-cAMP as a sole carbon source. “Ca. M. girerdii” does not metabolize pyruvate through the pyruvate dehydrogenase pathway that is used by M. genitalium or the other mycoplasma species that catabolize pyruvate to acetate. However, “Ca. M. girerdii” may utilize one or both of two alternate enzymes identified in the genome: pyruvate-formate lyase (MGM1_5430), which produces acetyl-CoA and formate from CoA and pyruvate, and/or pyruvate ferredoxin/flavodoxin oxidoreductase (MGM1_5310), which yields acetyl-CoA and carbon dioxide from the same substrates by reducing either ferredoxin or flavodoxin. Both of these enzymes seem to be unique to “Ca. M. girerdii” among the mycoplasmas. Acetyl-CoA may be converted to acetate by phosphate acetyltransferase (MGM1_0120) and acetate kinase (MGM1_2290), resulting in the production of ATP.
As with other mollicutes, “Ca. Mycoplasma girerdii” appears to have limited metabolic capabilities and imports much of what it needs from its environment or host. “Ca. M. girerdii” seems to lack gluconeogenesis and the TCA cycle like other mycoplasmas. It lacks enzymes for de novo purine or pyrimidine synthesis and amino acid synthesis, but appears to be capable of nucleotide salvage and amino acid transport. The genome encodes ∼40 genes associated with transport of various ions and substrates including amino acids, glucose, ribose/lactose, potassium ion, magnesium ion, calcium ion, ferrous iron, cobalt, phosphate and spermidine/putrescine (Table S2). An alcohol dehydrogenase (MGM1_5890) exhibiting homology to a butanol dehydrogenase and putative bifunctional aldehyde-alcohol dehydrogenase (MGM1_1150) were also annotated in the genome, thus ethanol may also be an end product of metabolism. Although “Ca. M. girerdii” is predicted to be able to convert butanol to butanoyl-CoA, it appears to lack other enzymes of butanoate metabolism. The predicted metabolic reconstruction may provide insight and help guide future cultivation attempts.
BspA-like proteins encoded in “Ca. M. girerdii”
Mycoplasma species contain surface proteins that exhibit high frequency antigenic variation [42]. Although these organisms exhibit a low level of horizontal gene transfer, expanded families of surface proteins are an exception [43]–[45]. In the reference genome of “Ca. M. girerdii”, we identified a family of 26 BspA-like proteins containing Treponema pallidum leucine rich repeat (TpLRR) domains with homology to the prototypical BspA virulence factor of Tannerella forsythia and a family of over 900 BspA-like proteins of T. vaginalis [46], [47]. BspA-like proteins from “Ca. M. girerdii” exhibit variable length ranging in length from 136 to 1481 amino acids (Figure 7). Twenty-three of the BspA-like proteins contained a predicted C-terminal transmembrane domain, and a signal peptide was detected for five of the BspA-like proteins.
Figure 7. Expanded “Ca. M. girerdii” BspA-like protein family.
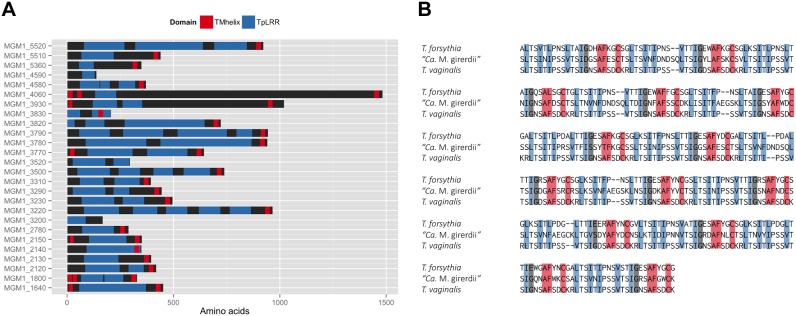
The diverse family of 26 BspA-like protein family members from the “Ca. M. girerdii” strain VCU_M1 is depicted in panel (A). Predicted transmembrane domains and TpLRR domains are represented. An alignment of TpLRR domains from Tannerella forsythia BspA (AAC82625.1, bases 382–1347), “Ca. M. girerdii” strain VCU_M1 MGM1_3780 (bases 449–782) and T.vaginalis BspA-like TVAG_495790 (XP_001327783.1, bases 112–1077) is shown in panel (B).
Members of this family stimulate a Toll-like receptor 2 (TLR2)-mediated host immune response. We also identified two other putative surface lipoproteins that lack the TpLRR domain, but exhibit homology to other mycoplasma proteins that stimulate a TLR-mediated innate immune response. It is intriguing to hypothesize that the expanded families of BspA-like proteins in the “Ca. M. girerdii” and T. vaginalis may represent a common host-adaptation strategy.
The BspA from T. forsythia is perhaps the best studied protein containing the TpLRR domain. This protein has been shown to mediate a host innate immune response through Toll-like receptor 2 (TLR2 [48]) by directly interacting with the receptor [49]. More recently, the protein has been shown to elicit a response through scavenger receptor gp340 [50]. T. forsythia BspA has also been shown to be required for host cell attachment and invasion [51] and co-aggregation with Fusobacterium nucleatum [51]. Thus, the “Ca. M. girerdii” BspA-like proteins may also mediate interactions with the host and contribute to virulence through induced host inflammation.
