

Abstract
Polypurine reverse Hoogsteen hairpins (PPRHs) are formed by two intramolecularly bound antiparallel homopurine domains linked by a five-thymidine loop. One of the homopurine strands binds with antiparallel orientation by Watson–Crick bonds to the polypyrimidine target sequence, forming a triplex. We had previously reported the ability of PPRHs to effectively bind dsDNA displacing the fourth strand away from the newly formed triplex. The main goal of this work was to explore the possibility of repairing a point mutation in mammalian cells using PPRHs as tools. These repair-PPRHs contain different combinations of extended sequences of DNA with the corrected nucleotide to repair the point mutation. As a model we used the dihydrofolate reductase gene. On the one hand, we demonstrate in vitro that PPRHs bind specifically to their polypyrimidine target sequence, opening the two strands of the dsDNA, and allowing the binding of a given repair oligonucleotide to the displaced strand of the DNA. Subsequently, we show at a cellular level (Chinese ovary hamster cells) that repair-PPRHs are able to correct a single-point mutation in a dihydrofolate reductase minigene bearing a nonsense mutation, both in an extrachromosomal location and when the mutated plasmid was stably transfected into the cells. Finally, this methodology was successfully applied to repair a single-point mutation at the endogenous locus, using the DA5 cell line with a deleted nucleotide in exon six of the dhfr gene.
Introduction
A large number of characterized genetic disorders are associated with a mutation in a single gene. Cystic fibrosis, sickle-cell anemia, Tay–Sachs disease, and different types of cancer are some examples of diseases caused by a point mutation. The repair of single-point mutations in a gene within its endogenous locus has been the focus of many researchers during the past decades. After all these efforts, still more methods are needed to improve gene correction. In this direction, an alternative method could be the usage of polypurine reverse Hoogsteen hairpins (PPRHs) that are formed by two antiparallel homopurine domains bound by reverse Hoogsteen bonds linked by a five-thymidine loop (Coma et al., 2005). One of the homopurine strands binds with antiparallel orientation and high affinity, by Watson–Crick bonds, to the polypyrimidine target sequence displacing the fourth strand away from the newly formed triplex (Coma et al., 2005).
We had previously described the design and usage of template-PPRHs that bind to the template DNA strand (de Almagro et al., 2009), and coding-PPRHs binding to the coding DNA strand (de Almagro et al., 2011), as a tool to decrease gene expression (Rodriguez et al., 2013) that shows high stability and low immunogenicity properties without the need for chemical modifications (Villalobos et al., 2014). In this work we explored a new application for PPRHs: the repair of point mutations, an ambitious gene therapy strategy.
Correction of a single nucleotide can be achieved by gene replacement or by gene repair, the latter consisting of targeting the genomic DNA with different types of oligonucleotides. In the last years, different approaches have emerged to correct a point mutation at the DNA level without provoking double-strand breaks (DSB): chimeric RNA-DNA oligonucleotides, which were first designed by Kmiec's group (Yoon et al., 1996); single-stranded oligonucleotides, studied for the first time in 1988 (Moerschell et al., 1988); bifunctional triple-helix-forming oligonucleotides, formed by two domains, a triplex forming oligonucleotide (TFO) domain and a repair domain (Chan et al., 1999; Culver et al., 1999); and several approaches of peptide nucleic acids (PNA) (Nielsen et al., 1991; Egholm et al., 1993), bis-PNA (Rogers et al., 2002; Chin et al., 2008), tc-PNA (Bentin et al., 2003; Kaihatsu et al., 2003), pc-PNA (Lohse et al., 1999) (Demidov et al., 2002; Lonkar et al., 2009), and more recently PNA- single-stranded oligodeoxynucleotides (ssODNs) (Kayali et al., 2010; Nik-Ahd and Bertoni, 2014).
Other approaches for gene repair involving DSB would be the nuclease-based methods, as homing endonucleases (Kleinstiver et al., 2012), zinc-finger nucleases (Urnov et al., 2005; Urnov et al., 2010), TALENs (Wood et al., 2011; Liu et al., 2012; Joung and Sander 2013), or CRISPR (Wiedenheft et al., 2012; Hruscha et al., 2013). One of the major hurdles in gene correction is that site-specific homologous recombination (HR) in mammalian cells occurs at a very low frequency (Hanson and Sedivy, 1995), although it can be enhanced by site-specific damage (Wang et al., 1988) and cleavage (Rouet et al., 1994; Bibikova et al., 2001). However, TFOs without DNA-damaging adducts and bis-PNAs have also been shown to stimulate HR in a site-specific manner (Rogers et al., 2002; Knauert et al., 2006) and to induce mutagenesis through a pathway dependent on nucleotide excision repair (NER) (Wang et al., 1996; Bailey and Weeks 2000). Based on these observations, we hypothesized that PPRHs could be an additional methodology for gene repair, since they also form triple-helix structures and consequently could induce recombination. It is also known by using chimeric RNA/DNA oligonucleotides that the DNA repair machinery of the cells recognizes a helical distortion, and the DNA sequence of the chimera can be used as a template to correct the mismatched base (Cole-Strauss et al., 1999). Thus, in this study we used PPRHs containing different combinations of extended sequences of DNA with the corrected nucleotide to repair a point mutation (repair-PPRHs).
As a model, we used the dihydrofolate reductase (dhfr) gene. We started analyzing in vitro the binding of regular PPRHs to their polypyrimidine target sequences, the opening of the two strands of the dsDNA, and the binding of a given oligonucleotide to the displaced strand of the DNA. Subsequently, we studied whether, at a cellular level (Chinese ovary hamster [CHO] cells), repair-PPRHs were able to correct a single-point mutation in three approaches: (1) in an extrachromosomal dhfr minigene bearing a nonsense mutation, (2) when the mutated dhfr plasmid was stably transfected into the cells, and (3) in a cell line containing a single-nucleotide deletion in the dhfr locus.
Materials and Methods
Oligodeoxynucleotides
Unmodified PPRHs and ODNs were designed according to the rules of Hoogsteen and Watson–Crick pairing, depending on the desired interaction. The repair-PPRHs used in the cellular transfections for repair purposes contained A's on the bases opposite to the purines interruptions in the polypyrimidine strand, thus conferring them an optimal binding to the DNA targets (de Almagro et al., 2009). However, when correcting the point mutation at the endogenous locus in DA5 cells, the complementary bases to the target interruptions were used in the PPRH. Lyophilized oligonucleotides (Sigma-Aldrich) were dissolved in sterile and RNase-free TE (1 mM EDTA and 10 mM Tris-HCl, pH 8.0) to attain a stock solution of 1 mM, and maintained at −20°C. The sequences of PPRHs and ODNs are listed in Table 1.
Table 1.
Sequence of PPRHs and Oligonucleotides Used in the Study
| Name | Sequence (5′ to 3′) Sequences used in vitro |
|---|---|
| Hp13 | GGAAAAAGGAGGA-(T)5-AGGAGGAAAAAGG |
| pPy13 | CCTTTTTCCTCCT |
| pPu13 | AGGAGGAAAAAGG |
| pPy25 | TTGATGCCTTTTTCCTCCTGGACTT |
| pPu25 | AAGTCCAGGAGGAAAAAGGCATCAA |
| RO10 | TTTTTCCTCC |
| pPy61 | TCTTTCTGTTAGCCTTTCTTCTCATAGACTTAAAATTTAT |
| ACTTGATGCCTTTTTCCTCCT | |
| pPu61 | AGGAGGAAAAAGGCATCAAGTATAAATTTTAAGTCTATGA GAAGAAAGGCTAACAGAAAGA |
| Hp23 | AGAAAGAAAAAAGGAAAGAAGAG-(T)5-GAGAAGAAAGGAAAAAAGAAAGA |
| RO20 | TAGACTTCAAATTTATACTT |
| Sequences used in in vitro and in cellular experiments | |
|---|---|
| HpE2rep | GGAGGAGGAGGAG-(T)5-GAGGAGGAGGAGGTCATTCTTTGGAAGTACTTGAATTC |
| HpE2link | GAATTCAAGTACTTCCAAAGTTTTTGGAGGAGGAGGAG-(T)5-GAGGAGGAGGAGG |
| HpE2link-ds | GAATTCAAGTACTTCCAATTTTTTTGGAGGAGGAGGAG-(T)5-GAGGAGGAGGAGG |
| RO18 | TTGGAAGTACTTGAATTC |
| HpE2ext | CCAAAGAATGTGGAGGAGGAGGAG-(T)5-GAGGAGGAGGAGG |
| RO24 | CATTCTTTGGAAGTACTTGAATTC |
| dHpE2link-rep | CTTTGGAAGTACTTGAATTCTTTTTGAATTCAAGTACTTCCAAAGTTTTTGGAGGAGGAGGAG-(T)5-GAGGAGGAGGAGG |
| p11Mut-pPy50 | GAACGAATTCAAGTAGTTCCAAAGAATGACCACCACCTCCTCAGTGGAAG |
| p11Mut-pPu50 | CTTCCACTGAGGAGGTGGTGGTCATTCTTTGGAACTACTTGAATTCGTTC |
| HpE2rep-NH | AAGAAGAAGAAGA-(T)5-GAGGAGGAGGAGGTCATTCTTTGGAAGTACTTGAATTC |
| HpE2rep-WC | CCTCCTCCTCCTC-(T)5-GAGGAGGAGGAGGTCATTCTTTGGAAGTACTTGAATTC |
| HpE6rep | CATCAAGTATAAATTTGAAGTCTATGAGAAGAAAGGCTAACAGAAAGA-(T)5-AGAAAGACAATCGGAAAGAAGAG |
PPRH, polypurine reverse Hoogsteen hairpins; ODNs, oligodeoxynucleotides.
