

Abstract
Glioblastoma multiforme patients have a poor prognosis due to therapeutic resistance and tumor relapse. It has been suggested that gliomas are driven by a rare subset of tumor cells known as glioma stem cells (GSCs). This hypothesis states that only a few GSCs are able to divide, differentiate, and initiate a new tumor. It has also been shown that this subpopulation is more resistant to conventional therapies than its differentiated counterpart. In order to understand glioma recurrence post therapy, we investigated the behavior of GSCs after primary chemotherapy. We first show that exposure of patient-derived as well as established glioma cell lines to therapeutic doses of temozolomide (TMZ), the most commonly used antiglioma chemotherapy, consistently increases the GSC pool over time both in vitro and in vivo. Secondly, lineage-tracing analysis of the expanded GSC pool suggests that such amplification is a result of a phenotypic shift in the non-GSC population to a GSC-like state in the presence of TMZ. The newly converted GSC population expresses markers associated with pluripotency and stemness, such as CD133, SOX2, Oct4, and Nestin. Furthermore, we show that intracranial implantation of the newly converted GSCs in nude mice results in a more efficient grafting and invasive phenotype. Taken together, these findings provide the first evidence that glioma cells exposed to chemotherapeutic agents are able to interconvert between non-GSCs and GSCs, thereby replenishing the original tumor population, leading to a more infiltrative phenotype and enhanced chemoresistance. This may represent a potential mechanism for therapeutic relapse.
Glioblastoma multiforme (GBM) is a heterogeneous, highly invasive brain tumor, which is treated with a multimodal approach that includes surgery followed by radio- and chemotherapy.1 Temozolomide (TMZ) is currently the best chemotherapeutic drug available on the market against malignant glioma because of its ability to cross the blood–brain barrier (BBB). Even after such an aggressive therapeutic intervention, disease relapse is inevitable due to GBM's infiltrative nature and ability to resist conventional therapies.2, 3 Thus, understanding the mechanisms of therapeutic escape and disease recurrence is crucial for developing more effective treatments against GBM.
GBMs are among the first solid tumors in which the discovery of stem-like tumor-initiating cells has suggested the existence of a hierarchical model of tumorigenesis. Such a dogma proposes that a distinct population of tumor cells, referred to as glioma stem cells (GSCs), are not only responsible for driving tumor growth, but also represent a population that can survive intensive oncological therapies and give rise to recurrent malignancies.4, 5 In the clinical setting, the presence of CD133+ GSCs correlates with a shorter overall survival as well as reduced progression-free survival and is considered a critical target for successful antiglioma therapies.6
The inability of conventional treatments, such as radio- and chemotherapies, to exterminate all infiltrative tumor foci is considered one of the main causes of therapeutic failure and malignant recurrence in GBM. Although the radio-resistance properties of glioma cells are fairly well established, the underlying molecular mechanisms of chemoresistance have been addressed only in a few studies.7, 8 On the basis of this, we set to investigate the biology of GSCs following TMZ therapy both in vitro and in vivo. We observed significant expansion of different GSC subpopulations after exposure to TMZ at the plasma (50 μM) and cerebral spinal fluid (CSF; 5 μM) concentrations detected in GBM patients.9, 10, 11, 12 This expansion arises from the high degree of plasticity that exists within glioma cell populations. After long-term exposure to therapeutic concentrations of TMZ, differentiated tumor cells convert into glioma stem-like cells. These newly formed GSCs acquire phenotypic and functional characteristics similar to those of native GSCs. Once implanted orthotopically in the animal brain, these newly converted GSCs demonstrate a very invasive characteristic similar to that of parental GSCs. In light of these findings, we propose that TMZ may induce specific changes in the tumor microenvironment, which facilitate a GSC-specific ‘niche', thereby providing the necessary contextual signals to initiate the interconversion between differentiated tumor cells and GSCs. Therefore, such cellular plasticity represents a new mechanism for therapeutic resistance in GBM, and understanding this may allow us to optimize TMZ-based antiglioma chemotherapy.
Results
Clinically relevant doses of TMZ cause an increase in the GSC pool in vitro
The clinically relevant regimen for TMZ consists of 150–200 mg/m2/day, via oral administration, on days 1–5 of a 28-day cycle.1 However, its peak concentration measured is only 50 μmol/l in patient's blood samples9, 10, 11, 12 and 5 μmol/l in the CSF.12 Thus, it has been proposed that the intratumoral concentration may not exceed 50 μmol/l.9 On the basis of this, we chose to use 5 and 50 μmol/l of TMZ throughout our experiments. The IC50 values for the GBM cell lines and xenografted specimens used throughout our experiments were significantly higher than the therapeutic concentrations of TMZ observed in patients (data not shown). All three xenografted specimens (GBM43, GBM6, and GBM26) and three GBM cell lines (U251, U87, and A172) were cultured as previously described. To identify the GSC population, based on published data, we considered the CD133 and CD15 cell markers in combination with other stem cell markers such as Nestin, Sox2, and Oct4.7, 13, 14, 15 To validate their use in our model, we first evaluated both in vivo and in vitro the functionality of the GSCs isolated utilizing these markers. The in vivo limiting dilution assay (Supplementary Figure S1A) showed that, independently of the GSC marker used in the sorting process (CD133 or CD15), mice intracranially implanted with GSCs (CD133 only or CD15 only) had a higher rate of tumor engraftment when compared with mice that received the double-negative (DN) non-GSC population (that is, 5/5 animals for the CD15+ and 4/5 animals for CD133+ subgroups versus 1/5 for the DN subgroup (**P<0.01) – 500 cells/animal). In addition, the group that received 500 GSCs/animal had a higher rate of engraftment when compared with the group that received 100 GSCs/animal (either 4/5 or 5/5 animals/group versus 2/5 animals/group (*P<0.05)). Furthermore, the analysis of in vitro neurosphere formation and clonogenicity assays (Supplementary Figures S1B-i and B-ii) using freshly sorted U251 GSCs (CD133+, CD15+, and DP) demonstrated that these populations are able to self-renew and effectively form neurospheres in the control group.
Our in vitro data indicated that therapeutic concentrations of TMZ consistently increased the GSC population over time in at least four out of six studied cell lines and xenografted specimens. Almost all of the patient-derived as well as established GBM lines treated with 50 μmol/l of TMZ for 8 days showed an increase (16% on average) in the number of GSCs (CD133+, CD15+, and CD133+CD15+ ***P<0.001, Figure 1 and Supplementary Figure S2). The biggest increase in the GSC pool was observed 8 days post-initial TMZ therapy (Figure 2a). Similar results were seen in a primary GBM sample treated with or without 50 μM of TMZ (Supplementary Figure S3). Within specific subpopulations, the analyzed cells also presented a significant increase in other GSC markers, such as Sox2, Oct4, and Nestin (11, 12, and 25% on average, respectively; ***P<0.001, Figure 1a and Supplementary Figure S2A). Additionally, they co-expressed markers of pluripotency and stemness – 14% of GSCs co-expressed SOX2, 9% co-expressed Oct4, and 10% co-expressed Nestin (***P<0.001, Figure 1b and Supplementary Figure S2B).
Figure 1.

Representative FACS plot characterizing our newly formed stem-like cells 8 days post-initial TMZ therapy in four human-derived as well as established glioma cell lines. (a) There is a consistent and significant increase of classical CSC markers (CD133, CD15, Sox2, and Oct4) post-TMZ therapy. These results are extendable to at least three out of four glioma cell lines and xenografted specimens studied, except Oct4, which is overexpressed in two out four cell lines and xenografts. (b) The expression of double-positive markers has been selected in order to allow a more reliable categorization. There was a significant increase in GSCs co-expressing both CD133 and CD15 markers. This increase is also observed for the co-expression of CD133 and Sox-2 and CD133 and Oct4. This last combination was not significantly increased in GBM43 xenografts (P: ns). The control group (CTRL) received DMSO. Error bars denote S.E.M. P: ns (P>0.05), *P<0.05, **P<0.01, ***P<0.001, one-way analysis of variance
Figure 2.

