

Abstract
Background
Water loss has significant effects on physiological performance and survival rates of algae. However, despite the prominent presence of aeroterrestrial algae in terrestrial habitats, hardly anything is known about the molecular events that allow aeroterrestrial algae to survive harsh environmental conditions. We analyzed the transcriptome and physiology of a strain of the alpine aeroterrestrial alga Klebsormidium crenulatum under control and strong desiccation-stress conditions.
Principal Findings
For comparison we first established a reference transcriptome. The high-coverage reference transcriptome includes about 24,183 sequences (1.5 million reads, 636 million bases). The reference transcriptome encodes for all major pathways (energy, carbohydrates, lipids, amino acids, sugars), nearly all deduced pathways are complete or missing only a few transcripts. Upon strong desiccation, more than 7000 transcripts showed changes in their expression levels. Most of the highest up-regulated transcripts do not show similarity to known viridiplant proteins, suggesting the existence of some genus- or species-specific responses to desiccation. In addition, we observed the up-regulation of many transcripts involved in desiccation tolerance in plants (e.g. proteins similar to those that are abundant in late embryogenesis (LEA), or proteins involved in early response to desiccation ERD), and enzymes involved in the biosynthesis of the raffinose family of oligosaccharides (RFO) known to act as osmolytes). Major physiological shifts are the up-regulation of transcripts for photosynthesis, energy production, and reactive oxygen species (ROS) metabolism, which is supported by elevated cellular glutathione content as revealed by immunoelectron microscopy as well as an increase in total antiradical power. However, the effective quantum yield of Photosystem II and CO2 fixation decreased sharply under the applied desiccation stress. In contrast, transcripts for cell integrative functions such as cell division, DNA replication, cofactor biosynthesis, and amino acid biosynthesis were down-regulated.
Significance
This is the first study investigating the desiccation transcriptome of a streptophyte green alga. Our results indicate that the cellular response is similar to embryophytes, suggesting that embryophytes inherited a basic cellular desiccation tolerance from their streptophyte predecessors.
Introduction
Poikilohydric plants such as algae do not have protective structures (such as a waxy cuticula) and cannot actively regulate the transpiration rate (e.g. by stomata), which can easily lead to desiccation under water-limited conditions. Desiccation tolerance of these organisms can be defined as the ability to survive drying to ∼10% remaining water content, which is equivalent to ∼ 50% relative air humidity (RH) at 20°C [1], [2]. Aeroterrestrial green algae are naturally exposed to this stress, which they must tolerate in order to survive and propagate under these conditions (for a recent summary see [3]). The mechanisms to tolerate dehydration might involve the formation of permanent stages (e.g. akinetes, zygospores; e.g. [4]), but dehydration might also be tolerated in the vegetative state. Desiccation-tolerant species of green algae have been found in phylogenetically distinct lineages, e.g. in the so-called lichen algae, Trebouxiophyceae (e.g. [5], [6]), but also in streptophyte green algae, which represents the sister lineage to land plants (e.g. [7]–[11]).
A primary target of dehydration in green algae is photosynthesis (for a summary see [3]). Early studies reported an effect on CO2 exchange in the Trebouxiophyte Apatococcus lobatus [12]. While the most favourable carbon assimilation was measured in A. lobatus at ∼98% RH, even at RH 90%, 50% of the maximum CO2 uptake still occurred. The lower limit of carbon assimilation was observed at 68% RH [12]. More recently, Gray et al. [13] used ambient air at 25% RH to desiccate various strains of algae isolated in deserts and their aquatic relatives from the Chlorophyceae (e.g. Bracteacoccus, Scenedesmus, Chlorogonium) and Trebouxiophyceae (e.g. Chlorella, Myrmecia). They found that the desert algae could survive 4 weeks of desiccation when dried in darkness, and recovered photosynthetic quantum yield within 1 h of rehydration. Resurrection kinetics were also studied in other desiccation-tolerant Chlorophyta (e.g. Desmococcus, Apatococcus, Trebouxia), where green biofilm samples underwent prolonged desiccation (up to 80 days) and upon rehydration recovered about half of the initial value prior to desiccation [6]. In searching for the mechanisms behind this tolerance, Wieners et al. [14] observed desiccation-induced non-radiative dissipation in isolated green lichen algae (Trebouxia). Dynamic photoinhibition has been confirmed for several species of desert and aquatic algae (e.g. Klebsormidium, Cylindrocystis, Stichococcus; [15]). Photoprotective mechanisms were also found in the investigated algal strains; however, lineage-specific modifications occurred [15].
In Klebsormidium crenulatum [16]–[18] and a related Klebsormidium species [19]–[20], the optimum quantum yield decreased to zero after the cells were desiccated at ambient room temperature and RH; the exact percentage decrease was not determined, but recovered to different extents quickly after rehydration [16], [19]. Differences in desiccation tolerance might be also important for intra-generic ecological differentiation. For example, Škaloud and Rindi [21] suggested the existence of an ecological differentiation in Klebsormidium, according to different water-holding capacities of the substrates. However, on the global scale populations of Klebsormidium were described as genetically homogenous, and the local genotypes may be caused by ecological factors such as habitat differentiation [22]. Phylogenetic relationships within the genus Klebsormidium were recently elucidated (e.g. [20]–[21], [23], [24]), and the strain of K. crenulatum investigated here has been characterized by its rbcL gene [18] and is belonging to clade F according to [24].
Although some information is available on physiological adaptations to desiccation in green algae, no molecular approach has been used so far to understand desiccation tolerance in these organisms. Proteome analyses of the Trebouxiophycean green alga Asterochloris erici, a lichen-forming organism, found that only a few proteins increase upon desiccation [25]. It was not possible to identify changes in protein content involved in the light reactions of photosynthesis; however, dehydration led to a decrease in proteins associated with the Calvin cycle, indicating a reduction in carbon fixation. Interestingly, A. erici is still able to maintain initial values of maximum and actual quantum yield until 20% relative water content is reached [5]. Presumably, the loss of certain proteins does not halt carbon fixation during drying [25].
Deep sequencing (Illumina RNA-seq) provides a fast and reliable approach to generate large expression databases for functional genomic analysis, and is particularly suitable for non-model species with unsequenced genomes (e.g. [26]–[28]). While the analysis of the plant transcriptomes has produced an unexpected abundance of data on transcript identity in vascular plants, including resurrection plants (e.g. [29]–[31]), little information is yet available on transcriptomes in algae. In the unsequenced microalga Chlorella vulgaris, triacylglycerol biosynthetic pathways were analyzed by a transcriptome approach [32]. In C. vulgaris Illumina sequencing produced 27 million reads, Velvet and Oasis program suites were utilized for de novo transcriptome assembly, yielding 29,237 transcripts with an average length of 970 b. The results suggested an up-regulation of fatty-acid and triacylglycerol-biosynthetic mechanisms under nitrogen limitation [32]. A study using brown algal kelp Saccarina latissima identified transcriptional changes upon temperature and light stress; 32% of the genes showed an altered expression under the experimental conditions used [33]. Heinrich et al. [33] also found that high temperatures had a stronger effect on gene expression than low temperatures. The molecular processes of acclimation included the adjustment of the primary metabolism, induction of several ROS scavengers and regulation of heat-shock proteins (HSPs) [33]. A combination of high temperatures and high light intensities proved to be most harmful to this kelp species. Another study on the kelp Saccharina japonica investigated the effect of blue light on the transcriptome [34]. Their RNA-seq analysis yielded more than 70,000 non-redundant unigenes with an average length of 538 bp, and 40.2% of these could be mapped to transcript-encoding regions. Upon blue light exposure, ∼7,800 unigenes were up-regulated and ∼3,850 down-regulated. The feasibility of next-generation sequencing in the green algae Coleochaete and Spirogyra has been shown by Timme and Delwiche [35], and more than 30 transcriptomes were sequenced in the 1KP Project (http://onekp.com/project.html). The sequenced charophyte green algal transcriptomes have been shown to be remarkably similar to land plants, which demonstrate the evolutionary significance of these studies. Currently about 60,000 expressed sequence tags (ESTs) are available in GenBank for Klebsormidium (NCBI, 22.07.2014), representing several different species and strains. However, so far no studies on the effect of desiccation on the transcriptome of a streptophyte green alga are available.
In the present study, we exposed laboratory-grown Klebsormidium crenulatum to a strong desiccation stress in order to study, at the transcriptome level, the key molecular processes required for desiccation tolerance of this alga. To corroborate our findings, we performed physiological measurements of CO2 assimilation rates, effective quantum yield and antiradical power, and determined the cellular glutathione levels by immunoelectron microscopy. This enabled us to discuss the transcriptome findings in the context of the cellular strategies against desiccation.
Material and Methods
Algal cultures
Klebsormidium crenulatum (Kützing) Lokhorst [36] SAG 2415, previously isolated from an alpine soil crust [16], were cultivated in modified Bold's basal medium (3NMBBM (triple nitrate concentration [37]) or Waris-medium [38]) at 30–25 µmol photons m−2s−1 in a light:dark regime of 16∶8 in an Intellus environmental controller (Percival Scientific, Perry, IA, USA) at 20°C as previously described [16], [18]. The phylogenetic position of this strain was determined by rbcL sequences, deposited in GenBank under accession number JN190354 [18].
Desiccation experiment
Two-week-old cultures of K. crenulatum were concentrated by vacuum filtration onto S&S mixed cellulose membrane filters (Sigma Aldrich Z 612383, pore size 0.45 µm) in triplicate (n = 3). The experiment was carried out 2 h after onset of light. The filters were then desiccated on top of 4 glass columns holding a perforated metal grid inside a 200 mL polystyrol box over ∼ 100 g silica gel (Silica Gel Orange, Carl Roth, Karlsruhe, Germany) for 2.5 h according to the methods of [39]. During this procedure, the temperature and the relative air humidity in the box were monitored with a PCE-MSR145S-TH mini data logger for air humidity and temperature (PCE Instruments, Meschede, Germany). The weight of the mixed cellulose membrane filters was determined prior to vacuum filtration. Then the weight of the fresh biomass (after filtration of the algae; cells and surrounding water film) and the weight of the desiccated biomass (after 2.5 h of desiccation over silica gel) were determined. The filter weight was subtracted to gain the net weight of the fresh or desiccated biomass, respectively. The reduction (%) of the water content of the fresh biomass was calculated following the equation:
The desiccated biomass was then dried at 100°C for 13 h. The relative water content (RWC; %) of the desiccated biomass was calculated following the equation:
Staining with FM 1–43
Control cells, cells desiccated for 2.5 h, and cells allowed to recover for 2 h in culture medium after desiccation were stained with a 40 µM solution of FM1–43 (green biofilm cell stain, Invitrogen Ltd., Paisley, UK, prepared from a 20 mM stock solution in DMSO). Cells were observed directly in the staining solution in a Zeiss Pascal confocal laser scanning microscope (Carl Zeiss AG, Jena, Germany). Samples were excited with an argon laser beam (488 nm), and the emission was collected in two separate channels at a wavelength band between 505–550 nm (false-colored green) and at wavelengths longer than 560 nm (false-colored red).
