

Abstract
Common pharmacological treatments of neuropathic and chronic inflammatory pain conditions generally lack efficacy and/or are associated with significant untoward side effects. However, recent preclinical data indicate that combined inhibition of cyclooxygenase (COX) and fatty acid amide hydrolase (FAAH), the primary catabolic enzyme of the endocannabinoid N-arachidonoylethanolamine (anandamide; AEA), produces enhanced antinociceptive effects in a variety of murine models of pain. Accordingly, the primary objective of the present study was to investigate the consequences of co-administration of the COX inhibitor diclofenac and the highly selective FAAH inhibitor PF-3845 in models of neuropathic pain (i.e., chronic constrictive injury of the sciatic nerve (CCI)) and inflammatory pain induced by an intraplantar injection of carrageenan. Here, we report that combined administration of subthreshold doses of these drugs produced enhanced antinociceptive effects in CCI and carrageenan pain models, the latter of which was demonstrated to require both CB1 and CB2 receptors. The combined administration of subthreshold doses of these drugs also increased AEA levels and decreased prostaglandin levels in whole brain. Together, these data add to the growing research that dual blockade of FAAH and COX represents a potential therapeutic strategy for the treatment of neuropathic and inflammatory pain states.
Keywords: endogenous cannabinoid (endocannabinoid), neuropathic pain, inflammatory pain, cyclooxygenase (COX), fatty acid amide hydrolase, anandamide, prostaglandins
INTRODUCTION
Neuropathic pain results from conditions that compromise or abolish nerve function, such as multiple sclerosis, diabetes, nerve injury, or other conditions, and remains among the most challenging pain-related disorders to treat (Campbell and Meyer 2006; Attal, Cruccu et al. 2010). A common symptom of neuropathic pain is allodynia, in which previously non-noxious stimuli are perceived as noxious following nerve injury (Treede, Jensen et al. 2008). Traditional analgesics used to treat neuropathic pain, such as nonsteroidal anti-inflammatory drugs (NSAIDs), anticonvulsants, and opioids lack efficacy and/or carry untoward side effects (Attal, Cruccu et al. 2010). Chronic inflammatory pain represents another challenging clinical problem which faces many of the same treatment challenges as neuropathic pain. A growing body of preclinical data suggests that fatty acid amide hydrolase (FAAH) (Cravatt, Giang et al. 1996; Cravatt, Demarest et al. 2001), the enzyme responsible for the hydrolysis of N-arachidonoylethanolamine (i.e., anandamide; AEA) (Devane, Hanus et al. 1992), represents a promising target for the treatment of neuropathic pain (Rahn and Hohmann 2009) as well as inflammatory pain (Naidu, Kinsey et al. 2010; Ahn, Smith et al. 2011). Genetic deletion (Cravatt, Demarest et al. 2001; Lichtman, Shelton et al. 2004) or pharmacological inhibition of FAAH (Lichtman, Leung et al. 2004; Lichtman, Shelton et al. 2004; Chang, Luo et al. 2006; Jayamanne, Greenwood et al. 2006; Russo, Loverme et al. 2007) produces antinociception in a wide range of pain models. FAAH not only plays a major role in AEA hydrolysis (Giang and Cravatt 1997; Cravatt, Demarest et al. 2001), but also metabolizes other fatty acid amides, including oleamide, oleoylethanolamide (OEA), and palmitoylethanolamide (PEA) (Lichtman, Hawkins et al. 2002) as well as N-acyl taurines (Saghatelian, McKinney et al. 2006). The findings that FAAH-compromised animals display antinociceptive phenotypes that generally require CB1 and/or CB2 receptors (Kinsey, Naidu et al. 2011; Ghosh, Wise et al. 2013) suggest that AEA plays a prominent role in these actions, since it is the only known substrate of this enzyme that binds cannabinoid receptors (Desroches, Charron et al. 2013).
FAAH inhibitors also attenuate allodynia and hyperalgesic responses in nerve injury models of pain (Russo, Loverme et al. 2007; La Rana, Russo et al. 2008; Clapper, Moreno-Sanz et al. 2010; Kinsey, Long et al. 2010) through a cannabinoid receptor mechanism of action, though these drugs generally show partial effectiveness in reversing the heightened nociceptive behavior. Also raising questions as to whether the antinociceptive actions of this class of enzyme inhibitors in preclinical studies will translate into the clinic is that the selective FAAH inhibitor PF04457845 did not reduce pain in osteoarthritis patients (Huggins, Smart et al. 2012). However, there are no published studies that investigated the effectiveness of this compound or other FAAH inhibitors in patients suffering from other types of inflammatory or neuropathic pain. Another strategy to treat pain is to combine different classes of analgesics, which can lead to enhanced antinociceptive effects, and reduced side effects by virtue of reducing the dose of each drug. In support of this idea, combined administration of FAAH inhibitors and cyclooxygenase (COX) inhibitors enhanced antinociception in several preclinical pain assays. Specifically, co-administration of the FAAH inhibitor URB597 and COX1/2 inhibitor diclofenac produced synergistic antinociceptive effects in the acetic acid abdominal stretching assay (Naidu, Booker et al. 2009). Likewise, combination of the peripherally restricted FAAH inhibitor URB937 and the COX1/2 inhibitor indomethacin significantly reduced nociceptive behavior in the carrageenan and CCI models in a synergistic fashion (Sasso, Bertorelli et al. 2012).
