

Abstract
Autophagy is a homeostatic degradation and recycling process that is also involved in defense against microbial pathogens and in certain forms of cellular suicide. Autophagy has been proposed to negatively regulate plant immunity-associated cell death related to the hypersensitive response (HR), as older autophagy-deficient mutants are unable to contain this type of cell death 5 to 10 d after infection. Such spreading cell death was found to require NPR1 (nonexpressor of PR genes 1), but surprisingly did not occur in younger atg mutants. In contrast, we find that npr1 mutants are not impaired in rapid programmed cell death activation upon pathogen recognition. Furthermore, our molecular evidence suggests that the NPR1-dependent spreading cell death in older atg mutants may originate from an inability to cope with excessive accumulation of ubiquitinated proteins and ER stress which derive from salicylic acid (SA)-dependent signaling (e.g., systemic acquired resistance). We also demonstrate that both senescence and immunity-related cell death seen in older atg mutants can be recapitulated in younger atg mutants primed with ER stress. We therefore propose that the reduction in SA signaling caused by npr1 loss-of-function is sufficient to alleviate the stress levels accumulated during aging in autophagy deficient cells which would otherwise become insurmountable and lead to uncontrolled cell death.
Keywords: age, atg, autophagy, cell death, ER stress, infection, npr1, senescence, ubiquitin
Introduction
Autophagy is a bulk degradation process involved in the recycling of nutrients, clearance of aggregates, and responses to various stresses.1-3 Autophagic recycling at basal levels maintains physiological concentrations of nutrients via protein and lipid recycling, and autophagic activity may be augmented to remove dysfunctional organelles.1,2 Several autophagic variants have been reported, including macroautophagy (hereafter termed autophagy), which is present in many organisms including animals and plants.2 This process relies on the concerted action of autophagy-related (ATG) genes to form double-membrane autophagosomes that fuse with vacuoles or lysosomes to degrade their internal cargo.2 Autophagosome assembly involves ATG8–phosphatidylethenolamine (ATG8–PE) conjugates, which are targeted to autophagosomal membranes, and levels of lipidated ATG8 can be proportional to the number of autophagosomes formed.2 In Arabidopsis thaliana, the protein NBR1 binds to autophagosomal-bound ATG8 and acts as a cargo receptor that targets ubiquitinated protein aggregates for autophagic degradation. NBR1 itself is also a selective substrate of autophagy.4,5
The early senescence phenotypes of loss-of-function atg mutants indicate that autophagy is required to maintain cellular homeostasis in response to internal and external cues (Fig. S1).6,7 However, the concurrent functions of autophagy complicate analyses of its contributions to responses to different stimuli. Nonetheless, it has been established in metazoans that autophagy, together with the endoplasmic reticulum (ER) stress response, is one of the earliest events of innate immune responses to combat intruders.8,9 In addition, autophagy is implicated as a mechanism for cellular suicide in several organisms.10-13
Plants rely on a multilayered defense system in which cytoplasmic receptors termed resistance (R) proteins recognize pathogen effectors or their virulence activities to trigger host immune responses, including rapid and restricted host cell death at the site of pathogen entry.14 This form of pathogen-induced cell death is known as the hypersensitive response (HR). Several reports have documented a gradual spread of cell death in atg mutants from infected to uninfected tissues which develops over several days following pathogen inoculation.6,15,16 However, it has also been shown that rapid development of HR cell death is suppressed in infected tissues of autophagy-deficient mutants,12 and that autophagy can promote HR cell death.17 More specifically, both Hofius et al.12 and Yoshimoto et al.6 note that cell death triggered by the resistance protein RPM1 does not spread beyond infection sites in younger atg mutants (4 to 5 wk, before the onset of early senescence),12 while spreading cell death occurs several days after infection in older atg mutants (7 to 8 wk, when early senescence is well established).6
Yoshimoto et al.6 have demonstrated that levels of the phytohormone SA increased along with transcripts of senescence-associated SAG12 and of the SA responsive markers PR1 and PR2 in atg mutants. These increases correlated with the onset of a visible early senescence phenotype compared with wild-type (WT) plants. Senescence-associated cell death and growth retardation of atg mutants could be reversed by expression of the bacterial SA hydroxylase NAHG, as well as by mutations in the SA biosynthetic gene SID2 (salicylic acid induction deficient 2), or in the SA receptor component NPR1 (Fig. S1).6,18 Importantly, suppression of the spreading lesions and apparently immunity-related cell death in atg mutants is also observed in SA- and NPR1-deficient backgrounds.6,18 In addition, Yoshimoto et al.6 have observed that application of the SA agonist BTH restores the premature senescence phenotype in atg5 nahG and in atg5 sid2 double mutants, but not in atg5 npr1. Concurrently, exogenous application of BTH also leads to NPR1-dependent accumulation of autophagosomal bodies in roots. This led the authors to propose that autophagy regulates a novel, negative feedback loop through NPR1, which modulates SA signaling and thereby limits senescence and immunity-associated cell death. This conclusion is surprising because npr1 mutants exhibit much stronger immunity-associated cell death than WT plants upon avirulent infection.19
Here we show that senescence or infection-induced accumulation of ubiquitinated proteins and ER stress are accelerated by NPR1 in autophagy-deficient mutants. We further demonstrate that both senescence and immunity-related cell death seen in older atg mutants can be recreated in younger atg mutants primed with ER stress. We therefore propose that rather than a negative regulation of cell death, the senescence-associated cell death and spreading lesions provoked by infections in old autophagy-deficient mutants are symptoms of perturbations in homeostasis.
