

Abstract
Mycoplasma gallisepticum, the most pathogenic mycoplasma in poultry, is able to glide over solid surfaces. Although this gliding motility was first observed in 1968, no specific protein has yet been shown to be involved in gliding. We examined M. gallisepticum strains and clonal variants for motility and found that the cytadherence proteins GapA and CrmA were required for gliding. Loss of GapA or CrmA resulted in the loss of motility and hemadsorption and led to drastic changes in the characteristic flask-shape of the cells. To identify further genes involved in motility, a transposon mutant library of M. gallisepticum was generated and screened for motility-deficient mutants, using a screening assay based on colony morphology. Motility-deficient mutants had transposon insertions in gapA and the neighbouring downstream gene crmA. In addition, insertions were seen in gene mgc2, immediately upstream of gapA, in two motility-deficient mutants. In contrast to the GapA/CrmA mutants, the mgc2 motility mutants still possessed the ability to hemadsorb. Complementation of these mutants with a mgc2-hexahistidine fusion gene restored the motile phenotype. This is the first report assigning specific M. gallisepticum proteins to involvement in gliding motility.
Electronic supplementary material
The online version of this article (doi:10.1186/s13567-014-0099-2) contains supplementary material, which is available to authorized users.
Introduction
Motility is regarded as a virulence factor in many pathogenic bacteria. The ability to move enables microorganisms to reach a specific niche or to leave hostile environments. Amongst motile bacteria, various mechanisms to create a momentum have evolved. In Bordetella bronchiseptica, Escherichia coli, and Salmonella enterica serovar Typhimurium flagellar motility has been shown to be crucial for the initial stages of infection, while in Legionella pneumophila motility is necessary to establish and maintain infection [1]. In contrast to these species, in which motility can be downregulated to favor a specific life-style, some bacteria, such as Helicobacter, Campylobacter, and Pseudomonas aeruginosa, depend on constitutive flagellar motility for successful infection [1]. Experiments showing that only motile bacteria can be reisolated after infection with a mixed population of motile and non-motile variants underline the importance of motility in the infection process [2].
Mycoplasmas lack a cell wall and are considered to be the smallest self-replicating microorganisms. They have limited biosynthetic capabilities as they are highly adapted to a parasitic life-style [3]. In spite of the many limitations that have resulted from their degenerative evolution, some mycoplasmas have the ability to travel over inert surfaces, like glass, plastic or over eukaryotic cells, even though they lack any obvious locomotory appendages such as flagella or pili [4].
Mycoplasma gallisepticum is an avian pathogen causing chronic respiratory disease in chickens and infectious sinusitis in turkeys, that is known to possess gliding motility. Like the majority of gliding mycoplasmas, M. gallisepticum belongs to the M. pneumoniae cluster [5], named after M. pneumoniae, the causative agent of human bronchitis and atypical pneumonia [6]. The mechanism that enables M. pneumoniae and other mycoplasmas to glide has been the subject of a number of studies [7].
The best studied gliding mechanism is that of M. mobile, isolated from the gills of a fresh-water fish [8], which is phylogenetically distant from the pneumoniae cluster. M. mobile can be cultivated at room temperature and its average gliding speed is 2 to 4.5 μm/s [9], thus visualization of the gliding process is not dependent on additional microscope equipment such as a plate heater or a computer-connected CCD video camera. Several proteins of M. mobile have been identified as motility proteins [10]. Centered at the neck region of the jellyfish shape-like M. mobile, the Gli349 leg protein binds to sialylated oligosaccharides on glass or animal cells. Together with the Gli521 gear protein and the Gli123 mount protein, a large number of legs may act in a continuous “bind, pull, and release” mode, thereby creating a continuous pull in the forward direction. The multiple legs involved suggested the term “centipede-like” locomotion [11].
However, no homologs of these M. mobile motility genes have been found in M. pneumoniae or M. gallisepticum, indicating that different mycoplasmas may have developed different gliding machineries. The motile members of the M. pneumoniae cluster share a characteristic morphological feature, cellular polarity. These mycoplasmas have a flask-shaped appearance, strengthened by a cytoskeleton, and have a differentiated tip structure, often called the attachment tip or terminal organelle (TO). In M. pneumoniae, the TO mediates adherence to the host respiratory epithelium, a prerequisite for successful colonization [12]. In addition, the TO is the leading end in gliding motility [13], as cells always glide in the direction of the tip structure.
Formation of the TO appears to be a complex process that has to be well orchestrated, chronologically and spatially [14]. The TO of M. pneumoniae consists of a network of cytadherence proteins, including P1, P30, the accessory proteins P65, B, C, and the structural proteins HMW1, HMW2, and HMW3 [15]. Mutations affecting cytadherence or the correct assembly of the TO have direct effects on gliding motility. Loss of proteins P1, P30, or P65 lead to a non-motile, as well as hemadsorption-negative, phenotype [16]. Similarly, mutations in the TO proteins P41 and P24 have an impact on the velocity and frequency of gliding [17]. Although several elements of the gliding machinery have been identified, it is still unclear how these motility-associated proteins work in concert to generate a propulsive force and move the cell forward.
Studies to elucidate the motility mechanisms of members of the pneumoniae cluster have also included M. genitalium, a close relative of M. pneumoniae. Their proteins share a high degree of homology [18]. Many of the proteins involved in M. pneumoniae motility have counterparts in M. genitalium [19]. Surprisingly, no protein involved in motility has yet been identified in M. gallisepticum, and although M. gallisepticum was included in a recent study of mycoplasma gliding [20], little is known about the proteins involved. Therefore, we examined the gliding ability of M. gallisepticum strain R and clonal variants of it, including a library of transposon insertion mutants. The aim of this study was to identify proteins that contribute to the motility process of M. gallisepticum, to investigate the molecular properties of such motility proteins, and to further refine the tools for screening and complementing motility mutants.
Materials and methods
Strains and growth conditions
M. gallisepticum strains Rlow, Rhigh [21], RCL1, RCL2, mHAD3 [22], motility mutants and complemented motility mutants were cultured in modified Hayflick medium [23] (HFLX) at 37 °C. To grow tetracycline- (TcR) or chloramphenicol- resistant (CmR) M. gallisepticum transformants, either Tc (4 μg mL−1; Roche Diagnostics, Penzberg, Germany) or Cm (17 μg mL−1; Carl Roth GmbH & Co KG, Karlsruhe, Germany) were added to HFLX medium. Escherichia coli DH10B (Invitrogen Corp., Carlsbad, CA, USA) was used for the propagation of plasmids used in this study.
Motility assays
To detect satellite growth of M. gallisepticum, a freshly grown culture was seeded in a 24-well microtiter plate at a concentration of 40 CFU per 400 μL of HFLX medium per well. After 2 h of attachment, the medium was replaced by HFLX containing 2% gelatin and, if transformants were to be analyzed, Tc was added to the HFLX-gelatin mixture. Colony morphology was examined after growth at 37 °C for five to seven days using an SMZ-U stereomicroscope (Nikon Corp., Tokyo, Japan).
Characterization of M. gallisepticum gliding motility was performed using a microcinematography motility assay (MMA). For this purpose, 100 μL of a culture freshly grown in HFLX medium was placed on a standard microscope glass slide (Thermo Fisher Scientific Inc., Waltham, MA, USA). After 1 h of incubation at 37 °C, attached cells were overlaid with 100 μL of fresh medium containing 2% gelatin. After 1.5 h of incubation, cell movement was examined using an Olympus AX70 microscope equipped with a heating plate set at 37 °C, and phase-contrast images were captured at 1-s intervals for a total of 180 s with a Color View CCD digital camera controlled using CellPlus (Olympus Soft Imaging Solutions GmbH, Muenster, Germany).
Computer-assisted qualitative analysis of motility was performed by overlaying 180 single frames of a 3 min microscope movie with the Z project tool of the Fiji image processing package [24], choosing “Minimum Intensity” as the critical parameter. Bacterial paths were highlighted by standard layer manipulations using Photoshop CS3 version 10.0 (Adobe Systems Inc., San Jose, CA, USA).