Pulse amplitude fluorescence (PAM) and gas exchange measurements
The effective quantum yield (ΔF/Fm’) of photosystem II (Yield II) was determined every 10 min during the dehydration period, using a pulse-amplitude modulated fluorimeter (PAM 2500, Heinz Walz GmbH, Effeltrich, Germany) according to the methods described by [39]. ΔF/Fm’ was calculated as (Fm’-F)/Fm’ with F as the fluorescence yield of light-exposed algal cells (40 µmol photons m−2 s−1) and Fm’ as the maximum light-adapted fluorescence yield after employing an 800 ms saturation pulse, as described by Schreiber and Bilger [40]. The PAM light probe was positioned outside the desiccation box at a distance of 2 mm. The measurements were carried out on 3 individual filters (n = 3) containing algal suspension, placed in individual desiccation boxes. At each filter 3 measurement points were recorded every 10 min. The CO2 assimilation rate of K. crenulatum was measured by a GFS-3000 portable gas exchange fluorescence system (Heinz Walz GmbH, Effeltrich, Germany) with a LED-Array/PAM-Fluorometer 3055-FL under strong desiccation stress at ∼20% RH (n = 3) for 2.5 h. 100 mL homogeneous algal culture was filtered on a ME25 membrane filter (mixed cellulose ester, Whatman GmbH, Germany). Cells were removed from the filter areas that were not placed inside the measurement chamber. The assimilation rate was calculated based on the area (8 cm2) of the measurement chamber. After a recovery period in culture medium overnight (21 h), the assimilation rate was monitored again. Standard conditions for all measurements were 20°C, 30 µmol photons m−2 s−1, 380 ppm absolute CO2 content and a molar flow rate at the inlet of the cuvette of 750 µmol s−1. Absolute water content was calculated with a Vaisala Humidity Calculator 1.3 (Vaisala, Helsinki, Finland) to adjust the required relative humidity in the GFS-3000 measurement chamber to 4919 ppm for 20% RH. Assimilation rate was measured at 1-min intervals, and in parallel Yield II was measured at 3-min interavls. After each measurement, the chlorophyll a content of the sample was determined. Chlorophyll a was extracted in dimethylformamide (DMF) and quantified according to Porra at al. [41]. Values of the initial 5 min of the desiccation period, of the desiccated sample, and of the initial 5 min of the recovery measurements were statistically analyzed with SPSS 15.0 for Windows (IBM Corporation, Somer, NY, USA), using one-way ANOVA followed by Tukeýs post-hoc test (p≤0.001).
RNA isolation
RNA was isolated from K. crenulatum cells either directly concentrated on mixed cellulose filters or desiccated as described above. The filters were swiped with 450 µL of Life Guard Soil Preservation solution (MO BIO Laboratories, Inc., Carlsbad, CA, USA, Cat No. 12868-100). The cells were frozen in liquid nitrogen, then ground with a mortar and pestle, followed by RNA extraction using a PEQLAB Gold Plant RNA isolation kit (PEQLAB, Erlangen, Germany) according to the manufacturer's instructions. For DNA removal the RNA was treated with DNaseI (Thermo Scientific, DNaseI, RNase free). A final cleanup was performed, again using the PEQLAB RNA column.
Expression analysis
Random primed cDNA libraries were prepared by GATC Biotech AG (Konstanz, Germany). The library preparations were sequenced on an Illumina HiSeq 2000 as single-reads to 100 bp in the same flow cell (first sequencing run). All sequences were analyzed using the CASAVA v1.7 (Illumina, USA). To establish a reference transcriptome, the same amounts of RNA from the control and desiccated samples were pooled and a normalized random primed cDNA library was prepared from the pooled samples by GATC Biotech AG (Konstanz, Germany). The reference library was sequenced on a Roche GS FLX (1/2 plate), yielding 1,553,953 sequenced reads (636,002,000 sequenced bases). Assembly and basic expression analysis were performed at GATC Biotech AG. Briefly, the reference genome library was assembled using the De Novo Assembler (Newbler) 2.8 software from Roche. For expression analysis, sequence reads were mapped to the reference sequence (unigene.highcov.fa) using BWA with the default parameters. Only uniquely mapped reads were considered for further processing. PCR duplicates were removed using PICARD. The resulting high-quality sequence alignments were taken for coverage determination and variant detection. The alignments were processed to compute the read counts for each exon/CDS/transcript. Read counts were then normalized to 1 million reads (RPM) by considering the total reads mapped in each sample, to remove the uneven sequencing coverage over multiplexed samples. Finally, RPM counts were normalized to 1 Kbp transcript length (RPKM) to remove the uneven gene/transcript length bias. The final table included all the transcripts present in the reference employed. The entries were filtered based on defined threshold before doing the expression analysis. Determining counts, RPM and RPKM computations were done using in-house scripts by GATC-Biotech.
For differential expression analysis, the DeSeq software [42] was used, following the program's vignette (DESeq version 1.12.0, Last revision 2013-02-24). Pathways and modules were analyzed using the KEGG module and pathway databases at http://www.genome.jp/kegg/. Local Blast analyses were performed using the NCBI BLASTX program and the protein databases from the following species obtained at www.phytozome.net: Chlamydomonas reinhardtii, Coccomyxa subellipsoidea, Micromonas pusilla, Ostreococcus lucimarinus, Physcomitrella patens, Selaginella italica, Zea mays, Oryza sativa, Aquilegia coerulea, Solanum tuberosum, Eucalyptus grandis, Theobroma cacao, Arabidopsis thaliana, Malus domesticus, Medicago trunculata and Populus trichocarpa RNA-Seq data and the assembled contigs have been deposited in the NCBI's Gene Expression Omnibus (GEO) [43] and are accessible through the NCBI Sequence Read Archive (http://trace.ncbi.nlm.nih.gov/Traces/sra/) under the accession numbers SRR1514242 (assembled reference library) and SRR1563130 to SRR1563134 (transcriptomes).
Cytohistochemical detection of glutathione
Preparation of samples for transmission electron microscopy and immunogold labeling of glutathione was performed as previously described [44], [45]. Briefly, algae were fixed in 2.5% paraformaldehyde and 0.5% glutardialdehyde in 0.06 M phosphate buffer (pH 7.2), rinsed in the buffer, dehydrated in increasing concentrations of acetone (50%, 70%, and 90%) and infiltrated with increasing concentrations of LR-White resin (30%, 60% and 100%; London Resin Company Ltd., Berkshire, UK). Samples were polymerized at 50°C. Ultrathin sections were blocked with 2% bovine serum albumin (BSA) in phosphate buffered saline (PBS, pH 7.2) and treated with the primary antibody (anti-glutathione rabbit polyclonal IgG, Millipore Corp., Billerica, MA, USA) diluted 1∶50 in PBS containing 1% goat serum. After three brief rinses in PBS, sections were incubated with 10 nm gold-conjugated secondary antibodies (goat anti-rabbit IgG for glutathione, British BioCell International, Cardiff, UK) 1∶50 in PBS and finally washed with distilled water. A minimum of 25 different cells were analyzed for gold particle density. The data obtained were presented as the number of gold particles per μm2.
DPPH-Test
For determination of the antiradical power of fresh and 2.5 h desiccated K. crenulatum cells, a 2,2-diphenyl-1-picrylhydrazyl (DPPH)-test using 6-hydroxy-2,5,7,8-tetramethylchroman-2-carbonacid (Trolox, Sigma Aldrich, Germany) as standard was used according to the methods of [46]. K. crenulatum cultures were filtered according to the above-mentioned procedure onto Whatman GF/F glass fiber filters (Whatman, Dassel, Germany) and wrapped in aluminum foil, either immediately as a control (n = 3) or after desiccation for 2.5 h over silica gel (n = 3). Filters were then frozen in liquid nitrogen, and stored at −80°C prior to lyophilization. Lyophilized algae were then extracted in plastic vials containing 5 mL of 70% acetone for 24 h at 4°C under continuous shaking in darkness. 200 µL of a 150 µM DPPH-solution in 80% ethanol was transferred into a 96-well microtiter plate. The DPPH solution was complemented with 22 µL of the respective samples or Trolox standards in 70% acetone (7.8 µM-1 mM). After 25 min the absorption was determined for each sample at 516 nm. The statistical significance of means was tested by two-sample t-test.
Results
Experimental setup and physiological response to desiccation stress in Klebsormidium crenulatum
To identify the molecular processes involved in the desiccation response in K. crenulatum, filaments were transferred to filter membranes and incubated for 2.5 h in a desiccation chamber over silica gel. At the end of the experiment, the relative humidity in the desiccation chamber was ∼10% (Fig. 1). The weight of the fresh biomass (algae and surrounding water film) was reduced by 95.16±1.41% during the desiccation process. The experimentally desiccated algae had a RWC of 6.54±1.89%.
Figure 1. Effective quantum yield of photosystem II (Yield II) of Klebsormidium crenulatum during desiccation (desiccated over 100 g of silica gel) monitored by PAM 2500.

Values represent means (n = 3), error bars show standard deviation (SD). The results of three filters containing similar amount of algal biomass are shown individually, to demonstrate that the decrease of Y II occurred within a short time frame (40–50 min). The relative air humidity (RH) inside the chamber (right y-axis) was monitored by a PCE sensor.
Within 40–50 min after exposure of the filters to silica gel, the effective quantum yield of K. crenulatum cells dropped to zero, suggesting complete inhibition of photosystem II (Fig. 1). When rehydrated with medium after the 2.5 h desiccation period, the effective quantum yield of photosystem II fluorescence did not recover for at least 2.5 h (not shown). Gas exchange measurements confirmed this observation (Fig. S1). The initial CO2 assimilation rate was ∼0.5 µmol CO2 m−2 s−1 at the beginning of the measurements (Fig. S1). CO2 assimilation of photosynthesis was inhibited immediately when the RH dropped (Fig. S1). At the end of the desiccation period, a net respiration was found. Cells did not recover completely when the filters were rewetted, and the measurements continued; after a 21-h recovery period, respiration still predominated, although at a lower level (−0.05 to −0.1 CO2 m−2 s−1). In agreement with the measurements from the desiccation chamber (Fig. 1), and also in the gas-exchange measurements, the effective quantum yield of photosystem II (Yield II, Fig. S1) did not recover.
To assess the cellular damage resulting from the desiccation stress, a cell viability test using the FM1-43 dye was performed. In control cells of K. crenulatum, FM1-43 dye stained the cell membrane predominantly (Fig. 2 A); however in some cases, the membranes of the vacuoles and chloroplast were also stained if the cells were exposed to the dye for a longer period (not shown). In contrast, dead cells, which are always present in small numbers in a cell culture, showed a bright fluorescence in the cell lumen (Fig. 2 D).
Figure 2. Klebsormidium crenulatum stained with FM 1–43.