Results
HR cell death in atg and npr1 mutants
Since spreading cell death in older atg mutants is first macroscopically visible several days post infection, and as NPR1 has previously been implicated in restricting HR cell death triggered by RPM1,19,20 we investigated the importance of NPR1-dependent HR cell death. We monitored cell death development in infected leaves of npr1 single and atg5 npr1 double mutants. Cell death induced by Pseudomonas syringae pv Tomato (Pst) DC3000 carrying AvrRpm1 (1 × 108 CFU mL−1) was quantified using an electrolyte leakage assay.21 As previously reported,19 hypersensitive cell death in npr1 exceeded the levels observed in WT controls (Fig. 1A). This confirms that NPR1 is not required for RPM1-triggered HR cell death. In contrast, HR cell death in atg5 mutants was significantly suppressed compared with WT, as previously observed.12 Surprisingly, as for npr1, HR cell death in the atg5 npr1 double mutant was also significantly higher than WT. This again indicates that NPR1 is not required for the promotion of HR cell death and in fact might suggest that it suppresses this type of cell death. To verify the effects of autophagy deficiency, we extended our analysis by measuring HR cell death in atg2 and in atg2 npr1 double mutants. atg2 mutants exhibit a more severe phenotype than atg5,6,18 which is in agreement with recent reports of residual autophagy activity in atg5 and atg7 mutants.22 As seen in Figure 1B, RPM1-triggered HR cell death was strongly suppressed in atg2 mutants compared with WT, but npr1 single mutants exhibited significantly more ion leakage than both WT and the single atg2 mutant. In contrast, the atg2 npr1 double mutant displayed an intermediate level of ion leakage. Since atg2 mutants are autophagy deficient, NPR1 must be needed to dampen autophagy-independent types of HR cell death because cell death in atg5 npr1 and atg2 npr1 double mutants exceeds cell death in atg5 and atg2 single mutants (Fig. 1A and B). We note that primary HR cell death is also similarly suppressed in older atg mutants, and that we observe no marked differences among genotypes if ion leakage measurements are extended to 12 h (data not shown). Nevertheless, these observations are also supported by microscopic analysis of HR cell death detected by trypan blue staining 12 h after infection with low doses of Pst DC3000 (AvrRpm1) (1 × 106 CFU mL−1).23 atg2 mutants exhibit the lowest visible level of HR cell death and atg2 npr1 exhibits WT-like levels of cell death, while most leakage is seen in npr1 single mutants (Fig. 1C). This is in agreement with the results of Rate and Greenberg,19 and is also supported by their finding that plants which overproduce NPR1 exhibit a weaker HR. More importantly, HR cell death triggered by RPM1 is both temporally and genetically discrete from the type of cell death seen in adjacent, uninfected tissues of older atg mutants, because autophagic cell death is NPR1-independent during the immediate HR (Fig. 1) but is NPR1-dependent outside of the initial HR lesion several days after infection.6 To exclude the possibility that major differences in bacterial growth may partially explain our observations, we assayed bacterial growth 6 h post infection (Pst DC3000 (AvrRpm1), 1 × 108 CFU mL−1). This revealed negligible bacterial growth during the first 6 h, and no significant difference in that time frame for all genotypes tested apart from the rpm1 knockout control compared with WT (Fig. S2). These results raise at least 2 further questions: 1) why is the early senescence phenotype of atg mutants suppressed by mutation of NPR1 (Fig. S1), and 2) why do cells in uninfected tissue of older atg mutants die some days after BTH treatment or RPM1 is triggered.

Figure 1. NPR1 is not required for the promotion of HR cell death. (A and B) Ion leakage assays of (A) 6-wk-old Col-0 WT, atg5, npr1, and atg5 npr1 plants and (B) 6-wk-old Col-0 WT, rpm1, atg2, npr1, and atg2 npr1 plants after inoculation with 1 x 108 CFU mL−1 Pst DC3000 (AvrRpm1). Mean and standard errors (SE) were calculated from 4 or 6 discs per treatment with 3 or 4 replicates within an experiment. Experiments were repeated with similar results. Pairwise comparisons at the last time point post infection for means vs. WT were performed with the one-way ANOVA test followed by the Holm-Šídák post-hoc test. a, P ≤ 0.05, b, P ≤ 0.01, c, P ≤ 0.0001. (C) Evaluation of tissue viability using lactophenol-trypan blue staining in leaves from Col-0 WT, npr1, atg2, atg2 npr1, and rpm1, 12 h after infection with 1 x 106 CFU mL−1 Pst DC3000 (AvrRpm1). Dead cells (labeled blue) are observed in leaves of all genotypes (except rpm1) treated with Pst DC3000 (AvrRpm1). No dead cells were observed in leaves infiltrated with MgCl2 (negative control) or in rpm1 mutants which are unable to trigger HR in the presence of the AvrRpm1 effector. Size bar: 2.5 mm.