For a quantitative analysis of motility, the ten mycoplasmas with the longest Z project paths in each field of view were selected, and their movements were tracked using the ImageJ/Fiji MTrackJ plugin [25]. Using the analysis tool of MTrackJ, the distance travelled and the overall speed of motility, including resting periods, were determined. The best three results of 5 independent experiments were chosen for graphical representation.
Results of quantitative MMAs were analyzed for statistical significance by using a two-tailed Student’s T test [26]. P values of ≤ 0.05 were considered to indicate significant differences between groups.
DNA isolation and sequencing reactions
Genomic DNA from mycoplasmas was isolated using the GenElute™ Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany). Plasmid DNA from E. coli cultures was purified using the PureYield™ Plasmid System Kit (Promega, Mannheim, Germany). Oligonucleotide synthesis was performed by either Microsynth (Microsynth AG, Balgach, Switzerland) or Invitrogen (Life Technologies GmbH, Darmstadt, Germany), and DNA sequencing was conducted by LGC Genomics (LGC Genomics GmbH, Berlin, Germany). If not otherwise mentioned, all enzymes used in this study were purchased from Promega. For DNA/PCR purification, the Wizard® SV Gel and PCR Clean-Up System (Promega) was used.
Construction of plasmids
Transposon Tn4001cam
To use transposon mutants in gentamicin-based cell invasion assays, we first had to replace the gentamicin resistance gene of Tn4001. The chloramphenicol-resistance cassette CmR of plasmid pACYC184 (Invitrogen) was amplified using primers Xcat5 and Xcat3, introducing BamHI and NarI cleavage sites, respectively. The purified amplicon was cloned into the corresponding sites of plasmid p5TlacZ + [27], thereby placing the CmR cassette under the control of tufPO in plasmid p5xCAT. Left (ISL) and right (ISR) IS256 elements of S. aureus transposon Tn4001mod were amplified from plasmid pISM2062 [28], using primers ISR-f and ISR-r, which introduced MluI and KasI cleavage sites, and ISL-f and ISL-r, which contained SacII and SalI cleavage sites. The amplicons ISL and ISR were cloned into the corresponding sites to the right and to the left end, respectively, of the CmR cassette on plasmid p5xCAT. Transformants of E. coli DH10B were selected on Luria-Bertani agar containing Cm (30 μg/mL). Transposon mutants of M. gallisepticum were stable for at least 20 passages without Cm and no re-transposition or excision of Tn4001cam could be detected (data not shown).
Integration plasmid p5Hmgc
Tn4001mod on plasmid pISM2062 [28] was modified by adding a 6xHis-tag and a multiple cloning site: a 51-bp DNA fragment, created by annealing oligonucleotides HisC-f and HisC-r, was inserted between the BamHI and SmaI cleavage sites of pISM2062, resulting in plasmid pTnHis. The M. gallisepticum gene mgc2 was then amplified by PCR using genomic DNA of strain Rlow as template and primers ISM-mgcF and ISM-mgcR, and subcloned into pTnHis using the BamHI and SphI cleavage sites. The resulting plasmid, pTHmgc, was linearized with NotI, treated with the Klenow fragment of DNA polymerase I (New England Biolabs GmbH, Frankfurt/Main, Germany) to fill in the 5′ overhang, and subsequently digested with BamHI. A 1093-bp fragment was gel-purified and ligated to a 3.5-kb fragment of plasmid pINT [27], obtained after digestion with BamHI and SfoI.
Transformation of mycoplasmas
M. gallisepticum transposon mutants were generated by electroporation of strain RCL1 with 3–5 μg of pTnC, as described previously [22]. For the transformation with integration plasmid p5Hmgc, 20–30 μg of plasmid DNA was used. Following electroporation, mycoplasma cells were cultured on HFLX plates containing either 17.5 μg chloramphenicol mL−1 or 4 μg tetracycline mL−1.
Ligation mediated PCR (LM-PCR)
Transposon insertion sites were determined by LM-PCR using the method of Sharma et al. [29], with modifications. Briefly, genomic DNA of M. gallisepticum transposon mutants was digested with BglII, and ligated to the BglII-adaptors Ad1B and Ad2B. Prior to ligation, the adaptor oligo nucleotides were dissolved separately in double distilled water at a concentration of 100 μM and equal volumes of both were mixed together. The mixture was incubated at 70 °C for 10 min, allowed to cool gradually to 40 °C, and then incubated at 40 °C for 10 min. The mixture was then cooled gradually to 25 °C and stored frozen in small aliquots until further usage. The ligation product was used as a template for PCR amplification using the adaptor-specific primer Bgl and primer IS-I, specific for IS256 of Tn4001mod. The PCR product was then used as template for a semi-nested PCR using primers Bgl and IS-N, and the gel-purified amplicons were sequenced (Microsynth).
Production of antibodies
MGC2
The full-length mgc2 gene was amplified using the LR-PCR kit (Roche) and primers mgc2_3 and mgc2_4 (Table 1), introducing EcoRI and HindIII cleavage sites at either end. The TGA codon was mutagenized to TGG using primer mgc_tga and the Site-directed Mutagenesis kit (Stratagene) according to the manufacturer’s instructions. The mutated mgc2 gene was then cloned between the EcoRI and HindIII cleavage sites in pRSET (Invitrogen) and introduced into E. coli BL21 (DE3)pLys Star (Invitrogen). Expression of MGC2 was induced by addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and the protein was purified using the MagneHis™ Protein Purification System (Promega) and immobilized metal affinity chromatography using ProBond™ nickel-chelating resin (Invitrogen) according to the manufacturers’ instructions. Purity of the MGC2 protein was confirmed by Western blotting using Anti-Xpress™ antibodies (1:5000) (Invitrogen). After elution and dialysis against phosphate-buffered saline (PBS), the protein was used for immunization of rabbits as described elsewhere [30].
Table 1.
Oligonucleotides used in this study
| Primer | Sequence (5′ to 3′) | Product (length [bp]) |
|---|---|---|
| Ad1B | CTCGTAGACTGCGTACC | LM-PCR (variable) |
| Ad2B | GATCGGTACGCAGTCTAC | |
| C’gapA5 | ATTAggatccAGTATTCAACGTTTCTAAG | MG gapA (911 bp) |
| C’gapA3 | TACGaagcttACCTTAATTATTCAATTTTC | |
| HisC-f | GATCCCTCGAGCCCGGGGCATGCCATCATCATCATCATCATTAATAGGG | synthetic 6xHis-tag |
| HisC-r | CGCCCTATTAATGATGATGATGATGATGGCATGCCCCGGGCTCGA | |
| ISL-f | TATAccgcggATAAAGTCCGTATAATTGTG | IS256L (1365 bp) |
| ISL-r | TATAccgcggATAAAGTCCGTATAATTGTG | |
| ISR-f | TATAacgcgtGATAAAGTCCGTATAATTGTG | IS256R (1342 bp) |
| ISR-r | ATTggcccgAAAATAATAAAGGAAGTGAGTC | |
| ISM-mgcF | ATAAggatccTGTTGAAAAGCGCTTAGC | MG mgc2 |
| ISM-mgcR | TTAAgcatgcTCTAGGTCCATTTTGTGG | (1001) |
| Bgl | TAGACTGCGTACCGATC | LM-PCR |
| IS-I | TGTACCGTAAAAGGACTG | products |
| IS-N | AAAGGACTGTTATATGGC | (variable) |
| mgc2_3 | ACGCAGgaattcATAACAATTATG | MG mgc2 |
| mgc2_4 | TTTACAaagcttGTCTTATCTAGG | (894) |
| mgc_tga | GAAAGATTACCTCCGAACCATGGTTTTATCCAGTAGTGGG | TGA > TGG |
| Xcat5 | TAGATGggatccATGGAGAAAAAAATCACTG | pACYC184 (751) |
| Xcat3 | ATAAATggcgccCGCTTATTATCACTTATTC | CmPO + Cm R |
C-GapA
The 3′-terminal part of the gapA gene was amplified using the LR-PCR system (Roche) and primers C’gapA5 and C’gapA3, introducing BamHI and HindIII cleavage sites, at either end. The gel-purified amplicon was ligated into plasmid pRSET-B (Invitrogen) and the resulting plasmid was introduced into E. coli BL21 (DE3)pLys Star (Invitrogen). The recombinant culture was grown at 28 °C, and gene expression was induced by addition of 0.3 mM IPTG at the early logarithmic growth phase. A protein of 35 kDa was retrieved from a sodium dodecyl sulfate polyacrylamide (SDS-PAA) gel after negative staining with a zinc stain and destain kit (BioRad Laboratories Inc., Hercules, CA, USA) and electroelution in an Electro-Eluter Model 422 (BioRad). The immunization of rabbits with the purified C-terminal part of GapA followed exactly the same procedures as for MGC2 antibodies.