The primary objective of the present study was to determine whether combined administration of the FAAH inhibitor PF-3845 (Ahn, Johnson et al. 2009) and the COX1/2 diclofenac sodium (Ku, Wasvary et al. 1975) would produce enhanced anti-allodynic effects in preclinical models of neuropathic pain (i.e., CCI) and inflammatory pain (i.e., intraplantar carrageenan). The rationale for using this drug combination approach was to provide further proof of principle that dual FAAH and COX inhibition represents a desirable strategy for pain reduction as it may allow for decreased doses of NSAIDs that would minimize serious gastric ulcer side effects and/or intestinal bleeding, while increasing efficacy of FAAH inhibitors. We also tested whether the anti-allodynic actions of diclofenac and PF-3845 given in combination required the activation of cannabinoid receptors by employing CB1 and CB2 receptors antagonists. Finally, brain levels of the endocannabinoids AEA and 2-arachidonoyl glycerol (2-AG), and the prostaglandins D2 and E2 were quantified following administration of PF-3845 and diclofenac alone and in combination to assess whether combined inhibition of FAAH and COX would alter relevant lipid substrates and products of these enzymes.
METHODS
Subjects
Male C57BL6/J mice purchased from Jackson Laboratories (Bar Harbor, ME) served as subjects. All mice were housed four per cage and allowed ad libitum access to food and water. The vivarium facilities were maintained at approximately 20–22°C and were maintained on a 12 h light/dark cycle. On the day prior to testing, mice were brought to the testing room and allowed at least 12 h to acclimate to the room. Prior to baseline testing, each mouse was placed upon the testing apparatus and allowed one hour to acclimate to the testing conditions, with sample sizes ranging from 6–16 mice. Following testing, mice were sacrificed humanely via CO2 asphyxiation followed by cervical dislocation. All procedures were approved by and executed in accordance with policies of the Institutional Animal Care and Use Committee (IACUC) of Virginia Commonwealth University.
Drugs
Diclofenac sodium, a COX1/2 inhibitor, was purchased from Sigma-Aldrich (Saint Louis, MO) and the FAAH inhibitor PF-3845 was synthesized by Organix, Inc. (Woburn, MA), according to previously published methods (Ahn, Johnson et al. 2009). PF-3845 was dissolved in vehicle consisting of 1:1:18 parts ethanol (Pharmco, Brookfield, CT), Alkamuls-620 (Rhodia, Cranbury, NJ), and saline (Ricca Chemical Co., Arlington, TX) and diclofenac was dissolved in a vehicle consisting of 1:1:8 parts of dimethyl sulfoxide (J.T. Baker, Phillipsburg), cremophor (Sigma-Aldrich, Saint Louis, MO), and saline. All injections were given via the intraperitoneal (i.p.) route of administration in a volume of 10 μl/g body weight. Drugs were administered one hour before behavioral testing.
Chronic constriction injury (CCI)
The sciatic nerve was ligated as previously described (Kinsey, Long et al. 2009). In brief, mice were anesthetized using 1.5% isoflurane in oxygen at a flow rate of 3 liters per min. The right hind leg was shaved, swabbed with betadine solution, and then swabbed again with alcohol. An incision was made posterior to the femur, the muscle were separated, and the sciatic nerve was exposed and ligated twice using 5-0 braided silk sutures 1 mm in diameter (Surgical Specialties Corporation, Reading, PA). The muscle and skin were sutured using 4-0 nylon monofilament suture, and mice were placed in heated cages and observed during the post-surgical recovery period, which was approximately 2 h. Acetaminophen was made freely available in the drinking water of all mice from 24 h before surgery to 48 h post-surgery. The contralateral paw was utilized as a paired control in each study. Performing CCI with braided silk suture causes allodynia in the hindpaw ipsilateral to the procedure, while having no effect on the paw contralateral to the procedure and does not differ from a sham paw (Kinsey, Wise et al. 2013). Accordingly, each mouse served as its own control, thus reducing the number of mice required for these experiments.
Carrageenan-induced allodynia
Subjects were given an intraplantar injection of 0.3% carrageenan (Sigma, St Louis) in 20 μl of distilled water using a 30 gauge needle. Mice were transported to the testing room, weighed, randomly assigned to the different treatment regimens, and allowed to acclimate for at least 1 h before injections. For consistency with our previous studies (Ghosh, Wise et al. 2013), mechanical allodynia was assessed 5 h post injection. Diclofenac (1, 3, 10, 30 mg/kg) and/or PF-3845 (1, 3, or 10 mg/kg) were injected 1 h prior to testing.
Experimental design
For the combination studies, subthreshold doses of PF-3845 and diclofenac were determined for each model. Based on the dose-response evaluation of these drugs in each assay, 5 mg/kg PF-3845 and 30 mg/kg diclofenac were used in the CCI model, whereas 3 mg/kg PF-3845 and 3 mg/kg diclofenac were used in the carrageen model. In experiments assessing cannabinoid receptor mechanism of action, the CB1 receptor antagonist rimonabant (1 mg/kg) or the CB2 receptor antagonist SR144528 (3 mg/kg) was administered 15 min prior to diclofenac (3 mg/kg) and/or PF-3845 (3 mg/kg) or vehicle. These doses of rimonabant (Lichtman et al., 1996; Lichtman and Martin, 1997, Lichtman et al., 2004) and SR144528 (Conti et al., 2002; Lichtman et al., 2004; Malan et al., 2002) have previously been shown to block the pharmacological effects of cannabinoid receptors.