Age- and immune-dependent accumulation of autophagic markers and ubiquitinated proteins are enhanced by NPR1
atg mutants are under high oxidative and metabolic stress,24 which may be further promoted by SA to induce premature senescence.25,26 To test if mutation of NPR1 suppressed stress responses as measured by autophagy induction, the accumulation of the autophagosomal membrane marker ATG8 was examined. Both nonlipidated and lipidated ATG8 accumulate in atg-deficient mutants for 2 reasons: i. autophagy is induced by the build-up of cellular ‘debris’, which triggers synthesis of components required for the autophagosomal membrane, such as ATG8, and ii. nucleation of the phagophore cannot be completed and autophagosome formation is arrested, leading to an accumulation of membrane-associated ATG8 that cannot be degraded. In line with this, marked accumulation of both isoforms of ATG8a were detected in 3-wk-old atg2 and atg2 npr1 mutants, but only trace amounts accumulated in npr1 (Fig. 2A). However, at 6 wk and more evidently at 9 wk, both isoforms of ATG8a were more abundant in atg2 than in atg2 npr1 mutants (Fig. 2A).
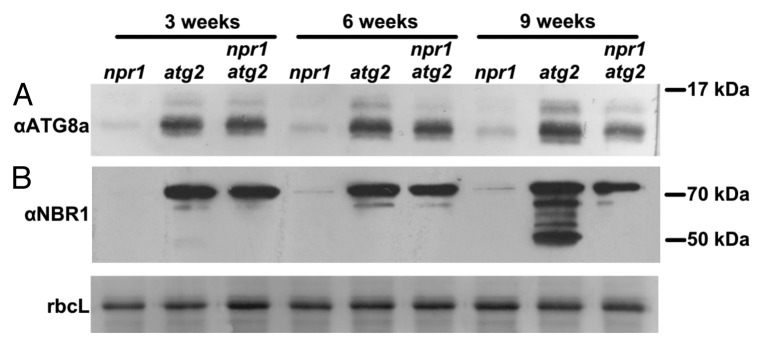
Figure 2. Loss of NPR1 function alleviates accumulation of autophagic components. Immunoblot detection of (A) ATG8A and (B) NBR1 accumulation in 3-, 6- and 9-wk-old npr1, atg2, and npr1 atg2 mutants. Coomassie Blue staining of the large subunit of RuBisCO serves as loading control.
We then assayed whether there was differential accumulation of the autophagy cargo adaptor NBR1, which contains a UBA domain capable of binding ubiquitinated substrates to target them for degradation through autophagy.4,5 Marked accumulation of endogenous NBR1 could be detected by immunoblotting with an anti-NBR1 antibody in 3-wk-old atg2 and atg2 npr1 mutants but not in npr1 (Fig. 2B). However, at 9 wk NBR1 forms recognized by the antibody were much more abundant in atg2 than atg2 npr1 double mutants (Fig. 2B). As in npr1, NBR1 did not accumulate in WT at any of the time points tested (see untreated samples in Fig. 3B).
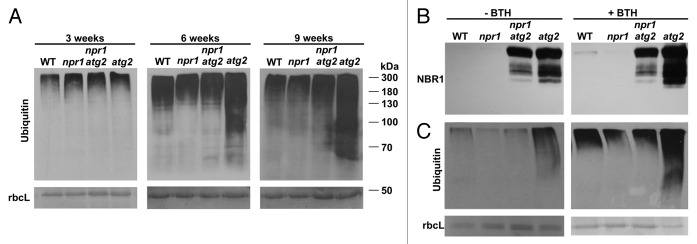
Figure 3. Absence of NPR1 and SA responses leads to a decrease in the accumulation of ubiquitinated proteins. (A) Immunoblot detection of ubiquitin (Ubq antibody Z0458 from Dako) in 3-, 6- and 9-wk-old WT, npr1, atg2 npr1, and atg2 plants. (B and C) Immunoblot detection of (B) NBR1 and (C) ubiquitin in 7-wk-old WT, npr1, atg2 npr1, and atg2 plants, after 24 h of treatment with mock or 100 μM BTH. Amido black or Coomassie Blue staining of the large subunit of RuBisCO serve as loading controls.
In addition, we assayed whether increased levels of NBR1 correlated with the levels of ubiquitinated proteins normally degraded via autophagy. Marked differences in the levels of ubiquitinated proteins were not detectable in 3-wk-old npr1, atg2, and atg2 npr1 mutants (Fig. 3A; Fig. S3A). Interestingly, in older plants there was a gradual accumulation of ubiquitinated proteins in atg2 that was more pronounced than in WT and npr1 (Fig. 3A). More importantly, this accumulation was greater in atg2 than in atg2 npr1, and the difference was more pronounced in 9-wk-old than in 6-wk-old plants (Fig. 3A; Fig. S3B) and also more pronounced in 8-wk-old plants compared with 4-wk-old plants (Fig S3C). As such, this age-dependent phenotype implicates NPR1-SA in the accumulation of ubiquitinated products. Our results also demonstrate that a steady build-up of ubiquitinated proteins in autophagy-deficient backgrounds can be alleviated by introducing loss-of-function mutations in the SA pathway, such as npr1.
