

Abstract
PPARγ, a ligand-activated nuclear receptor, regulates fundamental aspects of bone homeostasis and skeletal remodeling. PPARγ-activating anti-diabetic thiazolidinediones in clinical use promote marrow adiposity, bone loss, and skeletal fractures. As such, delineating novel regulatory pathways that modulate the action of PPARγ, and its obligate heterodimeric partner RXR, may have important implications for our understanding and treatment of disorders of low bone mineral density. We present data here establishing retinaldehyde dehydrogenase 1 (Aldh1a1) and its substrate retinaldehyde (Rald) as novel determinants of PPARγ-RXR actions in the skeleton. When compared to wild type (WT) controls, retinaldehyde dehydrogenase-deficient (Aldh1a1−/−) mice were protected against bone loss and marrow adiposity induced by either the thiazolidinedione rosiglitazone or a high fat diet, both of which potently activate the PPARγ-RXR complex. Consistent with these results, Rald, which accumulates in vivo in Aldh1a1−/− mice, protects against rosiglitazone-mediated inhibition of osteoblastogenesis in vitro. In addition, Rald potently inhibits in vitro adipogenesis and osteoclastogenesis in WT mesenchymal stem cells (MSCs) and hematopoietic stem cells (HSCs) respectively. Primary Aldh1a1−/− HSCs also demonstrate impaired osteoclastogenesis in vitro compared to WT controls. Collectively, these findings identify Rald and retinoid metabolism through Aldh1a1 as important novel modulators of PPARγ-RXR transactivation in the marrow niche.
Keywords: Retinaldehyde Dehydrogenase 1, Retinaldehyde, Retinoid, PPAR gamma
INTRODUCTION
Retinoids are a large, diverse family of vitamin A (retinol) metabolites that regulate fundamental cellular processes including differentiation, proliferation, and functional responses in a cell- and tissue-specific manner (1–3). A set of specific enzymes and proteins tightly govern the generation, catabolism, and transport of distinct, biologically active retinoid species. Retinoids like retinoic acid (RA) exert their effects mainly by modulating the activity of retinoid X receptors (RXR) and the retinoid activated receptors (RAR) – nuclear receptors (NRs) that function as ligand-activated transcription factors. RXR is particularly important given its role as the obligate heterodimeric partner for other NRs including the peroxisome proliferator-activated receptor-gamma (PPARγ) (4–6). In addition to its important role in adipogenesis and metabolism, PPARγ also regulates skeletal remodeling and marrow adipogenesis through fundamental effects on bone differentiation programs (6–8). PPARγ activation preferentially shifts mesenchymal stem cell (MSC) allocation toward an adipocyte lineage, thereby promoting bone marrow adiposity while also inhibiting osteoblastogenesis (9, 10). PPARγ also controls key transcriptional programs in osteoclastogenesis in hematopoietic stem cells (HSCs) through coordinated effects on nuclear factor-kappa β ligand signaling (11, 12). Consistent with these effects, both high fat diet (HFD), which can activate PPARγ, as well as PPARγ-activating anti-diabetic thiazolidinediones (TZDs) have been linked to bone loss, marrow adiposity, and fractures (13–17). The deleterious skeletal effects of TZDs have significantly limited their clinical use.
PPARγ and RXR belong to the same superfamily of ligand-activated NRs; however, important distinctions exist in their ligand selectivity and specificity. Although the endogenous ligands of PPARγ remain poorly defined, fatty acids and their metabolic derivatives bind and activate PPARγ. In contrast, RXR is activated by specific retinoids. The 9 cis isomer of retinoic acid (9cRA) retinoic acid is a putative RXR ligand, although it has only been demonstrated definitively in vivo in pancreatic islets (20). RXR functions as an obligate heterodimeric partner for multiple NRs including PPARγ. Importantly, selective synthetic RXR agonists can also modulate the PPARγ-RXR complex independent of exogenous PPARγ ligand stimulation (21–23). These observations combine to establish RXR and the pathways of retinoid metabolism that modulate RXR activity as critical nodal points for PPARγ transcriptional networks and metabolism in general. Despite the well-documented importance of PPARγ in bone metabolism and the central role of RXR in determining PPARγ responses, essentially nothing is known about how specific retinoid pathways modulate PPARγ-RXR action in bone and skeletal responses to other stimuli known to alter bone remodeling, like high fat diet (HFD) and TZDs.
Retinoic acid (RA) is formed by the enzymatic action of retinaldehyde dehydrogenases (Aldh1a1, Aldh1a2, Aldh1a3) on retinaldehyde (Rald). While global Aldh1a2 and Aldh1a3 deficiency cause RA deficiency and embryonic/perinatal lethality due to severe developmental defects (20, 21), Aldh1a1 deletion, a known model of high endogenous Rald levels, does not produce these abnormalities (27). Previously, we and others reported evidence that Rald may exert effects independent of its conversion to RA (28–33), including inhibition of the PPARγ-RXR complex and repression of adipogenesis (28). Recently, we demonstrated that Rald and Aldh1a1 modulate bone morphogenetic protein 2 (BMP2) expression in the skeleton, altering peak bone mass acquisition in vivo (24). Given these observations, we hypothesized that Rald and Aldh1a1 influence fundamental aspects of mesenchymal and hematopoietic stem cell differentiation through effects on PPARγ-RXR transactivation, thereby offsetting known effects of high fat diet HFD and TZDs on bone density and marrow adiposity. We show here that Aldh1a1-deficient (Aldh1a1−/−) mice are protected from the bone loss and marrow fat accumulation seen in WT mice when both are exposed to the TZD rosiglitazone or a HFD. Consistent with these findings, Rald stimulation and Aldh1a1 deficiency modulated key PPARγ-mediated effects on MSC and HSC differentiation in vitro and in vivo. Collectively, these findings establish Rald and Aldh1a1 as novel modulators of PPARγ activity in bone and provide insight into retinoid metabolism as a potentially novel therapeutic target for limiting untoward bone effects of PPARγ activation in the context of age-related bone loss, obesity, and treatment of diabetes.