Clinically relevant doses of temozolomide cause a significant increase in the GSC pool in vitro. Such an increase is originated from either the amplification of the GSC population or the conversion of non-GSCs into GSCs. FACS analysis of sorted and unsorted glioma cells treated with either DMSO or 50 μM of TMZ for 8 days. (a) The increase in the GSC population happens over time and in a time-dependent manner in all tested cell lines (figure depicting U251 cell line and GBM43-xenografted specimen). (b) This finding holds true for both unsorted and sorted (DN, CD15+, and DP) U251 populations. We observed a maximum increase in the GSC population 8 days post-initial TMZ therapy. There are both an amplification of GSCs and a conversion of DN non-GSCs into single-positive or double-positive GSCs. DN: FACS-sorted non-GSCs expressing double-negative markers (CD133−CD15−), CD15+: sorted GSCs expressing only the CD15 marker, DP: sorted GSCs expressing double-positive markers (CD133+CD15+). Error bars denote S.E.M. P: ns (P>0.05), *P<0.05, **P<0.01, ***P<0.001, one-way analysis of variance
TMZ induces expression of GSC phenotypic markers in the non-GSC subpopulation
On the basis of previously published reports as well as our observations, we postulated the following three scenarios: (1) selection, where TMZ exposure could selectively deplete the non-GSC population, thus increasing the percentage of the GSC pool within the total tumor cell population; (2) expansion, where TMZ can selectively expand the specific ‘pre-therapy' GSC pool by inducing proliferation, and finally (3) conversion, where differentiated non-GSCs can spontaneously convert into a stem-like state.16, 17 To examine these possibilities, both GSC-enriched (CD15+ and CD133+CD15+ (DP)) and non-GSC (CD133−CD15− (DN)) subpopulations were separated into three groups (CD15+, DP, and DN) by flow cytometry (FACS) sorting. Immediately post separation, U251 subpopulations were treated with or without 50 μmol/l of TMZ for 8 days. On days 2, 4, 6, and 8 post-TMZ therapy, cells were collected and analyzed by FACS for GSC markers (CD15 and CD133). As shown in Figure 2b, in the presence of TMZ, we observed an increased expression of GSC markers (CD15+CD133+) not only in previously positive GSC populations but also in non-GSCs (DN populations). Over time, whereas all control groups maintained the same baseline values (∼0.3% of positivity), all groups exposed to TMZ continually presented an increase in GSC markers up to day 8, when positivity levels reached their peak. The CD15+ group showed the highest increase in GSC expression after TMZ therapy (on average 12%), followed by unsorted (7.6%), DN (4.5%), and DP (3.9%) populations.
One possible shortcoming of the experiment described above is the fact that each subpopulation was cultured separately, which could have destroyed the equilibrium of a previously established tumor cell hierarchy. To exclude this possibility, we repeated the cell sorting explained above and each subpopulation was tagged separately with GFP by lentivirus-mediated stable introduction of GFP expression cassette (Figure 3). By mixing one GFP-tagged and two untagged subpopulations, we were able to maintain the same proportions found in the original unsorted group (2.1% CD15+, 1.3% DP, and 96.6% DN). Therefore, we finally obtained three mixtures of cells: MIX 1 (DN GFP+, CD15+ non-GFP, DP non-GFP), MIX 2 (DN non-GFP, CD15+GFP+, DP non-GFP), and MIX 3 (DN non-GFP, CD15+ non-GFP, DP GFP+). After seeding, all three mixtures were treated with DMSO or TMZ for up to 8 days. Cells were collected on days 4 and 8 post-TMZ treatment, stained for CD133 and CD15 markers, and analyzed using FACS. All mixtures treated with DMSO kept the same low levels of GSC markers, whereas all mixtures treated with TMZ, either 5 or 50 μmol/l, showed an increase in GSC markers both 4 and 8 days post therapy. Figures 3b and c display results from experiments using MIX 3 (DN non-GFP, CD15+ non-GFP, DP GFP+). We observed an increase in GSC phenotypic markers only in TMZ-treated cells, which increased 1.7-folds in 5 μmol/l treated cells and 19.6-folds in 50 μmol/l 8 days post therapy. This result was true for both CD133 (Figure 3b) and CD15 (Figure 3c) markers. In Figure 3d (representative FACS plots), we observe that both previous DP and DN populations present a significant increase in GSC markers (P<0.001). Thus, our results suggest that the increased pool of GSCs is a result of both the expansion of parental GSCs as well as conversion of non-GSCs into newly formed GSCs.
Figure 3.

Graphic representation of the increase in the GSC pool post-TMZ therapy in unsorted U251 glioma cells with GFP-tagged subpopulations (DN, CD15+, and DP). (a) Figure depicting the experiment set up with tagged GSC and non-GSC subpopulations. As there is no consensus on an optimal and most representative GSC marker, we used various single and combined markers to ratify our observations. (b) Plot depicting amplification of DP (GFP+) GSCs into CD133+ GSCs. TMZ triggered an amplification of the GFP-tagged CD133+ GSCs (GFP+CD133+) over time and in a dose-dependent manner (both with 5 and 50 μM of TMZ). Mock cells present a typical fluctuation in the GSC markers, with very low rates of GSCs (∼0.093%). The TMZ-triggered amplification of the GFP-tagged tumor cells aims to replenish the previously depleted GSC population, raising it to ∼1.16% at 8 days post-TMZ therapy (50 μM). (c) Similar representation showing the amplification of the DP GSC population (GFP-tagged). Now such amplification is measured by an increase in single-positive CD15 GSC markers. (d) Both unsorted DN (first column) and DP (second column) U251 GFP-tagged cells show an increase in the GSC population at 4 and 8 days post-initial TMZ therapy. Here we look for the presence of double-positive (CD133+CD15+) GSC markers. We see both a significant conversion of non-GSCs into DP GSCs (P<0.001) and an amplification of the DP GSCs (P<0.001). P-values represent the average of at least five samples, P: ns (P>0.05), *P<0.05, ***P<0.001, one-way analysis of variance
TMZ-mediated expansion of the GSC pool is driven by newly converted GSCs
Next, we investigated whether the observed increase in the GSC pool post-TMZ therapy was driven by an expansion of parental GSCs or conversion of non-GSCs into GSCs. We first FACS-sorted different GSC subpopulations and cultured them with or without TMZ in the presence of neurosphere formation media (Supplementary Figures S1 B-i and B-ii). In the control group, all sorted cells were able to self-renew and effectively form neurospheres. In the TMZ-treated group, the drug was shown to affect the ability of CD15+ and DP cells to form neurospheres (*P<0.05 and ***P<0.001, respectively), whereas it inhibited the ability of CD133+ cells to form neurospheres (***P<0.001). Next, we analyzed the death rate of glioma subpopulations in the GFP-tagged mixtures above described. The goal was to accurately analyze cell death in each specific compartment. We performed a FACS analysis of the previously described MIX 1 (a mixture of all U251 subpopulations containing GFP-tagged DN cells (non-GSCs)) and MIX 3 (a similar mixture with DP GSCs that are GFP-tagged) treated or not with TMZ for 4 and 8 days in two different concentrations (5 and 50 μmol/l; Figure 4). After TMZ treatment, both mixtures were labeled with anti-human CD133 and CD15 antibodies together with 7-AAD, a nucleic acid dye commonly used for the exclusion of non-viable cells in flow cytometry assays.