Plants respond to Pst DC3000 (AvrRpm1) and other avirulent pathogens by inducing SAR which leads to increased SA and defense gene transcript levels in uninfected tissues.27 NPR1, a central component of SA reception, controls the expression of PR genes and a large set of SAR-responsive, ER-resident proteins. These ER genes are upregulated to ensure proper folding and secretion of the high levels of PR proteins required to establish SAR.28,29 Autophagy is needed to counterbalance ER stress and to remove misfolded proteins.30 It is therefore probable that SAR induced by infection, or by treatment with the SA analog BTH, leads in atg mutants to deleterious hyperaccumulation of unprocessed PR proteins, and thus of NBR1 selective cargo and ubiquitinated proteins. To test this, we compared the accumulation of NBR1 and ubiquitinated proteins in 6-wk-old plants before and after BTH treatment. Similar to our earlier observations, NBR1 and ubiquitinated proteins were more abundant in untreated atg2 than in atg2 npr1, npr1, or WT (Fig. 3B and C). However, a dramatic accumulation of NBR1 and ubiquitinated proteins was observed in atg2 plants 24 h after treatment with BTH. More importantly, such BTH-induced accumulation was almost completely absent in atg2 npr1 (Fig. 3B and C). This demonstrates that NPR1 loss-of-function is also sufficient to abrogate accumulation of ubiquitinated proteins in autophagy-deficient mutants and to thus hinder disruption of cellular homeostasis.
Mutations in NPR1 dampen ER stress
SA treatment is sufficient to cause tissue collapse and cell death in bip2 mutants defective in ER protein folding and secretory capacity.29,31 Importantly, the SA-induced ER stress and leaf collapse phenotypes of bip2 mutants is NPR1-dependent. This rescue of leaf collapse is very similar to what has been described for atg mutants, as atg npr1 show no visible spreading lesions after avirulent infection.6 Autophagy-deficient mutants have also been shown in other model systems to be hypersensitive to ER stress, as autophagy is also required to remove the deleterious expansion of the endoplasmic reticulum.32 The susceptibility to ER stress displayed by atg mutants may explain why NPR1 mutation suppresses both the visible spread of lesions triggered by immune responses, as well as senescence-associated cell death in atg mutants: the protein flux through the endoplasmatic reticulum is significantly lowered in npr1 backgrounds. The increased expression of proteins destined for secretion during the immune response triggers the unfolded protein response in the ER.29 One induced component of this response is BZIP60, a transcription factor which regulates the expression of proteins involved in protein folding and degradation. BZIP60 is synthesized from mRNA spliced by PDI10/IRE1, and BZIP60 mRNA splicing is stimulated by ER stress inducers.33-35 Thus, in addition to a regular, unspliced BZIP60 amplicon a smaller spliced amplicon, BZIP60s can be detected by Q-PCR in ER stressed plants.33-35 To test if ER stress is lower in NPR1-deficient backgrounds, we assessed the expression of BZIP60s mRNA in 5- and 6-wk-old plants. As seen in Figure 4, the levels of spliced BZIP60s were similar in npr1, atg2 npr1, and in WT at these stages. However, atg2 mutants accumulated roughly 3-fold more BZIP60s in 5-wk-old plants and some 10-fold in 6-wk-old plants (Fig. 4). The comparably low levels of spliced BZIP60 in atg2 npr1 compared with atg2 indicate that the ER experiences a smaller flux of proteins in NPR1-deficient backgrounds, and thus much lower ER stress.

Figure 4. Induction of ER stress by NPR1-dependent responses. Q-PCR of spliced BZIP60 mRNA levels in 5- and 6-wk-old Col-0 WT, npr1, atg2 npr1, and atg2 plants. Expression was normalized to wild type and is presented in a log2 scale. ACT expression was used as a standard reference.
Combinations of ER stress and immune responses kill young autophagy-deficient mutants
Autophagy also functions as a pathway for the turnover of ER membranes and contents in response to ER stress in Arabidopsis.30 Thus, treatment with tunicamycin (TM), a potent inducer of ER-stress that inhibits protein N-glycosylation, leads to the accumulation of autophagosomes.30,33,36 Our findings that accumulation of autophagic markers and ubiquitinated proteins is reduced in npr1 backgrounds (Fig. 2; Fig. 3) indicate that compromised homeostasis in phenotypically presenescent and in ER-stressed atg mutants primes them to succumb to additional stress or injury. If so, then it should be possible to combine ER stress with an additional stress such as BTH treatment to produce lethal effects in younger atg mutants. To examine this, we germinated WT, atg2, npr1, and atg2 npr1 seedlings on MS plates containing TM and BTH. This revealed that young atg2 single mutants were unable to cope with these combined stresses and began to die within 4 wk (Fig. 5A). In contrast, loss of NPR1 function rescued these effects such that atg2 npr1 double mutants survived. Similar observations were made with atg5 and atg5 npr1 (Fig. S4A). Since atg2 seeds germinated to a lower extent than other genotypes on plates with both TM and BTH, it was possible that individual atg2 seedlings were exposed to higher TM and BTH concentrations in their immediate surroundings compared with the other genotypes (Fig. 5A). To clarify this, we seeded all genotypes on MS plates and subsequently transferred the germinated seedlings individually to MS plates containing TM and BTH. This demonstrates that atg2 mutants succumb to the same concentrations of TM and BTH tolerated by the other genotypes (Fig. S4B).