The names and sequences of the PPRHs and ODNs are indicated. Mismatches in the PPRHs compared with their targets are underlined. The nonsense mutation is in bold, and the corresponding corrected nucleotide in the repair-PPRHs is in italic.
Preparation of polypurine/polypyrimidine duplexes for binding assays
The target duplexes of the PPRHs were formed by mixing 25 μg of each single-stranded polypurine and polypyrimidine oligodeoxynucleotides in a 150 mM NaCl solution. After incubation at 90°C for 5 min, solutions were allowed to cool down slowly to room temperature. The target duplex Dup576 was prepared by PCR using dhfr-I1-Fw: TACTGGCTGGATTGGGTTAG and dhfr-E4-Rv: CGGAACTGCCTCCAACTATC as primers and p11Mut as the template. The duplexes were purified in a nondenaturing 20% polyacrylamide gel and quantified by absorbance at 260 nm at 25°C.
Oligodeoxynucleotide labeling
Single-stranded, double-stranded ODN and PPRHs (100 ng) were 5′-end-labeled with [γ-32P]-ATP (Perkin Elmer) by T4 polynucleotide kinase (New England BioLabs) in a 10 μl reaction mixture, according to the manufacturer's protocol. After incubation at 37°C for 1 hr, 15 μl of TE buffer (1 mM EDTA and 10 mM Tris, pH 8.0) was added to the reaction mixture, which was subsequently filtered through a Sephadex G-25 spin-column (Pharmacia) to eliminate the unincorporated [γ-32P]-ATP.
DNA-PPRH binding assays
Triplex formation was analyzed by incubating different radiolabeled PPRHs, dsDNA or ssDNA (20,000 cpm), in the presence or absence of unlabeled DNAs (10 nM), in a buffer containing 10 mM MgCl2, 100 mM NaCl, and 50 mM HEPES, pH 7.2. Binding reactions (20 μl) were incubated for 30 min at 37°C before running the electrophoresis. Unspecific DNA (Salmon Sperm DNA or poly-dI:dC) was included in each sample. Electrophoresis was performed on a nondenaturing 12% polyacrylamide gels containing 10 mM MgCl2, 5% glycerol, and 50 mM HEPES, pH 7.2. Gels were run for 3–4 hr at 220 V (12 V/cm) using a running buffer containing 10 mM MgCl2 and 50 mM HEPES, pH 7.2, at 4°C, dried, and analyzed on a Storm 840 Phosphorimager (Molecular Dynamics).
Construction of a mutated dhfr minigene
The mutated dhfr minigene (p11Mut) was derived from the starting plasmid pDCH1P11 (Noe et al., 1999), containing the six exons of the hamster dhfr gene, intron 1, about 400 bp of the 5′-flank, and the first of the three polyadenylation sites in exon 6. The p11Mut construct carries a nonsense mutation in exon 2, a stop codon, generated by PCR using the pDCH1P11 as a template and an oligonucleotide containing the desired mutation (G>C). The PCR fragment was digested with EcoRI and BamHI and cloned unidirectionally into pDCH1P11 digested with the same restriction enzymes. DNA sequencing was carried out to confirm the mutation.
Cell culture
DG44 CHO cells (Urlaub et al., 1986), lacking the dhfr gene, were used as a recipient cell line in the transfection experiments. The DG44-p11Mut cell line was obtained by transfecting the plasmid p11Mut into DG44 cells using the calcium phosphate method and by selection with 400 μg/ml of Geneticin (Sigma-Aldrich). The presence of the plasmid in this cell line was confirmed by PCR. In parallel, DG44 cells were transfected with plasmid pDCH1P11, but this time the selection and the growth were performed in selective medium lacking hypoxanthine (−H), one of the final products of DHFR activity, containing 7% of dialyzed fetal bovine serum (Invitrogen) to generate the cell line DG44-p11, used as a positive control. DA5 cells were used to repair the single-point mutation at the genomic level. DA5 is a CHO cell line, containing a single copy of the dhfr genomic gene bearing an endogenous point mutation in the exon 6 (GAA/G>-AAG) causing a frameshift in the sequence. UA21 is a CHO cell line containing one wild-type copy of the dhfr gene, and it was used as a positive control. Cells were grown in Ham's F12 medium containing 7% fetal bovine serum (Invitrogen), and incubated at 37°C in a humidified 5% CO2 atmosphere. For expansion and harvesting, cells were detached with 0.05% trypsin (Sigma).
PPRH/plasmid DNA binding before cellular transfection in DG44 cells
Three μg (0.9 pmol) of mutant pDCH1P11 plasmid (p11Mut) was incubated with a 233-fold molar excess of various PPRHs in a 10 μl final volume in the presence of binding buffer at 37°C for 2 hr. In experiments using additional repair oligonucleotides, the mix was heated to 90°C for 30 sec and annealed at room temperature. All control oligonucleotides were also subjected to the same heat-annealing protocol (Chan et al., 1999).
Transfection and selection
Transfection of repair-PPRH/p11Mut complexes was carried out using the calcium phosphate method (Wigler et al., 1979), which involved the addition of 250 mM CaCl2 to the DNA complex. DNA/CaCl2 solution was added dropwise to an equal volume of sterile twice-concentrated HEPES-buffered saline (2× HBS; 280 mM NaCl, 50 mM Hepes, and 1.5 mM sodium phosphate, pH-adjusted to 7.1). The calcium phosphate/DNA precipitate was allowed to form for 30 min at room temperature without agitation. An amount of 200 μl of the mix was added per well directly to the 1.8 ml of growth medium containing the recipient cells (DG44). After 5–6 hr of incubation at 37°C, the medium was replaced and the cells were incubated for an additional 24 hr. At that time, Ham's F-12 was changed to selective Ham's F-12 medium lacking hypoxanthine (−H medium), to initiate the selection of repaired cells. In the case of DG44-p11Mut, cells were transfected with the different repair-PPRHs in the absence of plasmid, as the p11Mut target sequence was already stably transfected into the cells. DA5 were also transfected with the repair-PPRH alone, since this cell line already contains a point mutation at the endogenous level.
A total of 41 colonies were randomly selected and grown in selective medium (−H). Each assay was conducted a minimum of three times, and for each repair-PPRH that succeeded in repairing the point mutation, a minimum of three colonies from three different replicates were analyzed for DNA sequencing, and for DHFR levels of mRNA, protein, and activity.
mRNA analysis
After selecting and growing the colonies, total RNA from repaired cells was extracted using Tri Reagent (Life Technologies), according to the manufacturer's specifications. The amount of RNA was determined by measuring its absorbance (260 nm) at 25°C in a Nanodrop ND-1000 spectrophotometer (Thermo Scientific). cDNA was synthesized in a 20 μl reaction mixture containing 500 ng of total RNA, 250 ng of random hexamers (Roche), 10 mM dithiothreitol, 20 units of RNasin (Promega), 0.5 mM of each dNTP (AppliChem), 4 μl of buffer (5×), and 200 units of Moloney murine leukemia virus reverse transcriptase (RT) (Invitrogen). The reaction mixture was incubated at 37°C for 1 hr. An amount of 3 μl of the cDNA mixture was used for PCR amplification by real time.