Figure 4.
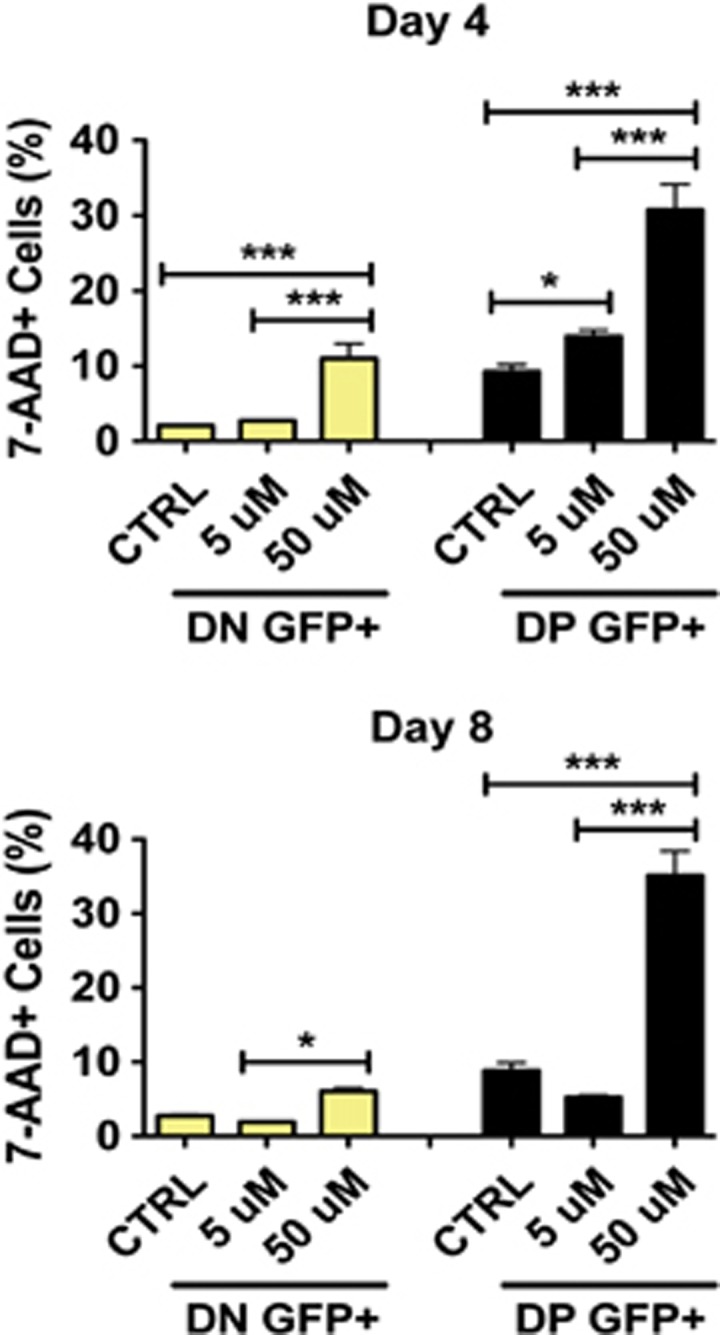
Therapeutic doses of temozolomide can deplete the pre-existing GSC pool. FACS analysis of the GFP-tagged (GFP+) DN (CD133−CD15−) non-GSC and DP (CD133+CD15+) GSC populations. Cell viability was assessed by 7-AAD staining. Although DN cells are killed by TMZ treatment, there is no preferential death of the DN non-GSC population upon long-term TMZ therapy (either on day 4 (∼12%) or 8 (∼7%)). DP GFP+ cells seem to be more vulnerable to TMZ therapy (∼33% of 7-AAD+ cells 8 days post-initial therapy). Error bars denote S.E.M. P: ns (P>0.05), *P<0.05, **P<0.01, ***P<0.001, one-way analysis of variance
At the dose of 50 μmol/l, the DP GFP+ (GSC) population presented significantly greater number of dying cells represented by 7-AAD-positive staining (Figure 4, P<0.0001). For the DP GSC population, on day 4 about 31%, and on day 8 about 35% of the cells were 7-AAD-positive, as compared with ∼11% (day 4) and ∼7% (day 8) for the non-GSC (DN) population. As shown in Figure 2b, the expansion rate of sorted DP cells on day 8 was 1–4% post-TMZ therapy, and for the DN cells it was 1–5%. From this we can conclude that the expansion rate post-TMZ therapy is roughly equal. However, the death rate of the DP population on day 8 was 28% greater than in the DN population after TMZ treatment. We found similar results using trypan blue exclusion method (data not shown). There was minimal difference in cell cycle profile among the studied cell lines and xenografted specimens (U251, GBM43, and GBM6), except for GBM26, which showed a cell cycle arrest on G2M upon TMZ treatment (Supplementary Figure S4A). Proliferation analysis by BrdU incorporation also did not show any difference in the U251 and GBM43 studied populations (P>0.05, ns) (Supplementary Figure S4C). Taken together, the above results imply that the observed increase of GSCs post-TMZ therapy is predominately a result of conversion of non-GSCs into GSCs.
In vivo evaluation of TMZ-induced expansion of the GSC pool
Next, we investigated whether TMZ-induced expansion of the GSC pool exists in vivo. We first implanted glioma xenografts by injecting GBM43 in the flank of athymic nude mice. When the tumor became palpable (∼1 cm3), mice were treated with three different doses of TMZ (2.5, 5, and 10 mg/kg/day) by intraperitoneal injection for 5 consecutive days, followed by a 5-day break (experimental schematic in Figure 5a). Tumor volumes were measured daily post-TMZ treatment. On day 10, animals were killed and tumors from each treatment group were harvested and weighed. As shown in Figures 5b and c, the group of mice that received the dose of 2.5 mg/kg of TMZ showed no difference with respect to the volume and weight of the treated tumor as compared with the control group. On the other hand, mice receiving 5 and 10 mg/kg of TMZ had, on average, ∼73% of reduction in their tumor volume. The GSC frequency in all tumors was analyzed using the FACS assay. We observed a significant increase in the GSC pool in the group treated with 2.5 mg/kg for all tested markers (CD133, CD15, and Sox2) compared with mock, indicating the in vivo amplification of the GSC pool at the dose of 2.5 mg/kg. In addition, there was a statistically significant increase in the mean fluorescence intensity (MFI) for CD133 and Sox2 markers in the flank tumors treated with 2.5 mg/kg (Figures 5c–e). These results were also confirmed with IF staining, which showed a considerable increase in CD133 and Sox2 markers in orthotopic tumors of mice that received 2.5 mg/kg of the drug (Figure 5f). Similar results are seen in Figure 5g using three other patient-derived glioma models, GBM12, GBM26, and GBM39. There was an increase in the CD133 population after TMZ treatment in all three GBM-xenografted specimens tested. Additionally, there was an increase in CD133+CD15+ and CD133+Sox2+ GSCs in two out of three GBM xenografts tested. Taken together, these data indicate that subtherapeutic doses of TMZ also induce amplification of the GSC subpopulations in vivo, similarly to the results observed in vitro. The mechanisms of such amplification remain to be investigated. However, our initial results reveal that DMSO-treated U251 cells, which mostly undergo asymmetric cell division within the CD133+ GSC subpopulation, dramatically shift to symmetric cell division upon TMZ therapy (Supplementary Figure S5), a mechanism reported to be required for neoplastic transformation, CSC maintenance, and cancer progression.18, 19, 20 Moreover, we observed significant epigenetic changes of the methylation status on the promoter region of four genes associated with stemness and self-renewal (FOS,21 Olig2,22 RUNX1,23 and SMAD224) in DN GBM cells treated with TMZ (Supplementary Figure S4B). These results provide new clues regarding the mechanisms of TMZ-mediated GSC amplification and require further investigation.