No TpLRR-containing proteins have been identified in U. parvum, U. urealyticum or M. genitalium and only one or two predicted BspA-like proteins were identified in the other related genomes examined: one in M. iowae (ZP_08916569.1), one in M. fermentans (YP_003922737.1 and YP_003922552.1), two in M. hominis (YP_003302763.1; YP_003303010.1) and one in M. penetrans (NP_757414.1). Although the role of the 26 member BspA-like gene family in “Ca. M. girerdii” is currently unclear, it is likely that they have important functions in host interactions. It is intriguing to hypothesize that the TpLRR domains of the BspA-like proteins from M. hominis, T. vaginalis, and “Ca. M. girerdii”, which appear to be absent in the Ureaplasma species and M. genitalium genomes, may interact with the human host cells in a similar manner via the TLR2 receptor.
Conclusions
In summary, we confirmed the identity of a recently described and still uncultivated species of mycoplasma, further documented its strong association with the presence of T. vaginalis, and comprehensively characterized the genomes of “Ca. M. girerdii” from four vaginal samples collected in the Vaginal Human Microbiome Project at VCU. The genomes of this potentially emerging pathogen provide insight into its metabolic strategies and reveal a potential for virulence and for triggering host inflammatory responses through innate immune mechanisms. This work lays the foundation for understanding the impact of “Ca. M. girerdii” on women’s urogenital health and the nature of its association with T. vaginalis.
Description of “Candidatus Mycoplasma girerdii”
“Candidatus Mycoplasma girerdii” [gir.erdii. N.L. gen. n. girerdii, named for P.H. Girerd, an American obstetrician and gynecologist, for his dedication to clinical practice and his contributions to the research of the vaginal microbiota].
Materials and Methods
Identification of “Ca. M. girerdii”
Mid-vaginal swab samples collected from women at 1,471 visits (1,361 outpatient visits, 110 visits to the labor and delivery unit) were assayed by 16S rRNA gene pyrosequencing according to the protocols of the Vaginal Human Microbiome Project at VCU [18], [52]. The team that performed the PCR was blinded to the BV and T. vaginalis diagnoses and results. Mid-vaginal pH, clinical diagnosis and health history were recorded. The teams that recorded and digitally entered this information were blinded to the PCR and RT-PCR results. Clinical diagnosis of trichomoniasis was based on identification of motile trichomonads in a saline wet mount preparation of vaginal discharge. BV was clinically diagnosed in women meeting at least three of Amsel’s four criteria [19]: characteristic BV discharge, clue cells on microscopy, vaginal pH >4.5 and positive whiff test. Consent was obtained from all participants in accordance with the study protocol (HM12169) as approved by the institutional review boards for human subjects protection at Virginia Commonwealth University and the Virginia Department of Health Raw sequence data from the project is available from the Short Read Archive at NCBI (projectID phs000256).
Fluorescence in situ hybridization
Vaginal swabs were resuspended in phosphate buffered saline (PBS) and incubated on poly L-lysine coverslips (BD Biosciences, San Jose, CA) in a 24-well tissue culture plate for 30 min at 37°C. Coverslips were washed with PBS, fixed in 4% paraformaldehyde/PBS for 1 h, and permeabilized with 0.2% TritonX-100/PBS for 10 min. Fifty nanograms of fluorescently labeled probe targeting bacterial 16S rRNA was added to 200 µl of pre-warmed hybridization buffer (0.9 M NaCl, 20 mM Tris-HCl (pH 7.2), 10% formamide, 0.01% SDS). Hybridization was carried out at 45°C for at least 2 h. Fluorescein-labeled broad range bacteria probe Eub338 (5′GCTGCCTCCCGTAGGAGT-3′) was used as a control. A Cy5-labeled specific probe (5′-TCCTCTTAGTGCCGTTCGTCC-3′) was used to detect “Ca. M. girerdii”. Following hybridization, coverslips were rinsed with pre-warmed hybridization wash buffer (0.45 M NaCl, 20 mM Tris-HCl (pH 7.2), 0.01% SDS) then incubated in the wash buffer for 15 min at 48°C. Immediately following, coverslips were washed with ice-cold distilled H2O, dried for 15 min at 48°C and mounted with ProLong Gold Antifade containing 4′,6-Diamidino-2-phenylindole (DAPI) (Invitrogen, Carlsbad, CA). Slides were visualized by laser scanning confocal microscopy using a Zeiss LSM 700.
Detection and genotyping of T. vaginalis
Detection of T. vaginalis by quantitative real-time RT-PCR was performed as described by Shirm et al. [53], and T. vaginalis genotyping was performed using three single-copy genes as described by Conrad et al. [23], [24].
Relative Risk Analysis
Bootstrap (n = 1,000) samples were selected from the outpatient clinic population to reflect the outpatient community composition. Samples from women enrolled in labor and delivery were not included in this analysis. Median relative risk and 95% bootstrap confidence intervals were calculated. A bacterial taxon was considered present in the mid-vaginal sample if at least 0.1% of the metagenomic 16S rRNA gene microbiome profile reads classified to the taxon.
Metagenomic assembly of “Ca. M. girerdii”
We selected one mid-vaginal sample (VCU_NT41; Table 1) with >90% of 16S rRNA reads classified to “Ca. Mycoplasma girerdii” from which to assemble the reference genome of the organism. The woman who provided this sample was in active preterm labor and also tested positive for group B Streptococcus (GBS), Chlamydia trachomatis and T. vaginalis.