Real-time PCR
The reaction was performed using the ABIPrism 7000 Sequence Detection System (Applied Biosystems) in a final volume of 20 μl, containing 3 μl of cDNA, 2× SYBR Select Master Mix (Applied Biosystems, Life Technologies), 0.125 μM of reverse and forward primers (Sigma-Aldrich), and H2O. Adenine phosphoribosyltransferase (APRT) mRNA was used to normalize the results. The primer sequences were as follows: Dhfr2-Fw, 5′-AAGAACGGAGACCTTCCCTG-3′; Dhfr2-Rv, 5′-CGGAACTGCCTCCAACTATC-3′; Aprt-Fw, 5′-GCATGGCGGCAAGATCGACT-3′; Aprt-Rv, 5′-GTTCTAAGGCGTCTTTCTGG-3′. PCR cycling conditions were 2 min at 50°C, 10 min denaturation at 95°C, followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C, and fourth stage of 15 sec at 95°C, 20 sec at 60°C, and 15 sec at 95°C. The mRNA amount of the target gene, normalized to APRT, was given by the ΔΔCT method, where CT is the threshold cycle indicating the fractional cycle number at which the amount of amplified mRNA reached the threshold of fluorescence.
Western blot analysis
Cells were harvested by trypsinization and treated with lysis buffer (50 mM HEPES, 0.5M NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, 1% Triton X-100, pH 7.2), supplemented with protease inhibitor mixture (Sigma-Aldrich). Total extracts were maintained at 4°C for 1 hr with vortexing every 15 min. Cell debris was removed by centrifugation (10,000×g for 10 min). Protein concentrations were determined by the Bio-Rad protein assay (based on the Bradford method, using bovine serum albumin as a standard; Sigma-Aldrich).
Total protein cell extracts (100 μg) were electrophoresed on SDS-12% polyacrylamide gels, and transferred to a polyvinylidene difluoride membrane (Immobilon P; Millipore) using a semidry electroblotter. The membranes were probed with antibodies against DHFR (1:250 dilution) and tubulin (1:800 dilution; CP06; Calbiochem, Merck) to normalize the results. Signals were detected with HRP-conjugated antibodies: anti-rabbit (1:2500 dilution; P0399; Dako) for DHFR, anti-mouse (1:2500 dilution; sc-2005; Santa Cruz Biotechnology) for tubulin, and enhanced chemiluminescence using ECL Prime Western Blotting Detection Reagent, as recommended by the manufacturer (GE Healthcare). Tubulin and total protein loading were both used to normalize the results. Chemiluminescence was detected with ImageQuant LAS 4000 Mini (GE Healthcare) and quantification was performed using the Image Quant 5.2 software.
DHFR activity
The method is based on the incorporation of radioactive deoxyuridine into cellular DNA, which depends on the generation of tetrahydrofolate by DHFR from folate supplied in the medium. Tetrahydrofolate is used in the reductive methylation of deoxyuridylate to thymidylate. The latter is subsequently incorporated into DNA, which is then isolated by a simple trichloroacetic acid precipitation procedure (Ciudad et al., 1988).
Dishes (35 mm) were seeded with 3×104 dhfr negative or repaired cells in 1 ml of selective medium lacking glycine, hypoxanthine, and thymidine (−GHT). After 16 hr, 2 μCi of 6-[3H] deoxyuridine (20 mCi/mmol; Moravek Biochemicals) was added for 24 hr. Cells were rinsed twice with phosphate-buffered saline and lysed in 100 μl of 0.1% sodium dodecyl sulfate. The lysate was harvested in 31ET paper (Whatman) and washed three times with 66% cold ethanol containing 250 mM NaCl, dried, and counted in a scintillation counter.
DNA sequencing
Total DNA was obtained using the Wizard Genomic DNA Purification Kit (Promega), according to the manufacturer's specifications. PCR was performed to amplify the specific region containing either the point mutation or the repaired nucleotide, using the Biotools DNA Polymerase (Biotools). The primer sequences were as follows: Dhfr2-Fw and Dhfr2-Rv for the DG44 and the DG44-p11Mut cell lines (as described in mRNA analysis), and Dhfr6-Fw (GTCATGTGTCTTCAATGGGTG) and Dhfr6-Rv (TCT AAAGCCAACACAAGTCCC) for the DA5 cell line. Sequencing was carried out by Macrogen.
Results
Before testing if repair-PPRHs were able to repair a point mutation in cells, we carried out different binding assays to explore the necessary conditions to open the double-stranded DNA target, using different combinations of PPRHs, for the subsequent binding of a putative repair oligonucleotide. To this aim, we used two assays: in the first one, we designed a simplified model using a fragment of the Chinese hamster dhfr gene, to study the potential of PPRHs to keep the dsDNA open. In the second, the repair-PPRHs to be used in DG44 cells were also studied for their ability to bind to the polypyrimidine target.
Design of PPRHs for binding assays
We searched for polypurine/polypyrimidine sequences within the Chinese hamster dhfr gene with a length ranging between 10 and 20 bases (Coma et al., 2005). Two sequences located within exon 6, separated from each other by 25 nucleotides, were selected. One of them contains 13 contiguous purines/pyrimidines, and the other one contains 23 nucleotides taking into account three interruptions. Two PPRHs were designed against these sequences, Hp13 and Hp23, respectively. Two oligonucleotides were also designed to bind to the displaced polypurine strand. All the sequences used in this study are listed in Table 1.
Binding assays
Three approaches were carried out to test the ability of PPRHs to bind to its target, displacing the polypurine strand of the dsDNA, and allowing the subsequent binding of a specific oligonucleotide to the displaced polypurine strand. This study was carried out by electrophoretic mobility shift assays.
In the first approach, all the oligonucleotides (PPRH, DNA duplex, and the specific oligonucleotide) had the same length of 13 nucleotides. The incubation of Hp13 with its radiolabeled polypurine/polypyrimidine dsDNA target (Dup13) resulted in the formation of two new bands, one corresponding to a triple helical structure between Hp13 and the polypyrimidine single strand of the duplex DNA, and the other corresponding to the displaced polypurine strand (Fig. 1, lane 3). Furthermore, the addition of a specific oligonucleotide with a sequence equivalent to the polypyrimidine single strand of the duplex (pPy13) resulted in the disappearance of the displaced polypurine strand, and an increase in the intensity of the duplex band, thus demonstrating that the specific oligonucleotide had bound to the polypurine strand, previously displaced by the PPRH. Proper controls were introduced to observe the mobility of the different single- and double-stranded DNAs, labeled either in the polypurine or polypyrimidine strand, or both.
FIG. 1.

Binding of PPRH to dsDNA. This binding occurs by triplex formation with the polypyrimidine strand, causing the displacement of the polypurine strand, allowing the binding of a specific oligonucleotide (RO13 or pPy13). Lane 1, labeled 32P-polypurine/32P-polypyrimidine duplex (Dup13) (1.2 nM); lane 2, 32P-polypurine/polypyrimidine duplex (1.33 nM); lane 3, 32P-Dup13 plus hairpin-13 (Hp13); lane 4, 32P-Dup13 plus Hp13 and RO13; lane 5, 32P-RO13 (13.14 nM) plus Hp13; lane 6, 32P-Dup13 plus RO13; lane 7, 32P-polypurine ssDNA (pPu13) (2.28 nM); lane 8, 32P-polypurine/polypyrimidine duplex plus Hp13; lane 9, 32P-polypurine/polypyrimidine duplex plus Hp13 and RO13; lane 10, polypurine/32P-polypyrimidine duplex (12.6 nM) plus Hp13; lane 11, polypurine/32P-polypyrimidine duplex; lane 12, 32P-RO13 plus Hp13 and Dup13; lane 13, 32P-RO13. All unlabeled species were used at 10 nM. Incubations were performed at 37°C for 30 min. As unspecific DNA, 1 μg of Salmon Sperm DNA was added. ssDNA targets, duplexes and triplexes are indicated by arrows. Labeled oligonucleotides are indicated as •. (+) indicate unlabelled oligonucleotides present in the reaction. (A) Triplex structure between the PPRH and the polypyrimidine strand (lanes 3, 4, 5, and 12). (B) Binding between the displaced polypurine strand and a specific polypyrimidine oligonucleotide (lanes 1, 2, 3, 4, 6, 9, 10, 11, and 12). (C) Displaced polypurine strand (lanes 3, 7, and 8). PPRH, polypurine reverse Hoogsteen hairpin. Color images available online at www.liebertpub.com/hgtb
In the second approach, the potency of Hp13 to open dsDNA was studied by increasing the length of the duplex dsDNA (Dup25). In this approach, the incubation of labeled Hp13 plus its Dup25 target originated a light-shifted band (Fig. 2, lane 4) of the same mobility as the triplex formed between Hp13 and its polypyrimidine ssDNA target (Fig. 2, lane 3). Once the strand displacement was observed, we performed the incubation of labeled Hp13 with Dup25 in the presence of a labeled specific polypyrimidine oligonucleotide RO10, corresponding to the complementary sequence of the polypurine strand of the duplex, which resulted in the appearance of three different bands in the electrophoresis (Fig. 2, lane 5). The lower one had the same mobility as RO10 (Fig. 2, lane 9) and the intermediate band corresponded to the binding between pPu25 and RO10 (Fig. 2, lane 7), confirming that a specific polypyrimidine oligonucleotide was able to bind to a polypurine-displaced ssDNA strand, and the upper band was attributed to the binding between Hp13 and RO10 (Fig. 2, lane 2), as they have complementary sequences. The required controls were included to confirm the results.