Figure 5.

The effects of temozolomide on GSCs in vivo (human-derived GBM43 flank model). (a) Figure describing in vivo experiment set up. GBM43 tumors were implanted in the flank of athymic nude mice and treated for 5 consecutive days with different doses of TMZ (2.5, 5, and 10 mg/kg/day) or DMSO. Five days post-therapy tumors were collected, measured, weighed, and analyzed using FACS and immunofluorescence staining. (b) Representative plot depicting the relative tumor volume over time (during the 10 days post-initial therapy). In addition, there is a representation of tumor measure post collection. TMZ treatment was able to cause tumor shrinkage under all conditions and in a dose-dependent manner. (c) FACS analysis of the GSC population in the collected tumors. Suboptimal doses of TMZ (2.5 mg/kg/day) consistently and significantly triggered the highest increase in the GSC population when compared with the control group. This observation was true for the CD133+, (d) CD15+, and (e) Sox-2+ GSC populations. (f) Immunofluorescence staining ( × 20 magnification) with double-positive GSC markers (CD133/Sox2) reaffirmed the above findings. (g) Frequency of CD133, CD133/CD15, and CD133/Sox2 markers in GBM12, GBM26, and GBM39 flank tumors treated with or without TMZ (2.5 mg/kg/day). Error bars represent S.D. from the average of at least five samples. Error bars denote S.E.M. P: ns (P>0.05), *P<0.05, **P<0.01, ***P<0.001, one-way analysis of variance
Next, to evaluate the tumorigenic potential of newly converted GSCs in vivo, a single cell suspension from DMSO and TMZ (2.5 mg/kg)-treated tumors was sorted with anti-human CD15 and CD133 antibodies. The sorted CD15+, DP, and DN subpopulations from both groups were orthotopically implanted into the brains of athymic nude mice (1000 cells/mouse, n=5; Figure 6a). The rate of tumor engraftment, survival patterns, and invasiveness were monitored in all groups. As shown in Figure 6c, three out of five CD15+ implanted GSCs (parental GSCs; from the DMSO-treated flank tumor) engrafted, whereas five out of five CD15+ GSCs (newly converted GSCs; from the TMZ-treated flank tumor) were tumorigenic (median survival of 36 days for both the groups). In the DP (CD133+CD15+) group, 100% of mice developed tumor engraftment. However, we observed a delayed tumor development in the TMZ-treated group as compared with DMSO-treated control (median survival 33 days for TMZ-treated group and 27 days for DMSO-treated control, P<0.05). These data corroborate our in vitro finding where we observed that the parental GSC population is more sensitive to TMZ therapy. For the DN non-GSC-implanted animals, we observed 60% of engraftment in the DMSO-treated group as compared with 100% of engraftment of DN non-GSCs previously treated with TMZ (Figure 6b; median survival of 33 days for both the groups). Most importantly, hematoxylin and eosin (H&E) analysis of DN glioma xenografts previously treated with TMZ showed a significantly increased number of invasive tumor foci in the DN non-GSC group treated with TMZ as compared with the DMSO-treated control (P<0.05). Such a property was very similar to DP GSC-implanted tumors that demonstrated invasive properties with or without TMZ treatment, a previously established characteristic of CSCs25, 26 (Figures 6d and f). The effects of TMZ on GSC frequency and malignant invasion can also be observed in Figure 6e and Supplementary Figure S6. H&E scans with higher magnification images (Figure 6e) display contained tumors with well-defined borders in animals that received DMSO, and scattered tumors with infiltrative borders in animals treated with TMZ. Additional immunofluorescence images (Supplementary Figure S6B) show an increase in both number and colocalization of GSC markers (CD133 and Sox2) and a decrease in differentiation markers (GFAP) in tumors treated with TMZ. Independent of the treatment received, CD133 and GFAP do not colocalize. Taken together, these results indicate that DN non-GSCs previously treated with suboptimal doses of TMZ start to behave as parental pre-existing GSCs with respect to expression of GSC-associated molecular markers, in vivo ability of tumor formation, and invasiveness.
Figure 6.

The effects of temozolomide on GSCs and tumor engraftment in vivo (human-derived GBM43 intracranial model). (a) Figure describing in vivo experiment set up. GBM43 tumors were implanted in the flank of athymic nude mice and treated for 5 consecutive days with 2.5 mg/kg/day of TMZ or DMSO (five animals/group). Five days post-therapy tumors were collected and disaggregated, and glioma cells were immediately FACS-sorted for DN (CD133−CD15−) non-GSCs, CD15+ GSCs, and DP (CD15+CD133+) GSCs. The sorted populations were orthotopically implanted in the brain of athymic nude mice and (b) frequency of tumor engraftment and (c) animal survival were observed. The Kaplan–Meier survival plots show that mice intracranially injected with DN non-GSCs previously treated with TMZ presented similar survival rates as those that received TMZ-treated GSCs. P-values were calculated using the log-rank test. (d and e) We also analyzed the histological characteristics and (f) the frequency of extra-tumor foci by H&E staining in both DP and DN populations. There was a consistently higher infiltrative and invasive phenotype in xenografts derived from both non-GSCs and GSCs previously treated with TMZ. The mice brains that received TMZ-treated cells displayed a significantly higher (P<0.05) number of extra-tumor foci when compared with the control groups. Error bars represent S.D. from the average of at least five samples. P: ns (P>0.05), *P<0.05, **P<0.01, ***P<0.001, unpaired t-test
TMZ-associated increment of hypoxia-inducible factors (HIFs)
It has been previously reported that HIF is accumulated in all tumor cells under hypoxic conditions16, 27 and is critical for the generation of a hypoxia-induced CSC niche.27, 28 On the basis of this, we investigated the role of HIF in the TMZ-induced conversion of non-GSCs to GSCs. We found that 8 days post-TMZ therapy there was a significantly higher expression of HIF2A in the GSC compartment of all four cell lines and xenografted specimens studied. Similarly, in three out of four glioma cell lines and xenografts, there was a greater accumulation of HIF1A and Ki67, a cell proliferation marker, in CD133+ GSCs (Figure 7a). In addition, we used hypoxyprobe assay to better identify hypoxic areas within brain tumors in an intracranial orthotopic GBM43 murine model. The ability of the hypoxyprobe to bind to hypoxic areas within the brain tumor was verified by immunostaining with hypoxyprobe antibody. Images of GBM43 brain tumors treated with or without 2.5 mg/kg/day of TMZ showed that animals that received TMZ presented more intratumoral hypoxic areas than the control group (Figure 7b). Moreover, in vivo IF staining of GBM43 flank tumors of mice previously treated either with DMSO or different doses of TMZ (2.5, 5, and 10 mg/kg/day) showed a significant increase in HIF1A and HIF2A in the tumors that were exposed to 2.5 mg/kg of the drug. The high expression of these markers also correlated with an elevated expression of Nestin. Interestingly, tumors from mice that received a dose that was able to shrink ∼80% of the tumor had very low HIF1A, HIF2A, and Nestin expression levels (Supplementary Figure S6A). These data suggest that suboptimal doses of TMZ (below the IC50) may trigger the overexpression of genes related with GSC maintenance, leading to an increased proliferation and renewal of tumor-initiating stem cells. Furthermore, we evaluated the expression of MGMT, a DNA repair enzyme associated with resistance to alkylating anticancer agents (such as TMZ), in the GSC compartment of all studied cell lines. We observed an increased MGMT expression in CD133+ GSCs in two out of four glioma cell lines, suggesting that TMZ may also induce chemoresistance, contributing to higher tumorigenesis and sustaining long-term tumor growth (Figure 7a).