Fifty nanograms of total DNA was used in a tagmentation reaction with a Nextera DNA Sample Prep Kit (Roche Titanium-compatible, Epicentre Biotechnologies) following the manufacturer’s protocol and sequenced in the Nucleic Acids Research Facilities at VCU. Titanium FLX pyrosequencing (Roche/454; 1/2 plate) yielded 793,732 reads and 241,486,162 bases. The raw data was pre-filtered to remove most human reads (55% of the total reads) using Bowtie 2 [54] with default parameters. The reads were then split into two bins using AbundanceBin [55]: (1) a bin containing abundant reads including those derived from “Ca. M. girerdii”, and (2) a bin containing less abundant reads derived from the minor components of the vaginal microbiome. Human-filtered reads from the bin of abundant sequences (i.e., 252,073 reads, 79,029,547 bases) were assembled using Newbler, resulting in 298 contigs larger than 500 bases and 1,966 contigs larger than 100 bases, with a total of 89.10% of reads aligned to a contig. Through a careful analysis of the single-end read flow information from Newbler, we inferred a circular scaffold for the organism that incorporated 19% (152,023 total reads) of the total unfiltered reads from the metagenomic sample. The scaffold incorporated eight of the largest ten contigs that ranged in size from 144,547 bases to 4,826 bases and exhibited 67.2-fold to 95-fold coverage. Sequence reads incorporated into the “Ca. M. girerdii” genome did not map to known Mycoplasma or Ureaplasma sequences, and no other unnamed mollicutes were detected in these samples by 16S rRNA analysis. The eighth-largest contig encoded a 16kb plasmid with an observed depth of 189-fold coverage. Because of its abundance, it is likely that this plasmid is from “Ca. M. girerdii” but the host of the plasmid cannot yet been unequivocally assigned. The ninth-largest contig aligned to T. vaginalis ribosomal RNA genes. An additional eight smaller contigs ranging in size from 161 bases to 814 bases were also incorporated into the scaffold, with two of the contigs incorporated twice. The majority of non-assembled contigs aligned to T. vaginalis (232 contigs), Homo sapiens (21 contigs), or other bacterial species; e.g., Gardnerella vaginalis (10 contigs). Physical gaps in the contig junctions were confirmed and closed by PCR across gaps and fluorescent chain termination sequence analysis on the AB3730 or AB3130 capillary sequencers (Applied Biosystems). These gaps commonly occurred either in genes exhibiting homology to type I restriction modification system proteins or those encoding BspA-like proteins, although one junction spanned a gene encoding a signal recognition particle protein, which was present in only one copy. PCR and sequencing primers are provided (Table S3). All physical gaps in the scaffold were closed by PCR-amplification and sequencing using the Sanger capillary methodology. The circularity of the 16kb plasmid was similarly confirmed.
Three additional mid-vaginal samples (VCU_CT62, VCU_NT44, VCU_NT71; Table 1) each containing more than 30% “Ca. M. girerdii” by metagenomic 16S rRNA gene microbiome analysis were also sequenced by whole metagenome shotgun sequencing using Titanium FLX pyrosequencing and the protocol described above. Each sample was run on approximately one eighth of a plate, yielded between 185,612 and 222,667 total reads and assembled using Newbler, and aligned to the reference strain. The genomes of “Ca. M. girerdii” have been deposited with NCBI under the Bioproject accession numbers PRJNA196996, PRJNA196997, PRJNA196998, and PRJNA196999. The complete genome of “Ca. M. girerdii” has been deposited at NCBI under accession number CP007711.
Genome annotation and metabolic reconstruction
Open reading frames (ORFs) greater than 100 nucleotides were predicted by Glimmer3 [56] and GeneMarkS [57] using translation table 4 and were manually examined. In most cases the start site predicted by Glimmer3 was chosen for genes that had the same predicted stop codon called by both Glimmer3 and GeneMarkS. Translated ORF predictions were searched against the non-redundant (nr) database from NCBI and a custom database of Mollicute proteins downloaded from NCBI using the blastp algorithm, and the gene products were manually annotated. Predicted gene products were compared to conserved domain databases (COGs and Pfam) by RPS-BLAST. Other annotation features were predicted using TMHMM 2.0c [58] for transmembrane domains and SignalP 4.0 for signal peptides. Although mollicutes have a unique membrane composition, SignalP has been previously validated on experimentally verified secreted proteins from mollicutes [59]. The hmmsearch program from HMMER3.0 [60] was used to search predicted proteins for the Treponema palladium family of leucine rich repeats (TpLRR) using the Pfam raw hidden markov model for the family (LRR_5; PF13306). Transfer RNA genes were predicted by tRNAscan-SE v 1.3 using the genetic code outlined in translation table 4. The tRNA-Ile, elongator tRNA-Met and initiator tRNA-fMet were distinguished by alignment with previously annotated tRNAs [61]. The CRISPR element containing four 34-nucleotide repeats and three 35-nucleotide spacers was identified in the genome with a consensus direct repeat sequence of 5′-AAGTATTAATATTCCAAGTAGTGTAACTAGTATT using the CRISPR recognition tool (CRT) [62]. Metabolic reconstruction and Gene Ontology classification assignments were performed using ASGARD [63] and the UniRef100 database.
Phylogenetic analysis
One reference genome was selected for each species in the Mollicutes class for which a completely sequenced genome is available. A total of 57 transitively closed orthologous clusters were retrieved from the RoundUp [64] database (release date: Dec. 23, 2011). “Ca. M. girerdii” orthologs were identified using blastp and confirmed using the Reciprocal Smallest Distance (RSD) algorithm. Orthologs were similarly identified for M. iowae for which only a draft genome is available. The maximum-likelihood tree was inferred by RAxML 7.2.74 [65] using the gamma-distributed heterogeneity rate categories with 1,000 bootstraps. The tree was rooted using Lactobacillus gasseri as the outgroup. Phylogenetic trees based on 16S rDNA gene sequences were similarly constructed. The 16S rRNA gene alignments were manually inspected and the maximum likelihood tree was inferred by RAxML 7.2.74 using the gamma-distributed heterogeneity rate categories with 1,000 bootstraps.