Allodynia Testing
Mice were assessed for mechanical allodynia as previously described (Kinsey et al. 2010) starting 2 weeks after surgery, once allodynia was confirmed in all mice, after which testing was performed for up to 4 months after surgery. In order to assess mechanical allodynia, von Frey filaments (0.16–6.0g, North Coast Medical, Morgan Hill, CA) were applied, using the up-and-down method (Chaplan, Bach et al. 1994), in which the 0.4g filament was first used to stimulate a given paw five times in succession. Next, higher weight filaments were applied in ascending order. Lifting of the paw in response to three out of five stimulations was coded as a positive response, after which the filaments were applied in descending order to determine the sensory threshold. The paws ipsilateral and contralateral to CCI were assessed for mechanical allodynia. All testing was completed by an experimenter who was blinded to the treatment groups.
Dosing procedure for brain lipids measurement
Using a two-factor design, naïve mice received 5 mg/kg PF-3845 or vehicle, and 30 mg/kg diclofenac or vehicle. At 90 min after dosing, mice were humanely euthanized via CO2 asphyxiation and decapitated. The brains were rapidly harvested, placed in liquid nitrogen, and stored in a −80 °C freezer until assay. This time point was selected to reflect an approximate midpoint in the duration of behavioral testing.
Measurement of brain lipids
Brain tissue was weighed and subsequently Dounce homogenized in 2:1:1 v/v/v CHCl3:MeOH:1% NaCl (8 ml) containing d5-2-arachidonoylglycerol (2-AG), d4-anandamide (AEA), d8-arachidonic acid (AA), and d9-prostaglandin E2 (PGE2). The mixture was vortexed and then centrifuged (1,400 x g, 3 min). The organic layer was removed, CHCl3 was added until the final volume was again 8 ml, and the extraction was repeated. The combined organic extracts were dried under a stream of N2 and resolubilized in 2:1 v/v CHCl3:MeOH (120 μl). 10 μL of resolubilized lipid were injected for positive mode (2-AG, AEA) and negative mode (AA, PGE2, PGD2) measurements, respectively. Lipid measurements were performed by LC-MS as detail below.
Biomarkers were quantified by selected reaction monitoring (SRM) of each metabolite using an Agilent G6460 Triple-Quad instrument. Liquid chromatography (LC) separation was achieved with a Kinetex reverse-phase C18 column (50 mm, 4.6 mm with 2.6 μm diameter particles, Phenomonex) together with a KrudKatcher Ultra HPLC in-line filter. Mobile phase A was made of 95:5 v/v H2O:MeOH, and mobile phase B was composed of 60:35:5 v/v/v i-PrOH:MeOH:H2O. Ammonium hydroxide (0.1%) and formic acid (0.1%) was included to assist in ion formation in negative and positive ionization modes, respectively. For targeted analysis in positive mode, the flow rate for each run started at 0.1 mL/min with 0% B. At 5 min, the solvent was changed immediately to 60% B with a flow rate of 0.4 mL/min and increased linearly to 100% B over 15 min. This was followed by an isocratic gradient of 100% B for 8 min at 0.5 mL/min before equilibrating for 3 min at 0% B at 0.5 mL/min. For targeted analysis in negative mode, the flow rate for each run started at 0.1 mL/min with 0% B. At 3 min, the flow rate was increased by 0.4 mL/min with a linear increase of solvent B to 100% over 17 min. This was followed by isocratic gradient of 100% B for 7 min at 0.5 mL/min before equilibrating for 3 min with 0% B at 0.5 mL/min. For measurement of hydrolysis products in enzyme substrate assays (positive mode), the flow rate for each run started at 0.1 ML/min with 0% B. At 5 min, the solvent was changed immediately to 100% B with a flow rate of 0.4 mL/min. This was followed by an isocratic gradient of 100% B for 5 min at 0.5 mL/min before equilibrating for 5 min with 0% B at 0.5 mL/min. The following parameters (MS) were used to measure the indicated metabolites by SRM (precursor ion, product ion, collision energy in V, polarity): C20:4 MAG or 2-AG (379, 287, 8, positive), d5-2-AG (384, 287, 5, positive), d4-AEA (352, 66, 11, positive), AEA (348, 62, 11, positive), arachidonic acid or AA (303, 303, 0, negative), d8-AA (311, 267, 5, negative), PGE2 (351, 271, 10, negative), PGD2 (351, 271, 10, negative), and d9-PGE2 (360.5, 121.9, 25, negative). MS analysis was performed with an electrospray ionization source with the following parameters: drying gas temperature = 350 °C, drying gas flow rate = 9 L/min, and the nebulizer pressure = 50 psi. Prostaglandin SRM parameters were based on previously reported methods and transitions. Metabolites targeted by SRM were quantified by measuring the area under the peak in comparison with the internal standards.
Data Analysis
One-way, between subjects ANOVA was used to analyze PF-3845 and diclofenac dose-response data. Two-way (diclofenac vs. PF-3845 treatments) between subjects ANOVA was used to analyze the drug combination behavioral and lipid quantification experiments. Dunnett’s test was used for post hoc analysis of the dose-response data (doses compared with vehicle) and Holm-Sidak test was used to analyze drug combination data and brain level data. To test for full or partial attenuation of allodynia, a paired t-test was conducted pairing ipsilateral and contralateral paw data for each treatment where appropriate. All animals were included in the analyses. P values of less than 0.05 were considered statistically significant.