Figure 7.

Temozolomide-associated increment of HIFs may control GSC maintenance and tumorigenesis. (a) Serial analysis of all four cell lines used throughout this study indicated significantly increased levels of HIF2α in CD133+ GSCs in four out of four glioma cell lines. The same was true for HIF1α and Ki67, a well-known marker of cell proliferation. Additional analysis revealed that only two out of four studied cell lines possessed CD133+ GSCs that expressed MGMT, a DNA repair protein. (b) Hypoxyprobe staining identifying hypoxic areas within GBM43 brain tumors. TMZ-treated tumors presented more hypoxic regions than the non-treated group. Fluorescent images were captured using the × 10 and × 40 objectives. Error bars denote S.E.M. P: ns (P>0.05), *P<0.05, **P<0.01, ***P<0.001, one-way analysis of variance
Discussion
CSCs have been held responsible for glioma initiation, progression, and therapeutic relapse. Thus, understanding the mechanisms underlying the emergence and maintenance of GSCs is critical for the development of new therapeutic approaches that may be able to target this specific population. In this study, we provide functional and some mechanistic evidence of the conversion of non-GSCs into GSCs both in vitro and in vivo after exposure to TMZ, one of the current standard of care treatments in antiglioma therapy.
The universal CSC marker remains a controversial topic due to high interpatient as well as intratumoral variability. Various surface markers such as CD133 and CD15 combined with other stem cell markers such as Nestin, SOX2, and Oct4 repeatedly reported to successfully identify GSC populations both in vitro and in vivo. Using these classical CSC markers, we demonstrated a consistent increase in multiple phenotypic and functional GSC markers across all studied glioma cell lines, xenografts, and primary specimen post-long-term exposure to clinically relevant doses of TMZ. This increase in the GSC population is time-dependent, being greatest at 8 days post-initial therapy. To our knowledge, this is the first time that such a finding has been reported in the literature, and it provides very important information concerning the origin, development, and maintenance of the GSC population post-TMZ treatment.
We next investigated whether the newly formed GSCs observed in this study are a result of selection, expansion, or conversion of glioma subpopulations. A number of experimental findings described in our report clearly argue against the concept that GSC populations are more chemoresistant than their non-GSC counterpart29 and also corroborate several recently published reports that show that GSCs can be quite sensitive to TMZ therapy.9, 30 On the contrary, we observed that non-GSCs are capable of converting into GSCs under the influence of TMZ and that the TMZ-mediated expansion of the GSC pool is driven by newly converted GSCs. These newly formed GSCs expressed molecular markers associated with the parental GSCs. Previous reports studying the dynamics of CSCs in breast cancer models have demonstrated that conversion of a non-CSC population into a CSC-like population happens spontaneously, which is also observed in our experiments where the sorted or GFP-labeled non-GSC pool became DP over time.17 However, the interconversion rate between non-GSC to GSC was significantly accelerated upon long-term TMZ exposure. Such a phenomenon has been widely postulated in the literature.16 Nevertheless, for the first time, we show that this interconversion occurs in glioblastoma models and, most importantly, can be modulated by a widely used anticancer chemotherapy.
We also demonstrate that the newly formed GSCs are functional, possess a high rate of tumor engraftment, and display a more invasive phenotype. Mice intracranially injected with newly formed GSCs presented similar survival rates to those that received parental GSCs. In addition, a histological analysis of parental GSC and newly formed GSC tumors revealed similar infiltrative and invasive phenotypes. Thus, we were able to functionally characterize these newly formed GSCs, showing an infiltrative phenotype, and a propensity for tumor relapse following chemotherapy.
Our results also reveal that the newly formed GSCs express high levels of both HIF1A and HIF2A, which are well-studied cellular factors important for maintaining the CSC niche.16, 27, 28 Similarly to us, Heddleston et al.16 showed that the HIF+ compartment possesses a higher self-renewal capability and induces an increase in typical CSC markers in both non-GSCs and GSCs. Therefore, there is a possibility that the conversion of non-GSCs into GSCs post-TMZ therapy observed in our experiments may be regulated by HIF. In addition, a recent report has shown that HIF is able to bind to the MGMT promoter of glioma cells.31 These results may explain the involvement of HIF in the mediation of glioblastoma resistance to TMZ. We observed MGMT expression in half of the CD133+ cell lines tested. However, the majority of these cell lines were resistant to TMZ. This suggests that alternative MGMT-independent mechanisms of therapeutic resistance may exist, and therefore HIF-regulated MGMT expression may be worthy of further investigation.
The balance of the interconversion between CSCs and non-CSCs could be shifted in one direction or another in response to specific niche factors, such as hypoxia, acidic stress, and metabolic stress. Recent studies have shown that mature CD133 negative non-CSCs are able to acquire CSC properties under intermittent hypoxic conditions.16, 32, 33 Their results suggest that such a mechanism is regulated by HIF. Similarly, our data show in an orthotopic xenograft glioma model that TMZ treatment significantly increases the number of intratumoral hypoxic foci. Acidic stress has also been linked to malignant transformation. Hjelmeland et al.34 have demonstrated that exposure of glioma cells to low pH environments promotes malignancy through the induction of a GSC phenotype.34 They found that the acquisition of HIF2A was an important contributor to this process. Metabolic reprogramming of cancer cells has also shown to trigger such an interconversion.35, 36 In keeping with our results, a recent study indicated that TMZ might induce mutations in pretreated tumors.37 Through the analysis of 4 938 362 mutations from 7042 human cancers, the authors extracted 21 distinct mutational signatures. Interestingly, signature 11 was found in malignant melanomas and GBMs pretreated with TMZ. The authors stated that the mutational features found in this signature were very similar to those previously reported in experimental studies of alkylating agents.38
In conclusion, the data presented here add details to mechanistic and functional evidence of conversion of non-GSCs into GSCs. We also show that chemotherapeutic agents such as TMZ can modulate this increased plasticity. We offer a new model that may be able to explain the high rates of tumor recurrence post-conventional therapy. This model opens doors for a better understanding of the mechanisms behind the maintenance and increase in GSCs post chemotherapy. Our results, along with other published data, question the rigid cellular hierarchy within a tumor proposed by the initial CSC theory, and suggest that the ‘stemness' of CSCs could be governed by cellular plasticity. Understanding how such plasticity can stimulate the dedifferentiation of non-CSCs and increase the overall ‘stemness' of the tumor will be critical for developing effective therapeutic strategies to target GSC populations.
Materials and Methods
Cell culture and propagation
U87MG, U251, and A172 human glioma cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA) and maintained according to the manufacturer's recommendations. U87MG cells were cultured in Minimum Essential Medium (MEM; HyClone, Thermo Fisher Scientific, San Jose, CA, USA), whereas U251MG and A172 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 2% penicillin–streptomycin antibiotic (Cellgro, Herdon, VA, USA; Mediatech, Herdon, VA, USA) and 10% fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA, USA). All cells were grown in a humidified atmosphere, with 5% CO2 and 37 °C conditions. All cell lines were enzymatically dissociated using 0.25% trypsin/2.21 mmol/1 EDTA solution (cat. no. 25-053-CI; Mediatech).