CrmA
The generation of CrmA-specific antibodies has been described previously [30].
Western blot analyses and tryptic digestion
Five millilitre aliquots of overnight cultures of mycoplasmas were centrifuged at 4200 g for 30 min, and the cell pellets washed once with PBS and resuspended in PBS containing 0.05% of a commercial trypsin/EDTA solution (Life Technologies, #15400). Control samples were treated in the same way but without trypsin. The samples were incubated at 37 °C, 1-mL samples removed after 10, 30, 40, 50, 70 and 90 min, and trypsin activity stopped by adding phenylmethylsulfonyl fluoride (PMSF) at a final concentration of 1 mM. Cells were then collected by centrifugation and the presence of MGC2 was assessed by Western blot analysis. The solubilization and separation of mycoplasma cell lysates using 10% SDS-PAA gel electrophoresis, and Western blot analysis is described elsewhere [31]. Membranes were probed with antibodies against MGC2 (1:500), GapA (1:6000), or CrmA (1:2000) using peroxidase-conjugated swine-anti-rabbit IgG (1:2000, Dako) as secondary antibody, or with anti-6xHis antibody (1:6000, Aviva Systems Biology Corp., San Diego, CA, USA) in combination with the AffiniPure Goat Anti-Mouse IgG, Fc Fragment specific (1:10 000, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA).
Hemadsorption assay
The ability of mycoplasma colonies grown on HFLX agar plates to hemadsorb was tested as described previously [30].
Scanning electron microscopy (SEM)
Mycoplasma samples for SEM were prepared as described previously [32], except that mycoplasma cultures were grown at 37 °C on glass coverslips, precoated with poly-L-lysine (Sigma-Aldrich) according to the manufacturer’s instructions.
Results
Identification of non-motile M. gallisepticum strains
To establish a method for screening for motility mutants of M. gallisepticum, we first identified motile and non-motile strains in our culture collection. Gliding of M. mobile and M. pneumoniae depends on cytadherence-associated components [16,33,34]. Therefore, we analyzed selected strains of M. gallisepticum that differed in hemadsorption (HA) (Table 2) using a qualitative microcinematography motility assay (MMA). The HA-positive (HA+) M. gallisepticum strain Rlow and its clonal variant RCL1, expressing the major cytadherence gene gapA as well as the cytadherence-related gene crmA, were capable of gliding. At any time, 60% of cells were moving, interrupted by short resting periods. Mycoplasma gliding paths were visualized by computer-generated overlay of all frames of a 3 min video and consisted mainly of circles and bends (Figure 1). In contrast, no gliding could be visualized for strains Rhigh, RCL2, or mHAD3, which lack either GapA and/or CrmA. These HA− strains appeared to have lost the ability to glide, as no moving cells were observed in MMAs, in spite of numerous trials under a variety of conditions.
Table 2.
Protein content, motility and hemadsorption ability of MG strain
| Strain | Hem-adsorption | Motility | Cell Shape | Presence of | Reference | ||
|---|---|---|---|---|---|---|---|
| MGC2 | GapA | CrmA | |||||
| Rlow | ++ | ++ | flask | + | + | + | [21] |
| Rhigh | - | - | round | + | - | - | [21] |
| RCL1 | ++ | ++ | flask | + | + | + | [22] |
| RCL2 | - | - | round | + | - | - | [22] |
| mHAD3 | - | - | round | + | (+) | - | [22] |
| T932A | + | - | rounded flask | - | + | + | this study |
| T932C | + | - | distorted flask | trc1 | + | + | this study |
1trc; truncated.
Figure 1.

Gliding paths of motile M. gallisepticum. A stack of phase-contrast pictures of M. gallisepticum RCL1, captured at 1-s intervals, was manipulated with Fiji by applying a Z projection method [24] to visualize the paths of gliding mycoplasmas. Differential colouring of mycoplasmas (red) and their paths (green) was done with Adobe Photoshop. This qualitative microcinematography motility assay allowed the rapid assessment of the motility of wild-type and mutant strains.
Colonies of motile M. gallisepticum form microsatellites
Formation of microsatellites around colonies grown on agar plates has been described for M. mobile [33] and M. pneumoniae [16], and appears to be an indicator of gliding motility in mycoplasmas. M. gallisepticum colonies have been reported to grow without satellite formation under the conditions that allow M. mobile to form microsatellites [33]. Conditions for microsatellite formation by M. gallisepticum were established using strains Rlow and Rhigh as prototypes of motile and non-motile strains (Table 2). Diffuse colonies spreading in all directions were observed for both strains when grown on HFLX medium solidified with a range of concentrations of agar (0.05 - 0.3%) (Figure 2). Higher concentrations of agarose have already been shown to allow only the formation of the typical fried-egg colonies and were therefore not tested.
Figure 2.
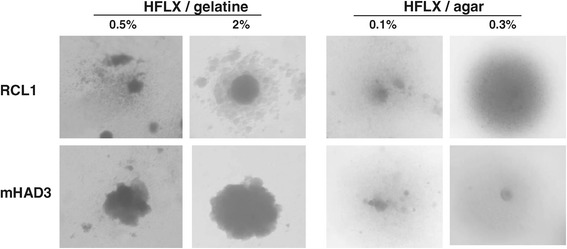
Morphology of M. gallisepticum colonies. Cultures of RCL1 and mHAD3 were either grown in HFLX medium solidified with 0.5 or 2% gelatin or on low-agar plates containing 0.1 or 0.3% agar. In gelatin-containing medium, the non-motile strain mHAD3 formed compact colonies, in contrast to the motile RCL1 which formed colonies surrounded by microsatellites.
When M. gallisepticum cells were first allowed to attach to the surface of a cell culture dish and then overlaid with HFLX medium containing gelatin, motile strains could be differentiated from non-motile strains (Figure 2). At 0.5% gelatin, Rlow and Rhigh were not able to form colonies on the bottom of the dish, but cloudy regions in the overlay medium indicated that mycoplasma cells had spread throughout the medium. With increasing gelatin concentrations, the number of colonies increased, while growth in the overlay medium decreased. At 2% gelatin, the motile strains Rlow and RCL1 formed round colonies with a smooth surface, surrounded by many satellites, while the non-motile strains Rhigh, RCL2, and mHAD3 formed colonies with a rough surface, uneven edges, and without satellite colonies. Higher concentrations of gelatin resulted in partial detachment of colonies and formation of microcolonies in the overlay medium. Therefore, for further experiments to detect colonies of M. gallisepticum with a satellite-growth altered (SGA) phenotype, HFLX medium solidified with 2% gelatin was used.
Generation of motility-deficient mutants and complementation
To identify proteins involved in the gliding motility of M. gallisepticum, RCL1 was transformed with transposon Tn4001cam and transformed colonies were screened for the SGA phenotype. In a proof-of-concept study, 4000 colonies were screened and 38 mutants were found to exhibit defects in satellite colony formation. Their ability to glide was further examined individually using MMAs. Eight mutants had a low proportion of motile cells (20%), so they were stored for later analysis. Southern blot analyses of thirty motility mutants found that the majority carried multiple Tn4001cam insertions within their genome. Only ten mutants had contained only one or two transposon insertions. They were subjected to LM-PCR to precisely determine the transposon insertion sites. In four mutants the transposon had integrated into the gapA gene, and in another four mutants Tn4001cam was inserted into the crmA gene (Figure 3). These mutants were not analyzed further, because the same genes were affected in the cytadherence-negative, non-motile strains Rhigh, RCL2, and mHAD3. However, in two motility-deficient mutants the transposon had integrated into different sites in the mgc2 gene (also known as the MGA_0932 coding region) and these mutant strains were designated T932A and T932C (Figure 3). After filter cloning, a LM-PCR analysis revealed that T932A contained the Tn4001cam at position 222 517 (numbering according to GenBank Accession AE015450.2) of the genomic DNA, 344 bp downstream of the initial coding nucleotide of the mgc2 gene (Figure 3). The ORF therefore terminated 119 amino acids (aa) after the start codon, whereby the last 5 aa were encoded by the transposon. Even after repeated filter cloning mutant T932C still appeared to contain two transposons, one within the mgc2 gene at position 222 749, which would allow for translation of 193 aa of the MGC2 protein. The second transposon was found in the CRISPR region at position 930 903.