Figure 5. Combinatory stress induced by tunicamycin and BTH leads to early senescence and uncontrolled cell death in autophagy-deficient mutants. (A) 4-wk-old Col-0 WT, npr1, atg2, and atg2 npr1 plants grown on MS plates supplemented with 0.00005% DMSO (Mock), 5 ng mL−1 TM, 50 μM BTH or 5 ng mL−1 TM and 50 μM BTH. Double treatment with BTH and TM decreases germination and induces early senescence in atg2 mutants but these features are rescued by NPR1 loss of function. (B) 4-wk-old WT, npr1, atg2 npr1, and atg2 leaves injected in one side with 100 ng mL−1 TM (“Tunicamycin” and “AvrRpm1/Tunicamycin”) or 0.00005% DMSO (“AvrRpm1”), and then injected 5 d later with Pst DC3000 (AvrRpm1) at 2 x 107 CFU mL−1 (“AvrRpm1” and “AvrRpm1/Tunicamycin”) or 10 mM MgCl2 (“Tunicamycin”), in the opposite side of the leaves. Priming of atg2 mutants with TM before infection leads to unrestricted HR cell death. Loss of NPR function in the atg2 background is sufficient to permit survival under combinatory stress and restrict HR cell death to the infection site. Pictures taken 4 d after Pst DC3000 (AvrRpm1) or MgCl2 injections.
Finally, we also assessed if it was possible to mimic the uncontrolled, age-dependent visible spread of cell death observed upon infection with avirulent pathogens.6,15,16 We therefore injected TM into one side of leaves of 4-wk-old WT, npr1, atg2 npr1, and atg2 plants, waited 5 d for the genotypes to cope with this stress, and then injected avirulent Pst DC3000 (AvrRpm1) (2 × 107 CFU mL−1) in the other side of the leaves. Surprisingly, while we observed contained HR-associated lesions develop in WT, npr1, and atg2 npr1 genotypes after 4 d, the atg2 genotype was unable to contain such cell death (Fig. 5B). This visible spread of cell death was not observed in young atg2 plants if Pst DC3000 (AvrRpm1) or TM were injected alone. Thus, when atg mutants reach high levels of ER stress, subsequent additive stressors, such as infection, will be detrimental and result in cell death and lesions (Fig. 5B).
Discussion
Autophagy has previously been implicated in HR-associated cell death, but conflicting reports have shown it to be required both during the immediate HR response and in containing HR cell death lesions.6,12,15,16 We also find that spreading cell death, observed upon pathogen infection, can be contained in old atg mutants by introduction of loss-of-function alleles of the central SA regulator NPR1, indicating that immune-related responses influence the activation of the autophagic pathway. In apparent confirmation of this hypothesis, we (Fig. 3B) and others also see indications of increased autophagy upon treatment with the SA-agonist BTH.6 However, we also observed accumulation of ubiquitinated proteins (Fig. 3) and increased ER stress (Fig. 4) in older atg mutants and these characteristics are enhanced by both infection and BTH treatment but suppressed by mutations in NPR1. We observed that while a combination of ER stress and BTH kills atg deficient seedlings, double atg npr1 mutants are able to survive this treatment (Fig. 5A; Fig. S4A). Furthermore, we were able to recreate in young atg deficient plants the spreading cell death induced by pathogen infection (as observed in older atg plants) by priming them with ER stress (Fig. 5B).
In agreement with previous reports,12,37 we also observed less HR cell death in atg mutants, upon pathogen infection. Interestingly, we observed strong HR cell death in npr1 plants but also in atg2 npr1 plants. Thus, NPR1 somehow suppresses cell death. Since atg2 npr1 mutants are also autophagy deficient (Fig. 3A), NPR1 must suppress autophagy-independent cell death mechanisms. Mutations in another important mediator of the SA signaling pathway, EDS1 (enhanced disease susceptibility 1), likewise accelerate HR cell death triggered by RPM1.12 While the molecular basis for these observations remains unclear, it is clear that HR cell death may be executed via more than one signaling pathway.12,38
We provide evidence that the kind of cell death outside of infected areas in older atg mutants can be reproduced in young atg2 mutants by a combination of ER stress and infection. Similarly, we find that a combination of ER stress and BTH causes lethality in very young, autophagy-deficient mutants. We therefore propose a scenario in which NPR1 loss-of-function may rescue the visible spread of cell death in uninfected tissue because defense gene products are not upregulated in npr1, thus minimizing pressure on the ER.29 In summary, we present an alternative model in which autophagy does not to regulate a negative feedback loop through NPR1 as previously proposed.6 Were the latter true, it would also imply that autophagy regulates cell death through NPR1 in one manner in the HR, a different manner in adjacent tissues, and differently again in younger plants compared with older plants. Instead, our data support a model in which autophagy-deficient mutants die before WT plants because of an inappropriate accumulation of ubiquitinated protein aggregates and increasing ER stress. This deficiency puts them at risk,5,6 and disruption of cellular homeostasis leads to SA buildup and NPR1-dependent accumulation of defense related transcripts6 in a deleterious cycle that is suppressed by mutations in NPR1. Thus, it appears to be problematic to draw conclusions about autophagic functions in developmentally older atg mutants, due to secondary effects of their long-term, compromised cellular homeostasis.