The patient-derived GBM43-, GBM12-, GBM6-, GBM26-, and GBM39-xenografted glioma specimens were provided by Dr. David James from the University of California San Francisco and maintained according to the published protocol.39, 40 Cells were propagated in vivo by serial passaging in the flank of nude mice and, for in vitro studies, cultured in DMEM 1% FBS containing 2% penicillin–streptomycin antibiotic (Cellgro; Mediatech). Concerning the MGMT methylation status, GBM12 and GBM39 tumors present a methylated MGMT, whereas GBM43, GBM6, and GBM26 xenografts have a nonmethylated MGMT promoter.39, 40, 41
The human glioblastoma sample was obtained from a consenting patient, as approved by the University of Chicago Institutional Review Board. The biopsy was identified as a primary glioblastoma based on neuropathological diagnosis. The tumor was washed, acutely dissociated, and then subjected to enzymatic dissociation. Harvested cells were then cultured in serum-free tumor sphere media (TSM) as previously described.13, 42
For experimental purposes, cells and primary neurospheres cultured in each appropriate cell culture medium were treated with either TMZ (Schering Plough; stock solution 50 mmol/l in DMSO) or DMSO control in the concentrations of 5 or 50 μmol/l over the periods indicated.
Flow cytometry analyses
For in vitro experiments, on serial time points post-TMZ or DMSO addition (days 2, 4, 6, and 8) cells were collected, labeled, and analyzed for both surface and intracellular markers as previously described.43 The following human-specific antibodies were used: anti-CD133-PE (2 : 100; Miltenyi Biotec, Auburn, CA, USA), anti-CD15-APC (5 : 100; BD Pharminogen, San Diego, CA, USA), anti-Oct4-FITC (5 : 100; Millipore, Billerica, MA, USA), anti-Ki67-FITC (4 : 100; Abcam, Cambridge, MA, USA), biotinylated anti-SOX2 (4 : 100; R&D Systems), purified rabbit anti-Nestin (4 : 100; Covance, Berkeley, CA, USA), purified monoclonal anti-HIF1A (hypoxia-inducible factor-1A; 4 : 100; Abcam), purified monoclonal anti-HIF2A (4 : 100; Millipore), purified polyclonal rabbit anti-MGMT (4 : 100; Cell Signaling, Beverly, MA, USA); and anti-mouse CD45 eFluor450 (eBioscience, San Diego, CA, USA). In addition, we used as secondary antibodies anti-rabbit IgGk1 FITC (1 : 500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-mouse IgGk1 PB (1 : 500; Abcam), and Streptavidin PB (1 : 500; Molecular Probes, Eugene, OR, USA). To label dead cells, 0.1 μg/ml of 7-Amino-Actinomycin D (7-AAD; BD Pharminogen) was applied for 10 min. Staining was quantified using flow cytometry (BD LSRII-Blue). Data analysis was carried on the FlowJo (TreeStar, Ashland, OR, USA) software. For the analysis of the ability of differentiation of cancer stem cells (CSCs; symmetric and asymmetric cell division) upon TMZ therapy, GBM cells were stained with anti-human CD133-PE (2 : 100; Miltenyi Biotec). Cell fluorescence data and images were acquired using ImageStreamX Mark II (Amnis, Seattle, WA, USA), and data were analyzed using the ImageStream Data Analysis and Exploration Software (IDEAS).
Cell cycle analysis
DNA content measurement utilized the Propidium Iodide/RNase Staining Buffer (BD Pharmingen, cat. no. 550825) with the addition of 0.1% Triton X-100. Cells treated with either DMSO or TMZ (50 μmol/l) for 8 days were detached with a cell scraper in PBS solution, fixed in 70% ethyl alcohol, stored at −20 °C for 24 h, and then stained. EdU incorporation was performed with the Click-iT EdU Flow Cytometry Assay kit Alexa Fluor 647 (Invitrogen, Carlsbad, CA, USA; cat. no. 10424), which was modified for usage with 70% ethyl alcohol fixation. Cells were incubated with 25 μM of EdU at 37 °C for 1 h before fixation and subsequent staining.
BrdU incorporation assay
Proliferation was assessed by examining bromodeoxyuridine (BrdU) incorporation 14 h after the addition of BrdU to glioma cells and xenografted specimens in culture. Analysis of BrdU labeling of cells was performed using the BrdU Flow kit from BD Biosciences (San Jose, CA, USA) according to the manufacturer's instructions.
Genomic DNA isolation and analysis of DNA methylation by methylation-specific digestion combined with qPCR
Genomic DNA was isolated from freshly sorted CD133−CD15− U251 non-GSCs treated for 8 days with or without 50 μmol/l of TMZ, using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA) following the manufacturer's instructions. For detection of promoter CpG island methylation sites on the analyzed genes, genomic DNA was subjected to four digestions (mock, methylation-sensitive, methylation-dependent, or both) by using the EpiTect Methyl DNA Restriction Kit (SABiosciences, Frederick, MD, USA) following the manufacturer's instructions. Products from the digestion were quantified for levels of methylation by qPCR using the Human Stem Cell Transcription Factors EpiTect DNA Methylation PCR Array (MeAH-511, SABiosciences). Results were analyzed using a software from SABiosciences (www.sabiosciences.com/dna_methylation_data_analysis.php).
GSC sorting and establishment of fluorescent-labeled GSC subpopulations
In order to track GSC fate and reliably detect proportions of specific stem cell populations, we generated GFP-labeled U251 GSC lines. We stained the cells as described above before sorting. CD15+, double-positive (DP), and double-negative (DN) cell fractions were sorted on BD FACSAria (Becton Dickinson, San Jose, CA, USA) and the following proportions were obtained: 2.1% CD15+, 1.3% DP (CD133+CD15+), and 96.6% DN (CD133−CD15−). Fractions were seeded at a density of 5 × 104 cells/well in six-well plastic culture dishes (Becton Dickinson). Cells were then incubated for 24 h with replication-deficient lentiviral vectors containing GFP expression cassettes.
To be able to reliably study GSC plasticity and track GSC fate in vitro, we labeled each GSC subpopulation with a GFP cassette (Lenitvirus–GFP) and recreated a heterogeneous tumor cell population, maintaining the original proportions of GSCs and DN subpopulations (above described). Freshly sorted populations were counted and mixed, forming three different groups with the following designs: (a) MIX 1: DN GFP+ (96.6%), CD15+ non-GFP (2.1%), DP non-GFP (1.3%); (b) MIX 2: DN non-GFP, CD15+ GFP+, DP non-GFP; and (c) MIX 3: DN non-GFP, CD15+ non-GFP, and DP GFP+.
Neurosphere culture assay
Freshly sorted U251 GSCs (DP, CD15+, and CD133+) were plated at a density of 10 cells/well in non-adherent 24-well plates. They were cultured for 14 days in Neurobasal media with B27 (without Vitamin A, Invitrogen), N2 (Invitrogen), basic fibroblast growth factor (10 ng/ml), and epidermal growth factor (10 ng/ml), as previously described.16
Clonogenicity assay
Freshly sorted U251 GSCs (DP, CD15+, and CD133+) were plated under optimal conditions for neurosphere formation, as described above. Each sorted population was treated with either TMZ 50 μM or DMSO. The capacity of these two main groups of enriching for neurosphere formation was then compared. After 14 days, the number of colonies were counted for all groups.
In vivo limiting dilution assay
GBM43 cells serially passaged in the flank of nude mice were collected, stained, and sorted for CD133 and CD15 GSC markers. CD133 only, CD15 only, and double-negative cells (CD133−CD15−) were then intracranially implanted in the right hemisphere of athymic nude mice. Two major groups were created: (a) animals that received 100 sorted GBM43 cells (CD133 only, CD15 only, and double-negative cells – five animals/subgroup) and (b) those that received 500 sorted GBM43 cells (CD133 only, CD15 only, and double-negative cells – five animals/subgroup). The rate of engraftment between these two major groups and sorted subgroups was then compared.