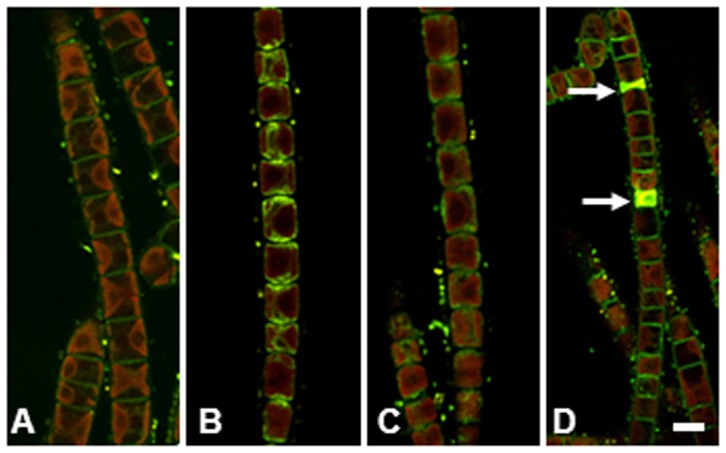
(A) Control cells. (B) Desiccated for 2.5 h. (C) 2 h recovery in culture medium after drying for 2.5 h. (D) Control filament with two dead cells (arrows). Bar 10 µm.
The cytoplasm of cells exposed to desiccation stress for 2.5 h was stained in a granular pattern; however, the plasma membrane was still visible and no labeling of the plastid was observed (Fig. 2 B). After a recovery period of ∼2 h the plasma membrane was stained more prominently as in the desiccated cells, although weaker than in the control cells, and there was still some cytoplasmic staining (Fig. 2 C). However, we rarely observed the bright fluorescence of the total cell volume, indicative of dead cells (Fig. 2 D).
Molecular analysis
To identify desiccation stress-regulated cellular processes and functions in K. crenulatum, we first established a reference transcriptome database. Algae were transferred to filter membranes and RNA was isolated either directly or after applying desiccation stress (2.5 h desiccation over silica gel). RNA was isolated from control and desiccated samples (each in triplicate). Control samples contained more RNA than desiccated samples (Fig. S2, compare the amount of 18S and 25S between control and desiccated samples), indicating a considerable reduction in the amount of RNA during desiccation stress in K. crenulatum. In addition, in the electropherograms of the desiccated samples, the 18S and 25S rRNA peaks were reduced and an increased baseline noise and lower RNA integrity number (RIN between 5.7 to 5.8 in the control samples and between 3.8 to 4.2 in the desiccated samples) were observed, indicative of RNA degradation, which is nearly absent from the control samples (see also discussion).
For the reference library, all samples were pooled (same amount of RNA from each sample), and a normalized random primed cDNA library was prepared and sequenced using 454 sequencing. Random primed cDNA libraries were also prepared from the individual samples and sequenced using the Illumina technology. All control samples and two desiccated samples yielded sufficient reads for further analyses. In the following sections, we will first describe the reference library and then present our results from the expression analysis.
Reference library
454 sequencing yielded 1,553,953 sequenced reads (average read length 409 b, GC-content 52.17%, N 0.02%), giving more than 138,213 contigs with a mean length of 563 b. However, 83,276 of the contigs represented short singletons; therefore a high-coverage reference database was constructed, which contained only contigs containing at least 5 reads. This database contained 24,183 contigs with a mean sequence length of 1,327 b (N50 = 1,463 b). All further analyses were performed using this high-coverage assembly.
BLAST-analyses
To identify transcripts of interest, we compared the deduced peptides of the high-coverage assembly with the proteomes of selected green algae and embryophytes (Table 1) using BLASTX. A total of 13,316 contigs (55.1%) showed similarity to the deduced protein sequences in the genomes (for simplicity, henceforth called proteins) of the selected viridiplants (chlorophytes and streptophytes, e-value <exp -10). 10,505 K. crenulatum contigs = 43.4% showed similarity to chlorophyte protein sequences, whereas 12,596 K. crenulatum contigs = 52.1% showed similarity to streptophyte protein sequences. However, only 4,752, 5,949 and 7,701 K. crenulatum contigs showed similarity to proteins in all investigated viridiplants, chlorophytes or streptophytes respectively. Overall, more sequences showed similarity to streptophyte genomes than to chlorophyte genomes (Table 1) and generally the conservation of the amino acid sequences was higher when the deduced peptide sequences of Klebsormidium contigs were compared with streptophyte proteins than when comparing with chlorophyte proteins (not shown). These results highlight the genetic diversity of chlorophytes and the large number of streptophyte-specific contigs (2,791 K. crenulatum contigs).
Table 1. Similarity of Klebsormidium crenulatum contigs to other organisms.
| Name | Growth type | Number of Blast hits | Up-regulated | Up-regulated (%) |
| Chlamydomonas reinhardtii | Unicellular Flagellate | 8727 | 1545 | 17.70 |
| Coccomyxa subellipsoidea | Unicellular Coccoid | 8856 | 1624 | 18.34 |
| Micromonas pusilla | Unicellular Flagellate | 7759 | 1367 | 17.62 |
| Ostreococcus lucimarinus | Unicellular Coccoid | 7180 | 1273 | 17.73 |
| Physcomitrella patens | Moss | 11842 | 2095 | 17.69 |
| Selaginella italica | Lycophyte | 10781 | 1886 | 17.49 |
| Zea mays | Grass | 10771 | 1884 | 17.49 |
| Oryza sativa | Grass | 10590 | 1879 | 17.74 |
| Aquilegia coerulea | Perennial Herb | 10995 | 1924 | 17.51 |
| Solanum tuberosum | Perennial Herb | 10127 | 1870 | 18.47 |
| Eucalyptus grandis | Tree | 10897 | 1875 | 17.21 |
| Theobroma cacao | Tree | 11137 | 1945 | 17.46 |
| Arabidopsis thaliana | Annual Herb | 10995 | 1931 | 17.56 |
| Malus domesticus | Tree | 10670 | 1890 | 17.71 |
| Medicago trunculata | Annual Herb | 9552 | 1742 | 18.24 |
| Populus trichocarpa | Tree | 11070 | 1931 | 17.44 |
| Ricinus communis | Shrub | 11015 | 1907 | 17.31 |
24,183 contigs were blasted against the protein database of the selected organisms listed (obtained from Phytozome). The number of BLASTX hits (e-value <exp -10) is shown. The number of hits of contigs that were up-regulated under desiccation stress (twofold or more) and the percentage of the total hits are also shown.
KEGG-Modules and Pathway analyses
To investigate whether the reference genome covers most cellular functions, we searched the KEGG databases using the high-coverage contigs as a query. As shown in Table S1, most basic cellular processes (e.g. carbohydrate metabolism, fatty acid metabolism, nucleotide metabolism, amino acid metabolisms, photosynthesis, and respiration) were completely or nearly completely represented in the deduced peptide sequences.
Expression analysis
Illumina sequencing of random primed cDNA libraries yielded between 12 and 30 million reads per sample. The read statistics are given in Table S2. Only high-quality reads (unique or duplicates) were used for further analyses. Table 2 gives an overview of the number of K. crenulatum contigs showing changes in transcript expression level. 10,493 K. crenulatum contigs (43.4%) showed at least a 2-fold change in their expression level, thus the majority of the transcriptome is apparently unaffected. Only a few K. crenulatum contigs were expressed exclusively under desiccation stress or control conditions (Table 2) and more transcripts were down-regulated than up-regulated (Table 2). Interestingly, the percentage of up-regulated transcripts showing similarity to viridiplant genomes is higher than the percentage of down-regulated transcripts showing similarity to viridiplant genomes. In Table 3 the 20 highest up-regulated K. crenulatum contigs are shown. The complete lists of all up- and down-regulated mRNAs including the most similar proteins in the 14 viridiplant proteomes searched, are presented in Table S3 and Table S4. Most of the highly up-regulated contigs do not show similarity to known viridiplant proteins and many show no similarity to any known protein at all. Thus a large number of regulated transcripts might represent specific adaptations of K. crenulatum to its environment. However, 100 of the 171 K. crenulatum contigs, up-regulated at least 10-fold, show similarity to proteins in viridiplant proteomes. Surprisingly, we could detect only 46 Arabidopsis proteins in this category. Interestingly, most of these Arabidopsis proteins are well conserved in embryophytes and 34 are also similar to proteins in chlorophytes, suggesting that these proteins represent basic cellular functions, generally up-regulated upon stress. In contrast, we could detect several additional K. crenulatum contigs (at least 10-fold up-regulated) that show similarity to other embryophytes. Overall there are 89 K. crenulatum contigs similar to proteins in Medicago and 61 to 71 K. crenulatum contigs similar to proteins from the trees investigated (except Eucalyptus, 49).
Table 2. Overview of changes in transcript expression during desiccation stress.
| Up-regulated | Down-regulated | |
| At least 2-fold change | 4129 (2264) | 5898 (2343) |
| •Significant (Pval * <0.05): | 3766 | 5011 |
| •Significant (Padj * <0.05): | 3642 | 4584 |
| •Number of KO-Terms | 736 | 616 |
| Expressed only in/upon controls/desiccation | 31 | 435 |
| •Significant (Pval * <0.05): | 4 | 136 |
| •Significant (Padj * <0.05): | 2 | 115 |
The number of Klebsormidium crenulatum contigs in each category is given except for Number of KO-terms, which gives the number of KO-Terms, obtained for up- and down-regulated K. crenulatum contigs with a Padj <0.05. Number in brackets gives the percentage of K. crenulatum contigs that show similarity to viridiplant genomes.
*obtained with the DeSeq program. Pval = p value for the statistical significance of this change.
Padj = p value adjusted for multiple testing with the Benjamini-Hochberg procedure, which controls the false discovery rate (FDR).
Table 3. Klebsormidium crenulatum contigs showing the largest changes upon desiccation stress.
| K. crenulatum -ID | Fold Change | padj | Function |
| UN027167 | Not expressed in controls | 0.03141657 | Plastid NADH:ubiquinone/plastoquinone oxidoreductase |
| UN034964 | Not expressed in controls | 0.00627352 | |
| UN022359 | 91.4439431 | 0.00021721 | |
| UN036634 | 60.5266278 | 8.9783E-16 | Similar protein in some plants |
| UN043434 | 52.011157 | 7.675E-16 | |
| UN036811 | 51.8868523 | 1.6101E-23 | Similar protein in some plants |
| UN012067 | 51.0430016 | 1.3086E-11 | Similar protein in some plants |
| UN034707 | 47.3497452 | 2.6678E-17 | Similar protein in some plants |
| UN020446 | 45.7941202 | 1.3302E-21 | Similar protein in some plants |
| UN000210 | 38.6089069 | 7.6031E-16 | |
| UN026331 | 38.1383995 | 1.679E-07 | Similar protein in some plants |
| UN028420 | 35.9012593 | 4.4486E-83 | ELIP1 early light inducible protein |
| UN031973 | 35.1401051 | 1.6143E-65 | NPQ4 (PSBS) |
| UN022671 | 33.789896 | 7.0903E-37 | |
| UN035517 | 30.9168329 | 5.324E-104 | Similar protein in some plants |
| UN052587 | 30.7842929 | 6.0114E-36 | |
| UN028622 | 30.4695717 | 1.9817E-51 | PHT6 phosphate transporter |
| UN026579 | 30.0812794 | 1.0935E-69 | GLT1 plastid glucose transporter |
| UN030270 | 29.7741941 | 5.6407E-34 | |
| UN029908 | 29.432577 | 2.1169E-81 | Catalase 2 |
A tentative function is given when possible.