RESULTS
In the CCI model of neuropathic pain, PF-3845 [F(3, 40) = 11.6, p < 0.0001] and diclofenac [F(3, 36) = 13.6, p < 0.0001] significantly attenuated mechanical allodynia (Fig. 1A–1B). Based on these dose-response studies, doses below the threshold of detection were selected for combination administration, in order to test for augmented anti-allodynia. The combination of 5 mg/kg PF-3845 and 30 mg/kg diclofenac produced enhanced attenuation of allodynia relative to the vehicle treatment or either drug alone [interaction between PF-3845 and diclofenac: F(1, 35)=11.0, p< 0.01; Fig. 1C].
Figure 1.

The fatty acid amide hydrolase (FAAH) inhibitor PF-3845 (Panel A: n = 11 mice, p<0.0001) or the cyclooxygenase (COX)1/2 inhibitor diclofenac (Panel B, n = 8–16 mice, p<0.0001) attenuates mechanical allodynia in the mouse CCI model of neuropathic pain. Panel C. Co-administration of inactive, sub-threshold doses both inhibitors (5 mg/kg PF-3845 and 30 mg/kg diclofenac) attenuates neuropathic allodynia (n = 8 mice/group; p<0.0001 vs. all other treatments). Paws ipsilateral to the sciatic injury displayed consistent allodynia throughout testing and did not vary from vehicle treated groups. Dotted lines indicate the average of contralateral paw measurements in all groups for each experiment. Values represent mean ± SEM. **p<0.01, ****p<0.0001
Similarly, both PF-3845 [F(3, 20) = 6.6, p <0.01] and diclofenac [F(4, 21)=8.8, p<0.001] significantly attenuated mechanical allodynia elicited by carrageenan microinjection (Fig. 2A–B). Sub-threshold doses of PF-3845 (3 mg/kg) or diclofenac (3 mg/kg) were administered alone and in combination. Dual administration of these drugs significantly attenuated mechanical allodynia, as compared with all other treatments [interaction between PF-3845 and diclofenac: F(1, 20) = 7.8, p < 0.05; Fig. 2C]. To test cannabinoid receptor mechanism of action, rimonabant or SR144528 was administered 15 min prior to the PF-3845/diclofenac combination. Both rimonabant (p < 0.05) and SR144528 (p < 0.05) attenuated the anti-allodynic effects produced by tandem FAAH and COX inhibition (Fig. 2D).
Figure 2.

PF-3845 (Panel A, p<0.01) or diclofenac (Panel B, p<0.001) attenuated carrageenan-induced mechanical allodynia in a dose-related fashion. Combination of sub-threshold doses of PF-3845 (3 mg/kg) and diclofenac (3 mg/kg) attenuated mechanical allodynia (Panel C, p<0.0001). Panel D. The CB1 receptor antagonist rimonabant (SR1) or the CB2 receptor antagonist SR144528 (SR2) significantly reduced the antinociceptive effects of the FAAH and COX inhibitors given in combination. Dotted lines indicate the average of contralateral paw measurements in all groups for each experiment. *p<0.05, **p<0.01, ***p<0.001#p<0.05 vs. appropriate vehicle control; n = 8 mice/group. Values represent mean ± SEM.
To investigate the consequences of dual COX and FAAH inhibition on biomarkers, we quantified whole brain levels of arachidonic acid, AEA, 2-AG, PGD2, and PGE2 following administration of 5 mg/kg PF-3845 and 30 mg/kg diclofenac given separately and in combination. PF-3845 (5 mg/kg) significantly elevated AEA levels [F(1, 8) = 338, p < 0.0001] regardless of diclofenac treatment (Fig. 3A), but none of the treatments affected 2-AG (Fig. 3B, p = 0.25). PGD2 levels were elevated relative to the vehicle control in the absence of diclofenac (Fig. 3D, p < 0.05). However, neither arachidonic acid (Fig. 3C, p = 0.95) nor PGE2 (Fig. 3E, p = 0.07) brain levels were affected by PF-3845. Diclofenac significantly reduced PGD2 (Fig. 3D) and PGE2 (Fig 3E) levels (p < 0.001) compared with vehicle-vehicle or PF-3845-vehicle groups. Finally, diclofenac given alone did not affect AEA, 2-AG, or arachidonic acid levels, but markedly reduced prostaglandins levels, regardless of PF-3845 administration.
Figure 3.
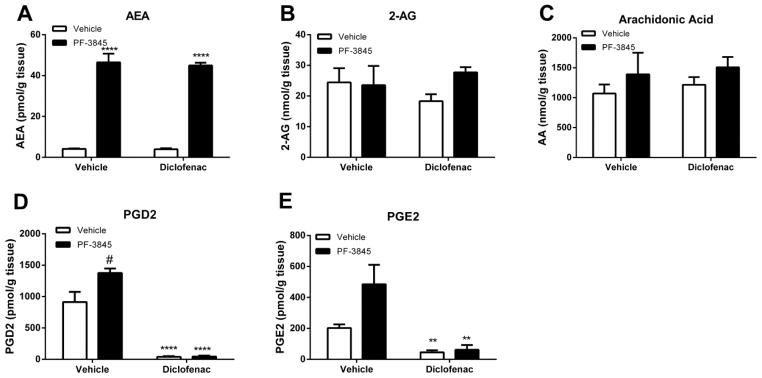
Brain levels of relevant lipids following separate or combined administration of PF-3845 (5 mg/kg) and diclofenac (30 mg/kg). Panel A. PF-3845 significantly elevated brain AEA levels regardless of diclofenac. Neither PF-3845 nor diclofenac given alone or in combination altered 2-AG levels (Panel B) or arachidonic acid levels (Panel C). Panel D. Diclofenac significantly reduced PGD2 levels when given either alone or in combination with PF-3845. PF-3845 administered alone elevated PGD2 levels. Panel E. Diclofenac significantly reduced PGD2 levels when given either alone or in combination with PF-3845. n = 3 mice/group; ** p < 0.01, ****p<0.0001, #p<0.05 vs. vehicle treatment. Values represent mean ± SEM.