Figure 3.
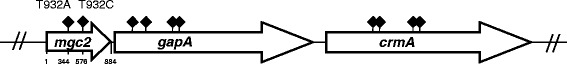
Insertion sites of Tn 4001 cam transposon in motility mutants. Non-motile RCL1 mutants harbored transposons in the mgc cytadherence locus, consisting of mgc2, gapA (formerly mgc1), and crmA (formerly mgc3). In mutants T932A and T932C Tn4001cam integrated 344 and 576 bp after the translational initiation nucleotide of mgc2.
To confirm that disruption of the mgc2 ORF by transposition was responsible for the loss of motility, mutants T932A and T932C were complemented with an mgc2-6xHis fusion gene. For this purpose, the fusion gene was subcloned into a derivative of plasmid pINT [27], which integrates into the oriC region of M. gallisepticum by homologous recombination. The resulting plasmid p5Hmgc was introduced into the mutants by electroporation, and the integration into the genomic oriC locus was proven by Southern blot analyses.
Characterization of mgc2 mutants and complemented mutants
Expression and surface localization of MGC2
The effect of transposon integration on expression of mgc2 and the gapA and crmA genes immediately downstream of it was investigated by Western blot analyses. For this purpose, polyvalent rabbit antisera against MGC2, GapA and CrmA were produced. MGC2 was equally well detected in HA+ and HA−M. gallisepticum strains (Table 2). In contrast, no MGC2 could be detected in T932A lysates (Figure 4A, lane 2), and only a truncated MGC2, with an apparent size of 19 kDa, was detected in T932C (Figure 4A, lane 3). When complemented with p5Hmgc, both T932A and T932C expressed full-length MGC2 at concentrations comparable to RCL1 (Figure 4A, lanes 4–5). The C-terminal 6xHis-tag did not appear to influence the stability of the recombinant MGC2.
Figure 4.

Absence of MGC2 protein in gliding mutants. Immunoblot analysis of RCL1 (lane 1), motility mutants T932A (lane 2) and T932C (lane 3), and complemented mutants T932A::p5Hmgc (lane 4) and T932C::p5Hmgc (lane 5) with antibodies specific for MGC2 (A), GapA (B), and CrmA (C). No MGC2 was detected in T932A, and T932C weakly displayed a truncated MGC2′, while RCL1 and complemented mutants produced full-length MGC2. Immunoblot analysis of whole cell lysates of RCL1 and mgc2-complemented mutants T932A::p5Hmgc and T932C::p5Hmgc (D) after tryptic digest for 0 (T0), 40 (T1), and 90 min (T2); M, molecular weight marker.
As it has been suggested that the gapA transcript initiates in the 3′ region of mgc2 [35], we analyzed the expression of gapA and crmA. Western blot analyses revealed that expression of these genes did not appear to be affected by the Tn4001cam insertion into mgc2, as GapA and CrmA were detected at wild-type levels in both the mgc2 mutants (Figures 4B and 4C).
MGC2 has been identified by immunoelectron microscopy on the mycoplasma cell surface [36]. To confirm this finding and to assess the size of the surface-exposed portion of MGC2, whole M. gallisepticum RCL1 cells were incubated with trypsin for different time periods and subjected to Western blot analyses with anti-MGC2 antiserum. Tryptic digestion produced a shorter fragment of MGC2, migrating at an apparent molecular mass of 31 kDa (Figure 4D), while untreated MGC2 migrated at 33 kDa. The 31-kDa band was first detected after 10 min of trypsin treatment (not shown), and became more prominent with longer periods of digestion. After 90 min of trypsin digestion the 33-kDa protein was no longer detectable (Figure 4D), indicating the degree of surface accessibility of MGC2 to trypsin. The fact that trypsin digestion reduced the molecular weight of MGC2 by only 2 kDa suggested that only a small region of MGC2 was exposed on the surface. Trypsin digestion patterns for the mgc2-complemented mutants T932A::p5Hmgc and T932C::p5Hmgc were comparable to those seen with RCL1 (Figure 4D), indicating the same degree of surface localization of the 6xHis-tagged MGC2 protein.
Hemadsorption activity
The hemadsorptive activity of wild-type strains, and cytadherence and motility mutants was analyzed using a standard HA assay. In contrast to the non-motile strains Rhigh, RCL2 and mHAD3, which have previously been reported to be deficient in cytadherence [22], the colonies of the motility-impaired mgc2 mutants were able to bind erythrocytes and thus these strains were HA+ (Table 2 and Additional file 1).
Cell morphology
Scanning electron microscopy revealed that the typical flask shape of the wild-type strain Rlow (Figure 5A) was altered in the non-motile, GapA and CrmA-lacking strains Rhigh, RCL2 and mHAD3, which consisted mainly of enlarged, rounded cells (Figures 5D-F). The mgc2 mutant T932A appeared to have a smaller, coccoid morphology (Figure 5B). However, flask-shaped T932A cells with a slightly swollen body, but otherwise a well-defined TO, were also present (Figure 5B, arrow and Additional file 2). T932C had a wild-type flask shape except for a distorted bulging middle region (Figure 5C). For both mutants large spheroidal cells were also observed. The TOs of the more rounded GapA/CrmA mutants were generally not as well defined as in Rlow and T932C.
Figure 5.
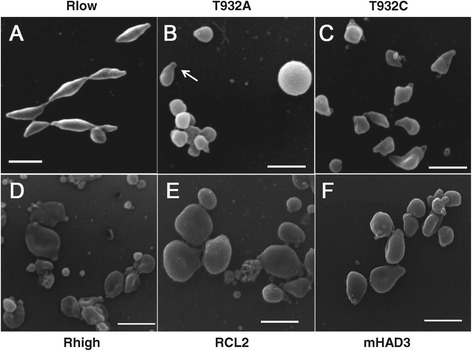
Morphology of M. gallisepticum strains. Scanning electron microscopy revealed the typical M. gallisepticum flask-shape morphology with a pronounced TO (A , Rlow), while CrmA- (F, mHAD3) or GapA-deficient strains (D, Rhigh; E, RCL2) tended to have an enlarged, round-shaped appearance with a less defined TO. Loss of MGC2 (B, T932A) or the presence of a truncated MGC2 (C, T932C) resulted in an overall smaller appearance with either a rounded body or a flask shape-like morphology with bulges and distortions. Scale bars, 1 μm.
Gliding motility
Motility of T932A and T932C was assessed in at least 5 independent MMAs. Qualitative MMAs demonstrated the impaired motility of the mgc2 mutants (Additional file 3) compared to RCL1. In contrast to the large number of long gliding tracks seen with RCL1 (Figure 1), only a very small proportion of the mgc2 mutants produced gliding paths and these were very short. When complemented with mgc2-6xHis, these mutants were able to glide again over long distances, comparable in number and length to those of RCL1 (Additional file 3).
A quantitative MMA revealed that the mean gliding distance for wild-type M. gallisepticum RCL1 was about 27.5 μm over a 3 min observation interval. The gliding distances of the mutants T932A and T932C were significantly reduced to 12.1 and 15.9% of this, respectively (Figure 6A). The complemented strain T932A::p5Hmgc reached 82.7% of the gliding distance of RCL1, while T932C::p5Hmgc reached 63.9% of that of RCL1 (Figure 6A). Statistical analysis revealed that the mean gliding distance of complemented mutants was not significantly different from that of RCL1.