Materials and Methods
Plant material and growth conditions
Plants grown on soil were kept in environmental chambers under 8 h of light (150 mE/m2/s) at 21 °C and 70% relative humidity after seed surface sterilization with 70% ethanol. Seedlings grown on solid MS medium (0.44% w/v agar, 1% w/v sucrose, pH5.7) were kept under 16 h of light (150 mE/m2/s) at 21 °C after seed surface sterilization with 1.3% v/v bleach followed by 70% ethanol. Seeds germinated on MS plates were supplemented with 0.00005% (v/v) DMSO (Mock), 0.005 μg mL−1 TM (Sigma, T7765), 50 μM BTH (Syngenta, UN 3077) or 5 ng mL−1 TM and 50 μM BTH. Arabidopsis knockout mutants were rpm1,39 npr1,27 atg2, and atg2 npr16,18 and atg5 and atg5 npr1.6 All data results described, were obtained from at least 3 independent experiments.
Bacterial assays
Leaves of wt, npr1, atg5, atg5 npr1, atg2, atg2 npr1, and rpm1 were syringe infiltrated with the avirulent Pst DC3000 (AvrRpm1) strain for ion leakage assay (1 × 108 CFU mL−1) and trypan blue staining (1 × 106 CFU mL−1) as previously described.12 Bacterial growth assays following syringe-infiltration with avirulent Pst DC3000 (AvrRpm1) were performed at 1 × 108 CFU mL−1, as previously described.21
Lesion development
One side of leaves from 4-wk-old plants was infiltrated with 100 ng mL−1 TM, followed by an injection in the opposite side of the leaf 5 d later with avirulent Pst DC3000 (AvrRpm1) at 2 × 107 CFU mL−1. Representative pictures were taken 4 d after the last injection.
Immunoblotting
Total proteins were extracted in urea buffer (4 M urea, 100 mM DTT [Applichem, A3668], 1% Triton X-100 [Applichem, A1388]) using a mortar and pestle. Samples were centrifuged 10 min at 15,000 g and 3× SDS loading buffer (30% glycerol, 3% SDS, 94 mM Tris, pH 6.8, 75 mM DTT) was added to the supernatant fraction.
For anti-ubiquitin immunoblots, total protein extracts were separated by 10% SDS-PAGE, blocked with 5% high grade BSA (Applichem, A1391) or 2% gelatin (Merck, 104070) for 1 h and subsequently probed with anti-ubiquitin antibody (Fig. 3, Dako, Z0458; Fig. S3, Agrisera, AS08 307; 0.02% sodium azide) followed by anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (Promega, W4028). For anti-NBR1 immunoblots, extracts were separated in 10% SDS or 12% 6 M urea gels by SDS-PAGE, blocked with 5% milk or 5% high-grade BSA for 1 h, and probed with anti-NBR1 antibody (kindly provided by T Johansen, Tromsø, Norway; 1% BSA, 0.02% sodium azide) followed by anti-rabbit HRP-conjugated antibody (Promega, W4018). For anti-ATG8A immunoblots, extracts were separated on 6 M 12% urea gels using SDS-PAGE, blocked with 5% milk for 1 h, and probed with anti-ATG8A antiserum (kindly provided by Y Ohsumi, Okazaki, Japan; 1% BSA, 0.02% sodium azide) followed by anti-rabbit alkaline phosphatase-conjugated antibody (Promega, S3738; 1% milk). Amido black or Coomassie Blue staining of the large subunit of RuBisCO served as loading controls.
Quantitative (Q) RT-PCR
Total RNA was extracted using the Omegas Plant RNA kit (Omega Biotech, R6837) and cDNA synthesized using the Life SuperScript III Reverse Transcriptase kit (Thermo Fisher, 18080). For Q-PCR, transcript abundance was quantified using BZIP60us- and BZIP60s-specific primers (BZIP60s reverse GAACCCAACA GCAGACTCCTG and BZIP60us forward GTATGCTTGA GTGCTTCGTT GC) using the Luminaris color probe Q-PCR kit (Thermo Scientific, K0351) with actin (ACT) expression as standard reference.34,35
Statistics
Statistical analyses were performed with an ANOVA with the post-hoc Holm-Šídák test, using GraphPad Prism 6. Significance was accepted at the level of P ≤ 0.05.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dingzhong Tang and Kokhi Yoshimoto for the atg2 and atg2 npr1 knockout seeds, and Kokhi Yoshimoto for the atg5 and atg5 npr1 knockout seed lines. We thank Terje Johansen for the NBR1 antibody, and Yoshinori Ohsumi for the ATG8A antiserum. We also thank Suksawad Vongvisuttikun for technical assistance. This work was funded by grants to MP from the Danish Council for Independent Research (11-106302) and to ER by the Portuguese Foundation for Science and Technology (FCT) (SFRH/BPD/75696/2011).