KEGG analysis of desiccation induced changes
To gain more insight into the molecular mechanisms of desiccation tolerance, we searched the KEGG database for pathways (Table 4) and modules (Table S1), using the complete, the up-regulated and the down-regulated datasets. K. crenulatum contigs could be assigned to 782 different KO functions using the pathway searching tool, and transcripts representing 229 KO functions were up-regulated and transcripts representing 186 KO functions were down-regulated, respectively. The majority of the deduced KEGG-pathways (for simplicity called pathways in the following sections) showed complex regulation patterns with several contigs up- and down-regulated (e.g. Figs. 3 and 4). The 15 pathways which were overall most strongly up- and down-regulated are shown in Table 4 and Table 5. As is evident from Table 4, contigs involved in energy production (glycolysis, TCA cycle, respiration, photosynthesis etc.) were mainly up-regulated. Other important up-regulated pathways were galactose, ascorbate (Fig. 3 A) and glutathione (Fig. 3 B) metabolism; and plant hormone and calcium signaling. No contigs showed similarity to proteins of the auxin and gibberellin signal transduction pathways. In contrast, homologues for all components of the cytokinin signaling pathway (CRE1, down-regulated; AHP, up-regulated; B-ARR, down-regulated and A-AAR, up-regulated) and 3 of the 4 components of the abscisic acid signaling pathway (PP2C, SnRK2, ABF, all up-regulated) were found. Surprisingly, putative homologues of the ethylene and jasmonic acid receptors (ETR, JAR1) are also present. However, putative homologues of other components of these signaling pathways are missing, except for (CTR1 and EIN3) for which we also detected putative homologues.
Table 4. Effect of desiccation stress on transcript expression in Klebsormidium crenulatum: strongly up-regulated pathways.
| KO number | Cellular Function | All contigs | Up-regulated contigs | % up-regulated | Down-regulated contigs | % down-regulated |
| ko00195 | Photosynthesis | 32 | 30 | 93.8 | 1 | 3.1 |
| ko03010 | Ribosome | 124 | 114 | 91.9 | 4 | 3.2 |
| ko00196 | Photosynthesis - antenna proteins | 12 | 9 | 75.0 | 1 | 8.3 |
| ko00710 | Carbon fixation in photosynthetic organisms | 21 | 14 | 66.7 | 2 | 9.5 |
| ko00190 | Oxidative phosphorylation | 79 | 48 | 60.8 | 5 | 6.3 |
| ko04075 | Plant hormone signal transduction | 14 | 8 | 57.1 | 5 | 35.7 |
| ko00030 | Pentose phosphate pathway | 18 | 10 | 55.6 | 4 | 22.2 |
| ko00020 | Citrate cycle (TCA cycle) | 20 | 11 | 55.0 | 1 | 5.0 |
| ko00630 | Glyoxylate and dicarboxylate metabolism | 29 | 15 | 51.7 | 4 | 13.8 |
| ko00052 | Galactose metabolism | 15 | 7 | 46.7 | 4 | 26.7 |
| ko00071 | Fatty acid degradation | 13 | 6 | 46.2 | 4 | 30.8 |
| ko04020 | Calcium signaling pathway | 11 | 5 | 45.5 | 3 | 27.3 |
| ko00010 | Glycolysis/Gluconeogenesis | 32 | 14 | 43.8 | 8 | 25.0 |
| ko00053 | Ascorbate and aldarate metabolism | 15 | 6 | 40.0 | 2 | 13.3 |
| ko00500 | Starch and sucrose metabolism | 36 | 14 | 38.9 | 15 | 41.7 |
The KEGG ontology (KO) numbers were retrieved using the KAAS annotation server. Using the KO numbers, pathway maps were constructed using the KEGG pathway reconstruction tool. The pathways with the largest percentage of up-regulated KO functions are shown. Human disease-related pathways and overview maps are not shown.
Figure 3. Ascorbate (A) and glutathione (B) metabolism in Klebsormidium crenulatum.

(A) Biosynthesis of ascorbate via the D-galacturonate pathway is upregulated, as are ascorbate-consuming and recycling enzymes. (B) The biosynthesis of glutathione is up-regulated except for glutathione synthase. Glutathione degradation is down-regulated, whereas glutathione-consuming enzymes are up-regulated.
Figure 4. Biosynthesis of carotenoids in Klebsormidium crenulatum.

(A) Terpenoid backbone synthesis, the cytosolic mevalonate pathway is inhibited. (B) Carotenoid biosynthesis is up-regulated.
Table 5. Effect of desiccation stress on transcript expression in Klebsormidium crenulatum: strongly down-regulated pathways.
| KO number | Function | All contigs | Up-regulated contigs | % up-regulated | Down-regulated contigs | % down-regulated |
| ko03450 | Non-homologous end joining | 9 | 2 | 22.2 | 6 | 66.7 |
| ko04140 | Regulation of autophagy | 11 | 1 | 9.1 | 7 | 63.6 |
| ko00970 | Aminoacyl-tRNA biosynthesis | 27 | 3 | 11.1 | 15 | 55.6 |
| ko03420 | Nucleotide excision repair | 35 | 5 | 14.3 | 19 | 54.3 |
| ko03030 | DNA replication | 28 | 1 | 3.6 | 15 | 53.6 |
| Ko03430 | Mismatch repair | 19 | 1 | 5.3 | 10 | 52.6 |
| ko04142 | Lysosome | 43 | 7 | 16.3 | 21 | 48.8 |
| ko00310 | Lysine degradation | 19 | 5 | 26.3 | 9 | 47.4 |
| ko04150 | mTOR signaling pathway | 13 | 3 | 23.1 | 6 | 46.2 |
| ko04111 | Cell cycle - yeast | 48 | 6 | 12.5 | 22 | 45.8 |
| Ko00670 | One carbon pool by folate | 11 | 1 | 9.1 | 5 | 45.5 |
| ko00513 | Various types of N-glycan biosynthesis | 20 | 2 | 10.0 | 9 | 45.0 |
| ko04113 | Meiosis - yeast | 38 | 3 | 7.9 | 17 | 44.7 |
| ko00640 | Propanoate metabolism | 23 | 6 | 26.1 | 10 | 43.5 |
| ko00910 | Nitrogen metabolism | 14 | 4 | 28.6 | 6 | 42.9 |
The KEGG ontology (KO) numbers were retrieved using the KAAS annotation server. Using the KO numbers, pathway maps were constructed using the KEGG pathway reconstruction tool. The pathways with the largest percentage of down-regulated KO functions are shown. Human disease-related pathways and overview maps are not shown.
Desiccation induced up-regulation of transcripts
Only for six of the top 20 up-regulated contigs it is possible to infer a putative function. Four of these transcripts are similar to plastidic proteins: two up-regulated transcripts are possibly involved in non-photochemical quenching (UN028420, ELIP1, 35.9-fold up-regulated; UN031973, NPQ4 (PSBS), 35.1-fold up-regulated); another interesting contig shows similarity to a plastidic glucose transporter (UN026579, GLT1, 30.1-fold up-regulated). Transcripts for sucrose phosphate synthase (2 contigs: UN036592, 3.6-fold; UN039619, 2.8-fold) and sucrose synthase (3 contigs: UN023940, 2.1-fold, UN038702 3.2-fold; UN039004 3.8-fold) are also up-regulated, whereas transcripts for cellulose synthase and some enzymes of the starch and trehalose metabolism are down-regulated. We also found a contig displaying strong similarity to AT2G35840, a sucrose-6-phosphate phosphohydrolase family protein (e-value: 3e-104). This transcript is 4-fold up-regulated; however, the pathway reconstruction tool at KEGG failed to recognize this transcript as a putative SPSP.
Several transcripts for putative enzymes of the galactinol/raffinose metabolism were found to be up-regulated, including a transcript for galactinol synthase (UN010053, BLASTX result: AT1G60450.1, AtGolS7, e-value 5 exp-30), the key enzyme for the biosynthesis of the raffinose family of oligosaccharides (RFO). Ascorbate is probably synthesized from GDP-Man as a transcript similar to GDP-D-mannose 3′, 5′-epimerase (UN025544, similar to AT5G28840.2, GME, e-value: 3E-49) is under desiccation stress 26-fold up-regulated, as are several contigs with similarity to enzymes using ascorbate as reductant. Similarly to ascorbate, glutathion plays an important role in reactive oxygen metabolism. Several transcripts coding for enzymes in the glutathion metabolism are up-regulated (Fig. 3 B). For example, contigs with similarity to enzymes involved in the first step of glutathion biosynthesis, glutathione-S-transferase and glutathione peroxidase are up-regulated, while transcripts with similarity to glutathione-degrading enzymes and the glutathione-disulfide reductase are down-regulated. The importance of reactive oxygen species (ROS) protection is also highlighted by the up-regulation of a transcript similar to catalase 2 (UN029908, 29.4-fold up-regulation). To corroborate these findings, we investigated changes in the total cellular glutathione content, using an immune gold glutathione-labeling approach (Fig. 5), and the cellular antiradical power of K. crenulatum. The distribution of glutathione-specific gold labeling in K. crenulatum grown under control conditions revealed that the highest glutathione labeling density was found in mitochondria, followed by nuclei, chloroplasts and the cytosol (Fig. 5 A and Table 6). Due to ultrastructural changes induced by desiccation, gold particles could not be clearly assigned to the individual cell compartments of desiccation-stressed K. crenulatum cells (Fig. 5 B). Thus, the total amount of glutathione labeling was calculated for both control and desiccation-stressed K. crenulatum cells (Table 7). The glutathione labeling density increased in desiccation-stressed K. crenulatum cells to about 177% of the value in control cells (Table 6). The total cellular antiradical power was determined using the DPPH-test (see material and methods). The cellular antiradical power was significantly higher (p <0.01) in 2.5 h desiccated samples than in control cells (Fig. 6).
Figure 5. Immunogold labeling with 10 nm gold particles, of glutathione in transmission electron microscopic sections of Klebsormidium crenulatum.

(A) Control cell. (B) Desiccated cell (2.5 h at ∼ 10% RH). Bar 1 µm.
Table 6. Glutathione content of Klebsormidium crenulatum cells.
| Gold particles per μm2 | ||
| control | desiccated | |
| Mitochondria | 168±8a | n.d. |
| Chloroplasts | 28±1d | n.d. |
| Nuclei | 42±2 c | n.d. |
| Cytosol | 29±1d | n.d. |
| Whole cell | 30±1e | 53±2b |
| Cell without vacuole | 32±1e | n.d. |
Values are means with standard errors, and document the total amount of gold particles bound to glutathione per μm2 in different cell compartments and whole cells of Klebsormidium crenulatum grown under control and desiccation conditions. n = 40 for the individual cell structures and n = 25 for the whole cell. Different lowercase letters indicate significant differences (P<0.05) analyzed with the Kruskal-Wallis test followed by post-hoc comparison according to Conover. n.d. = not determined.