Figure 6.
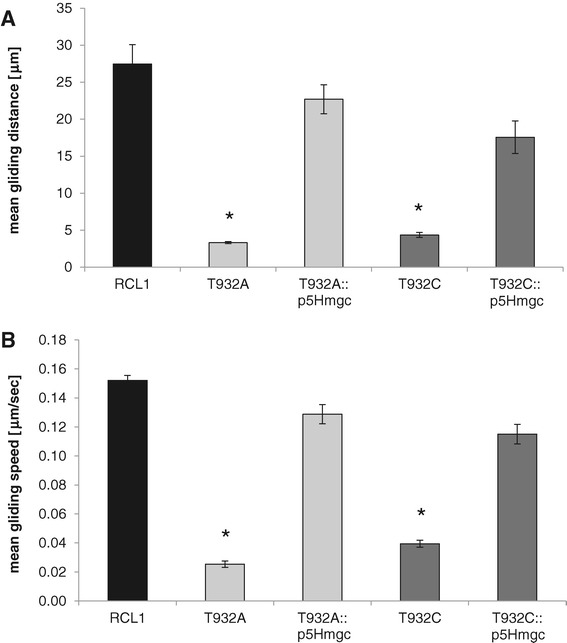
Quantitative analysis of gliding motility. Time-lapse cinematography was performed for cultures of parental strain RCL1 and the gliding-deficient transposon mutants (T932A, T932C) and mgc2-complemented mutants (T932A::p5Hmgc, T932C::p5Hmgc). Using the image processing Fiji package, movies were analyzed to determine the gliding distance (A) and the average speed (B) of mycoplasmas. The mean values from three independent experiments are shown. Statistically significant differences to RCL1 (P ≤ 0.05) are marked by asterisks.
A similar relationship was seen when the mean gliding velocity over the same time interval was calculated. RCL1 glided with a mean speed of 150 nm s−1. The mutants T932A and T932C had a mean gliding velocity that was significantly reduced to 16.7 and 25.9% of that of RCL1, respectively, while mgc2-complementation restored the mean velocity of the mutants back to 84.6 and 75.6% of that of RCL1 (Figure 6B).
Discussion
Not all bacteria are able to move. However, motile bacteria have a competitive advantage over their sessile relatives: motility enables bacteria to reach and remain in individual niches where they may find nutrients and/or shelter from the host’s defense mechanisms. Various motility mechanisms have evolved to allow bacteria to swim or float through liquid media, or to swarm, crawl, twitch or glide over solid surfaces. Many phylogenetically unrelated bacteria have been shown to be able to glide, some, like Neisseria and Pseudomonas, use surface appendices, while others, like Flavobacterium and Myxococcus, glide without any obvious locomotive structures [4]. Mycoplasmas are capable of gliding as well. In spite of the degenerative evolution process which shaped the Mycoplasma genomes to a minimum size, gliding motility seems to be essential for the parasitic life-style of some mycoplasmas. Of the currently described 132 Mycoplasma species [37], 14 motile species are listed: M. agassizii, M. amphoriforme, M. gallisepticum, M. genitalium, M. imitans, M. insons, M. iowae, M. mobile, M. penetrans, M. pirum, M. pneumoniae, M. pulmonis, M. testudineum, and M. testudinis. Interestingly, most of these mycoplasmas were either originally isolated from the human or animal respiratory tract, or they were at least occasionally recovered from such samples. As the respiratory tract is well protected against incoming particles by a thick layer of mucus and underlying epithelial cells covered with constantly beating cilia, gliding motility might be essential to overcome this mechanical barrier. The human pathogen M. pneumoniae has been shown to bind initially to the apical surface of ciliated human bronchial epithelial cells in vitro, then to move down towards the base of ciliated cells before spreading [38]. M. pneumoniae mutants that are defective in motility, but not in cytadherence, have impaired capacity to colonize differentiated bronchial epithelium in vitro [39], and these mutants cannot be recovered from the lung tissue four days after inoculation, whereas motile strains can be [40]. Gliding motility, therefore, seems to be essential for spreading of this pathogen in the respiratory tract, a first step in successful colonization of the host.
Contributing to mycoplasma pathogenesis, and being at the same time a possible target for the development of antimycoplasmal drugs, the elucidation of the mycoplasma motility mechanism is of major importance. Even though the gliding ability of the avian pathogen M. gallisepticum was already observed in the 1970s [13], little was known about the molecular basis of its motility. Here, we report for the first time the involvement of three proteins, MGC2, GapA and CrmA in the gliding motility of M. gallisepticum.
Both, GapA and CrmA have been shown before to be essential for cytadherence, colonization of the chicken trachea and induction of host responses [41-43], while mgc2 has mainly been used to differentiate M. gallisepticum strains [44]. When M. gallisepticum strains from our culture collection were analyzed for motility, gapA mutants such as Rhigh and RCL2 were found to be non-motile. The finding that GapA is involved in the gliding mechanism of M. gallisepticum concords with data obtained for M. pneumoniae. Addition of a monoclonal antibody against the major cytadhesin of M. pneumoniae, P1, a homolog of GapA, removed gliding cells from the glass, but did not interfere with the binding of non-moving cells [34]. These data suggest that P1 has a crucial role in gliding motility of M. pneumoniae, independent of its adhesion properties, but that adhesion is a prerequisite for gliding [16,33,34,45]. Hasselbring et al. found that all motility and HA mutant strains of M. pneumoniae were able to bind to a glass surface [16]. Current models of the motility mechanism of M. pneumoniae suggest that P1 might serve as leg proteins that attach to sialylated oligosaccharides on glass or animal cells [46,47]. After binding to a solid support, with the energy provided by ATP hydrolysis, the legs might repeatedly bind, pull, and release surface structures, thus generating a continuous drag force that propels the cell forward. Addition of free sialylated oligosaccharides inhibits the motility of M. pneumoniae cells, but not the binding of non-gliding cells [47]. In our study, we similarly observed that GapA-deficient strains of M. gallisepticum lacked hemadsorption and motility, but were still able to attach to glass (data not shown), indicating that M. gallisepticum might be equipped with adhesion molecules of differently functionality.
M. gallisepticum strain mHAD3, which is HA− and non-motile, carries a transposon in crmA, which lies directly downstream of gapA. The involvement of CrmA in motility is in concord with findings on M. pneumoniae, in which the orf6 gene, a homolog of crmA [48], has been shown to be involved in motility. Mutant III-4, which lacks the ORF6 cleavage products P40 and P90, is non-motile [16]. Interestingly, P40 and P90 have been shown to complex with P1 in chemical cross-linking studies [49] and purified P1 and P90 have been found to form complexes in vitro [50], suggesting that P1 physically interacts with P90 in the mycoplasma membrane. Similarly, the M. genitalium proteins P110 and P140, homologs of GapA and CrmA, have been shown to be cytadhesins required for TO development and to be reciprocally dependent on each other for posttranslational stability [51]. Such mutual dependence has also been reported for GapA and CrmA in M. gallisepticum [22]. Mutations in gapA seem to have a polar effect on expression of crmA, as no CrmA is found in Rhigh or RCL2 [22,43]. A reason for this might be the operon structure of gapA/crmA (previously known as mgc1/mgc3) as suggested by Keeler et al., who mapped the transcriptional start site for these genes to the end of upstream mgc2 [35]. However, loss of CrmA has a direct negative impact on the level of GapA. In mHAD3, the amount of GapA produced was greatly reduced [22], possibly a result from accelerated turnover of GapA due to the absence of its binding partner CrmA. However, a mutual dependence of GapA/CrmA is not certain, because some transposon mutants in crmA were reported to express GapA [41,52]. On the other hand, these transposon mutants were not analyzed for C-terminally truncated CrmA fragments, which might be sufficient to stabilize GapA. Knock-out of either gapA or crmA, therefore, might have an impact on expression of both proteins, and, as a consequence, it might be difficult to dissect whether the loss of motility in Rhigh, RCL2 or mHAD3 is attributable to the loss of GapA or CrmA.