FIG. 2.
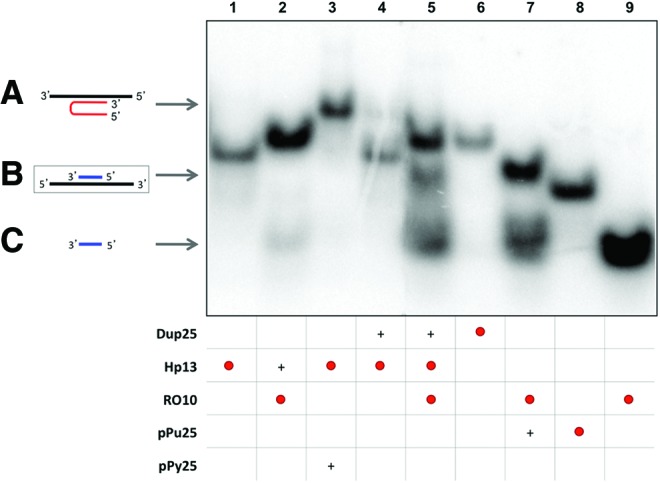
Binding between the displaced polypurine strand of the duplex (pPu25) and a specific polypyrimidine oligonucleotide (RO10). Lane 1, labeled 32P-Hp13 (2.9 nM); lane 2, specific polypyrimidine 32P-oligonucleotide (RO10) (17.1 nM) plus Hp13; lane 3, 32P-Hp13 plus polypyrimidine ssDNA (pPy25); lane 4, 32P-Hp13 plus polypurine/polypyrimidine duplex (Dup25); lane 5, 32P-Hp13 plus 32P-RO10 plus dup25; lane 6, 32P-dup25 (3.7 nM); lane 7, 32P-RO10 plus polypurine ssDNA (pPu25); lane 8, 32P-pPu25 (3.2 nM); lane 9, 32P-RO10. All unlabeled probes were used at 10 nM except for the dup25 in lane 5, which was used at 1 μM to provide an excess of duplex to be bound by the PPRH and prevent that the PPRH would sequester RO, since RO10 is also complementary to the PPRH sequence. The whole mixture of all samples was heated at 50°C before binding reaction. Incubations were performed at 37°C for 30 min. An amount of 0.5 μg of poly-dI:dC was added to each sample. Oligonucleotides are indicated by arrows. Labeled oligonucleotides are indicated as •. (+) indicate unlabelled oligonucleotides present in the reaction. (A) Triplex structure between PPRH and polypyrimidine strand (lanes 3 and 4). (B) Binding between the displaced polypurine strand and a specific polypyrimidine oligonucleotide (lanes 5 and 7). (C) Repair-oligonucleotide (RO10) (lanes 2, 5, 7, and 9). Color images available online at www.liebertpub.com/hgtb
The third approach was designed with an interruption in the duplex (61bp, Dup61) to simulate a point mutation. We incubated the labeled dsDNA duplex with both Hp13 and Hp23 either in the absence (Fig. 3, lane 3) or in the presence (Fig. 3, lane 5) of a longer specific oligonucleotide (RO20). As shown in Fig. 3, the band corresponding to pPu61 was shifted upward (compare lanes 3 and 5), originating a band of the same mobility as the binding between pPu61 and RO20 (lane 6), which indicated that RO20 was able to bind to polypurine ssDNA after the opening of the duplex. The two PPRHs were also incubated separately and at lower concentrations with the radiolabeled dsDNA. Hp23 alone and the combination of Hp13 and Hp23 were able to displace the polypurine strand even at 0.3 nM, while Hp13 alone was needed at least at 3 nM (data not shown). Unrelated competitors were unable to interfere with targets recognition (data not shown). In previous works, we had demonstrated the ability of PPRHs to bind to different concentrations of longer dsDNA sequences (152 and 227 bp) (Coma et al., 2005).
FIG. 3.

Binding between the displaced polypurine strand of the duplex (pPu61) and a specific polypyrimidine oligonucleotide (RO20). Lane 1, labeled 32P-polypyrimidine ssDNA (pPy61) (0.99 nM); lane 2, 32P-pPy61 plus hairpin-13 (Hp13) and hairpin-23 (Hp23); lane 3, 32P-polypurine/32P-polypyrimidine dsDNA (Dup61) (0.19 nM) plus Hp13 and Hp23; lane 4, 32P-Dup61 (pPu-61/pPy-61 duplex); lane 5, 32P-Dup61 plus Hp13, Hp23 and RO20; lane 6, specific 32P-RO20 (0.82 nM) plus polypurine ssDNA (pPu61); lane 7, 32P-pPu61 (0.66 nM). The entire mixture of all samples was heated at 75°C before the binding reaction. Incubations were performed at 37°C for 30 min. An amount of 1 μg of poly-dI:dC was added to each sample. Oligonucleotides are indicated by arrows. Labeled oligonucleotides are indicated as •. (+) indicate unlabelled oligonucleotides present in the reaction. (A) Triplex structure between the two PPRHs and polypyrimidine strand (lanes 2, 3, and 5). (B) Binding between displaced the polypurine strand and a specific polypyrimidine oligonucleotide (lanes 5 and 6). (C) Displaced polypurine strand (lanes 3 and 7). Color images available online at www.liebertpub.com/hgtb
Design of repair-PPRHs for cellular transfection in DG44 and DG44-p11Mut
To design the repair-PPRHs used in the repair experiments at the dhfr minigene level, we searched in the p11Mut sequence for a polypyrimidine domain near to the stop codon introduced in the exon 2 of the dhfr plasmid. A sequence of 13 nucleotides downstream of the nonsense mutation was selected, containing two purine interruptions. We designed different repair-PPRHs, all containing the same Hoogsteen hairpin core, substituting the two interrupting pyrimidines for adenines (“A”) (de Almagro et al., 2009). This core was extended on one of its ends with a 25 nt tail homologous to the point mutation region of the target, except for the position of the mutated nucleotide, which was corrected. The extended tails were added to provide the PPRHs with the ability to repair the point mutation and were either directly attached to the hairpin core (HpE2rep) (Fig. 4C), or connected through thymidine linkers of different extensions (5–7 nt) (HpE2link and HpE2link-ds).
FIG. 4.

Scheme of the p11Mut and the different PPRHs designs. (A) Scheme representing the p11Mut plasmid containing the nonsense mutation in the dhfr minigene. (B) The dhfr minigene contained the six exons of the hamster dhfr gene and its intron 1. The point mutation is localized in exon 2, where the repair-PPRHs are targeted. (C) Five repair-PPRHs were designed against the exon 2 of the dhfr gene. Two of them (HpE2rep and HpE2link) contained only a single extended sequence of DNA bearing the repaired nucleotide; HpE2link-ds/RO18 was complemented with an extra oligonucleotide hybridized to the extension of the repair-PPRH; dHpE2link-rep carried an oligonucleotide covalently bound to the extended tail by a loop, and the last repair-PPRH (HpE2ext/RO24) contained an extension tail until 2nt before the point mutation, and carried an extra oligonucleotide that did contain the repaired nucleotide. Color images available online at www.liebertpub.com/hgtb
As a negative control, we used a PPRH with an extension 2 nt shorter than the location of the mutated base in the DNA (HpE2ext). Some of these repair-PPRHs were used either alone or hybridized to a repair oligonucleotide (RO18 or RO24) complementary to the extended tail of the repair-PPRH. A last design (dHpE2link-rep) contained the repair oligonucleotide covalently bound to the extended tail of the repair-PPRH by a loop. As negative controls, we designed HpE2rep-NH, unable to form the intramolecular Hoogsteen bonds, and HpE2rep-WC, forming Watson–Crick bonds between the two domains of the hairpin core. To confirm the specificity of the sequences, all repair-PPRHs were subjected to BLAST analyses, using the sequence corresponding to the arm of the PPRH binding to the DNA plus the repair tail, with a total length ranging from 24 to 48 nucleotides. The dhfr gene target obtained an E-value of 3×10−9, while the first unspecific target achieved an E-value of 0.68, thus showing high specificity of the repair-PPRH and minimizing the possible off-target effects. The sequences of the repair-PPRHs and ODNs are listed in Table 1.