Attempts to cultivate “Ca. M. girerdii”
Frozen vaginal swab samples were incubated on A8 and SP4 agar (Hardy Diagnostics). When these plates did not yield colonies, the frozen samples were cultured on PPLO broth base containing 10% horse or 10% human serum, 10% yeast extract, 1% arginine and 1.5% Bacto agar [66]. The samples were also incubated on supplemented BHI agar [67] containing 10% human blood. All solid media was supplemented with 100 µg ampicillin/mL and plates were incubated at 37°C under anaerobic conditions and in air supplemented with 5% CO2. None of these efforts yielded detectable growth.
Ethics Statement
Consent was obtained from all participants in accordance the study protocol as approved by the institutional review boards for human subjects protection at Virginia Commonwealth University and the Virginia Department of Health. All enrolled subjects were 18 years of age or older and provided written informed consent.
Supporting Information
Cluster analysis of mid-vaginal samples with a clinical diagnosis of trichomoniasis. Relative abundance of microbial taxa in mid-vaginal bacterial communities of 63 women with clinically diagnosed trichomoniasis is shown. The dendrogram was generated using Ward’s method with Manhattan distance. Presence of “Ca. M. girerdii” as determined by 16S rDNA profiling is indicated in the top bar. Samples that contained “Ca. M. girerdii” at less than 0.1% abundance are indicated as ambiguous (AMB) in light blue.
(PDF)
Detection of “ Ca . M. girerdii” in mid-vaginal samples. Fluorescence in situ hybridization detection of bacteria in mid-vaginal samples from two participants with clinically diagnosed trichomoniasis (subject 1, panels A–O); subject 2, panels P–T) by confocal laser scanning microscopy. The merged photomicrographs are also depicted in Figure 1. Nuclear DNA was detected with 4′6′-diamidine-2-phenylindole, dehydrochloride (DAPI, blue) as shown in panel B, G, L and Q. Most bacteria were detected with fluorescein-labeled broad-range bacteria probe Eub338 (green) as shown in panels C, H, M and R. “Ca. M. girerdii” was also stained with a Cy5-labeled probe targeting 16S rDNA (red) as shown in panels D, I, N and S.
(PDF)
“Ca. M. girerdii ” coexists with both genotypes of T. vaginalis. The maximum likelihood tree was constructed using concatentated, aligned partial protein sequences from three single-copy orthologs (CRN, PMS1, Mlh1a). Isolates indicated as type 1 or type 2 were previously typed using microsatellite markers. Strains from 43 clinically diagnosed cases of trichomoniasis from this study are indicated with the prefix “VCU”. The type 1 cluster is shaded blue and contains eight “Ca. M. girerdii” positive cases, the type 2 cluster is shaded green and contains ten “Ca. M. girerdii” positive cases and the ambiguous cluster is shaded gray and contains five “Ca. M. girerdii” positive cases. In this analysis, the ambiguous cluster groups with type 2 T. vaginalis, but the subgroup contains isolates that were differentially classified as type 1 using microsatellite markers. T. vaginalis strains from “Ca. M. girerdii” positive cases as determined by 16S rRNA microbiome profiling (0.1% threshold) are indicated with red boxes. Ambiguous cases that were detected at less than 0.1% threshold are denoted with pink boxes. Blue dots denote branches with bootstrap values greater than 50.
(PDF)
Conserved synteny among “Ca. M. girerdii” strains not shared with related species. Panel (A) shows dot plot nucleotide-based alignments of the reference “Ca. M. girerdii” strain VCU_M1 with contigs from three different “Ca. M. girerdii” strains (VCU_PA1, VCU_JB1, VCU_G1). Panel (B) shows dot plot amino-acid based alignments of the reference with three closely related species (M. iowae, M. penetrans and U. parvum). Horizontal grid lines delineate contigs. Nucleotide-based and protein-based alignments were performed using Nucmer and Promer respectively.
(PDF)
Distribution of Clusters of Orthologous Groups (COGS).
(DOCX)
Putative transporters in “ Ca . M. girerdii” strain VCU_M1.
(DOCX)
List of primers for circularization of “ Ca . M. girerdii” genome.
(DOCX)
Hominis Group orthologs.
(DOCX)
Pneumoniae and Hemoplasma Group orthologs.
(DOCX)
Spiroplasma, Phytoplasma and Outgroup orthologs.
(DOCX)
Acknowledgments
We acknowledge Frances White and Scott Henderson for guidance with microscopy, and Jennifer I. Drake for comments on the manuscript. We thank Robert P. Hirt and Pier Luigi Fiori for advice on protocols for detection of T. vaginalis. Sequencing was performed in the Nucleic Acids Research Facilities, and bioinformatics analysis was supported by the Bioinformatics Computational Core Laboratories at VCU. Analysis was performed on the servers in the Center for High Performance Computing at VCU.
Vaginal Microbiome Consortium at VCU
All authors are members of the Vaginal Microbiome Consortium (VMC) at VCU (vmc.vcu.edu). Additional members of the Vaginal Microbiome Consortium who have contributed to this study are listed in alphabetical order: Steven P. Bradley1, Robert A. Duckworth III1, David J. Edwards2, Michael D. Harwich Jr.1, Vishal N. Koparde3, Melissa A. Prestosa1, Federico A. Puma1, Mark A. Reimers3,4, and Aaron R. Wolen4. Correspondence to the VMC should be addressed to Gregory A. Buck (gabuck@vcu.edu).
1Department of Microbiology and Immunology, Virginia Commonwealth University, Richmond, VA, USA.
2Department of Statistical Sciences and Operations Research, Virginia Commonwealth University, Richmond, VA, USA.