To add another level of complexity, mutations in gapA or crmA have been shown to affect the morphology of M. gallisepticum. Our electron microscopy studies revealed that the typical flask-shaped appearance of M. gallisepticum, presenting a defined single knob-like structure at one polar end, changed to a rounder, bulkier morphology, with less defined tip structures in strains lacking GapA and CrmA. Similarly, M. pneumoniae mutant M5 which had lost the homolog of CrmA exhibits a perfect round cell shape, but has lost the tip-like structure [53]. Mutants of M. pneumoniae lacking homologs of GapA and CrmA also have lost the elongated flask-shape and display a branched cell morphology [54]. Our findings support a direct link between the major adhesin and the gliding mechanism. However, the question remains whether loss of GapA leads to a loss of motility because GapA cannot act any longer as the “leg” adhesin for the “bind-and-release”-cycles of gliding, or because loss of GapA leads to a drastically changed morphology with conceivable consequences on the correct positioning of any locomotive regions. As the correct morphology may be a strict requirement for the gliding process, future work should focus on the creation of defined M. gallisepticum mutants with modified variants of GapA or CrmA that have no mutual interference on expression of each other and a defined effect on only morphology, cytadherence, or motility.
To identify other genes involved in motility, we constructed a transposon by exchanging the gentamicin resistance gene in Tn4001mod [28] with the gene for chloramphenicol resistance, envisaging future needs such as assessing transposon mutants in gentamicin-based cell invasion assays. The chloramphenicol resistance gene of plasmid pACYC184 was effective when placed behind the MG tufPO, which has previously been shown to function as an effective transcriptional promoter [27]. Stability assays showed that transposon mutants of M. gallisepticum could be cultivated for 20 passages without antibiotic selection pressure. No re-transposition or excision of Tn4001cam could be detected (data not shown). However, a drawback of this, and presumably any Tn4001-based transposon strategy, was the transposon’s tendency to integrate into multiple genomic sites simultaneously. Around 60% of our mutants were not further analyzed because they carried multiple insertions of Tn4001cam. The limited number of mutants analyzed in this study suggests that other genes involved in motility might be identified using a similar, more optimized approach. Recently, a mariner-based transposon was reported to produce mutants in M. hyopneumoniae with stable single insertions [55]. Use of a similar transposon would ensure that each mutant could be used for analysis.
Screening of a small transposon mutant library led to the identification of mgc2 as a major motility gene. Loss of MGC2 resulted in a drastic reduction in motility (Figure 6) that could be restored by complementation of the mutants with a recombinant mgc2-6xHis gene. In contrast to the non-motile gapA/crmA mutants Rhigh, RCL2 and mHAD3, the mgc2 mutation in T932A or T932C did not influence the presence of GapA or CrmA, and the cellular morphology was not as drastically altered as in the GapA/CrmA-deficient mutants. After carefully analyzing many electron micrographs, it seems that both mgc2 mutants had a flask-shaped morphology similar to that of RCL1, characterized by the presence of a short TO (Figure 5). The majority of T932A cells appeared as coccoid cells, possibly a consequence of T932A binding almost exclusively via the TO to the glass slide surface. The flask-shaped T932A is then viewed along its longitudinal axis, with the distal end of the body orientated to the viewer and only virtually pretending a coccoid morphology. Occasionally, a TO became visible below the spherical body, bent towards the glass surface (Additional files 2A, B; indicated by arrows).
The ability to cytadhere, as measured by the HA assay, was not affected by the loss of MGC2 in T932A or T932C. This is in contrast to the first report about MGC2, which was classified as a cytadhesin [36], primarily based on the strong homology between MGC2 and the M. pneumoniae cytadhesin P30, and on attachment inhibition assays. Composed of an N-terminal domain I which is likely to be localized in the cytoplasm, a transmembrane region, a surface-exposed domain II, and highly repetitive, proline-rich domain III [56], P30 has been shown to be a membrane protein that co-localizes with the major cytadhesin P1 to the TO of M. pneumoniae [57]. MGC2 shares with P30 the same overall domain architecture [36,56], a similar size, and 40.9% amino acid sequence identity [36]. Specifically the transmembrane region and domain II are highly conserved between M. pneumoniae P30 and M. gallisepticum MGC2 [56]. Another coincidence is that P30 mutations have effects on cell morphology, cytadherence, motility and virulence [46]. The P30 null mutant II-3 has an ovoid, branched shape, has no ability to hemadsorb [58] nor glide [16], although all other cytadherence-related proteins, such as P1 and the cytadherence accessory proteins, are synthesized as usual [59]. Complementation of the II-3 mutant with the gene encoding P30 rescued the wild-type phenotype [46]. P30 mutations have an impact on the stability of P65 which is located at the distal end of the TO. P65 is involved in cytadherence and motility, and is thought to form a complex with P30 [60]. It would be interesting to investigate whether the reported reciprocal requirement for stabilization between P65 and P30 also exists between MGC2 and PlpA, the ortholog of P65 in M. gallisepticum.
A striking difference between P30 and MGC2 is its impact on cytadherence. In contrast to the HA− P30 mutants of M. pneumoniae, the MGC2-deficient M. gallisepticum mutants were still able to hemadsorb. The binding of erythrocytes seemed to primarily depend on the presence of GapA or CrmA (Table 2). As the mgc2 mutations did not have any polar effects on the expression of gapA or crmA, there was no influence on hemadsorption. However, the first description of MGC2 reported that MGC2-specific antiserum was able to reduce the attachment of M. gallisepticum to CEF cells by 30 to 48% [36]. The lack of complete inhibition was attributed to the existence of additional adhesins such as hemagglutinin VhlA (pMGA) or GapA (MGC1). Although HA has been widely used as an assay for cytadherence [3], the inhibition of attachment to CEF cells by MGC2-specific antibodies might not necessarily prove that MGC2 is a cytadhesin. This protein, as shown by our trypsinization assays, has a very small extracellular domain, and we hypothesize that MGC2 is rather linking internal components of the locomotive machinery to another external adhesion component. GapA could be the external adhesion component in analogy to the proposed leg protein P1 of M. pneumoniae [47], and antibody against MGC2 might affect GapA functions due to the close connection between MGC2 and the putative leg protein, thus causing only moderate inhibition of attachment.
We have shown the genes of the mgc locus [35], mgc2, gapA, and crmA, to be involved in motility of M. gallisepticum. It is of particular importance that mgc2 is not involved in hemadsorption. This will enable us to study these two mechanisms, hemadsorption and motility, independently in M. gallisepticum.
Acknowledgements
This work was supported by the PostDoc Programme of the University of Veterinary Medicine Vienna, Austria. We thank F. Hilbert for critical review of this manuscript, and K. Siebert-Gulle for excellent technical assistance.
Additional files
Qualitative HA assessment of wild-type and gliding mutants. Colonies of RCL1 and motility mutants T932A and T932C were grown on agar plates and overlaid with sheep erythrocytes. No remarkable difference in hemadsorption was seen between wild-type RCL1 and mgc2 mutant strains.
SEM pictures of M. gallisepticum motility mutants. Mutant T932A (A, B) partly appeared as small spheres without a TO. In some cases TOs seemed to be placed between the glass surface and the main body of the mycoplasma cell (arrows), indicating that T932A had attached to the glass via the TO, therefore appearing spherical. Interestingly, in T932C (C) large spheroid cells with TO structures were also seen (triangle).
Gliding paths of M. gallisepticum motility mutants before and after complementation. Only short gliding paths were seen, if any, in mgc2 motility mutants (T932A and T932C), while the complementation of the mutants with mgc2 (T932A::p5Hmgc and T932C::p5Hmgc) restored the gliding motility to almost wild-type levels (see Figure 1).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: II, MPS; performed experiments: II, MV, MPS; performed motility analyses and data analysis: II, MPS; wrote the manuscript: II, MS. All authors read and approved the manuscript.
Contributor Information
Ivana Indikova, Email: ivana.indikova@vetmeduni.ac.at.
Martin Vronka, Email: martin_vronka2@yahoo.de.
Michael P Szostak, Email: michael.szostak@vetmeduni.ac.at.