Glossary
Abbreviations:
- ACT
actin
- ATG
autophagy-related
- ATG8–PE
ATG8–phosphatidylethenolamine
- avrRPM1
Type III effector protein AvrRpm1
- BIP2
luminal binding protein 2
- BTH
benzothiadiazole
- BZIP60
basic region/leucine zipper motif 60
- CFU
colony forming units
- ER
endoplasmic reticulum
- HR
hypersensitive response
- HRP
horseradish peroxidase
- NBR1
next to BRCA1 gene 1
- NPR1
nonexpressor of PR genes 1
- Pst
Pseudomonas syringae pv. Tomato
- R
resistance
- RuBisCO
ribulose-1,5-bisphosphate carboxylase/oxygenase
- SA
salicylic acid
- SAG12
senescence-associated gene 12
- SAR
systemic acquired resistance
- SID2
salicylic acid induction deficient 2
- TM
tunicamycin
- WT
wild type
References
- 1.Bassham DC, Laporte M, Marty F, Moriyasu Y, Ohsumi Y, Olsen LJ, Yoshimoto K. Autophagy in development and stress responses of plants. Autophagy. 2006;2:2–11. doi: 10.4161/auto.2092. [DOI] [PubMed] [Google Scholar]
- 2.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 3.Hofius D, Munch D, Bressendorff S, Mundy J, Petersen M. Role of autophagy in disease resistance and hypersensitive response-associated cell death. Cell Death Differ. 2011;18:1257–62. doi: 10.1038/cdd.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Svenning S, Lamark T, Krause K, Johansen T. Plant NBR1 is a selective autophagy substrate and a functional hybrid of the mammalian autophagic adapters NBR1 and p62/SQSTM1. Autophagy. 2011;7:993–1010. doi: 10.4161/auto.7.9.16389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou J, Wang J, Cheng Y, Chi Y-J, Fan B, Yu J-Q, Chen Z. NBR1-mediated selective autophagy targets insoluble ubiquitinated protein aggregates in plant stress responses. PLoS Genet. 2013;9:e1003196. doi: 10.1371/journal.pgen.1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshimoto K, Jikumaru Y, Kamiya Y, Kusano M, Consonni C, Panstruga R, Ohsumi Y, Shirasu K. Autophagy negatively regulates cell death by controlling NPR1-dependent salicylic acid signaling during senescence and the innate immune response in Arabidopsis. Plant Cell. 2009;21:2914–27. doi: 10.1105/tpc.109.068635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson AR, Doelling JH, Suttangkakul A, Vierstra RD. Autophagic nutrient recycling in Arabidopsis directed by the ATG8 and ATG12 conjugation pathways. Plant Physiol. 2005;138:2097–110. doi: 10.1104/pp.105.060673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Travassos LH, Carneiro LAM, Ramjeet M, Hussey S, Kim Y-G, Magalhães JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 9.Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity. 2013;39:211–27. doi: 10.1016/j.immuni.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 10.Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2008;15:105–12. doi: 10.1038/sj.cdd.4402231. [DOI] [PubMed] [Google Scholar]
- 11.Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19:1741–6. doi: 10.1016/j.cub.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hofius D, Schultz-Larsen T, Joensen J, Tsitsigiannis DI, Petersen NHT, Mattsson O, Jørgensen LB, Jones JDG, Mundy J, Petersen M. Autophagic components contribute to hypersensitive cell death in Arabidopsis. Cell. 2009;137:773–83. doi: 10.1016/j.cell.2009.02.036. [DOI] [PubMed] [Google Scholar]
- 13.Kwon SI, Cho HJ, Jung JH, Yoshimoto K, Shirasu K, Park OK. The Rab GTPase RabG3b functions in autophagy and contributes to tracheary element differentiation in Arabidopsis. Plant J. 2010;64:151–64. doi: 10.1111/j.1365-313X.2010.04315.x. [DOI] [PubMed] [Google Scholar]
- 14.Jones JDG, Dangl JL. The plant immune system. Nature. 2006;444:323–9. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Schiff M, Czymmek K, Tallóczy Z, Levine B, Dinesh-Kumar SP. Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005;121:567–77. doi: 10.1016/j.cell.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 16.Patel S, Dinesh-Kumar SP. Arabidopsis ATG6 is required to limit the pathogen-associated cell death response. Autophagy. 2008;4:20–7. doi: 10.4161/auto.5056. [DOI] [PubMed] [Google Scholar]
- 17.Kwon SI, Cho HJ, Kim SR, Park OK. The Rab GTPase RabG3b positively regulates autophagy and immunity-associated hypersensitive cell death in Arabidopsis. Plant Physiol. 2013;161:1722–36. doi: 10.1104/pp.112.208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Nishimura MT, Zhao T, Tang D. ATG2, an autophagy-related protein, negatively affects powdery mildew resistance and mildew-induced cell death in Arabidopsis. Plant J. 2011;68:74–87. doi: 10.1111/j.1365-313X.2011.04669.x. [DOI] [PubMed] [Google Scholar]
- 19.Rate DN, Greenberg JT. The Arabidopsis aberrant growth and death2 mutant shows resistance to Pseudomonas syringae and reveals a role for NPR1 in suppressing hypersensitive cell death. Plant J. 2001;27:203–11. doi: 10.1046/j.0960-7412.2001.1075umedoc.x. [DOI] [PubMed] [Google Scholar]