3Center for Study of Biological Complexity, Virginia Commonwealth University, Richmond, VA USA.
4Virginia Institute for Psychiatric and Behavioral Genetics, Virginia Commonwealth University, Richmond, VA, USA.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. The complete genome has been deposited at NCBI under accession number CP007711.
Funding Statement
This work was supported by several grants from the National Institutes of Health: Grant NHGRI/NIAID 4UH3AI083263, The Vaginal Microbiome: Disease, Genetics and the Environment, Grant NHGRI/NIDCR/NICHD/NCCAM/ORWH 1U54DE023786-01/8U54HD080784-02 and Grant NIH P60MD002256, VCU NIMHD Comprehensive Center of Excellence. Sequencing was performed in the Next Generation Sequencing Core of the Nucleic Acids Research Facilities at VCU, supported in part by funding from NIH/NCI 2P30 CA16059. Sequence analysis was performed with infrastructure and support from the Bioinformatics Computational Core Laboratories and the Center for High Performance Computing at VCU. Microscopy was performed at the VCU Department of Neurobiology and Anatomy Microscopy Facility, supported, in part, with funding from NIH-NINDS Center core grant (5P30NS047463). ARW was supported by NIH Grant MH-20030. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. The Human Microbiome Project Consortium (2012) Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin DH, Zozaya M, Lillis RA, Myers L, Nsuami MJ, et al.. (2013) Unique Vaginal Microbiota That Includes an Unknown Mycoplasma-Like Organism Is Associated With Trichomonas vaginalis Infection. J Infect Dis. doi:10.1093/infdis/jit100. [DOI] [PMC free article] [PubMed]
- 3. Taylor-Robinson D, Lamont RF (2011) Mycoplasmas in pregnancy. BJOG Int J Obstet Gynaecol 118: 164–174 10.1111/j.14710528.2010.02766.x [DOI] [PubMed] [Google Scholar]
- 4.Larsen B, Hwang J (2010) Mycoplasma, Ureaplasma, and adverse pregnancy outcomes: a fresh look. Infect Dis Obstet Gynecol 2010. Available: http://www.ncbi.nlm.nih.gov/pubmed/20706675. Accessed 18 April 2012. [DOI] [PMC free article] [PubMed]
- 5. Kacerovsky M, Pliskova L, Bolehovska R, Musilova I, Hornychova H, et al. (2011) The microbial load with genital mycoplasmas correlates with the degree of histologic chorioamnionitis in preterm PROM. Am J Obstet Gynecol 205: 213.e1–e7 10.1016/j.ajog.2011.04.028 [DOI] [PubMed] [Google Scholar]
- 6. DiGiulio DB, Romero R, Kusanovic JP, Gómez R, Kim CJ, et al. (2010) Prevalence and diversity of microbes in the amniotic fluid, the fetal inflammatory response, and pregnancy outcome in women with preterm pre-labor rupture of membranes. Am J Reprod Immunol N Y N 1989 64: 38–57 10.1111/j.16000897.2010.00830.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Djigma F, Ouedraogo C, Sagna T, Ouermi D, Sanogo K, et al. (2011) HIV-infected women of Burkina Faso: a “reservoir” of mycoplasma infection. J Infect Dev Ctries 5: 176–181. [DOI] [PubMed] [Google Scholar]
- 8. Haggerty CL, Taylor BD (2011) Mycoplasma genitalium: an emerging cause of pelvic inflammatory disease. Infect Dis Obstet Gynecol 2011: 959816 10.1155/2011/959816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diaz N, Dessì D, Dessole S, Fiori PL, Rappelli P (2010) Rapid detection of coinfections by Trichomonas vaginalis, Mycoplasma hominis, and Ureaplasma urealyticum by a new multiplex polymerase chain reaction. Diagn Microbiol Infect Dis 67: 30–36 10.1016/j.diagmicrobio.2009.12.022 [DOI] [PubMed] [Google Scholar]
- 10. Van der Schee C, Sluiters HJ, van der Meijden WI, van Beek P, Peerbooms P, et al. (2001) Host and pathogen interaction during vaginal infection by Trichomonas vaginalis and Mycoplasma hominis or Ureaplasma urealyticum. J Microbiol Methods 45: 61–67. [DOI] [PubMed] [Google Scholar]
- 11.World Health Organization (2001) Global programme of AIDS. Geneva, Switzerland: World Health Organization, Geneva, Switzerland.