References
- 1.Josenhans C, Suerbaum S. The role of motility as a virulence factor in bacteria. Int J Med Microbiol. 2002;291:605–614. doi: 10.1078/1438-4221-00173. [DOI] [PubMed] [Google Scholar]
- 2.Eaton KA, Morgan DR, Krakowka S. Campylobacter pylori virulence factors in gnotobiotic piglets. Infect Immun. 1989;57:1119–1125. doi: 10.1128/iai.57.4.1119-1125.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Razin S, Yogev D, Naot Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol Mol Biol Rev. 1998;62:1094–1156. doi: 10.1128/mmbr.62.4.1094-1156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jarrell KF, McBride MJ. The surprisingly diverse ways that prokaryotes move. Nat Rev Microbiol. 2008;6:466–476. doi: 10.1038/nrmicro1900. [DOI] [PubMed] [Google Scholar]
- 5.Johansson KE, Pettersson B. Taxonomy of Mollicutes. In: Razin S, Hermann R, editors. Molecular Biology and Pathogenicity of Mycoplasmas. New York: Kluwer Academic/Plenum Publishers; 2002. pp. 1–30. [Google Scholar]
- 6.Waites KB, Talkington DF. Mycoplasma pneumoniae and its role as a human pathogen. Clin Microbiol Rev. 2004;17:697–728. doi: 10.1128/CMR.17.4.697-728.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyata M. Centipede and inchworm models to explain Mycoplasma gliding. Trends Microbiol. 2008;16:6–12. doi: 10.1016/j.tim.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Kirchhoff H, Rosengarten R. Isolation of a motile mycoplasma from fish. J Gen Microbiol. 1984;130:2439–2445. doi: 10.1099/00221287-130-9-2439. [DOI] [PubMed] [Google Scholar]
- 9.Miyata M, Ryu WS, Berg HC. Force and velocity of Mycoplasma mobile gliding. J Bacteriol. 2002;184:1827–1831. doi: 10.1128/JB.184.7.1827-1831.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uenoyama A, Miyata M. Gliding ghosts of Mycoplasma mobile. Proc Natl Acad Sci U S A. 2005;102:12754–12758. doi: 10.1073/pnas.0506114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyata M. Unique centipede mechanism of Mycoplasma gliding. Annu Rev Microbiol. 2010;64:519–537. doi: 10.1146/annurev.micro.112408.134116. [DOI] [PubMed] [Google Scholar]
- 12.Powell DA, Hu PC, Wilson M, Collier AM, Baseman JB. Attachment of Mycoplasma pneumoniae to respiratory epithelium. Infect Immun. 1976;13:959–966. doi: 10.1128/iai.13.3.959-966.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bredt W. Motility and multiplication of Mycoplasma pneumoniae. A phase contrast study. Pathol Microbiol (Basel) 1968;32:321–326. doi: 10.1159/000162074. [DOI] [PubMed] [Google Scholar]
- 14.Krause DC, Balish MF. Structure, function, and assembly of the terminal organelle of Mycoplasma pneumoniae. FEMS Microbiol Lett. 2001;198:1–7. doi: 10.1111/j.1574-6968.2001.tb10610.x. [DOI] [PubMed] [Google Scholar]
- 15.Balish MF, Krause DC. Mycoplasma Attachment Organelle And Cell Division. In: Blanchard A, Browning GF, editors. Mycoplasmas: Molecular Biology, Pathogenicity and Strategies for Control. Norfolk, UK: Horizon Scientific Press; 2005. pp. 189–237. [Google Scholar]
- 16.Hasselbring BM, Jordan JL, Krause DC. Mutant analysis reveals a specific requirement for protein P30 in Mycoplasma pneumoniae gliding motility. J Bacteriol. 2005;187:6281–6289. doi: 10.1128/JB.187.18.6281-6289.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasselbring BM, Krause DC. Proteins P24 and P41 function in the regulation of terminal-organelle development and gliding motility in Mycoplasma pneumoniae. J Bacteriol. 2007;189:7442–7449. doi: 10.1128/JB.00867-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herrmann R, Reiner B. Mycoplasma pneumoniae and Mycoplasma genitalium: a comparison of two closely related bacterial species. Curr Opin Microbiol. 1998;1:572–579. doi: 10.1016/S1369-5274(98)80091-X. [DOI] [PubMed] [Google Scholar]
- 19.Hasselbring BM, Page CA, Sheppard ES, Krause DC. Transposon mutagenesis identifies genes associated with Mycoplasma pneumoniae gliding motility. J Bacteriol. 2006;188:6335–6345. doi: 10.1128/JB.00698-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakane D, Miyata M. Cytoskeletal asymmetrical dumbbell structure of a gliding mycoplasma, Mycoplasma gallisepticum, revealed by negative-staining electron microscopy. J Bacteriol. 2009;191:3256–3264. doi: 10.1128/JB.01823-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin MY, Kleven SH. Evaluation of attenuated strains of Mycoplasma gallisepticum as vaccines in young chickens. Avian Dis. 1984;28:88–99. doi: 10.2307/1590131. [DOI] [PubMed] [Google Scholar]
- 22.Winner F, Markova I, Much P, Lugmair A, Siebert-Gulle K, Vogl G, Rosengarten R, Citti C. Phenotypic switching in Mycoplasma gallisepticum hemadsorption is governed by a high-frequency, reversible point mutation. Infect Immun. 2003;71:1265–1273. doi: 10.1128/IAI.71.3.1265-1273.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wise KS, Watson RK. Mycoplasma hyorhinis GDL surface protein antigen p120 defined by monoclonal antibody. Infect Immun. 1983;41:1332–1339. doi: 10.1128/iai.41.3.1332-1339.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meijering E, Dzyubachyk O, Smal I. Methods for cell and particle tracking. Methods Enzymol. 2012;504:183–200. doi: 10.1016/B978-0-12-391857-4.00009-4. [DOI] [PubMed] [Google Scholar]
- 26.Simple interactive statistical analysis. [http://www.quantitativeskills.com/sisa/]
- 27.Nieszner I, Vronka M, Indikova I, Szostak MP. Development of a site-directed integration plasmid for heterologous gene expression in Mycoplasma gallisepticum. PLoS One. 2013;8:e81481. doi: 10.1371/journal.pone.0081481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knudtson KL, Minion FC. Construction of Tn4001lac derivatives to be used as promoter probe vectors in mycoplasmas. Gene. 1993;137:217–222. doi: 10.1016/0378-1119(93)90009-R. [DOI] [PubMed] [Google Scholar]
- 29.Sharma VM, Chopra R, Ghosh I, Ganesan K. Quantitative target display: a method to screen yeast mutants conferring quantitative phenotypes by ‘mutant DNA fingerprints’. Nucleic Acids Res. 2001;29:E86–86. doi: 10.1093/nar/29.17.e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Indiková I, Much P, Stipkovits L, Siebert-Gulle K, Szostak MP, Rosengarten R, Citti C. Role of the GapA and CrmA cytadhesins of Mycoplasma gallisepticum in promoting virulence and host colonization. Infect Immun. 2013;81:1618–1624. doi: 10.1128/IAI.00112-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wise KS, Kim MF, Watson-McKown R. Variant Membrane Proteins. In: Razin S, Tully JG, editors. Molecular and Diagnostic Procedures in Mycoplasmology. New York, N. Y: Academic; 1995. pp. 227–241. [Google Scholar]
- 32.Vogl G, Plaickner A, Szathmary S, Stipkovits L, Rosengarten R, Szostak MP. Mycoplasma gallisepticum invades chicken erythrocytes during infection. Infect Immun. 2008;76:71–77. doi: 10.1128/IAI.00871-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyata M, Yamamoto H, Shimizu T, Uenoyama A, Citti C, Rosengarten R. Gliding mutants of Mycoplasma mobile: relationships between motility and cell morphology, cell adhesion and microcolony formation. Microbiology. 2000;146:1311–1320. doi: 10.1099/00221287-146-6-1311. [DOI] [PubMed] [Google Scholar]