Binding of repair-PPRHs for cellular transfection
The binding between the different repair-PPRHs and their target sequence was analyzed by labeling the double-stranded p11Mut target sequence. After incubation of the repair-PPRH with its dsDNA target (Dup50), a clear retarded band, corresponding to the triplex structure between four of the repair-PPRHs and the polypyrimidine strand, was observed (Fig. 5A, lanes 4, 6, 8, and 12). The binding of hairpin HpE2ext was not so clear, but it was definitively different from the probe alone and produced the strand displacement of the polypurine strand (pPu50). Strand displacement was also observed for HpE2rep and dHpE2link-rep (lanes 4 and 12). As controls, each repair-PPRH was also incubated with its labeled polypyrimidine single strand (pPy50), in the absence of the double-strand sequence target (lanes 5, 7, 9, 11, and 13), to confirm that the band with the lowest mobility corresponded to the triplex between Hp/pPy50 and that the band with the highest mobility corresponded to the displaced pPu50. HpE2Link-ds and HpE2ext carrying their repair oligonucleotides RO18 and RO24, respectively, were also incubated with the p11Mut target, to confirm that the presence of the repair oligonucleotide did not affect the binding of the repair-PPRHs to their target (data not shown). Since genomic DNA is longer in length, we also performed binding assays with a dsDNA sequence of 576 bp with the repair-PPRHs target site centrally located. As it can be observed in Fig. 5B, the incubation of radiolabeled HpE2rep with its dsDNA target (Dup576) originated an upper band corresponding to the binding between the repair-PPRH and its polypyrimidine target.
FIG. 5.

Repair-PPRHs binding to their target sequence. (A) Binding between the radiolabeled dsDNA duplex (Dup50) and the different repair-PPRHs. Lane 1, labeled 32P-polypurine/32P-polypyrimidine dsDNA (Dup50) (0.67 nM); lane 2, 32P-polypyrimidine ssDNA (pPy50) (0.27 nM); lane 3, 32P-polypurine ssDNA (pPu50) (0.42 nM); lane 4, 32P-Dup50 plus HpE2rep; lane 5, 32P-pPy50 plus HpE2rep; lane 6, 32P-Dup50 plus HpE2link; lane 7, 32P-pPy50 plus HpE2link; lane 8, 32P-Dup50 plus HpE2link-ds; lane 9, 32P-pPy50 plus HpE2link-ds; lane 10, 32P-Dup50 plus HpE2ext; lane 11, 32P-pPy50 plus HpE2ext; lane 12, 32P-Dup50 plus dHpE2link-rep; lane 13, 32P-pPy50 plus dHpE2link-rep; lane 14, 32P-Dup50 plus RO18; lane 15, 32P-Dup50 plus RO24. (B) Binding of a longer dsDNA duplex (Dup575) and the radiolabeled HpE2rep. Lane 1, labeled 32P-HpE2rep (0.35 nM); lane 2, 32P-HpE2rep plus Dup576. (+) indicate unlabelled oligonucleotides present in the reaction. Color images available online at www.liebertpub.com/hgtb
Correction of a point mutation in DG44 cells
In an effort to demonstrate the ability of PPRHs to repair a point mutation in a cellular setting, 225,000 DG44 cells were seeded and transfected with repair-PPRHs plus p11Mut, previously incubated with each other to increase the likelihood of union and recognition of the specific sequence target. After transfection using calcium phosphate, cells were grown in selective medium (−H). Cell colonies were obtained with HpE2rep, HpE2link, and HpE2link-ds/RO18 (Table 1) at a repair frequency ranging between 1.5 and 1.7×10−3 (Fig. 8A). To calculate the repair frequency, the number of colonies appearing in each well were counted and divided by the number of cells initially plated. HpE2ext/RO24 did not succeed in correcting the point mutation, as no colonies survived in selective medium. As negative controls we used either HpE2ext without its hybridized repair oligonucleotide, which did not originate repaired colonies, or the repair oligonucleotides RO18 and RO24 alone that gave a very extremely low frequency of repair, in the order of 10−6. HpE2rep-NH and HpE2rep-WC were also used as negative controls, and did not give rise to any repair colony. Spontaneous rate of correction was not observed in our assays. Repair of the sequence was confirmed by DNA sequencing.
FIG. 8.

Repair frequencies and DHFR activity. (A) Repair frequencies. Each repair-PPRH, previously incubated with the p11Mut plasmid in the DG44 cell line approach, was transfected into CHO cells. After 29–30 hr, cells were grown in selective medium to initiate the selection of repaired cells. The bars represent the frequency of cell colonies out of total cells seeded. Error bars indicate standard errors. Each assay was carried out a minimum of three times. (B) DHFR activity in repaired colonies from DG44, DG44p11Mut, and DA5 cells. Thirty thousand cells from each repair-PPRH approach were plated in 35 mm dishes and their DHFR activity was determined by the incorporation of 2 μCi of 6-[3H] deoxyuridine to the DNA. Cells were collected and lysed with SDS after 24 hr. Radioactivity was counted in a scintillation counter. The activity was determined in repaired individual clones obtained by the transient transfection into DG44, in stably DG44-p11Mut cells and in DA5 cell line. *p<0.05, **p<0.01, ***p<0.001 compared with the corresponding control.
DHFR mRNA analysis
Random surviving cell colonies resulting from independent experiments upon transfection of DG44 cells with PPRH/p11Mut were analyzed for DHFR mRNA levels. Because of the nonsense-mediated mRNA decay mechanism, by which mRNA bearing premature termination codons is selectively degraded, it was expected that DHFR mRNA encoded by p11Mut would be low and that repaired cell colonies would recover higher levels of mRNA. Indeed, the results confirmed in all cases higher levels of DHFR mRNA in repaired cell colonies than those in DG44-p11Mut. DHFR mRNA levels in DG44 and DG44-p11 cells were used as negative and positive controls, respectively (Fig. 6A).
FIG. 6.

Levels of DHFR mRNA and protein in repaired cells. (A) DHFR mRNA in repaired DG44 cells. RNA was extracted from repaired colonies of DG44 cells upon transfection with p11Mut and PPRHs. DHFR mRNA levels were determined using qRT-PCR and referred to the levels of endogenous APRT control. DHFR mRNA levels of the repaired colonies were referred to the mRNA levels of the positive control p11 (DG44-p11). (B) DHFR protein levels in repaired DG44 cells. Total protein extracts were obtained from the repaired DG44 colonies upon transfection with p11Mut and PPRHs and analyzed by Western blot. Protein levels were normalized using tubulin. Protein levels of the repaired colonies were referred to the protein levels of the positive control p11 (DG44-p11). (C) DHFR mRNA levels in repaired DG44-p11Mut cells. RNA was extracted from surviving DG44-p11Mut cell colonies upon transfection with PPRHs. DHFR mRNA levels were determined using qRT-PCR and referred to the levels of endogenous APRT control. DHFR mRNA levels of the repaired colonies were referred to the mRNA levels of the positive control p11 (DG44-p11). (D) DHFR protein levels in repaired DG44-p11Mut cells. Total protein extracts were obtained from DG44-p11Mut upon transfection with PPRHs and analyzed by Western blot. Protein levels were normalized using tubulin. Protein levels of the repaired colonies were referred to the protein levels of the positive control p11 (DG44-p11).
DHFR protein levels
Given that DHFR mRNA levels of repaired cells were recovered, we further explored the ability of the surviving cell colonies to produce DHFR protein. In all cases, the protein levels were restored. DG44-p11Mut and DG44 cells were used as negative controls, and DG44-p11 cells were used as a positive control (Fig. 6B and Fig. 9C.1).