DISCUSSION
The results of the present study support the notion that tandem COX and FAAH inhibition produced enhanced reductions in mechanical allodynia in the CCI and carrageenan pain models. These data extend published findings of COX/FAAH analgesic synergy in a visceral pain model (Naidu, Booker et al. 2009) and demonstrate efficacy of this combination therapy in neuropathic and inflammatory pain models. These results also corroborate the report of Sasso et al. (2012) who found that the brain-impermeant FAAH inhibitor, URB937, given in combination with the COX inhibitor indomethacin elicited antinociceptive effects in the carrageenan and CCI assays in a synergistic fashion. Additionally, we showed that the anti-allodynic actions of combined PF-3845 and diclofenac administration in the carrageenan assay required both CB1 and CB2 receptors. The results of these studies taken together provide strong support for the idea that dual FAAH and COX inhibition represents a promising approach to reduce neuropathic and inflammatory pain. An added benefit of this strategy is that reducing the dose of NSAIDs reduces the likelihood of their untoward side effects, such as gastric ulcers and increased risk of cardiac events.
The present study also quantified relevant lipid signaling molecules in whole brain, which contribute to the observed anti-allodynic effects of PF-3845 and diclofenac. Lipidomic analysis indicated that PF-3845 (5 mg/kg) elevated AEA with no effect on arachidonic acid or 2-AG, as previously reported (Ahn, Johnson et al. 2009). Curiously, PF-3845 caused a significant elevation of PGD2 as well as producing a trend to increase PGE2, consistent with findings that FAAH inhibition may elicit a pro-nociceptive phenotype (Ligresti, Martos et al. 2013). Nonetheless, diclofenac abolished prostaglandin levels regardless of PF-3845 administration, consistent with its actions as a COX inhibitor. A limitation of the present study is that the prostaglandin and lipid biomarker assays were conducted on whole brain samples of naïve mice. Given that CB1 receptors, COX and FAAH are expressed differentially throughout the rodent brain (Breivogel, Sim et al. 1997; Egertova, Cravatt et al. 2003), there are likely to be differences in the expression of AEA and prostaglandins in discrete brain or spinal cord regions. Future studies may investigate discrete neural tissues relating to pain, such as the cingulate cortex, thalamus, periaqueductal gray, ventromedial medulla, lumbar spinal cord, and DRGs.
The brain levels of the measured lipids, as well as the antagonism studies, grant insight into possible mechanisms of action for the endocannabinoid-prostaglandin interactions. The enhanced anti-allodynic effects of subthreshold doses of PF-3845 and diclofenac given in combination were attenuated by either rimonabant or SR144528, implicating the involvement of CB1 and CB2 receptors. While the present study did not assess whether cannabinoid receptors play a role in the anti-allodynic effects of this combination in the CCI model, previous reports indicate that both cannabinoid receptors contribute to the anti-allodynic effects of FAAH inhibitors in both the carrageenan and CCI assays (Russo, Loverme et al. 2007; Kinsey, Long et al. 2009; Kinsey, Long et al. 2010; Ghosh, Wise et al. 2013). CB1 receptors are present in the periaqueductal grey (Wilson-Poe, Morgan et al. 2012), as well as other areas involved in pain including cingulate cortex, rostral ventromedial medulla, dorsal horn, and dorsal root ganglia (Herkenham, Lynn et al. 1990). CB2 receptors are known to be associated with immune tissue, microglia and play a role in inflammation (Wotherspoon, Fox et al. 2005; Racz, Nadal et al. 2008; Romero-Sandoval, Nutile-McMenemy et al. 2008; Rom and Persidsky 2013). Thus, FAAH blockade enhances AEA levels, which could reduce allodynia by activating CB1 receptors at different levels of nociceptive processing within the CNS and peripheral nervous system), as well as dampening inflammatory-induced nociception elicited by CB2 receptor activation. In addition, stimulation of CB1 receptors on peripheral nociceptors (Hohmann and Herkenham 1999) may reduce neuronal firing. In the present study, we report that carrageenan-induced allodynia is attenuated by the combination treatment, and that this anti-allodynic effect was blocked by selective antagonism of either CB1 or CB2 receptor. As there is no evidence that diclofenac acts on either cannabinoid receptor, it is reasonable to conclude that PF-3845 reduces allodynia by prolonging AEA activity at each cannabinoid receptors. Although we did not assess cannabinoid receptor involvement in mice subjected to CCI and administered the combination treatment in the present study, previous research using a variety of FAAH inhibitors including URB597, OL-135 (Kinsey, Long et al. 2009), ST4070 (Caprioli, Coccurello et al. 2012), and PF-3845 (Kinsey, Long et al. 2010) were found to reduce CCI-induced allodynia via a CB1 and CB2 receptor mechanism. However, mice subjected to CCI and administered the combination treatment in the present study were not challenged with selective cannabinoid receptor antagonists in the present study. Thus, stimulation of CB1 and CB2 receptors at multiple sites in the nervous system and periphery are likely to contribute to the overall antinociceptive phenotype.