Table 7. Plant desiccation/dehydration/drought stress-related contigs in Klebsormidium crenulatum.
| Name | Arabidopsis Gene ID | Up/down-regulation | Padj | Similarity to the Ath gene |
| Late embryogenesis abundant (LEA) proteins | ||||
| LEA-related | AT5G60530.1 | 0.23991374 | 2.5624E-05 | 6E-18 |
| LEA_2 | AT2G44060.2 | 1.86413136 | 0.00014204 | 1E-60 |
| LEA-related | AT1G54890.1 | 0.29400396 | 6.7547E-07 | 5E-13 |
| LEA-related | AT5G54370.1 | 1.22790739 | 0.53229844 | 4E-16 |
| LEA_4 | AT5G44310.2 | 3.76509689 | 1.5892E-16 | 1E-11 |
| LEA-related Hydroxyproline-rich glycoprotein | AT3G44380.1 | 4.68751795 | 1.8879E-16 | 1E-17 |
| LEA_4 | AT5G44310.2 | 4.25267018 | 1.2762E-18 | 4E-13 |
| LEA_4 | AT5G44310.2 | 4.25267018 | 1.2762E-18 | 4E-13 |
| LEA-related | AT3G62580.1 | 3.00106737 | 8.899E-13 | 8E-41 |
| LEA-related Hydroxyproline-rich glycoprotein | AT2G01080.1 | 3.882294126 | 0.00435072 | 3E-11 |
| LEA-related Hydroxyproline-rich glycoprotein | AT2G01080.1 | 4.659043737 | 4.6237E-05 | 2e-11 |
| LEA-related | AT3G62580.1 | 3.993800234 | 0.39220796 | 4E-17 |
| Early responsive to dehydration stress (ERD) genes | ||||
| ERD-family protein | AT4G22120.6 | 0.40330981 | 0.00264642 | eE-35 |
| ERD-family PM protein | AT4G04340.1 | 1.914357304 | 0.00119979 | 3E-29 |
| ERD-family PM protein | AT4G04340.1 | 0.719668846 | 0.54074033 | 4E-16 |
| ERD4 | AT1G30360.1 | 3.326696704 | 0.09599311 | 3E-46 |
| ERD4 | AT1G30360.1 | 2.282664946 | 0.00333271 | 1E-148 |
| ERD4 | AT1G30360.1 | 4.305228158 | 7.4444E-15 | 4E-22 |
| ERD4 | AT1G30360.1 | 4.069239817 | 3.643E-21 | 1E-169 |
| ERD4 | AT1G30360.1 | 2.682142777 | 0.54503736 | 5E-61 |
| ERD-family PM protein | AT4G04340.1 | 1.200513952 | 0.40890632 | 1E-121 |
| ERD4 | AT1G30360.1 | 0.347378285 | 1.6875E-06 | 5E-166 |
| ERD-family protein, localized in endomembrane system | AT4G02900.1 | 0.851420448 | 0.49554025 | 1E-100 |
| Other | ||||
| Chloroplast drought-induced stress protein of 23 kDa, CIDSP32, thioredoxin | AT1G76080.1 | 1.71514379 | 0.00189019 | 5E-74 |
| Drought-sensitive 1, DRS1, | AT1G80710.1 | 1.574535846 | 0.17300482 | 3E-64 |
| Drought-sensitive 1, DRS1, | AT1G80710.1 | 1.722093198 | 0.03356124 | 3e-63 |
Significantly up-regulated genes are in bold.
Figure 6. Antiradical power (Trolox equivalents g−1 dry weight (DW) of Klebsormidium crenulatum control samples (n = 3) and samples (n = 3) desiccated above silica gel for 2.5 h.

The values are expressed in mg Trolox equivalent.g dry weight (DW)−1. Means and standard deviations are shown. The statistical siginficance was thested by two sample t-test and demonstrated that the antiradical power of the desiccated samples is significantly higher (p <0.01).
Desiccation induced down-regulation of transcripts and complex regulation patterns
Pathways, which were strongly down-regulated, were mainly involved in integrative cellular functions, such as cell division and DNA repair mechanisms. Purine-, pyrimidine-, most cofactors, and many amino acid biosyntheses were down-regulated as well. However, as mentioned above, most pathways showed complex regulation patterns. Fig. 4 illustrates this for the terpenoid backbone (Fig. 4 A) and carotenoid biosynthesis (Fig. 4 B). Contigs for the cytosolic mevalonate pathway are down-regulated, but contigs for the plastidic MEP/DOXP-pathway are not regulated, and many contigs of the carotenoid pathways are up-regulated and none down-regulated. In agreement with this, we also observed up-regulation of contigs coding for most light-harvesting complex proteins psbS and elip (see above), supporting the idea that non-photochemical quenching plays an important role in protection from ROS during desiccation stress.
Protein degradation using the ubiquitin/proteasome system is another interesting example with a complex regulation pattern; contigs for all core subunits are up-regulated, as well as some contigs for the enzymes of the ubiquitination mechanism. However, transcripts for some other components of the ubiquitination mechanism and some components of the regulatory proteasome lid and were down-regulated. In protein synthesis, contigs for nearly all ribosomal proteins were up-regulated, while contigs for some components of the nuclear ribosome processing pathway and some aminoacyl-tRNA synthases were down-regulated.
Embryophyte desiccation/dehydration/drought stress-related proteins
Several K. crenulatum contigs showed similarity to embryophyte proteins implicated in the desiccation response (Table 7). Generally, more than one contig showed similarity to a certain Arabidopsis gene, suggesting the presence of small protein families. Most contigs were significantly up-regulated, however a few contigs were not-regulated or down-regulated.
Discussion
In the present study we investigated the transcriptome and physiology of K. crenulatum under strong desiccation stress, including tests of cell viability. Cell viability in K. crenulatum was previously tested by using the amphiphilic styril dye FM 1–43 [17]. The present study corroborated the previous results, as we found the plasma membranes stained in control cells, whereas in desiccated cells, only slight uptake of the dye was observed. FM dyes only fluorescence under hydrophobic conditions, and mechanisms of uptake into embryophyte plant cells have been discussed [47], [48]. However, even under the strong desiccation conditions used, the total cell volume was not stained, suggesting that the damage was not as extensive as reported in cells that were desiccated for 7 days [17]. In contrast, initially dead cells of control filaments show a bright FM 1–43 fluorescence throughout the whole cell lumen. We cannot fully exclude the presence of damage to the cell membrane or changes in the cell membrane composition caused by our desiccation treatment; however, these results indicate that cells survive strong desiccation over silica gel. To further analyze the physiological state of the desiccated K. crenulatum cells, photosynthetic parameters were measured. The CO2 assimilation rate of K. crenulatum cells was determined for the first time in the present study. By gas exchange measurements it is possible to determine CO2 assimilation rates of K. crenulatum under defined RH leading to desiccation under controlled conditions (temperature, CO2 and H2O content, light). Until now most measurements of photosynthetic parameters in green algae were performed in the fluid phase. Therefore, so far only chlorophyll fluorescence parameters could be determined in the desiccated state (e.g. [3]), but no information on assimilation rates was available.
During the desiccation period of 2.5 h under low light conditions (30 µmol photons) the assimilation rate dropped below zero, indicating that the cells shut down their photosynthetic activity; the still-measurable respiration rate might be attributed to failure to reach equilibrium to 20% RH in the chamber of the GFS-3000 (it is not possible to reach RHs as low as obtained in the desiccation box over silica gel); however after 21 h recovery, both the assimilation rate and Yield II were near zero, indicating drastic perturbation of the cells. Suppression of photosynthesis in green algae during dehydration has been previously described [13], [16], [19], [49]. In an earlier study using K. crenulatum, short-term desiccation for 3 hours at ambient room temperature and humidity was performed [16]. There, the Fv/Fm value recovered after 30 min rehydration to 48% of the initial value, and after 2 h the cells were fully recovered [16]. In a study using K. dissectum desiccated under different RH (100, 55 and 5%) for 1 or 3 weeks, the cells recovered within 1 or 2 weeks respectively, indicating that some cells have the ability to survive these harsh treatments [19].
Transcriptome analysis
High-quality RNA is crucial for any transcriptome analysis. RNA agarose gels and Bioanalyzer electropherogram profiles indicated a reduced amount of RNA and some RNA degradation in our desiccated samples, but little or no degradation in our control samples (Fig. S2). We are convinced that the reduced RNA amount and decreased RNA integrity number are not experimental artifacts, but rather were caused by the applied stress conditions, for the following reasons: (1) The same protocol was used for all samples and samples were processed in parallel. (2) Similar results have been obtained for other systems, e.g. Tortula ruralis gametophytes [50], pea seeds [51] and yeast [52]. Particularly in the study by Chen et al. [51], a detailed analysis of the RNA integrity numbers (RIN, [53]) is given, and similar values to those obtained in this study were found in artificially aged pea seeds, which were considered as ‘partially degraded’ but still usable for the analysis.
To our knowledge this is the first broad-scale expression study of a Klebsormidium species in response to abiotic stress. ESTs have been sequenced for Klebsormidium flaccidum and Klebsormidium subtile, yielding about 60,000 ESTs (NCBI, 22.07.2014). As no reference data were available for K. crenulatum, we first established a reference transcriptome based on the pooled RNA samples. This reference transcriptome contains over 24,000 contigs. About 60% are similar to proteins in sequenced viridiplant genomes, with about 10% apparently present only in streptophytes, supporting the close relationship of Klebsormidium with embryophytes. However, currently the number of genomes from chlorophytes is limited. Therefore it seems possible that the addition of further genomes will increase the total number of K. crenulatum contigs that are similar to chlorophyte proteins. The established reference transcriptome covers all major cellular functions and metabolic pathways. However, for more than 30% of the contigs, no similar protein was found in any database, suggesting the existence of a large number of genus- or species-specific genes, which is very similar to the situation observed in other organisms [54].
Expression Analysis
Our expression analysis revealed that a large number of contigs are up- or down-regulated. Given the large number of contigs that show no similarity to any organism and which are strongly up-regulated in K. crenulatum, we did not perform any GO or KEGG-ontology enrichment study, but instead we focused on KEGG pathway analyses. There are three major responses to desiccation in K. crenulatum. First, contigs related to energy metabolism (glycolysis, TCA cycle, respiration) are up-regulated. The role of these pathways during desiccation tolerance is not clear. In actively growing roots and whole plants water stress inhibits mitochondrial respiration; however, in leaves the response is more variable, ranging from reduction in most cases over no change even to increase under severe water stress e.g. in Arnica alpina, Triticum aestivum or Helianthus annuus [55]. As the algal cell is more comparable with a plant leaf than with plant roots or seeds, we suggest that similar to the above mentioned examples, Klebsormidium cells try to increase respiration for energy production, which might also help to explain the high respiration rate in the desiccated state, however it seems also possible that the cells just prepare for a fast energy production upon rehydration.
Photosynthesis is inhibited very rapidly upon desiccation (see above). Klebsormidium responds by up-regulation of genes involved in photosynthesis, obviously preparing for rehydration. Finally, the cells show strong protection against light damage and ROS stress, by increasing carotenoid and LHC biosynthesis as well as up-regulating glutathione and ascorbic-acid metabolism. The observed up-regulation of the pentose-phosphate cycle is probably required to provide the cells with the reducing agent NADPH. Overall these responses are very similar to typical embryophyte responses [56]. Interestingly, we found in K. crenulatum contigs that are similar to well-known plant proteins involved in the desiccation/drought response, e.g. LEA proteins [57] and genes involved in early response to drought [58] further emphasizing the similarities between the streptophyte algal and plant desiccation responses.