FIG. 9.

DNA sequencing and DHFR protein of repaired cell colonies. (A) Sequences of the nonsense mutant of the dhfr minigene p11Mut, and those of repaired cells using different repair-PPRHs upon transfection in either DG44 or DG44p11Mut cell lines. (B) Repaired cell colony upon transfection of DG44 cells with the mutated version of the dhfr minigene (p11Mut) and HpE2rep, and selection in selective medium (w/o hypoxanthine). (C) Representative image of a Western blot showing the DHFR protein levels of DG44-p11 (positive control), DG44-p11Mut (negative control), and three repaired colonies obtained from DG44 upon transfection with p11Mut and HpE2rep, and three colonies from DG44-p11Mut transfected with HpE2rep. Color images available online at www.liebertpub.com/hgtb
Correction of a point mutation in DG44-p11Mut
Once we demonstrated that a point mutation was repaired in DG44 cells, we further examined if the same result could be achieved in DG44-p11Mut cells. The stable presence of p11Mut in these cells could represent an obstacle for the repair-PPRHs to recognize and bind to its target sequence, but on the other hand it would resemble more closely to the goal of repairing a point mutation of a gene in its endogenous locus. In total, 150,000 cells were seeded and transfected with the different repair-PPRHs, and then grown in −H selective medium. HpE2rep, HpE2link, and dHpE2link-rep succeeded in correcting the point mutation with a frequency of about 1.5×10−3 (number of colonies/number of cells seeded) (Fig. 8A). The levels of DHFR mRNA and protein were determined in random surviving colonies. In all cases, repair-PPRHs achieved to recover DHFR mRNA levels compared with those in DG44-p11Mut (Fig. 6C). Regarding protein levels, all repair-PPRHs were able to produce DHFR (Fig. 6D and Fig. 9C.2), and the restored dhfr sequence was confirmed by DNA sequencing. HpE2ext, RO18, RO24, HpE2rep-NH, and HpE2rep-WC were used as negative controls. Whereas HpE2ext, HpE2rep-NH, HpE2rep-WC, and RO18 were unable to repair the point mutation, RO24 alone did achieve to correct it, although in a much lower efficiency (1.5×10−4). Spontaneous corrections were not observed in our assays.
Correction of a point mutation at the endogenous locus
As a further step, we studied if repair-PPRHs were also able to repair a point mutation in an endogenous locus, using the DA5 cell line bearing a deletion of a guanine in exon 6 of the dhfr gene. We selected a polypyrimidine sequence of 23 nucleotides downstream of the G deletion site, containing three purine interruptions in the target. We designed a repair-PPRH similar to HpE2rep, but in this case the interruptions were not substituted by adenines and the repair-PPRH contained three pyrymidines in the hairpin core, to minimize off-targets effects (Fig. 7A). In this approach, 150,000 cells were seeded and transfected with HpE6rep. After selecting cells in −H medium, surviving cell colonies were obtained at a frequency of 1×10−4 (Fig. 8A). The repair frequency was lower than in the other approaches, since in this case the repair of the point mutation was achieved at the endogenous locus and DA5 cells carry only one copy of the dhfr gene.
FIG. 7.

Repair of a point mutation in an endogenous locus (DA5 cell line). (A) Scheme of HpE6rep. One repair-PPRH (HpE6rep) was designed against the exon 6 of the dhfr gene. HpE6rep contained a single extended sequence of DNA bearing the deleted nucleotide, and three pyrymidine interruptions in the hairpin core complementary to the target sequence. (B) DHFR mRNA levels in repaired DA5 cells. RNA was extracted from surviving DA5 cell colonies upon transfection with HpE6rep. DHFR mRNA levels were determined using qRT-PCR and were normalized to APRT. DHFR mRNA levels of the repaired colonies were referred to the positive control UA21. (C) DHFR protein levels in repaired DA5 cells. Total protein extracts were obtained from surviving DA5 cell colonies upon transfection with HpE6rep. Western blots were performed with 100 μg of total extracts and were normalized to tubulin levels. Protein levels of the repaired colonies were referred to the protein levels of the positive control UA21. A representative image of the Western analyses is shown. Color images available online at www.liebertpub.com/hgtb
Six random cell colonies from different experiments were analyzed at DHFR mRNA and protein levels. mRNA levels of the repaired colonies were recovered compared with the negative control (DA5 cells) (Fig. 7B), and DHFR protein was restored in all cell colonies (Fig. 7C). In this approach, the point mutation could not activate NMD, since it is localized in the last exon of the dhfr gene and there is no downstream exon–exon junction (Maquat 2005; Neu-Yilik et al., 2011). Thus, negative control DA5 can show a very low level of DHFR protein. The dhfr DNA of these cells was also sequenced to verify the restoration/introduction of the guanine.
Since the human recombinase Rad51 has been shown to stimulate the formation of the D-loop via Rad51-mediated strand invasion (Papaioannou et al., 2012), it is required for triplex helix-induced recombination (Gupta et al., 1997; Datta et al., 2001), and it is a central player in HR (Krejci et al., 2012) as a strand-exchange protein, we performed different assays with DA5 cells in the presence of Rad51. Following the same protocol, 150,000 cells were seeded and transfected with HpE6rep plus the Rad51 plasmid. The results showed a frequency of repair of 10−3, which represents an increase of 10-fold compared with the transfection of HpE6rep alone (Fig. 8A).
DHFR activity
Although the repaired colonies had been selected in DHFR selective medium (−H), we wanted to corroborate that the DHFR protein present in those colonies showed DHFR activity. Thus, we selected a representation of one repaired colony recovered from each of the approaches using the different PPRHs. These cells were incubated with 6-[3H] deoxyuridine that was incorporated into DNA. The degree of incorporation depended on the rate-limiting enzyme DHFR when the cells were grown in selective −GHT medium. As it can be observed in Fig. 8B, the repaired cells showed a high degree of DHFR activity compared with the negative control cells DG44, DG44-p11Mut, or DA5.
DNA sequencing
The presence of the corrected nucleotide, containing the same sequence as pDCH1P11 or UA21, using the three cell lines approaches (DG44, DG44-p11Mut, and DA5), was confirmed by DNA sequencing in randomly selected colonies. In all cases, the point mutation was corrected, thus demonstrating the effectiveness of the repair-PPRHs (Fig. 9A). The two-purine interruptions in the hairpin core (HpE2) did not cause a change in the DNA sequence of the target in the repaired colonies (data not shown).
Effect of aphidicolin and hydroxyurea incubation on gene repair by repair-PPRHs
We incubated two cell lines (DG44 and DG44-p11Mut) with 2 mM of hydroxyurea (HU) or 5 μg/ml of aphidicolin for 3 hr before transfection of the PPRHs. To carry out these experiments, we used HpE2rep/p11Mut in DG44 and HpE2rep in DG44-p11Mut. In these conditions, the frequency of repair increased by twofold (Fig. 8A). Since both aphidicolin and HU are known to increase the frequency of HR, our results are consistent with the hypothesis that repair-PPRHs promote repair of point mutations through this type of mechanism. Besides, aphidicolin is known to block the action of DNA polymerase δ and enzyme required for mismatch repair (MMR) synthesis in human cells (Papadopoulos et al., 1994), and so we hypothesize that MMR mechanism could also be involved in the correction of a point mutation by repair-PPRHs.
Discussion
The main purpose of this work was to explore the possibility of repairing point mutations in mammalian cells using PPRHs. We previously described the use of PPRHs as a silencing tool in both in vitro and in vivo approaches (de Almagro et al., 2009; Almagro et al., 2011; Rodriguez et al., 2013), by decreasing the expression of different targets. In this work we present evidence that PPRHs can also be used as a tool for gene repair. Repair-PPRHs are PPRH bearing an extended tail homologous to the point mutation region of the target, except for the nucleotide to be altered. The model we used to validate the effect of these repair-PPRHs was the repair of a nonsense mutation in exon 2 of a dhfr minigene (p11-Mut) in CHO cells, and a deletion point mutation in exon 6 of the genomic sequence in the dhfr endogenous locus (DA5 cells).
As a first step of this study, we demonstrated by binding assays the capacity of regular PPRHs to bind to dsDNA forming triplex structures, displacing the polypurine strand of the dsDNA, extending previous observations from our laboratory (Coma et al., 2005). Then, we further explored the experimental conditions to maintain the dsDNA opened for the subsequent binding of a given repair oligonucleotide. Repair-PPRHs were also analyzed by binding assays before being transfected to the cells, demonstrating their ability to bind to its polypurine/polypyrimidine dsDNA target.