While the results of the present study implicate a significant role of AEA at cannabinoid receptors, other substrates of FAAH (e.g., PEA and OEA) may contribute to the anti-allodynic actions through noncannabinoid receptor mechanisms of action, such as peroxisome proliferator-activated receptor γ (PPARγ) (Lo Verme, Fu et al. 2005; LoVerme, Russo et al. 2006). Diclofenac most likely produced its anti-allodynic actions through as COX inhibitor, thereby preventing prostaglandin synthesis. It is noteworthy that higher doses of diclofenac were required to reduce CCI-elicited allodynia than carrageenan-induced allodynia, consistent with the known reduced efficacy of NSAIDs in reducing neuropathic pain, as compared with inflammatory pain (Negus, Vanderah et al. 2006). Likewise, 3–30 mg/kg diclofenac did not alter nociceptive behavior in a mouse spinal nerve ligation model (Kiso, Watabiki et al. 2008). Thus, the combined administration of subthreshold doses of PF-3845 and diclofenac was sufficient to produce anti-allodynic effects through the activation of multiple cannabinoid receptor pathways, inhibition of prostaglandin synthesis, and potentially other substrates and products of FAAH acting at noncannabinoid sites.
A curious finding was that PF-3845 significantly elevated PGD2 and trended towards increasing PGE2 in brain relative to a vehicle control, effects that were occluded upon co-administration of diclofenac. Inhibiting FAAH may lead to elevated prostaglandin levels through an alternative AEA degradative pathway that could lead to the production of PGD2-ethanolamide, which might then be converted into PGD2 (Koda, Tsutsui et al. 2004). This increase in prostaglandins may work in opposition to the CB1 and CB2 receptor-mediated anti-allodynic effects of PF-3845, which would be abolished by COX inhibition. As central and peripheral components mediate inflammatory and neuropathic pain states, teasing out the exact interplay of the determinants of this pain-evoked behavior remains challenging. Nonetheless, the fact that URB937, a brain-impermeant FAAH inhibitor, produced synergistic anti-allodynic effects when given in combination with indomethacin argues for an important peripheral locus of action (Sasso, Bertorelli et al. 2012). Moreover, the observation that both CB1 and CB2 receptors play a role, suggests the involvement of AEA.
In addition to effects on the endocannabinoid system, combined FAAH and COX inhibition produced notable effects on the production and maintenance of prostaglandins. Currently three cyclooxygenase (COX) isoforms are known: COX1, which is constitutively expressed in a variety of tissues and is important for various physiological processes; COX2, which is induced during the inflammatory immune response (Simmons, Botting et al. 2004); and COX3, which is unlikely to play a significant role in pain and inflammation due to its relative paucity in vivo (Chandrasekharan, Dai et al. 2002). COX enzymes convert arachidonic acid into prostaglandins, which in turn bind to G-protein coupled receptors potentially evoking an inflammatory response. Downstream effects of prostaglandins include inducing the perception of pain and inflammation, both of which can be attenuated by selectively abating prostaglandin synthesis, via COX1/2 inhibition or through other mechanisms. COX2 also metabolizes endocannabinoids, though with substantially decreased efficacy relative to FAAH, implicating them as a possible secondary pathway of AEA degradation (Weber, Ni et al. 2004). Prostamides are the resulting major products (Kozak, Crews et al. 2002; Ross, Craib et al. 2002) and can produce pro-nociceptive effects in vivo (Gatta, Piscitelli et al. 2012). Additionally, blockade of nociceptive behavior may be achieved using antagonists of the putative prostamide F2α receptor (Ligresti, Martos et al. 2013).
Another possible advantage of simultaneously inhibiting COX and FAAH is reducing the prevalence of gastric ulcergenic effects of NSAIDs by virtue of lowering the NSAID dose. Moreover, previous studies have demonstrated that FAAH inhibitors have protective effects on the formation of NSAID-induced gastric ulcers in mice (Naidu, Booker et al. 2009; Kinsey, Nomura et al. 2011; Sasso, Bertorelli et al. 2012). These findings suggest that combination of COX and FAAH inhibitors may not only produce increased antinociceptive effects, but also may decrease the incidence of untoward side effects. Taken together, the results of the present study contribute to the growing body of evidence suggesting that dual inhibition of COX and FAAH enzymes offers a potential therapeutic strategy to attenuate inflammatory and neuropathic pain states (Cipriano, Bjorklund et al. 2013). Specifically, combination of sub-threshold doses of PF-3845 and diclofenac produced marked anti-allodynic effects that required both cannabinoid receptors. These drugs respectively increased AEA and decreased prostaglandins in whole brain, a pattern of results that is generally consistent with known effects of these drugs. In conclusion, the development of dual FAAH-COX inhibitors would be of great potential value as a therapeutic strategy to treat different types of pain. Indeed, efforts are underway to synthesize such compounds (Favia, Habrant et al. 2012; Cipriano, Bjorklund et al. 2013).
PERSPECTIVE.
Tandem inhibition of FAAH and COX attenuates inflammatory and neuropathic pain states, which may avoid potentially harmful side effects of other therapeutic options, such as NSAIDs or opioids.
Highlights.
Combination of diclofenac and PF-3845 attenuates both neuropathic and inflammatory pain.
CB1 and CB2 antagonists block the anti-inflammatory effects of tandem inhibition fatty acid hydrolase and cyclooxygenase enzymes.
Administration of PF-3845 elevates prostaglandins in whole brain, which is abolished by co-administration of diclofenac.