- 12. Brotman RM, Bradford LL, Conrad M, Gajer P, Ault K, et al. (2012) Association between Trichomonas vaginalis and vaginal bacterial community composition among reproductive-age women. Sex Transm Dis 39: 807–812 10.1097/OLQ.0b013e3182631c79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hay P, Czeizel AE (2007) Asymptomatic trichomonas and candida colonization and pregnancy outcome. Best Pract Res Clin Obstet Gynaecol 21: 403–409 10.1016/j.bpobgyn.2007.02.002 [DOI] [PubMed] [Google Scholar]
- 14. Fichorova RN (2009) Impact of T. vaginalis infection on innate immune responses and reproductive outcome. J Reprod Immunol 83: 185–189 10.1016/j.jri.2009.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krieger JN, Poisson MA, Rein MF (1983) Beta-hemolytic activity of Trichomonas vaginalis correlates with virulence. Infect Immun 41: 1291–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Costello EK, Carlisle EM, Bik EM, Morowitz MJ, Relman DA (2013) Microbiome assembly across multiple body sites in low-birthweight infants. mBio 4: e00782–00713 10.1128/mBio.0078213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hyman RW, Fukushima M, Jiang H, Fung E, Rand L, et al. (2014) Diversity of the vaginal microbiome correlates with preterm birth. Reprod Sci Thousand Oaks Calif 21: 32–40 10.1177/1933719113488838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fettweis JM, Serrano MG, Girerd PH, Jefferson KK, Buck GA (2012) A new era of the vaginal microbiome: advances using next-generation sequencing. Chem Biodivers 9: 965–976 10.1002/cbdv.201100359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amsel R, Totten PA, Spiegel CA, Chen KC, Eschenbach D, et al. (1983) Nonspecific vaginitis. Diagnostic criteria and microbial and epidemiologic associations. Am J Med 74: 14–22. [DOI] [PubMed] [Google Scholar]
- 20. Sha BE, Chen HY, Wang QJ, Zariffard MR, Cohen MH, et al. (2005) Utility of Amsel criteria, Nugent score, and quantitative PCR for Gardnerella vaginalis, Mycoplasma hominis, and Lactobacillus spp. for diagnosis of bacterial vaginosis in human immunodeficiency virus-infected women. J Clin Microbiol 43: 4607–4612 10.1128/JCM.43.9.46074612.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nugent RP, Krohn MA, Hillier SL (1991) Reliability of diagnosing bacterial vaginosis is improved by a standardized method of gram stain interpretation. J Clin Microbiol 29: 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hillier SL, Krohn MA, Nugent RP, Gibbs RS (1992) Characteristics of three vaginal flora patterns assessed by gram stain among pregnant women. Vaginal Infections and Prematurity Study Group. Am J Obstet Gynecol 166: 938–944. [DOI] [PubMed] [Google Scholar]
- 23. Conrad M, Zubacova Z, Dunn LA, Upcroft J, Sullivan SA, et al. (2011) Microsatellite polymorphism in the sexually transmitted human pathogen Trichomonas vaginalis indicates a genetically diverse parasite. Mol Biochem Parasitol 175: 30–38 10.1016/j.molbiopara.2010.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Conrad MD, Gorman AW, Schillinger JA, Fiori PL, Arroyo R, et al. (2012) Extensive genetic diversity, unique population structure and evidence of genetic exchange in the sexually transmitted parasite Trichomonas vaginalis. PLoS Negl Trop Dis 6: e1573 10.1371/journal.pntd.0001573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Conrad MD, Kissinger P, Schmidt N, Martin DH, Carlton JM (2013) Genetic diversity of Trichomonas vaginalis reinfection in HIV-positive women. Sex Transm Infect 89: 473–478 10.1136/sextrans-2013051053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. York TP, Strauss JF, Neale MC, Eaves LJ (2010) Racial Differences in Genetic and Environmental Risk to Preterm Birth. PLoS ONE 5: e12391 10.1371/journal.pone.0012391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fettweis JM, Brooks JP, Serrano MG, Sheth NU, Girerd PH, et al.. (2014) Differences in vaginal microbiome in African American women versus women of European ancestry. Microbiol Read Engl. doi:10.1099/mic.0.081034–0. [DOI] [PMC free article] [PubMed]
- 28. Cotch MF, Pastorek JG 2nd, Nugent RP, Hillier SL, Gibbs RS, et al. (1997) Trichomonas vaginalis associated with low birth weight and preterm delivery. The Vaginal Infections and Prematurity Study Group. Sex Transm Dis 24: 353–360. [DOI] [PubMed] [Google Scholar]
- 29. Dessì D, Rappelli P, Diaz N, Cappuccinelli P, Fiori PL (2006) Mycoplasma hominis and Trichomonas vaginalis: a unique case of symbiotic relationship between two obligate human parasites. Front Biosci J Virtual Libr 11: 2028–2034. [DOI] [PubMed] [Google Scholar]
- 30. Lo S-C, Hayes MM, Tully JG, Wang RY-H, Kotani H, et al. (1992) Mycoplasma penetrans sp. nov., from the Urogenital Tract of Patients with AIDS. Int J Syst Bacteriol 42: 357–364 10.1099/00207713-42-3-357 [DOI] [PubMed] [Google Scholar]
- 31.Fiori PL, Diaz N, Cocco AR, Rappelli P, Dessì D (2013) Association of Trichomonas vaginalis with its symbiont Mycoplasma hominis synergistically upregulates the in vitro proinflammatory response of human monocytes. Sex Transm Infect. doi:10.1136/sextrans-2012–051006. [DOI] [PubMed]
- 32. Volokhov DV, Simonyan V, Davidson MK, Chizhikov VE (2012) RNA polymerase beta subunit (rpoB) gene and the 16S–23S rRNA intergenic transcribed spacer region (ITS) as complementary molecular markers in addition to the 16S rRNA gene for phylogenetic analysis and identification of the species of the family Mycoplasmataceae. Mol Phylogenet Evol 62: 515–528 10.1016/j.ympev.2011.11.002 [DOI] [PubMed] [Google Scholar]