In cell culture experiments, correction of the nonsense mutation in p11Mut was attempted in two different settings. In the first approach, repair was accomplished by the co-incubation of the mutated target plasmid with three different repair-PPRHs (HpE2rep, HpE2link, and HpE2link-ds/RO18) in DG44 cells. Repair-PPRHs would open the dsDNA forming a triplex and increasing HR, and at the same time they could convey the repair tail to the mutated region producing a mismatched base, thus activating DNA repair systems. It is important to note that PPRHs can inhibit transcription (de Almagro et al., 2009), which in turn could trigger error-prone repair. To deepen and broaden the knowledge of repair-PPRHs, a second approach was carried out in cells stably transfected with the p11Mut plasmid (DG44-p11Mut), trying to hinder the recognition and binding of the repair-PPRH to the target sequence. The fact that HpE2link, HpE2rep, and dHpE2link-rep corrected the point mutation demonstrate the capacity of repair-PPRHs to act even when the hairpin is not preincubated with its target sequence. One of the repair oligonucleotides alone (RO24) did achieve some gene correction in DG44-p11Mut, as expected, since it is known that short oligonucleotides can induce recombination (Campbell et al., 1989; Lin et al., 1990). Nevertheless, the repair frequency of this oligonucleotide (RO24) was 10 times less than the repair-PPRHs.
The hairpin core of the repair-PPRHs (HpE2) contained two adenines in front of the purine interruptions in the polypyrimidine target sequence. This fact might originate two possible problems: (1) a reduction in the binding specificity of the PPRH because of its mismatches, and (2) the possibility that those interruptions would provoke a change in the nucleotides of the target sequence. None of these two possibilities constituted a real drawback, since we demonstrated the binding of the repair-PPRHs to their target, and no mutations in the PPRH target sequence appeared when DNA sequencing of repaired colonies was carried out.
The incubation of both cell lines, DG44 and DG44-p11Mut, with the recombination inducers HU or aphidicolin (Saintigny et al., 2001) before transfection increased the frequency of correction by twofold. This fact suggests that HR might play a role in the mechanism for gene correction exerted by repair-PPRHs, simultaneously with MMR, since both a single-nucleotide change and an insertion took place, and aphidicolin is an inhibitor of the DNA polymerase δ, an enzyme required for MMR (Papadopoulos et al., 1994). Since aphidicolin and HU are also known to stall replication, our results are also consistent with the possibility that unwound conformation of duplex DNA during replication might be favorable to triplex formation by PPRHs. Interestingly, the only repair-PPRH that did not succeed in repairing the point mutation was HpE2ext/RO24, which is the only hairpin whose extended tail did not reach the mutated base, and the hybridized repair oligonucleotide (RO24) did not form a full double-strand molecule. This might mean that to accomplish gene repair of the point mutation, the corrected nucleotide needs to be a part of the repair-PPRH sequence. Besides, the absence of repair colonies using HpE2rep-NH that is not able to form the intramolecular Hoogsteen bonds, and HpE2rep-WC forming Watson–Crick bonds between the two domains of the hairpin core, demonstrates that the formation of the triplex structure is essential for repairing point mutations using repair-PPRHs. At the endogenous level, a deletion in the exon 6 of the dhfr gene was also corrected using HpE6rep, thus demonstrating the capacity of repair-PPRHs to recover a deleted nucleotide in an actual genomic environment.
In this approach we also study the importance of Rad51 protein in the methodology of repair-PPRHs. Rad51 is one of the major factors in HR (Liu et al., 2002), it is required for triplex helix-induced recombination, and it can boost invasion or binding of the ssODN to its homologous target region in a structure similar to a D-loop (Symington, 2005). The results are consistent with our hypothesis that Rad51 and consequently HR are involved in correcting point mutations using repair-PPRHs, since the transfection of HpE6rep with pRad51 increased the repair frequency by 10-fold. It is worth noting that the repair at the DNA level accomplished by these repair-PPRH was verified by DNA sequencing and functionally validated at the DHFR mRNA, protein, and activity levels.
PPRHs have advantages when compared with other approaches for gene repair. They can be easily synthesized since they do not bear chemical modifications, which is an advantage in terms of toxicity (Rodriguez et al., 2013) and economy. At the same time, due to their hairpin structure, PPRHs are known to be highly resistant to endogenous nucleases (Villalobos et al., 2014). Also, they do not induce mutations in the target gene of the repaired colonies, which is an important difference with TFOs (Wang et al., 1996). As a matter of fact, this absence of mutations in the target sequence argues against a mechanism of repair during replication and in favor of a mechanism of HR played by the repair-PPRHs. It is also important to note that repair-PPRHs act without damaging the DNA with chemical agents, and without the requirement of DSB that is likely to result in an increase in non-HR (Vasquez et al., 2001).
In summary, in this work we present an innovative technology for gene repair applying a strategy derived from PPRHs. The obtained results suggest that repair-PPRHs could provide the basis for a new therapeutic alternative to repair single-point mutation diseases that also works at the endogenous gene locus.
Acknowledgments
We acknowledge Dr. Silvia Coma for helpful suggestions.
This work was supported by “Plan Nacional de Investigación Científica” (Spain), Grant SAF2011-23582. Our group holds the Quality Mention from the “Generalitat de Catalunya” SGR2009-118. A.S. is the recipient of a fellowship (Formació d'Investigadors) from the “Generalitat de Catalunya.” X.V. is a recipient of an APIF fellowship from the University of Barcelona.
Author Disclosure Statement
The authors declare that they do not have competing financial interests.
References
- Almagro D., Agramonte O., Castillo D., et al. (2011). Experience with a single dose of recombinant activated factor VII for the management of mild-to-moderate bleeds in haemophilia. Haemophilia 17, 322–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey C., and Weeks D.L. (2000). Understanding oligonucleotide-mediated inhibition of gene expression in Xenopus laevis oocytes. Nucleic Acids Res. 28, 1154–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentin T., Larsen H.J., and Nielsen P.E. (2003). Combined triplex/duplex invasion of double-stranded DNA by “tail-clamp” peptide nucleic acid. Biochemistry 42, 13987–13995 [DOI] [PubMed] [Google Scholar]
- Bibikova M., Carroll D., and Segal D.J., et al. (2001). Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol. Cell Biol. 21, 289–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C.R., Keown W., Lowe L., et al. (1989). Homologous recombination involving small single-stranded oligonucleotides in human cells. New Biol. 1, 223–227 [PubMed] [Google Scholar]
- Chan P.P., Lin M., Faruqi A.F., et al. (1999). Targeted correction of an episomal gene in mammalian cells by a short DNA fragment tethered to a triplex-forming oligonucleotide. J. Biol. Chem. 274, 11541–11548 [DOI] [PubMed] [Google Scholar]
- Chin J.Y., Kuan J.Y., Lonkar P.S., et al. (2008). Correction of a splice-site mutation in the beta-globin gene stimulated by triplex-forming peptide nucleic acids. Proc. Natl. Acad. Sci. USA 105, 13514–13519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciudad C.J., Urlaub G., and Chasin L.A. (1988). Deletion analysis of the Chinese hamster dihydrofolate reductase gene promoter. J. Biol. Chem. 263, 16274–16282 [PubMed] [Google Scholar]
- Cole-Strauss A., Gamper H., Holloman W.K., et al. (1999). Targeted gene repair directed by the chimeric RNA/DNA oligonucleotide in a mammalian cell-free extract. Nucleic Acids Res. 27, 1323–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coma S., Noe V., Eritja R., and Ciudad C.J. (2005). Strand displacement of double-stranded DNA by triplex-forming antiparallel purine-hairpins. Oligonucleotides 15, 269–283 [DOI] [PubMed] [Google Scholar]
- Culver K.W., Hsieh W.T., Huyen Y., et al. (1999). Correction of chromosomal point mutations in human cells with bifunctional oligonucleotides. Nat. Biotechnol. 17, 989–993 [DOI] [PubMed] [Google Scholar]