In vivo xenotransplantation assay – flank model
The patient-derived glioma xenograft GBM43 was established as described previously.40 GBM43 glioma cells were bilaterally implanted in the flank of 6- to 10-week-old athymic nude mice. Three weeks post implantation, when the tumors were visible and palpable, mice were divided into four groups, five mice/group: (A) DMSO; (B) TMZ 2.5 mg/kg/day; (C) TMZ 5 mg/kg/day; and (D) TMZ 10 mg/kg/day. Mice were treated intraperitoneally either with TMZ or DMSO for 5 consecutive days. Five days post therapy mice were killed and tumors were collected, weighed, disaggregated as described above, and the single-cell suspensions were subjected to the following analysis:
To characterize the in vivo effect of different TMZ concentrations on the GSC subpopulations, GBM43 cells were labeled with human-specific antibodies for two cancer stem cell markers (CD15 and CD133) together with a broad stem cell marker (SOX2). To improve the exclusion of non-glioma cells, GBM43 cells were put through negative selection with anti-mouse CD45 (mouse-derived hematopoietic cell marker) followed by FACS analyses. Within the 10-day therapeutic window (between day 0 of TMZ therapy and day 10 when the tumor was collected), flank tumor volume was measured daily. As this same experiment was repeated twice, flank tumors from the second experiment were collected and frozen on OCT (Cryo-OCT Compound; Andwin Scientific Tissue-Tek) and dry ice (−70 °C). Tumors were serially sliced and analyzed using H&E and immunostaining.
In vivo xenotransplantation assay – intracranial model. Freshly sorted CD15+, DP (CD133+CD15+), and DN (CD133−CD15−) GBM43 from the in vivo flank model were intracranially implanted in male athymic nude mice. Three microliters of cell suspension (1000 cells/mouse) were injected (by a 5-μl Hamilton syringe) into the striatum using a stereotactic apparatus. Injection coordinates were 2 mm lateral to bregma and 3 mm deep. Five days later, mice were randomly divided into six groups (five animals/group): (a) DMSO-treated flank-derived CD15+ GBM43 GSCs; (b) TMZ-treated 2.5 mg/kg/day flank-derived CD15+ GBM43 GSCs; (c) DMSO flank-derived DP GBM43 GSCs; (d) TMZ 2.5 mg/kg/day flank-derived DP GBM43 GSCs; (e) DMSO flank-derived DN GBM43 non-GSCs; and (f) TMZ 2.5 mg/kg/day flank-derived DN GBM43 non-GSCs. When symptoms were observed (≥30% of weight loss, hunched back, lethargy, or paralysis), animals were killed. Tumor implantation was confirmed using immunohistochemical analysis and H&E staining.
Selection of TMZ in vivo dose
In order to choose the TMZ dose used throughout our in vivo experiments, we performed an allometric scaling conversion.44 We found that the initial dose used in humans (75 mg/m2/day) is equivalent to 25 mg/kg/day in a mouse. Our goal was to establish an in vivo model to study the behavior of GSCs after primary chemotherapy. Owing to the differential lifespan, metabolism, and brain size between mice and humans, a TMZ dose of 25 mg/kg/day cleared the disease burden in all the tested xenograft models, thus making it impossible to study the effects of chemotherapy on GSCs.45 Therefore, to select a more conducive dose we performed a TMZ dose–response experiment that evaluated serial in vivo doses of TMZ, which included 2.5, 5, and 10 mg/kg/day. In all experiments the dose of 2.5 mg/kg/day was the only one that was able to reduce tumor size and did not cause selection/death of specific GBM subpopulations. Therefore, we chose this specific dose to study the impact of TMZ on tumor cell populations in vivo.
Hypoxyprobe, immunofluorescence, and histology
The anti-human hypoxyprobe kit was purchased from Hypoxyprobe (Burlington, MA, USA). Mice intracranially injected with GBM43 cells (five animals/group) were intraperitoneally treated 2 weeks post-tumor injection with either DMSO or TMZ 2.5 mg/kg/day for 5 consecutive days. When symptoms were observed (≥30% of weight loss, hunched back, lethargy, or paralysis), all mice received a tail veil injection of hypoxyprobe as per the manufacturer's published protocol. Mice were euthanized 30 min after tail vein injection. The brains were harvested, flash-frozen (in 62.5% n-Butyl Bromide (Fisher Scientific, Pittsburg, PA, USA)+37.5% 2-methylbutane (Fisher Scientific) surrounded by crushed dry ice), and sectioned at 8-mm intervals. Frozen sections were post-fixed with 4% paraformaldehyde and blocked for nonspecific staining with 10% bovine serum albumin (BSA; A4503; Sigma-Aldrich, St. Louis, MO, USA) in PBS for 1 h. Sections were stained with hypoxyprobe antibody as per the manufacturer's instructions, anti-human CD133 (1 : 100, Millipore), anti-human Sox2 (1 : 100, Millipore), and anti-human GFAP (1 : 100, Abcam) antibodies.
Human GBM43 GSCs were identified in frozen flank sections using anti-human CD133 (1 : 50, Millipore) alone or in combination with CD15 (1 : 50, BD Pharminogen). The influence of TMZ on hypoxia-inducible factors was also analyzed by immunostaining with human-specific HIF1A (1 : 100, Abcam), HIF2A (1 : 100, Millipore), Nestin (Covance), and anti-human MGMT (1 : 50, Cell Signaling). Fluorescent images were analyzed and rendered for publication using Openlab v5.0 (Improvision, Coventry, England).
Statistical analysis
All statistical analyses were performed using the GraphPad Prism Software v4.0 (GraphPad Software, San Diego, CA, USA). Where applicable, one-way ANOVA, unpaired t-test, and log-rank test were applied. Survival distributions were estimated with the Kaplan–Meier method. A P-value <0.05 was considered statistically significant.
Acknowledgments
We thank Dr. David James for providing GBM43, GBM12, GBM6, GBM26, and GBM39 xenografts, and Ms. Lingjiao Zhang for her assistance on statistical analysis. This work was supported by the NCI (K99CA160775, R00CA160775, American Cancer Society (IL) 299503, R01CA138587).