Acknowledgments
We would like to thank the National Institute for Drug Abuse for funding from the following sources: P01-DA009789, P01-DA017259, T32-DA007027, and K99 DA035864 as well as other support through the Tony Rosenburg Fellowship.
Footnotes
DISCLOSURES: The authors state no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn K, Johnson DS, et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009;16(4):411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, Smith SE, et al. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther. 2011;338(1):114–124. doi: 10.1124/jpet.111.180257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attal N, Cruccu G, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol. 2010;17(9):1113–e1188. doi: 10.1111/j.1468-1331.2010.02999.x. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Sim LJ, et al. Regional differences in cannabinoid receptor/G-protein coupling in rat brain. J Pharmacol Exp Ther. 1997;282(3):1632–1642. [PubMed] [Google Scholar]
- Campbell JN, Meyer RA. Mechanisms of neuropathic pain. Neuron. 2006;52(1):77–92. doi: 10.1016/j.neuron.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprioli A, Coccurello R, et al. The novel reversible fatty acid amide hydrolase inhibitor ST4070 increases endocannabinoid brain levels and counteracts neuropathic pain in different animal models. J Pharmacol Exp Ther. 2012;342(1):188–195. doi: 10.1124/jpet.111.191403. [DOI] [PubMed] [Google Scholar]
- Chandrasekharan NV, Dai H, et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A. 2002;99(21):13926–13931. doi: 10.1073/pnas.162468699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Luo L, et al. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br J Pharmacol. 2006;148(1):102–113. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, et al. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53(1):55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cipriano M, Bjorklund E, et al. Inhibition of fatty acid amide hydrolase and cyclooxygenase by the N-(3-methylpyridin-2-yl)amide derivatives of flurbiprofen and naproxen. Eur J Pharmacol. 2013 doi: 10.1016/j.ejphar.2013.09.065. [DOI] [PubMed] [Google Scholar]
- Clapper JR, Moreno-Sanz G, et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci. 2010;13(10):1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A. 2001;98(16):9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, et al. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384(6604):83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Desroches J, Charron S, et al. Endocannabinoids decrease neuropathic pain-related behavior in mice through the activation of one or both peripheral CB and CB receptors. Neuropharmacology. 2013 doi: 10.1016/j.neuropharm.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258(5090):1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Egertova M, Cravatt BF, et al. Comparative analysis of fatty acid amide hydrolase and cb(1) cannabinoid receptor expression in the mouse brain: evidence of a widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience. 2003;119(2):481–496. doi: 10.1016/s0306-4522(03)00145-3. [DOI] [PubMed] [Google Scholar]
- Favia AD, Habrant D, et al. Identification and characterization of carprofen as a multitarget fatty acid amide hydrolase/cyclooxygenase inhibitor. J Med Chem. 2012;55(20):8807–8826. doi: 10.1021/jm3011146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatta L, Piscitelli F, et al. Discovery of prostamide F2alpha and its role in inflammatory pain and dorsal horn nociceptive neuron hyperexcitability. PLoS One. 2012;7(2):e31111. doi: 10.1371/journal.pone.0031111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Wise LE, et al. The monoacylglycerol lipase inhibitor JZL184 suppresses inflammatory pain in the mouse carrageenan model. Life Sci. 2013;92(8–9):498–505. doi: 10.1016/j.lfs.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giang DK, Cravatt BF. Molecular characterization of human and mouse fatty acid amide hydrolases. Proc Natl Acad Sci U S A. 1997;94(6):2238–2242. doi: 10.1073/pnas.94.6.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, et al. Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A. 1990;87(5):1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Herkenham M. Cannabinoid receptors undergo axonal flow in sensory nerves. Neuroscience. 1999;92(4):1171–1175. doi: 10.1016/s0306-4522(99)00220-1. [DOI] [PubMed] [Google Scholar]
- Huggins JP, Smart TS, et al. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153(9):1837–1846. doi: 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Jayamanne A, Greenwood R, et al. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br J Pharmacol. 2006;147(3):281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, et al. Fatty acid amide hydrolase and monoacylglycerol lipase inhibitors produce anti-allodynic effects in mice through distinct cannabinoid receptor mechanisms. J Pain. 2010;11(12):1420–1428. doi: 10.1016/j.jpain.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, et al. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther. 2009;330(3):902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Naidu PS, et al. Fatty acid amide hydrolase blockade attenuates the development of collagen-induced arthritis and related thermal hyperalgesia in mice. Pharmacol Biochem Behav. 2011;99(4):718–725. doi: 10.1016/j.pbb.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Nomura DK, et al. Inhibition of monoacylglycerol lipase attenuates nonsteroidal anti-inflammatory drug-induced gastric hemorrhages in mice. J Pharmacol Exp Ther. 2011;338(3):795–802. doi: 10.1124/jpet.110.175778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Wise LE, et al. Repeated low-dose administration of the monoacylglycerol lipase inhibitor JZL184 retains cannabinoid receptor type 1-mediated antinociceptive and gastroprotective effects. J Pharmacol Exp Ther. 2013;345(3):492–501. doi: 10.1124/jpet.112.201426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiso T, Watabiki T, et al. Pharmacological characterization and gene expression profiling of an L5/L6 spinal nerve ligation model for neuropathic pain in mice. Neuroscience. 2008;153(2):492–500. doi: 10.1016/j.neuroscience.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Koda N, Tsutsui Y, et al. Synthesis of prostaglandin F ethanolamide by prostaglandin F synthase and identification of Bimatoprost as a potent inhibitor of the enzyme: new enzyme assay method using LC/ESI/MS. Arch Biochem Biophys. 2004;424(2):128–136. doi: 10.1016/j.abb.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Kozak KR, Crews BC, et al. Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J Biol Chem. 2002;277(47):44877–44885. doi: 10.1074/jbc.M206788200. [DOI] [PubMed] [Google Scholar]