- 33. Inamine JM, Ho KC, Loechel S, Hu PC (1990) Evidence that UGA is read as a tryptophan codon rather than as a stop codon by Mycoplasma pneumoniae, Mycoplasma genitalium, and Mycoplasma gallisepticum. J Bacteriol 172: 504–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cho I, Blaser MJ (2012) The human microbiome: at the interface of health and disease. Nat Rev Genet 13: 260–270 10.1038/nrg3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li M, Zhou M, Adamowicz E, Basarab JA, Guan LL (2012) Characterization of bovine ruminal epithelial bacterial communities using 16S rRNA sequencing, PCR-DGGE, and qRT-PCR analysis. Vet Microbiol 155: 72–80 10.1016/j.vetmic.2011.08.007 [DOI] [PubMed] [Google Scholar]
- 36. Shinzato N, Muramatsu M, Matsui T, Watanabe Y (2005) Molecular phylogenetic diversity of the bacterial community in the gut of the termite Coptotermes formosanus. Biosci Biotechnol Biochem 69: 1145–1155 10.1271/bbb.69.1145 [DOI] [PubMed] [Google Scholar]
- 37. Hongoh Y, Ohkuma M, Kudo T (2003) Molecular analysis of bacterial microbiota in the gut of the termite Reticulitermes speratus (Isoptera; Rhinotermitidae). FEMS Microbiol Ecol 44: 231–242 10.1016/S01686496(03)000266 [DOI] [PubMed] [Google Scholar]
- 38. Yang H, Schmitt-Wagner D, Stingl U, Brune A (2005) Niche heterogeneity determines bacterial community structure in the termite gut (Reticulitermes santonensis). Environ Microbiol 7: 916–932 10.1111/j.14622920.2005.00760.x [DOI] [PubMed] [Google Scholar]
- 39. Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, et al. (2008) Evolution of mammals and their gut microbes. Science 320: 1647–1651 10.1126/science.1155725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Glasfeld A, Leanz GF, Benner SA (1990) The stereospecificities of seven dehydrogenases from Acholeplasma laidlawii. The simplest historical model that explains dehydrogenase stereospecificity. J Biol Chem 265: 11692–11699. [PubMed] [Google Scholar]
- 41. Trülzsch K, Roggenkamp A, Pelludat C, Rakin A, Jacobi CA, et al. (2001) Cloning and characterization of the gene encoding periplasmic 2′,3′-cyclic phosphodiesterase of Yersinia enterocolitica O:8. Microbiology 147: 203–213. [DOI] [PubMed] [Google Scholar]
- 42. Browning GF, Marenda MS, Noormohammadi AH, Markham PF (2011) The central role of lipoproteins in the pathogenesis of mycoplasmoses. Vet Microbiol 153: 44–50 10.1016/j.vetmic.2011.05.031 [DOI] [PubMed] [Google Scholar]
- 43. Treangen TJ, Rocha EPC (2011) Horizontal Transfer, Not Duplication, Drives the Expansion of Protein Families in Prokaryotes. PLoS Genet 7: e1001284 10.1371/journal.pgen.1001284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nouvel LX, Sirand-Pugnet P, Marenda MS, Sagné E, Barbe V, et al. (2010) Comparative genomic and proteomic analyses of two Mycoplasma agalactiae strains: clues to the macro- and micro-events that are shaping mycoplasma diversity. BMC Genomics 11: 86 10.1186/14712164-11-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sirand-Pugnet P, Lartigue C, Marenda M, Jacob D, Barré A, et al. (2007) Being Pathogenic, Plastic, and Sexual while Living with a Nearly Minimal Bacterial Genome. PLoS Genet 3: e75 10.1371/journal.pgen.0030075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Noël CJ, Diaz N, Sicheritz-Ponten T, Safarikova L, Tachezy J, et al. (2010) Trichomonas vaginalis vast BspA-like gene family: evidence for functional diversity from structural organisation and transcriptomics. BMC Genomics 11: 99 10.1186/1471-2164-11-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hirt RP, Noel CJ, Sicheritz-Ponten T, Tachezy J, Fiori P-L (2007) Trichomonas vaginalis surface proteins: a view from the genome. Trends Parasitol 23: 540–547 10.1016/j.pt.2007.08.020 [DOI] [PubMed] [Google Scholar]
- 48. Onishi S, Honma K, Liang S, Stathopoulou P, Kinane D, et al. (2008) Toll-like receptor 2-mediated interleukin-8 expression in gingival epithelial cells by the Tannerella forsythia leucine-rich repeat protein BspA. Infect Immun 76: 198–205 10.1128/IAI.0113907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Myneni SR, Settem RP, Sojar HT, Malone JP, Loimaranta V, et al. (2012) Identification of a unique TLR2-interacting peptide motif in a microbial leucine-rich repeat protein. Biochem Biophys Res Commun 423: 577–582 10.1016/j.bbrc.2012.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Loimaranta V, Hytönen J, Pulliainen AT, Sharma A, Tenovuo J, et al. (2009) Leucine-rich repeats of bacterial surface proteins serve as common pattern recognition motifs of human scavenger receptor gp340. J Biol Chem 284: 18614–18623 10.1074/jbc.M900581200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Inagaki S, Onishi S, Kuramitsu HK, Sharma A (2006) Porphyromonas gingivalis vesicles enhance attachment, and the leucine-rich repeat BspA protein is required for invasion of epithelial cells by “Tannerella forsythia.”. Infect Immun 74: 5023–5028 10.1128/IAI.0006206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fettweis JM, Serrano MG, Sheth NU, Mayer CM, Glascock AL, et al. (2012) Species-level classification of the vaginal microbiome. BMC Genomics 13: S17 10.1186/1471-2164-13-S8-S17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schirm J, Bos PAJ, Roozeboom-Roelfsema IK, Luijt DS, Möller LV (2007) Trichomonas vaginalis detection using real-time TaqMan PCR. J Microbiol Methods 68: 243–247 10.1016/j.mimet.2006.08.002 [DOI] [PubMed] [Google Scholar]