- 20.Fu ZQ, Yan S, Saleh A, Wang W, Ruble J, Oka N, Mohan R, Spoel SH, Tada Y, Zheng N, et al. NPR3 and NPR4 are receptors for the immune signal salicylic acid in plants. Nature. 2012;486:228–32. doi: 10.1038/nature11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackey D, Belkhadir Y, Alonso JM, Ecker JR, Dangl JL. Arabidopsis RIN4 is a target of the type III virulence effector AvrRpt2 and modulates RPS2-mediated resistance. Cell. 2003;112:379–89. doi: 10.1016/S0092-8674(03)00040-0. [DOI] [PubMed] [Google Scholar]
- 22.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–8. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 23.Torres MA, Dangl JL, Jones JDG. Arabidopsis gp91phox homologues AtrbohD and AtrbohF are required for accumulation of reactive oxygen intermediates in the plant defense response. Proc Natl Acad Sci U S A. 2002;99:517–22. doi: 10.1073/pnas.012452499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiong Y, Contento AL, Nguyen PQ, Bassham DC. Degradation of oxidized proteins by autophagy during oxidative stress in Arabidopsis. Plant Physiol. 2007;143:291–9. doi: 10.1104/pp.106.092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson MD, Chen Z, Klessig DF. Possible involvement of lipid peroxidation in salicylic acid-mediated induction of PR-1 gene expression. Phytochemistry. 1998;47:555–66. doi: 10.1016/S0031-9422(97)00726-7. [DOI] [Google Scholar]
- 26.Chaouch S, Queval G, Vanderauwera S, Mhamdi A, Vandorpe M, Langlois-Meurinne M, Van Breusegem F, Saindrenan P, Noctor G. Peroxisomal hydrogen peroxide is coupled to biotic defense responses by ISOCHORISMATE SYNTHASE1 in a daylength-related manner. Plant Physiol. 2010;153:1692–705. doi: 10.1104/pp.110.153957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maleck K, Levine A, Eulgem T, Morgan A, Schmid J, Lawton KA, Dangl JL, Dietrich RA. The transcriptome of Arabidopsis thaliana during systemic acquired resistance. Nat Genet. 2000;26:403–10. doi: 10.1038/82521. [DOI] [PubMed] [Google Scholar]
- 28.Cao H, Bowling SA, Gordon AS, Dong X. Characterization of an Arabidopsis Mutant That Is Nonresponsive to Inducers of Systemic Acquired Resistance. Plant Cell. 1994;6:1583–92. doi: 10.1105/tpc.6.11.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang D, Weaver ND, Kesarwani M, Dong X. Induction of protein secretory pathway is required for systemic acquired resistance. Science. 2005;308:1036–40. doi: 10.1126/science.1108791. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, Burgos JS, Deng Y, Srivastava R, Howell SH, Bassham DC. Degradation of the endoplasmic reticulum by autophagy during endoplasmic reticulum stress in Arabidopsis. Plant Cell. 2012;24:4635–51. doi: 10.1105/tpc.112.101535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299–304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iwata Y, Koizumi N. An Arabidopsis transcription factor, AtbZIP60, regulates the endoplasmic reticulum stress response in a manner unique to plants. Proc Natl Acad Sci U S A. 2005;102:5280–5. doi: 10.1073/pnas.0408941102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagashima Y, Mishiba K, Suzuki E, Shimada Y, Iwata Y, Koizumi N. Arabidopsis IRE1 catalyses unconventional splicing of bZIP60 mRNA to produce the active transcription factor. Sci Rep. 2011;1:29. doi: 10.1038/srep00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng Y, Humbert S, Liu J-X, Srivastava R, Rothstein SJ, Howell SH. Heat induces the splicing by IRE1 of a mRNA encoding a transcription factor involved in the unfolded protein response in Arabidopsis. Proc Natl Acad Sci U S A. 2011;108:7247–52. doi: 10.1073/pnas.1102117108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koizumi N, Ujino T, Sano H, Chrispeels MJ. Overexpression of a gene that encodes the first enzyme in the biosynthesis of asparagine-linked glycans makes plants resistant to tunicamycin and obviates the tunicamycin-induced unfolded protein response. Plant Physiol. 1999;121:353–61. doi: 10.1104/pp.121.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hackenberg T, Juul T, Auzina A, Gwizdz S, Malolepszy A, Van Der Kelen K, Dam S, Bressendorff S, Lorentzen A, Roepstorff P, et al. Catalase and NO CATALASE ACTIVITY1 promote autophagy-dependent cell death in Arabidopsis. Plant Cell. 2013;25:4616–26. doi: 10.1105/tpc.113.117192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coll NS, Vercammen D, Smidler A, Clover C, Van Breusegem F, Dangl JL, Epple P. Arabidopsis type I metacaspases control cell death. Science. 2010;330:1393–7. doi: 10.1126/science.1194980. [DOI] [PubMed] [Google Scholar]
- 39.Grant MR, Godiard L, Straube E, Ashfield T, Lewald J, Sattler A, Innes RW, Dangl JL. Structure of the Arabidopsis RPM1 gene enabling dual specificity disease resistance. Science. 1995;269:843–6. doi: 10.1126/science.7638602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.