- Ku EC, Wasvary JM, et al. Diclofenac sodium (GP 45840, Voltaren), a potent inhibitor of prostaglandin synthetase. Biochem Pharmacol. 1975;24(5):641–643. doi: 10.1016/0006-2952(75)90186-0. [DOI] [PubMed] [Google Scholar]
- La Rana G, Russo R, et al. AM404, an anandamide transport inhibitor, reduces plasma extravasation in a model of neuropathic pain in rat: role for cannabinoid receptors. Neuropharmacology. 2008;54(3):521–529. doi: 10.1016/j.neuropharm.2007.10.021. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Hawkins EG, et al. Pharmacological activity of fatty acid amides is regulated, but not mediated, by fatty acid amide hydrolase in vivo. J Pharmacol Exp Ther. 2002;302(1):73–79. doi: 10.1124/jpet.302.1.73. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Leung D, et al. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol Exp Ther. 2004;311(2):441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Shelton CC, et al. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain. 2004;109(3):319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Ligresti A, Martos J, et al. Prostamide F receptor antagonists with inhibitory activity at FAAH: a way to prevent the confounding effects of pro-inflammatory mediators formed following selective FAAH inhibition? Br J Pharmacol. 2013 doi: 10.1111/bph.12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Verme J, Fu J, et al. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67(1):15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- LoVerme J, Russo R, et al. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol Exp Ther. 2006;319(3):1051–1061. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- Naidu PS, Booker L, et al. Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J Pharmacol Exp Ther. 2009;329(1):48–56. doi: 10.1124/jpet.108.143487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidu PS, Kinsey SG, et al. Regulation of inflammatory pain by inhibition of fatty acid amide hydrolase. J Pharmacol Exp Ther. 2010;334(1):182–190. doi: 10.1124/jpet.109.164806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Vanderah TW, et al. Preclinical assessment of candidate analgesic drugs: recent advances and future challenges. J Pharmacol Exp Ther. 2006;319(2):507–514. doi: 10.1124/jpet.106.106377. [DOI] [PubMed] [Google Scholar]
- Racz I, Nadal X, et al. Crucial role of CB(2) cannabinoid receptor in the regulation of central immune responses during neuropathic pain. J Neurosci. 2008;28(46):12125–12135. doi: 10.1523/JNEUROSCI.3400-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn EJ, Hohmann AG. Cannabinoids as pharmacotherapies for neuropathic pain: from the bench to the bedside. Neurotherapeutics. 2009;6(4):713–737. doi: 10.1016/j.nurt.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rom S, Persidsky Y. Cannabinoid receptor 2: potential role in immunomodulation and neuroinflammation. J Neuroimmune Pharmacol. 2013;8(3):608–620. doi: 10.1007/s11481-013-9445-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Sandoval A, Nutile-McMenemy N, et al. Spinal microglial and perivascular cell cannabinoid receptor type 2 activation reduces behavioral hypersensitivity without tolerance after peripheral nerve injury. Anesthesiology. 2008;108(4):722–734. doi: 10.1097/ALN.0b013e318167af74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA, Craib SJ, et al. Pharmacological characterization of the anandamide cyclooxygenase metabolite: prostaglandin E2 ethanolamide. J Pharmacol Exp Ther. 2002;301(3):900–907. doi: 10.1124/jpet.301.3.900. [DOI] [PubMed] [Google Scholar]
- Russo R, Loverme J, et al. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J Pharmacol Exp Ther. 2007;322(1):236–242. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- Saghatelian A, McKinney MK, et al. A FAAH-regulated class of N-acyl taurines that activates TRP ion channels. Biochemistry. 2006;45(30):9007–9015. doi: 10.1021/bi0608008. [DOI] [PubMed] [Google Scholar]
- Sasso O, Bertorelli R, et al. Peripheral FAAH inhibition causes profound antinociception and protects against indomethacin-induced gastric lesions. Pharmacol Res. 2012;65(5):553–563. doi: 10.1016/j.phrs.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DL, Botting RM, et al. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56(3):387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- Treede RD, Jensen TS, et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. 2008;70(18):1630–1635. doi: 10.1212/01.wnl.0000282763.29778.59. [DOI] [PubMed] [Google Scholar]
- Weber A, Ni J, et al. Formation of prostamides from anandamide in FAAH knockout mice analyzed by HPLC with tandem mass spectrometry. J Lipid Res. 2004;45(4):757–763. doi: 10.1194/jlr.M300475-JLR200. [DOI] [PubMed] [Google Scholar]
- Wilson-Poe AR, Morgan MM, et al. Distribution of CB1 cannabinoid receptors and their relationship with mu-opioid receptors in the rat periaqueductal gray. Neuroscience. 2012;213:191–200. doi: 10.1016/j.neuroscience.2012.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotherspoon G, Fox A, et al. Peripheral nerve injury induces cannabinoid receptor 2 protein expression in rat sensory neurons. Neuroscience. 2005;135(1):235–245. doi: 10.1016/j.neuroscience.2005.06.009. [DOI] [PubMed] [Google Scholar]