Osmolytes
It has been previously reported that in Klebsormidium flaccidum, sucrose is increased upon cold stress [59], and a similar reaction could be expected after desiccation stress. Indeed, we observed an up-regulation of sucrose synthase (K00695) and sucrose phosphate synthase (K00696). Nagao and Uemura [60] found that sucrose-phosphate phosphatase is present in K. flaccidum, but has a different structure, lacking an extensive C-terminal domain. In agreement with this, we observed a transcript similar to a plant sucrose-phosphate-phosphohydrolase family protein, which could not be mapped on the sucrose pathway by the reconstruct-pathway tool at KEGG. In addition to sucrose, members of the raffinose family of oligosaccharides (RFO) have been shown to be compatible solutes involved in tolerance of stresses such as desiccation and cold in plants [61]. Previously raffinose has been detected in K. crenulatum as one of the major soluble carbohydrates [18]. Contigs for several enzymes of the galactinol/raffinose metabolism were up-regulated in desiccated samples, suggesting that RFOs might function as compatible solutes in K. crenulatum as well.
Phytohormones
Several phytohormones have been implicated in signaling abiotic stress responses (recently reviewed [62]). ABA has been implicated in signaling of cold and desiccation responses [63] and we could detect contigs similar to all components of the ABA signaling pathway except the ABA receptor (PYR/PYL) in K. crenulatum, suggesting that this pathway might have evolved in streptophyte algae and facilitated colonization of the terrestrial habitat. It has been suggested that jasmonic acid acts even before ABA in the stress response [64]. Interestingly, a contig similar to JAR1 (the embryophyte jasmonic acid receptor) was also found, but no other contig showed similarity to other components of the jasmonic acid signaling pathway. This raises the possibility that the jasmonic acid receptor might function as an ABA receptor in K. crenulatum. Further work will be required to clarify this possibility.
Lipids
Air drying has recently been shown to increase the synthesis of triacylglycerol in the green alga Chlorella kessleri [65]. The authors subjected their samples to ambient air conditions on glass filters for 11 h, and obtained an increase of triacylglycerols from 0.3 to 15.3% (w/w), which corresponds to an increase from 4.7 to 70.3 mole% of fatty acids; the cell weight increased 2.7-fold in 96 h [65]. Our data also suggest that an increase in fatty acid production can be expected from the up-regulation of the respective enzymes. However, for acquiring desiccation tolerance, the composition of the biomembranes was found to be more crucial, as they are the initial targets in the desiccation process [66]. Moreover, chloroplast membranes are protected, as these authors reported the removal of the thylakoid lipid monogalactosyldiacylglycerol (MGDG), which was hydrolyzed and converted into diacylglycerol (DAG). It was then further converted into phosphatidylinositol (PI) in the resurrection plant Craterostigma plantagineum [66]. Interestingly, we also find that in K. crenulatum the CDP-diacylglyerol-inositol 3-phosphatidyltransferase (EC 2.7.8.11) is up-regulated, which is responsible for the conversion of CDP-diacylglycerol into phosphatidyl D myo-inositol. Moreover, enzymes in the diacylglycerol pathway such as digalactosyldiacyl-glycerol synthase (K09480) or enzymes leading to triacylglycerol [e.g. diacylglycerol O-acyltransferase (K00635) and phospholipid: diacylglycerol acyltransferase (K00679)] are up-regulated.
Antioxidants
Antioxidant protection has been studied extensively in lichens and photobionts of lichens [67]–[69]. It has been found that the green photobionts contain protective enzymes such as superoxide dismutase, catalase, peroxidases, glutathione reductase and ascorbate peroxidase, in combination with non-enzymatic substances such as glutathione, α-tocopherol and ascorbic acid (recently summarized [4]). Increased glutathione levels have been determined to be a consequence of supplemental UV irradiation in Chlorococcum infusionum and Chlorogonium elongatum [70]. In Trebouxia excentrica, the photobiont of the lichen Cladonia vulcani, the reduced form of glutathione (GSH) is progressively oxidized to glutathione disulfide (GSSG) during a desiccation period of 20 days [68].
Our transcriptome data showed that contigs with similarity to the first step of glutathion biosynthesis, glutathione-S-transferase and glutathione peroxidase are up-regulated. The latter are involved in the protection of the cells from oxidative damage by reducing lipid hydroperoxides to alcohols and free hydrogen peroxide to water. In contrast, contigs with similarity to glutathione-degrading enzymes are down-regulated. However, we currently cannot explain why the glutathione-disulfide reductase which is responsible for reduction of GSSG to GSH is also down-regulated. To corroborate these findings, we have tested if elevated levels of glutathione could be found in K. crenulatum. We found a marked elevation (to 177% of the original value) as demonstrated by immunoelectron microscopy with polyclonal antibodies against glutathione [45]. It would have been interesting to determine the compartments which show the largest increases in glutathione levels. Unfortunately, the subcellular distribution, which showed the highest concentrations in mitochondria in control cells, was virtually impossible to determine in desiccated cells of K. crenulatum. The reason for this is probably that the cytoplasm appears condensed in desiccated K. crenulatum cells [17], as well as in other desiccated green algae (for a summary see [3]).
In concordance with the above findings, we measured a significant increase in the total antiradical power of K. crenulatum (this study, DPPH-test). This test is commonly used to determine the total antioxidant activity (e.g. [71], [72]). For example, in Saccharina latissima, threefold higher level of antioxidants has been detected in generative (sorus) versus vegetative tissue [71]. In the case of the brown macroalga, this was attributed to a high phlorotannin content, which was considered responsible for protection of the reproductive tissue. In the present study we found evidence for a rapid adjustment of free-radical scavengers such as ascorbate and glutathione, induced by desiccation stress.
Additional potent antioxidants are pigments such as the secondary carotenoid astaxanthin, which has been found to be accumulated, e.g. in the green alga Haematococcus pluvialis [73]. However, based on our transcriptome data, K. crenulatum apparently cannot synthesize astaxanthin. Nevertheless we observed a strong up-regulation of enzymes for other carotenoids, which might also have antioxidant activity.
Conclusion
The present study revealed that a transcriptome approach is feasible for investigating a strong stress as imposed by the desiccation of K. crenulatum. Our results indicate that cells react to the desiccation stress, in attempting to re-establish their energy metabolism (up-regulation of contigs related to respiration and photosynthesis). Furthermore the response is surprisingly similar to that found in embryophytes (LEA proteins, ERD proteins, ROS protection, possibly accumulation of RFO osmolytes) and might also be regulated by the abscisic acid signaling pathway, highlighting the close relationship of streptophyte algae to land plants and supporting an important role for streptophyte algae in the adaptation process that eventually led to colonization of the terrestrial habitat.
Supporting Information
A CO2 assimilation rate, filter temperature and effective quantum yield at different relative air humidity levels during a desiccation period of 2.5 h (RH 20%). Measurements were continued after a 21-h recovery period in culture medium. B CO2 assimilation rate and effective quantum yield of photosystem II at the beginning of the desiccation period (n = 5, 5 min of initial desiccation period, blue bars), at the end of the desiccation period of 2.5 h (n = 5, purple bars) and after a recovery period of 21 h in culture medium (n = 5, green bars). Statistical analyses were carried out by one-way ANOVA with Tukey test (p≤0.001), C Effective quantum yield (Yield II) at the same time points as in B.
(PPTX)
RNA integrity: A Gel electrophoretic separation of isolated total RNA from Klebsormidium crenulatum control cells (K1, K2, K3) and 2.5 h over silica gel-desiccated samples (T1, T2, T3). 18 S and 25 S RNA bands are marked. The same percentage of the isolated total RNA was loaded for each sample. B Electropherograms of corresponding total RNA samples as shown in (A). The 18 S and 25 S bands were marked when applicable. RNA integrity (RIN) numbers were calculated with Agilent 2100 Expert software and ranged between RIN 5.7 and RIN 5.8 in the control samples and between RIN 3.8 and RIN 4.2 in the desiccated samples. RNA quality was considered appropriate when two distinct peaks were visible (K1 to K3); samples with elevated baselines (T1 to T3), where two peaks were still visible, were considered partially degraded, but were also used for the analysis. Although sample T3 had the highest RIN of the desiccated samples (RIN 4.2), no cDNA library could be constructed from this sample.
(PPTX)
Module reconstruction. KO identifiers were retrieved from the KEGG orthology database for the complete, up-regulated and down-regulated dataset used to reconstruct KEGG modules. Column A, B and C show the results for the complete (all), the up-regulated and the down-regulated data set, respectively. Column D displays the KO identifier and column E the name of the pathway or enzyme.
(XLSX)
Read statistics for NG-6357 K1 lib21278,NG-6357 K2 lib21279,NG-6357 K3 lib21280, NG-6357 T2 lib21282, NG-6357 T3 lib21283.
(DOCX)
Klebsormidium crenulatum contigs up-regulated upon strong desiccation. The white region displays the result of the expression analysis using the DeSeq program. The green and blue region give the accession number of the most similar protein in streptophytes (green) and chlorophytes (blue). Kcr-ID, sequence identifier; baseMean, mean normalized counts, averaged over all samples from both conditions; baseMeanA, mean normalized counts from condition A; baseMeanB, mean normalized counts from condition B; foldChange, fold change from condition A to B; log2FoldChange, the logarithm (to basis 2) of the fold change; pval, p value for the statistical significance of this change; padj; p value adjusted for multiple testing with the Benjamini-Hochberg procedure (see the R function p.adjust), which controls false discovery rate (FDR). Ath-ID Aradopsis thaliana; Mtr-ID, Medicago trunculata; Aca-ID, Aquilegia coerulea; Stu-ID, Solanum tuberosum; Mdo-ID, Malus domesticus; Ptr-ID, Populus trichocarpa; Egr-ID, Eucalyptos grandiflora; Tca-ID, Theobroma cacao; Ppa-ID, Physcomitrella patens; Sit-ID Selaginella italica; Cre_ID, Chlamydomonas reinhardtii; Csu_ID, Cocomycxa subellipsoidea; Mpu_ID, Micromonas pusilla; Olu_ID; Ostreococcus lucimarinus.
(XLSX)
Klebsormidium crenulatum contigs down-regulated upon strong desiccation. The white region displays the result of the expression analysis using the DeSeq program. The green and blue region give the accession number of the most similar protein in streptophytes (green) and chlorophytes (blue). Kcr-ID, sequence identifier; baseMean, mean normalized counts, averaged over all samples from both conditions; baseMeanA, mean normalized counts from condition A; baseMeanB, mean normalized counts from condition B; foldChange, fold change from condition A to B; log2FoldChange, the logarithm (to basis 2) of the fold change; pval, p value for the statistical significance of this change; padj; p value adjusted for multiple testing with the Benjamini-Hochberg procedure (see the R function p.adjust), which controls false discovery rate (FDR). Ath-ID Aradopsis thaliana; Mtr-ID, Medicago trunculata; Aca-ID, Aquilegia coerulea; Stu-ID, Solanum tuberosum; Mdo-ID, Malus domesticus; Ptr-ID, Populus trichocarpa; Egr-ID, Eucalyptos grandiflora; Tca-ID, Theobroma cacao; Ppa-ID, Physcomitrella patens; Sit-ID Selaginella italica; Cre_ID, Chlamydomonas reinhardtii; Csu_ID, Cocomycxa subellipsoidea; Mpu_ID, Micromonas pusilla; Olu_ID; Ostreococcus lucimarinus.