- 34.Seto S, Kenri T, Tomiyama T, Miyata M. Involvement of P1 adhesin in gliding motility of Mycoplasma pneumoniae as revealed by the inhibitory effects of antibody under optimized gliding conditions. J Bacteriol. 2005;187:1875–1877. doi: 10.1128/JB.187.5.1875-1877.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keeler CL, Jr, Hnatow LL, Whetzel PL, Dohms JE. Cloning and characterization of a putative cytadhesin gene (mgc1) from Mycoplasma gallisepticum. Infect Immun. 1996;64:1541–1547. doi: 10.1128/iai.64.5.1541-1547.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hnatow LL, Keeler CL, Jr, Tessmer LL, Czymmek K, Dohms JE. Characterization of MGC2, a Mycoplasma gallisepticum cytadhesin with homology to the Mycoplasma pneumoniae 30-kilodalton protein P30 and Mycoplasma genitalium P32. Infect Immun. 1998;66:3436–3442. doi: 10.1128/iai.66.7.3436-3442.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.List of Prokaryotic names with Standing in Nomenclature. [http://www.bacterio.net/index.html]
- 38.Krunkosky TM, Jordan JL, Chambers E, Krause DC. Mycoplasma pneumoniae host-pathogen studies in an air-liquid culture of differentiated human airway epithelial cells. Microb Pathog. 2007;42:98–103. doi: 10.1016/j.micpath.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 39.Jordan JL, Chang HY, Balish MF, Holt LS, Bose SR, Hasselbring BM, Waldo RH, 3rd, Krunkosky TM, Krause DC. Protein P200 is dispensable for Mycoplasma pneumoniae hemadsorption but not gliding motility or colonization of differentiated bronchial epithelium. Infect Immun. 2007;75:518–522. doi: 10.1128/IAI.01344-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szczepanek SM, Majumder S, Sheppard ES, Liao X, Rood D, Tulman ER, Wyand S, Krause DC, Silbart LK, Geary SJ. Vaccination of BALB/c mice with an avirulent Mycoplasma pneumoniae P30 mutant results in disease exacerbation upon challenge with a virulent strain. Infect Immun. 2012;80:1007–1014. doi: 10.1128/IAI.06078-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mudahi-Orenstein S, Levisohn S, Geary SJ, Yogev D. Cytadherence-deficient mutants of Mycoplasma gallisepticum generated by transposon mutagenesis. Infect Immun. 2003;71:3812–3820. doi: 10.1128/IAI.71.7.3812-3820.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shil PK, Kanci A, Browning GF, Marenda MS, Noormohammadi AH, Markham PF. GapA + Mycoplasma gallisepticum ts-11 has improved vaccine characteristics. Microbiology. 2011;157:1740–1749. doi: 10.1099/mic.0.046789-0. [DOI] [PubMed] [Google Scholar]
- 43.Papazisi L, Frasca S, Jr, Gladd M, Liao X, Yogev D, Geary SJ. GapA and CrmA coexpression is essential for Mycoplasma gallisepticum cytadherence and virulence. Infect Immun. 2002;70:6839–6845. doi: 10.1128/IAI.70.12.6839-6845.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lysnyansky I, Garcia M, Levisohn S. Use of mgc2-polymerase chain reaction-restriction fragment length polymorphism for rapid differentiation between field isolates and vaccine strains of Mycoplasma gallisepticum in Israel. Avian Dis. 2005;49:238–245. doi: 10.1637/7285-10020R. [DOI] [PubMed] [Google Scholar]
- 45.Burgos R, Pich OQ, Querol E, Pinol J. Functional analysis of the Mycoplasma genitalium MG312 protein reveals a specific requirement of the MG312 N-terminal domain for gliding motility. J Bacteriol. 2007;189:7014–7023. doi: 10.1128/JB.00975-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Relich RF, Balish MF. Insights into the function of Mycoplasma pneumoniae protein P30 from orthologous gene replacement. Microbiology. 2011;157:2862–2870. doi: 10.1099/mic.0.052464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kasai T, Nakane D, Ishida H, Ando H, Kiso M, Miyata M. Role of binding in Mycoplasma mobile and Mycoplasma pneumoniae gliding analyzed through inhibition by synthesized sialylated compounds. J Bacteriol. 2013;195:429–435. doi: 10.1128/JB.01141-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshida S, Fujisawa A, Tsuzaki Y, Saitoh S. Identification and expression of a Mycoplasma gallisepticum surface antigen recognized by a monoclonal antibody capable of inhibiting both growth and metabolism. Infect Immun. 2000;68:3186–3192. doi: 10.1128/IAI.68.6.3186-3192.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Layh-Schmitt G, Herrmann R. Spatial arrangement of gene products of the P1 operon in the membrane of Mycoplasma pneumoniae. Infect Immun. 1994;62:974–979. doi: 10.1128/iai.62.3.974-979.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakane D, Adan-Kubo J, Kenri T, Miyata M. Isolation and characterization of P1 adhesin, a leg protein of the gliding bacterium Mycoplasma pneumoniae. J Bacteriol. 2011;193:715–722. doi: 10.1128/JB.00796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burgos R, Pich OQ, Ferrer-Navarro M, Baseman JB, Querol E, Pinol J. Mycoplasma genitalium P140 and P110 cytadhesins are reciprocally stabilized and required for cell adhesion and terminal-organelle development. J Bacteriol. 2006;188:8627–8637. doi: 10.1128/JB.00978-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tseng C-W, Kanci A, Citti C, Rosengarten R, Chiu C-J, Chen Z-H, Geary SJ, Browning GF, Markham PF. MalF is essential for persistence of Mycoplasma gallisepticum in vivo. Microbiology. 2013;159:1459–1470. doi: 10.1099/mic.0.067553-0. [DOI] [PubMed] [Google Scholar]
- 53.Layh-Schmitt G, Harkenthal M. The 40- and 90-kDa membrane proteins (ORF6 gene product) of Mycoplasma pneumoniae are responsible for the tip structure formation and P1 (adhesin) association with the Triton shell. FEMS Microbiol Lett. 1999;174:143–149. doi: 10.1111/j.1574-6968.1999.tb13561.x. [DOI] [PubMed] [Google Scholar]
- 54.Seto S, Miyata M. Attachment organelle formation represented by localization of cytadherence proteins and formation of the electron-dense core in wild-type and mutant strains of Mycoplasma pneumoniae. J Bacteriol. 2003;185:1082–1091. doi: 10.1128/JB.185.3.1082-1091.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maglennon GA, Cook BS, Deeney AS, Bosse JT, Peters SE, Langford PR, Maskell DJ, Tucker AW, Wren BW, Rycroft AN. Transposon mutagenesis in Mycoplasma hyopneumoniae using a novel mariner-based system for generating random mutations. Vet Res. 2013;44:124. doi: 10.1186/1297-9716-44-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang HY, Jordan JL, Krause DC. Domain analysis of protein P30 in Mycoplasma pneumoniae cytadherence and gliding motility. J Bacteriol. 2011;193:1726–1733. doi: 10.1128/JB.01228-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krause DC, Balish MF. Cellular engineering in a minimal microbe: structure and assembly of the terminal organelle of Mycoplasma pneumoniae. Mol Microbiol. 2004;51:917–924. doi: 10.1046/j.1365-2958.2003.03899.x. [DOI] [PubMed] [Google Scholar]
- 58.Romero-Arroyo CE, Jordan J, Peacock SJ, Willby MJ, Farmer MA, Krause DC. Mycoplasma pneumoniae protein P30 is required for cytadherence and associated with proper cell development. J Bacteriol. 1999;181:1079–1087. doi: 10.1128/jb.181.4.1079-1087.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dallo SF, Lazzell AL, Chavoya A, Reddy SP, Baseman JB. Biofunctional domains of the Mycoplasma pneumoniae P30 adhesin. Infect Immun. 1996;64:2595–2601. doi: 10.1128/iai.64.7.2595-2601.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hasselbring BM, Sheppard ES, Krause DC. P65 truncation impacts P30 dynamics during Mycoplasma pneumoniae gliding. J Bacteriol. 2012;194:3000–3007. doi: 10.1128/JB.00091-12. [DOI] [PMC free article] [PubMed] [Google Scholar]