- 54. Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu Y-W, Ye Y (2011) A novel abundance-based algorithm for binning metagenomic sequences using l-tuples. J Comput Biol J Comput Mol Cell Biol 18: 523–534 10.1089/cmb.2010.0245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Delcher AL, Bratke KA, Powers EC, Salzberg SL (2007) Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinforma Oxf Engl 23: 673–679 10.1093/bioinformatics/btm009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Besemer J, Lomsadze A, Borodovsky M (2001) GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29: 2607–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sonnhammer EL, von Heijne G, Krogh A (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol ISMB Int Conf Intell Syst Mol Biol 6: 175–182. [PubMed] [Google Scholar]
- 59. Bai X, Correa VR, Toruño TY, Ammar E-D, Kamoun S, et al. (2009) AY-WB phytoplasma secretes a protein that targets plant cell nuclei. Mol Plant-Microbe Interact MPMI 22: 18–30 10.1094/MPMI-22-1-0018 [DOI] [PubMed] [Google Scholar]
- 60. Eddy SR (2011) Accelerated Profile HMM Searches. PLoS Comput Biol 7: e1002195 10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Silva FJ, Belda E, Talens SE (2006) Differential annotation of tRNA genes with anticodon CAT in bacterial genomes. Nucleic Acids Res 34: 6015–6022 10.1093/nar/gkl739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, et al. (2007) CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8: 209 10.1186/1471-2105-8-209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Alves JMP, Buck GA (2007) Automated system for gene annotation and metabolic pathway reconstruction using general sequence databases. Chem Biodivers 4: 2593–2602 10.1002/cbdv.200790212 [DOI] [PubMed] [Google Scholar]
- 64. DeLuca TF, Wu I-H, Pu J, Monaghan T, Peshkin L, et al. (2006) Roundup: a multi-genome repository of orthologs and evolutionary distances. Bioinformatics 22: 2044–2046 10.1093/bioinformatics/btl286 [DOI] [PubMed] [Google Scholar]
- 65. Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinforma Oxf Engl 22: 2688–2690 10.1093/bioinformatics/btl446 [DOI] [PubMed] [Google Scholar]
- 66. Harwich MD, Alves JM, Buck GA, Strauss JF, Patterson JL, et al. (2010) Drawing the line between commensal and pathogenic Gardnerella vaginalis through genome analysis and virulence studies. BMC Genomics 11: 375 10.1186/14712164-11-375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Henrich B, Feldmann RC, Hadding U (1993) Cytoadhesins of Mycoplasma hominis. Infect Immun 61: 2945–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cluster analysis of mid-vaginal samples with a clinical diagnosis of trichomoniasis. Relative abundance of microbial taxa in mid-vaginal bacterial communities of 63 women with clinically diagnosed trichomoniasis is shown. The dendrogram was generated using Ward’s method with Manhattan distance. Presence of “Ca. M. girerdii” as determined by 16S rDNA profiling is indicated in the top bar. Samples that contained “Ca. M. girerdii” at less than 0.1% abundance are indicated as ambiguous (AMB) in light blue.
(PDF)
Detection of “ Ca . M. girerdii” in mid-vaginal samples. Fluorescence in situ hybridization detection of bacteria in mid-vaginal samples from two participants with clinically diagnosed trichomoniasis (subject 1, panels A–O); subject 2, panels P–T) by confocal laser scanning microscopy. The merged photomicrographs are also depicted in Figure 1. Nuclear DNA was detected with 4′6′-diamidine-2-phenylindole, dehydrochloride (DAPI, blue) as shown in panel B, G, L and Q. Most bacteria were detected with fluorescein-labeled broad-range bacteria probe Eub338 (green) as shown in panels C, H, M and R. “Ca. M. girerdii” was also stained with a Cy5-labeled probe targeting 16S rDNA (red) as shown in panels D, I, N and S.
(PDF)
“Ca. M. girerdii ” coexists with both genotypes of T. vaginalis. The maximum likelihood tree was constructed using concatentated, aligned partial protein sequences from three single-copy orthologs (CRN, PMS1, Mlh1a). Isolates indicated as type 1 or type 2 were previously typed using microsatellite markers. Strains from 43 clinically diagnosed cases of trichomoniasis from this study are indicated with the prefix “VCU”. The type 1 cluster is shaded blue and contains eight “Ca. M. girerdii” positive cases, the type 2 cluster is shaded green and contains ten “Ca. M. girerdii” positive cases and the ambiguous cluster is shaded gray and contains five “Ca. M. girerdii” positive cases. In this analysis, the ambiguous cluster groups with type 2 T. vaginalis, but the subgroup contains isolates that were differentially classified as type 1 using microsatellite markers. T. vaginalis strains from “Ca. M. girerdii” positive cases as determined by 16S rRNA microbiome profiling (0.1% threshold) are indicated with red boxes. Ambiguous cases that were detected at less than 0.1% threshold are denoted with pink boxes. Blue dots denote branches with bootstrap values greater than 50.
(PDF)
Conserved synteny among “Ca. M. girerdii” strains not shared with related species. Panel (A) shows dot plot nucleotide-based alignments of the reference “Ca. M. girerdii” strain VCU_M1 with contigs from three different “Ca. M. girerdii” strains (VCU_PA1, VCU_JB1, VCU_G1). Panel (B) shows dot plot amino-acid based alignments of the reference with three closely related species (M. iowae, M. penetrans and U. parvum). Horizontal grid lines delineate contigs. Nucleotide-based and protein-based alignments were performed using Nucmer and Promer respectively.
(PDF)
Distribution of Clusters of Orthologous Groups (COGS).
(DOCX)
Putative transporters in “ Ca . M. girerdii” strain VCU_M1.
(DOCX)
List of primers for circularization of “ Ca . M. girerdii” genome.
(DOCX)
Hominis Group orthologs.
(DOCX)
Pneumoniae and Hemoplasma Group orthologs.
(DOCX)
Spiroplasma, Phytoplasma and Outgroup orthologs.
(DOCX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. The complete genome has been deposited at NCBI under accession number CP007711.