Glossary
- GBM
Glioblastoma multiforme
- GSCs
Glioma stem cells
- TMZ
Temozolomide
- BBB
Blood–brain barrier
- CSF
Cerebral spinal fluid
- MEM
Minimum essential medium
- DMEM
Dulbecco's modified Eagle's medium
- FBS
Fetal bovine serum
- MGMT
O(6)-Methylguanine-DNA-methyltransferase
- TSM
Tumor sphere media
- FACS
Flow cytometry
- CSCs
Cancer stem cells
- BrdU
Bromodeoxyuridine
- 7-AAD
7-aminoactinomycin D
- qPCR
Quantitative polymerase chain reaction
- CpG
Phosphodiester bond between cytosine and guanine
- DP
Double-positive (CD133+CD15+)
- DN
Double-negative (CD133-CD15-)
- GFP
Green-fluorescent protein
- DMSO
Dimethyl sulfoxide
- OCT
Cryo-OCT Compound
- H&E
Hematoxylin and eosin
- ANOVA
Analysis of variance
- GFAP
Glial fibrillary acidic protein
- HIFs
Hypoxia-inducible factors
- Oct4
Octamer-binding transcription factor 4
- SOX2
(Sex determining region Y)-box 2
- Ki67
Antigen KI-67
- CD133
Prominin 1
- CD15
3-fucosyl-N-acetyl-lactosamine
- BSA
Bovine serum albumin
- IC50
Half maximal inhibitory concentration
- MFI
Mean fluorescence intensity
- IF
Immunofluorescence
- DNA
Deoxyribonucleic acid
- FOS
Proto-oncogene C-Fos
- Olig2
Oligodendrocyte transcription factor 2
- RUNX1
Runt-related transcription factor 1
- SMAD2
Mothers against decapentaplegic homolog 2
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Edited by R De Maria
Supplementary Material
References
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Hau P, Koch D, Hundsberger T, Marg E, Bauer B, Rudolph R, et al. Safety and feasibility of long-term temozolomide treatment in patients with high-grade glioma. Neurology. 2007;68:688–690. doi: 10.1212/01.wnl.0000255937.27012.ee. [DOI] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4:226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier D, Schulz JB, Beier CP. Chemoresistance of glioblastoma cancer stem cells—much more complex than expected. Mol cancer. 2011;10:128. doi: 10.1186/1476-4598-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeppernick F, Ahmadi R, Campos B, Dictus C, Helmke BM, Becker N, et al. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res. 2008;14:123–129. doi: 10.1158/1078-0432.CCR-07-0932. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, et al. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006;13:1238–1241. doi: 10.1038/sj.cdd.4401872. [DOI] [PubMed] [Google Scholar]
- Beier D, Rohrl S, Pillai DR, Schwarz S, Kunz-Schughart LA, Leukel P, et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res. 2008;68:5706–5715. doi: 10.1158/0008-5472.CAN-07-6878. [DOI] [PubMed] [Google Scholar]
- Rosso L, Brock CS, Gallo JM, Saleem A, Price PM, Turkheimer FE, et al. A new model for prediction of drug distribution in tumor and normal tissues: pharmacokinetics of temozolomide in glioma patients. Cancer Res. 2009;69:120–127. doi: 10.1158/0008-5472.CAN-08-2356. [DOI] [PubMed] [Google Scholar]
- Brada M, Judson I, Beale P, Moore S, Reidenberg P, Statkevich P, et al. Phase I dose-escalation and pharmacokinetic study of temozolomide (SCH 52365) for refractory or relapsing malignancies. Br J Cancer. 1999;81:1022–1030. doi: 10.1038/sj.bjc.6690802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermann S, Csajka C, Buclin T, Leyvraz S, Lejeune F, Decosterd LA, et al. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin Cancer Res. 2004;10:3728–3736. doi: 10.1158/1078-0432.CCR-03-0807. [DOI] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822–829. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- Son MJ, Woolard K, Nam DH, Lee J, Fine HA. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. 2009;4:440–452. doi: 10.1016/j.stem.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle. 2009;8:3274–3284. doi: 10.4161/cc.8.20.9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. 2011;108:7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiarto S, Persson AI, Munoz EG, Waldhuber M, Lamagna C, Andor N, et al. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell. 2011;20:328–340. doi: 10.1016/j.ccr.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enderling H, Hlatky L, Hahnfeldt P. Cancer stem cells: a minor cancer subpopulation that redefines global cancer features. Front Oncol. 2013;3:76. doi: 10.3389/fonc.2013.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Hitomi M, Gallagher J, Gadani SP, Adkins J, Vasanji A, et al. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis. 2011;2:e200. doi: 10.1038/cddis.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolou P, Toloudi M, Ioannou E, Chatziioannou M, Kourtidou E, Vlachou I, et al. AP-1 gene expression levels may be correlated with changes in gene expression of some stemness factors in colon carcinomas. J Signal Transduction. 2013;2013:497383. doi: 10.1155/2013/497383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–517. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoda L, Osato M, Yamashita N, Jacob B, Chen LQ, Yanagida M, et al. Runx1 protects hematopoietic stem/progenitor cells from oncogenic insult. Stem Cells. 2007;25:2976–2986. doi: 10.1634/stemcells.2007-0061. [DOI] [PubMed] [Google Scholar]
- Miyazono K, Ehata S, Koinuma D. Tumor-promoting functions of transforming growth factor-beta in progression of cancer. Upsala J Med Sci. 2012;117:143–152. doi: 10.3109/03009734.2011.638729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu B, Zhang D, Tao J, Tie X, Wu A, Wang Y. Human brain glioma stem cells are more invasive than their differentiated progeny cells in vitro. J Clin Neurosci. 2012;19:130–134. doi: 10.1016/j.jocn.2011.06.014. [DOI] [PubMed] [Google Scholar]
- Wakimoto H, Kesari S, Farrell CJ, Curry WT, Jr, Zaupa C, Aghi M, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69:3472–3481. doi: 10.1158/0008-5472.CAN-08-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel S, Garvalov BK, Wirta V, von Stechow L, Schanzer A, Meletis K, et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain. 2010;133 (Pt 4:983–995. doi: 10.1093/brain/awq042. [DOI] [PubMed] [Google Scholar]
- Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riganti C, Salaroglio IC, Caldera V, Campia I, Kopecka J, Mellai M, et al. Temozolomide downregulates P-glycoprotein expression in glioblastoma stem cells by interfering with the Wnt3a/glycogen synthase-3 kinase/beta-catenin pathway. Neuro-oncology. 2013;15:1502–1517. doi: 10.1093/neuonc/not104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persano L, Pistollato F, Rampazzo E, Della Puppa A, Abbadi S, Frasson C, et al. BMP2 sensitizes glioblastoma stem-like cells to Temozolomide by affecting HIF-1alpha stability and MGMT expression. Cell Death Dis. 2012;3:e412. doi: 10.1038/cddis.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wang D, Liu Y, Su Z, Zhang L, Chen F, et al. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int. 2013;13:119. doi: 10.1186/1475-2867-13-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed AU, Auffinger B, Lesniak MS. Understanding glioma stem cells: rationale, clinical relevance and therapeutic strategies. Exp Rev Neurother. 2013;13:545–555. doi: 10.1586/ern.13.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD, et al. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011;18:829–840. doi: 10.1038/cdd.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez JA, Joven J, Cufi S, Corominas-Faja B, Oliveras-Ferraros C, Cuyas E, et al. The Warburg effect version 2.0: metabolic reprogramming of cancer stem cells. Cell Cycle. 2013;12:1166–1179. doi: 10.4161/cc.24479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Martin A, Oliveras-Ferraros C, Cufi S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle. 2010;9:3807–3814. [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita-Mitchell A, Kat AG, Marcelino LA, Li-Sucholeiki XC, Goodluck-Griffith J, Thilly WG. Mismatch repair deficient human cells: spontaneous and MNNG-induced mutational spectra in the HPRT gene. Mut Res. 2000;450:125–138. doi: 10.1016/s0027-5107(00)00020-8. [DOI] [PubMed] [Google Scholar]
- Kitange GJ, Carlson BL, Mladek AC, Decker PA, Schroeder MA, Wu W, et al. Evaluation of MGMT promoter methylation status and correlation with temozolomide response in orthotopic glioblastoma xenograft model. J Neuro-oncol. 2009;92:23–31. doi: 10.1007/s11060-008-9737-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitange GJ, Carlson BL, Schroeder MA, Grogan PT, Lamont JD, Decker PA, et al. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro-oncology. 2009;11:281–291. doi: 10.1215/15228517-2008-090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad G, Sottero T, Yang X, Mueller S, James CD, Weiss WA, et al. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro-oncology. 2011;13:384–392. doi: 10.1093/neuonc/noq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- Tyler MA, Ulasov IV, Sonabend AM, Nandi S, Han Y, Marler S, et al. Neural stem cells target intracranial glioma to deliver an oncolytic adenovirus in vivo. Gene Ther. 2009;16:262–278. doi: 10.1038/gt.2008.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Tobias AL, Thaci B, Auffinger B, Rincon E, Balyasnikova IV, Kim CK, et al. The timing of neural stem cell-based virotherapy is critical for optimal therapeutic efficacy when applied with radiation and chemotherapy for the treatment of glioblastoma. Stem Cells Transl Med. 2013;2:655–666. doi: 10.5966/sctm.2013-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.