(XLSX)
Acknowledgments
We would like to thank O. Buchner, University of Innsbruck, for help in gas exchange measurements.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. RNA-Seq data and the assembled contigs have been deposited in the NCBI's Gene Expression Omnibus (GEO)and are accessible through the NCBI Sequence Read Archive (http://trace.ncbi.nlm.nih.gov/Traces/sra/) under the accession numbers SRR1514242 (assembled reference library) and SRR1563130 to SRR1563134 (transcriptomes).
Funding Statement
This work was supported by FWF grant P24242-B16 (http://www.fwf.ac.at/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Alpert P (2006) Constraints of tolerance: why are desiccation tolerant organisms so small or rare? J Exp Bot 209: 1575–1584. [DOI] [PubMed] [Google Scholar]
- 2.Oliver MJ, Cushman JC, Koster KL (2010) Dehydration tolerance in plants. In: Sunkar R, editor. Plant Stress Tolerance, Methods in Molecular Biology 639 , (New York: Springer) pp. 3–24. [DOI] [PubMed] [Google Scholar]
- 3. Holzinger A, Karsten U (2013) Desiccation stress and tolerance in green algae: Consequences for ultrastructure, physiological and molecular mechanisms. Frontiers in Plant Science 4 327 10.3389/fpls.2013.00327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Agrawal SC (2012) Factors controlling induction of reproduction in green algae – review: the text. Folia Microbiol 57: 387–407. [DOI] [PubMed] [Google Scholar]
- 5. Gasulla F, de Nova PG, Esteban-Carrasco A, Zapata JM, Barreno E, et al. (2009) Dehydration rate and time of desiccation affect recovery of the lichen alga Trebouxia erici: alternative and classical protective mechanisms. Planta 231: 195–208. [DOI] [PubMed] [Google Scholar]
- 6. Lüttge U, Büdel B (2010) Resurrection kinetics of photosynthesis in desiccation-tolerant terrestrial green algae (Chlorophyta) on tree bark. Plant Biol 123: 437–444. [DOI] [PubMed] [Google Scholar]
- 7. Becker B, Marin B (2009) Streptophyte algae and the origin of embryophytes. Ann Bot-London 103: 999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wodniok S, Brinkmann H, Glöckner G, Heidel AJ, Philippe H, et al. (2011) Origin of land plants: Do conjugating green algae hold the key? BMC Evol Biol 11: 104 10.1186/1471-2148-11-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Turmel M, Otis C, Lemieux C (2013) Tracing the evolution of streptophyte algae and their mitochondrial genome. Genome Biol Evol 5: 1817–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Becker B (2013) Snowball earth and the split of Streptophyta and Chlorophyta. Trends Plant Sci 18: 180–183. [DOI] [PubMed] [Google Scholar]
- 11. Zhong B, Xi Z, Goremykin VV, Fong R, McLenachan PA, et al. (2014) Streptophyte algae and the origin of land plants revisited using heterogeneous models with three new algal chloroplast genomes. Mol Biol Evol 31: 177–183. [DOI] [PubMed] [Google Scholar]
- 12. Bertsch A (1966) CO2 Gaswechsel der Grünalge Apatococcus lobatus . Planta 70: 46–72. [DOI] [PubMed] [Google Scholar]
- 13. Gray DW, Lewis LA, Cardon ZG (2007) Photosynthetic recovery following desiccation of desert green algae (Chlorophyta) and their aquatic relatives. Plant Cell Environ 30: 1240–1255. [DOI] [PubMed] [Google Scholar]
- 14. Wieners PC, Mudimu O, Bilger W (2012) Desiccation-induced non-radiative dissipation in isolated green lichen algae. Photosynth Res 113: 239–247. [DOI] [PubMed] [Google Scholar]
- 15. Lunch CK, LaFountain AM, Thomas S, Frank HA, Lewis LA, et al. (2013) The xanthophyll cycle and NPQ in diverse desert and aquatic green algae. Photosynth Res 115: 139–151. [DOI] [PubMed] [Google Scholar]
- 16. Karsten U, Lütz C, Holzinger A (2010) Ecophysiological performance of the aeroterrestrial green alga Klebsormidium crenulatum (Charophyceae, Streptophyta) isolated from an alpine soil crust with an emphasis on desiccation stress. J Phycol 46: 1187–1197. [DOI] [PubMed] [Google Scholar]
- 17. Holzinger A, Lütz C, Karsten U (2011a) Desiccation stress causes structural and ultra-structural alterations in the aeroterrestrial green alga Klebsormidium crenulatum (Klebsormidiophyceae, Streptophyta) isolated from an alpine soil crust. J Phycol 47: 591–602. [DOI] [PubMed] [Google Scholar]
- 18. Kaplan F, Lewis LA, Wastian J, Holzinger A (2012) Plasmolysis effects and osmotic potential of two phylogenetically distinct alpine strains of Klebsormidium (Streptophyta). Protoplasma 249: 789–804. [DOI] [PubMed] [Google Scholar]
- 19. Karsten U, Holzinger A (2012) Light, temperature and desiccation effects on photosynthetic activity and drought-induced ultrastructural changes in the green alga Klebsormidium dissectum (Streptophyta) from a high alpine soil crust. Microb Ecol 63: 51–63. [DOI] [PubMed] [Google Scholar]
- 20. Karsten U, Pröschold T, Mikhailyuk T, Holzinger A (2013) Photosynthetic performance of different genotypes of the green alga Klebsormidium sp. (Streptophyta) isolated from biological soil crusts of the Alps. Algol Stud 142: 45–62. [Google Scholar]
- 21. Škaloud P, Rindi F (2013) Ecological differentiation of cryptic species within an asexual protist morphospecies: A case study of filamentous green alga Klebsormidium (Streptophyta). J Eukar Microbiol 60: 350–362. [DOI] [PubMed] [Google Scholar]
- 22.Ryšánek D, Hrcková K, P ?kaloud (2014) Global ubiquity and local endemism of free-living terrestrial protists: phylogeographic assessment of the streptophyte alga Klebsormidium. Environm Microbiol doi:10.1111/1462–2920.12501 [DOI] [PubMed]
- 23. Rindi F, Guiry MD, López-Bautista JM (2008) Distribution, morphology, and phylogeny of Klebsormidium (Klebsormidiales, Charophyceae) in urban environments in Europe. J Phycol 44: 1529–1540. [DOI] [PubMed] [Google Scholar]
- 24. Rindi F, Mikhailyuk TI, Sluiman HJ, Friedl T, López-Bautista JM (2011) Phylogenetic relationships in Interfilum and Klebsormidium (Klebsormidiophyceae, Streptophyta). Mol Phylogenet Evol 58: 218–231. [DOI] [PubMed] [Google Scholar]
- 25. Gasulla F, Jain R, Barreno E, Guéra A, Balbuena TS, et al. (2013a) The response of Asterochloris erici (Ahmadjian) Skaloud et Peksa to desiccation: a proteomic approach. Plant Cell Environ 36: 1363–1378. [DOI] [PubMed] [Google Scholar]
- 26. Nowrousian M (2010) Next-generation sequencing techniques for eukaryotic microorganisms: sequencing-based solutions to biological problems. Eukaryot Cell 9: 1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malone JH, Oliver B (2011) Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biology 9: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McIntyre LM, Lopiano KK, Morse AM, Amin V, Oberg AL, et al. (2011) RNA-seq: technical variability and sampling. BMC Genomics 12: 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rodriguez MC, Edsgärd D, Hussain SS, Alquezar D, Rasmussen M, et al. (2010) Transcriptomes of the desiccation-tolerant resurrection plant Craterostigma plantagineum . Plant J 63: 212–228. [DOI] [PubMed] [Google Scholar]
- 30. Usadel B, Fernie AR (2013) The plant transcriptome – from integrating observations to models. Front Plant Sci 4: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dinakar C, Bartels D (2013) Desiccation tolerance in resurrection plants: new insights from transcriptome, proteome, and metabolome analysis. Front Plant Sci 4: 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guarnieri MT, Nag A, Smolinski SL, Darzins A, Seibert M, et al. (2011) Examination of triacylglycerol biosynthetic pathways via de novo transcriptomic and proteomic analyses in an unsequenced microalga. PLoS ONE 6: e25851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heinrich S, Valentin K, Frickenhaus S, John U, Wiencke C (2012) Transcriptomic analysis of acclimation to temperature and light stress in Saccharina latissima (Phaeophyceae). PLoS ONE 7: e44342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Deng Y, Yao J, Wang X, Guo H, Duan D (2012) Transcriptome sequencing and comparative analysis of Saccharina japonica (Laminariales, Phaeophyceae) under blue light induction. PLoS ONE 7: e39704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Timme RE, Delwiche CF (2010) Uncovering the evolutionary origin of plant molecular processes: comparison of Coleochaete (Coleochaetales) and Spirogyra (Zygnematales) transcriptomes. BMC Plant Biology 10: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lokhorst GM, Star W (1985) Ultrastructure of mitosis and cytokinesis in Klebsormidium mucosum nov. comb., formerly Ulothrix verrucosa (Chlorophyta). J Phycol 21: 466–476. [Google Scholar]
- 37. Starr RC, Zeikus JA (1993) UTEX – the culture collection of algae at the University of Texas at Austin 1993 List of cultures. J Phycol 29: 1–106. [Google Scholar]
- 38. McFadden GI, Melkonian M (1986) Use of HEPES buffer for microalgal culture media and fixation for electron microscopy. Phycologia 25: 551–557. [Google Scholar]
- 39.Karsten U, Herburger K, Holzinger A (2014) Dehydration, temperature and light tolerance in members of the aeroterrestrial green algal genus Interfilum (Streptophyta) from biogeographically different temperate soils. J Phycol DOI: 10.1111/jpy.12210. [DOI] [PMC free article] [PubMed]
- 40. Schreiber U, Bilger W (1993) Progress in chlorophyll fluorescence research: major developments during the past years in retrospect. Progr Bot 54: 151–173. [Google Scholar]
- 41. Porra RJ, Thompson WA, Kriedmann PE (1989) Determination of accurate extinction coefficients and simultaneous equations for assaying chlorophylls a and b extracted with four different solvents: verification of the concentration of chlorophyll standards by atomic absorption spectroscopy. Biochim Biophys Acta 975: 384–394. [Google Scholar]
- 42. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biology 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Edgar R, Domrachev M, Lash AE (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zechmann B, Müller M (2010) Subcellular compartmentation of glutathione in dicotyledonous plants. Protoplasma 246: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zechmann B, Koffler BE, Russell SD (2011) Glutathione synthesis is essential for pollen germination in vitro. BMC Plant Biol 11: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cruces E, Huovinen P, Gómez I (2012) Phlorotannin and antioxidant responses upon short-term exposure to UV radiation and elevated temperature in three South Pacific kelps. Photochem Photobiol 88: 58–66. [DOI] [PubMed] [Google Scholar]
- 47. Emans N, Zimmermann S, Fischer R (2002) Uptake of a fluorescent marker in plant cells is sensitive to brefeldin A and wortmannin. Plant Cell 14: 71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bolte S, Talbot C, Boutte Y, Catrice C, Read ND, et al. (2004) FM dyes as experimental probes for dissecting vesicle trafficking in living plant cells. J Microsc Oxford 214: 159–173. [DOI] [PubMed] [Google Scholar]