- Datta H.J., Chan P.P., Vasquez K.M., et al. (2001). Triplex-induced recombination in human cell-free extracts. Dependence on XPA and HsRad51. J. Biol. Chem. 276, 18018–18023 [DOI] [PubMed] [Google Scholar]
- De Almagro M.C., Coma S., Noe V., and Ciudad C.J. (2009). Polypurine hairpins directed against the template strand of DNA knock down the expression of mammalian genes. J. Biol. Chem. 284, 11579–11589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Almagro M.C., Mencia N., Noe V., and Ciudad C.J. (2011). Coding polypurine hairpins cause target-induced cell death in breast cancer cells. Hum. Gene Ther. 22, 451–463 [DOI] [PubMed] [Google Scholar]
- Demidov V.V., Protozanova E., Izvolsky K.I., et al. (2002). Kinetics and mechanism of the DNA double helix invasion by pseudocomplementary peptide nucleic acids. Proc. Natl. Acad. Sci. USA 99, 5953–5958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egholm M., Buchardt O., Christensen L., et al. (1993). PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature 365, 566–568 [DOI] [PubMed] [Google Scholar]
- Gupta R.C., Bazemore L.R., Golub E.I., and Radding C.M. (1997). Activities of human recombination protein Rad51. Proc. Natl. Acad. Sci. USA 94, 463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson K.D., and Sedivy J.M. (1995). Analysis of biological selections for high-efficiency gene targeting. Mol. Cell Biol. 15, 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruscha A., Krawitz P., Rechenberg A., et al. (2013). Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 140, 4982–4987 [DOI] [PubMed] [Google Scholar]
- Joung J.K., and Sander J.D. (2013). TALENs: a widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 14, 49–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaihatsu K., Shah R.H., Zhao X., and Corey D.R. (2003). Extending recognition by peptide nucleic acids (PNAs): binding to duplex DNA and inhibition of transcription by tail-clamp PNA-peptide conjugates. Biochemistry 42, 13996–14003 [DOI] [PubMed] [Google Scholar]
- Kayali R., Bury F., Ballard M., and Bertoni C. (2010). Site-directed gene repair of the dystrophin gene mediated by PNA-ssODNs. Hum. Mol. Genet. 19, 3266–3281 [DOI] [PubMed] [Google Scholar]
- Kleinstiver B.P., Wolfs J.M., Kolaczyk T., et al. (2012). Monomeric site-specific nucleases for genome editing. Proc. Natl. Acad. Sci. USA 109, 8061–8066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauert M.P., Kalish J.M., Hegan D.C., and Glazer P.M. (2006). Triplex-stimulated intermolecular recombination at a single-copy genomic target. Mol. Ther. 14, 392–400 [DOI] [PubMed] [Google Scholar]
- Krejci L., Altmannova V., Spirek M., and Zhao X. (2012). Homologous recombination and its regulation. Nucleic Acids Res. 40, 5795–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F.L., Sperle K., and Sternberg N. (1990). Repair of double-stranded DNA breaks by homologous DNA fragments during transfer of DNA into mouse L cells. Mol. Cell Biol. 10, 113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Cheng S., Van Brabant A.J., and Kmiec E.B. (2002). Rad51p and Rad54p, but not Rad52p, elevate gene repair in Saccharomyces cerevisiae directed by modified single-stranded oligonucleotide vectors. Nucleic Acids Res. 30, 2742–2750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Li C., Yu Z., et al. (2012). Efficient and specific modifications of the Drosophila genome by means of an easy TALEN strategy. J. Genet. Genomics 39, 209–215 [DOI] [PubMed] [Google Scholar]
- Lohse J., Dahl O., and Nielsen P.E. (1999). Double duplex invasion by peptide nucleic acid: a general principle for sequence-specific targeting of double-stranded DNA. Proc. Natl. Acad. Sci. USA 96, 11804–11808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonkar P., Kim K.H., Kuan J.Y., et al. (2009). Targeted correction of a thalassemia-associated beta-globin mutation induced by pseudo-complementary peptide nucleic acids. Nucleic Acids Res. 37, 3635–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat L.E. (2005). Nonsense-mediated mRNA decay in mammals. J. Cell Sci. 118, 1773–1776 [DOI] [PubMed] [Google Scholar]
- Moerschell R.P., Tsunasawa S., and Sherman F. (1988). Transformation of yeast with synthetic oligonucleotides. Proc. Natl. Acad. Sci. USA 85, 524–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu-Yilik G., Amthor B., Gehring N.H., et al. (2011). Mechanism of escape from nonsense-mediated mRNA decay of human beta-globin transcripts with nonsense mutations in the first exon. RNA 17, 843–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen P.E., Egholm M., Berg R.H., and Buchardt O. (1991). Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 254, 1497–1500 [DOI] [PubMed] [Google Scholar]
- Nik-Ahd F., and Bertoni C. (2014). Ex vivo gene editing of the dystrophin gene in muscle stem cells mediated by peptide nucleic acid single stranded oligodeoxynucleotides induces stable expression of dystrophin in a mouse model for Duchenne muscular dystrophy. Stem Cells 32, 1817–1830 [DOI] [PubMed] [Google Scholar]
- Noe V., Ciudad C.J., and Chasin L.A. (1999). Effect of differential polyadenylation and cell growth phase on dihydrofolate reductase mRNA stability. J. Biol. Chem. 274, 27807–27814 [DOI] [PubMed] [Google Scholar]
- Papadopoulos N., Nicolaides N.C., Wei Y.F., et al. (1994). Mutation of a mutL homolog in hereditary colon cancer. Science 263, 1625–1629 [DOI] [PubMed] [Google Scholar]
- Papaioannou I., Simons J.P., and Owen J.S. (2012). Oligonucleotide-directed gene-editing technology: mechanisms and future prospects. Expert Opin. Biol. Ther. 12, 329–342 [DOI] [PubMed] [Google Scholar]
- Rodriguez L., Villalobos X., Dakhel S., et al. (2013). Polypurine reverse Hoogsteen hairpins as a gene therapy tool against survivin in human prostate cancer PC3 cells in vitro and in vivo. Biochem. Pharmacol. 86, 1541–1554 [DOI] [PubMed] [Google Scholar]
- Rogers F.A., Vasquez K.M., Egholm M., and Glazer P.M. (2002). Site-directed recombination via bifunctional PNA-DNA conjugates. Proc. Natl. Acad. Sci. USA 99, 16695–16700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet P., Smih F., and Jasin M. (1994). Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell Biol. 14, 8096–8106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saintigny Y., Delacote F., Vares G., et al. (2001). Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 20, 3861–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington L.S. (2005). Focus on recombinational DNA repair. EMBO Rep. 6, 512–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urlaub G., Mitchell P.J., Kas E., et al. (1986). Effect of gamma rays at the dihydrofolate reductase locus: deletions and inversions. Somat Cell Mol. Genet. 12, 555–566 [DOI] [PubMed] [Google Scholar]
- Urnov F.D., Miller J.C., Lee Y.L., et al. (2005). Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435, 646–651 [DOI] [PubMed] [Google Scholar]
- Urnov F.D., Rebar E.J., Holmes M.C., et al. (2010). Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 11, 636–646 [DOI] [PubMed] [Google Scholar]
- Vasquez K.M., Marburger K., Intody Z., and Wilson J.H. (2001). Manipulating the mammalian genome by homologous recombination. Proc. Natl. Acad. Sci. USA 98, 8403–8410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalobos X., Rodriguez L., Prevot J., et al. (2014). Stability and immunogenicity properties of the gene-silencing polypurine reverse Hoogsteen hairpins. Mol. Pharm. 11, 254–264 [DOI] [PubMed] [Google Scholar]
- Wang Y.Y., Maher V.M., Liskay R.M., and Mccormick J.J. (1988). Carcinogens can induce homologous recombination between duplicated chromosomal sequences in mouse L cells. Mol. Cell Biol. 8, 196–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Seidman M.M., and Glazer P.M. (1996). Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science 271, 802–805 [DOI] [PubMed] [Google Scholar]
- Wiedenheft B., Sternberg S.H., and Doudna J.A. (2012). RNA-guided genetic silencing systems in bacteria and archaea. Nature 482, 331–338 [DOI] [PubMed] [Google Scholar]
- Wigler M., Pellicer A., Silverstein S., et al. (1979). DNA-mediated transfer of the adenine phosphoribosyltransferase locus into mammalian cells. Proc. Natl. Acad. Sci. USA 76, 1373–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood A.J., Lo T.W., Zeitler B., et al. (2011). Targeted genome editing across species using ZFNs and TALENs. Science 333, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K., Cole-Strauss A., and Kmiec E.B. (1996). Targeted gene correction of episomal DNA in mammalian cells mediated by a chimeric RNA.DNA oligonucleotide. Proc. Natl. Acad. Sci. USA 93, 2071–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]