- 49. Häubner N, Schumann R, Karsten U (2006) Aeroterrestrial algae growing in biofilms on facades – response to temperature and water stress. Microb Ecol 51: 285–293. [DOI] [PubMed] [Google Scholar]
- 50. Oliver MJ, Bewley JD (1984) Plant Desiccation and Protein Synthesis V. Stability of Poly (A)− and Poly (A)+ RNA during desiccation and their synthesis upon rehydration in the desiccation-tolerant moss Tortula ruralis and the intolerant moss Cratoneuron filicinum . Plant Physiol 74: 917–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen H, Osuna D, Colville L, Lorenzo O, Graeber K, et al. (2013) Transcriptome-wide mapping of pea seed ageing reveals a pivotal role for genes related to oxidative stress and programmed cell death. PLoS ONE 8: e78471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rapoport AI, Beker ME (1986) Ribonucleic acid degradation during dehydration of yeast cells. Microbiol 55: 689–691. [Google Scholar]
- 53. Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, et al. (2006) The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 7: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Adams M (2013) Open questions: genomics and how far we haven’t come. BMC Biol 11: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Atkin OK, Mahcerel D (2009) The crucial role of plant mitochondria in orchestrating drougth tolerance. Ann Bot 103: 581–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dinakar C, Djilianov D, Bartels D (2012) Photosynthesis in desiccation tolerant plants: energy metabolism and antioxidative stress defense. Plant Sci 182: 29–41. [DOI] [PubMed] [Google Scholar]
- 57. Shi C-Y, Yang H, Wei C-L, Yu O, Zhang Z-Z, et al. (2011) Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genomics 12: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shinozaki K, Yamaguchi-Shinozaki K (2000) Molecular responses to dehydration and low temperature: differences and cross-talk between two stress signalling pathways. Curr Opin Plant Biol 3: 217–223. [PubMed] [Google Scholar]
- 59. Nagao N, Matsui K, Uemura M (2008) Klebsormidium flaccidum, a charophycean green alga, exhibits cold acclimation that is closely associated with compatible solute accumulation and ultrastructural changes. Plant Cell Environ 31: 872–885. [DOI] [PubMed] [Google Scholar]
- 60. Nagao M, Uemura M (2012) Sucrose phosphate phosphatase in the green alga Klebsormidium flaccidum (Streptophyta) lacks an extensive C-terminal domain and differs from that of land plants. Planta 235: 851–861. [DOI] [PubMed] [Google Scholar]
- 61. ElSayed AI, Rafudeen MS, Golldack D (2014) Physiological aspects of raffinose family oligosaccharides in plants: protection against abiotic stress. Plant Biol 16: 1–8. [DOI] [PubMed] [Google Scholar]
- 62.Ahmad P, Bhardwaj R, Tuteja N (2012) ‘Plant Signaling Under Abiotic Stress Environment’. In: P Ahmad, MNV Prasad, editors. Environmental Adaptations and Stress Tolerance of Plants in the Era of Climate Change. Springer New York, pp. 297–323. [Google Scholar]
- 63. Osakabe Y, Osakabe K, Shinozaki K, Tran L-SP (2014) Response of plants to water stress. Front Plant Sci 5: 86 10.3389/fpls.2014.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Djilianov D, Dobrev P, Moyankova D, Vaňková R, Georgieva D, et al. (2013) Dynamics of endogenous phytohormones during desiccation and recovery of the resurrection plant species Haberlea rhodopensis . J Plant Growth Reg 32: 564–574. [Google Scholar]
- 65. Shiratake T, Sato A, Minoda A, Tsuzuki M, Sato N (2013) Air-drying of cells, the novel conditions for simulated synthesis of triacylglycerol in green alga, Chlorella kessleri . PLoS ONE 8: e79630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gasulla F, Vom Dorp K, Dombrink I, Zähringer U, Gisch N, et al. (2013b) The role of lipid metabolism in the acquisition of desiccation tolerance in Craterostigma plantagineum: a comparative approach. Plant J 75: 726–741. [DOI] [PubMed] [Google Scholar]
- 67. Kranner I, Birtic F (2005) A modulation role for antioxidants in desiccation tolerance. Integr Comp Biol 45: 734–740. [DOI] [PubMed] [Google Scholar]
- 68. Kranner I, Cram WJ, Zorn M, Wornik S, Yoshimura I, et al. (2005) Antioxidants and photoprotection in a lichen as compared with its isolated symbiotic partners. Proc Natl Acad Sci USA 102: 3141–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Weissman L, Garty J, Hochman A (2005) Characterization of enzymatic antioxidants in the lichen Ramalina lacera and their response to rehydration. Appl Environ Microbiol 71: 6508–6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Agrawal SC (1992) Effects of supplemental UV-B radiation on photosynthetic pigment, protein and glutathione contents in green algae. Environ Exp Bot 32: 137–143. [Google Scholar]
- 71. Holzinger A, Di Piazza L, Lütz C, Roleda MY (2011b) Sporogenic and vegetative tissues of Saccharina latissima (Laminariales, Phaeophyceae) exhibit distinctive sensitivity to experimentally enhanced ultraviolet radiation: photosynthetically active radiation ratio. Phycol Res 59: 221–235. [Google Scholar]
- 72. Plank DW, Szpylka J, Sapirstein H, Woollard D, Zapf CM, et al. (2012) Determination of antioxidant activity in foods and beverages by reaction with 2,2′-Diphenyl-1-Picrylhydrazyl (DPPH): Collaborative study first action 2012.04. J AOAC Internat 95: 1562–1569. [DOI] [PubMed] [Google Scholar]
- 73. Borowitzka MA, Huisman JM, Osborn A (1991) Culture of the astaxanthin-producing green alga Haematococcus pluvialis 1. Effects of nutrients on growth and cell type. J Appl Phycol 3: 295–304. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A CO2 assimilation rate, filter temperature and effective quantum yield at different relative air humidity levels during a desiccation period of 2.5 h (RH 20%). Measurements were continued after a 21-h recovery period in culture medium. B CO2 assimilation rate and effective quantum yield of photosystem II at the beginning of the desiccation period (n = 5, 5 min of initial desiccation period, blue bars), at the end of the desiccation period of 2.5 h (n = 5, purple bars) and after a recovery period of 21 h in culture medium (n = 5, green bars). Statistical analyses were carried out by one-way ANOVA with Tukey test (p≤0.001), C Effective quantum yield (Yield II) at the same time points as in B.
(PPTX)
RNA integrity: A Gel electrophoretic separation of isolated total RNA from Klebsormidium crenulatum control cells (K1, K2, K3) and 2.5 h over silica gel-desiccated samples (T1, T2, T3). 18 S and 25 S RNA bands are marked. The same percentage of the isolated total RNA was loaded for each sample. B Electropherograms of corresponding total RNA samples as shown in (A). The 18 S and 25 S bands were marked when applicable. RNA integrity (RIN) numbers were calculated with Agilent 2100 Expert software and ranged between RIN 5.7 and RIN 5.8 in the control samples and between RIN 3.8 and RIN 4.2 in the desiccated samples. RNA quality was considered appropriate when two distinct peaks were visible (K1 to K3); samples with elevated baselines (T1 to T3), where two peaks were still visible, were considered partially degraded, but were also used for the analysis. Although sample T3 had the highest RIN of the desiccated samples (RIN 4.2), no cDNA library could be constructed from this sample.
(PPTX)
Module reconstruction. KO identifiers were retrieved from the KEGG orthology database for the complete, up-regulated and down-regulated dataset used to reconstruct KEGG modules. Column A, B and C show the results for the complete (all), the up-regulated and the down-regulated data set, respectively. Column D displays the KO identifier and column E the name of the pathway or enzyme.
(XLSX)
Read statistics for NG-6357 K1 lib21278,NG-6357 K2 lib21279,NG-6357 K3 lib21280, NG-6357 T2 lib21282, NG-6357 T3 lib21283.
(DOCX)
Klebsormidium crenulatum contigs up-regulated upon strong desiccation. The white region displays the result of the expression analysis using the DeSeq program. The green and blue region give the accession number of the most similar protein in streptophytes (green) and chlorophytes (blue). Kcr-ID, sequence identifier; baseMean, mean normalized counts, averaged over all samples from both conditions; baseMeanA, mean normalized counts from condition A; baseMeanB, mean normalized counts from condition B; foldChange, fold change from condition A to B; log2FoldChange, the logarithm (to basis 2) of the fold change; pval, p value for the statistical significance of this change; padj; p value adjusted for multiple testing with the Benjamini-Hochberg procedure (see the R function p.adjust), which controls false discovery rate (FDR). Ath-ID Aradopsis thaliana; Mtr-ID, Medicago trunculata; Aca-ID, Aquilegia coerulea; Stu-ID, Solanum tuberosum; Mdo-ID, Malus domesticus; Ptr-ID, Populus trichocarpa; Egr-ID, Eucalyptos grandiflora; Tca-ID, Theobroma cacao; Ppa-ID, Physcomitrella patens; Sit-ID Selaginella italica; Cre_ID, Chlamydomonas reinhardtii; Csu_ID, Cocomycxa subellipsoidea; Mpu_ID, Micromonas pusilla; Olu_ID; Ostreococcus lucimarinus.
(XLSX)
Klebsormidium crenulatum contigs down-regulated upon strong desiccation. The white region displays the result of the expression analysis using the DeSeq program. The green and blue region give the accession number of the most similar protein in streptophytes (green) and chlorophytes (blue). Kcr-ID, sequence identifier; baseMean, mean normalized counts, averaged over all samples from both conditions; baseMeanA, mean normalized counts from condition A; baseMeanB, mean normalized counts from condition B; foldChange, fold change from condition A to B; log2FoldChange, the logarithm (to basis 2) of the fold change; pval, p value for the statistical significance of this change; padj; p value adjusted for multiple testing with the Benjamini-Hochberg procedure (see the R function p.adjust), which controls false discovery rate (FDR). Ath-ID Aradopsis thaliana; Mtr-ID, Medicago trunculata; Aca-ID, Aquilegia coerulea; Stu-ID, Solanum tuberosum; Mdo-ID, Malus domesticus; Ptr-ID, Populus trichocarpa; Egr-ID, Eucalyptos grandiflora; Tca-ID, Theobroma cacao; Ppa-ID, Physcomitrella patens; Sit-ID Selaginella italica; Cre_ID, Chlamydomonas reinhardtii; Csu_ID, Cocomycxa subellipsoidea; Mpu_ID, Micromonas pusilla; Olu_ID; Ostreococcus lucimarinus.
(XLSX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. RNA-Seq data and the assembled contigs have been deposited in the NCBI's Gene Expression Omnibus (GEO)and are accessible through the NCBI Sequence Read Archive (http://trace.ncbi.nlm.nih.gov/Traces/sra/) under the accession numbers SRR1514242 (assembled reference library) and SRR1563130 to SRR1563134 (transcriptomes).