MATERIALS AND METHODS
Mice
Aldh1a1−/− mice have been previously described (27, 28, 31). For rosiglitazone experiments, 18 week-old WT and Aldh1a1−/− females (n = 5 per group) were fed 10% kcal % fat diet (D12450B, Research Diets Inc.) supplemented with rosiglitazone (Cayman Chemicals) at a dose of 20 mg/kg/day for 12 weeks. For HFD experiments, 12 week-old WT and Aldh1a1−/− females (n = 9 per group) were fed a 60% kcal % HFD (D12492, Research Diets Inc.) for 18 weeks. All experiments and animal use were in accordance with NIH guidelines for the use of laboratory animals and were approved by the Harvard Medical School IACUC.
Histology
Static histomorphometry was performed as previously described (36, 37). Briefly, tibias were collected, fixed in 70% ethanol, dissected, and embedded in methyl methacrylate. Longitudinal sections that were 5 μm thick were prepared using a Micron microtome (Richard-Allan Scientific) and then stained with toluidine blue. Quantification of marrow fat was performed as previously described (38). The terminology and units used are in accordance with guidelines established by the Histomorphometry Nomenclature Committee of the American Society for Bone and Mineral Research (39).
Dual-energy x-ray absorptiometry (DXA)
Dual-energy x-ray absorptiometry (DXA) scanning was performed using the PIXImus system (GE-Lunar) as previously described (36). The PIXImus was used to assess femoral bone mineral density (BMD), and bone mineral content (BMC) in WT and Aldh1a1−/− mice after rosiglitazone treatment and high fat diet. A phantom standard provided by the manufacturer was assessed each day for instrument calibration.
Micro-computed tomography (μCT)
Microarchitecture of the trabecular bone and midshaft cortical bone of the femur was analyzed by μCT (resolution 10 μm, VivaCT-40; Scanco Medical AG). Bones were scanned at an energy level of 55 kVp and intensity of 145 μA. Trabecular bone volume fraction and microarchitecture were evaluated starting approximately at 0.6 mm proximal to the distal femoral growth plate and extending proximally 1.5 mm. Measurements included bone volume/total volume (BV/TV), trabecular number (Tb.N.), trabecular thickness (Tb.Th.), and trabecular spacing (TbS). Scans for the cortical region were measured at the midpoint of each femur, with an isotropic pixel size of 21 μm and slice thickness of 21 μm, and used to calculate the average BA, total cross-sectional area, BA/total cross-sectional area, and Ct.Th. All scans were analyzed using manufacturer software (version 4.05; Scanco Medical AG).
MRI of Marrow Fat
Mice were scanned for quantification of marrow adiposity by spectroscopy of marrow fat using a BRUKER PharmaScan 7 T magnet (3D RARE32 method respiration gated with fat suppression off; TE=6.15ms; TR=600ms; 4averages; FOV=4×4×1cm; axial slice placed over right kidney slice thickness=10 mm; Matrix= 256×256× 256; resolution 156 microns per pixel). Ex vivo T1-weighted imaging and MR spectral peak analysis of marrow tibial fat was performed in WT and Aldh1a1−/− mice that were 30 weeks of age after 12 weeks of rosiglitazone treatment, and 30 weeks of age after 18 weeks of HFD.
Bone Marrow Isolation and in vitro Differentiation Assays
Bone marrow was isolated from 12–16 week-old female WT and Aldh1a1−/− mice as previously described (40). Briefly, tissue was dissected away from femurs and tibiae of age and sex matched WT and Aldh1a1−/− mice. Bone marrow was then flushed from the bones with αMEM media (Life Technologies) supplemented with 10% lot-selected Hyclone fetal bovine serum (FBS) (VWR International) and 1% penicillin-streptomycin (Life Technologies). Cells were then strained with 70μM filter and then seeded for differentiation assays as outlined below.
In vitro Osteoblastogenesis Culture
In vitro osteoblastogenesis assays were performed by plating marrow stromal cells isolated as described above at a density of 10 × 106 cell/well in 6 well dishes (1 × 106 cells/cm2). The cells were grown in basal media consisting of αMEM media (Life Technologies) supplemented with 10% Hyclone fetal bovine serum (VWR International) and 1% penicillin-streptomycin (Life Technologies) for 7 days, and then osteogenic media (basal media supplemented with ascorbic acid [AA] at 50 μg/mL and beta-glycerol phosphate [BGP] at 10 mM) either in the presence or absence of rosiglitazone (1 μm) for another 7 days. Alkaline phosphatase (ALP) staining (Sigma 86R-KT) was then performed.
In vitro Marrow Adipogenesis Culture
For in vitro adipogenesis differentiation assays, WT marrow cells were seeded in 24 well dishes (1 × 106 cells/cm2) and grown in basal media for 10 days. Treatment with either DMSO or Rald 1 μM (Sigma Aldrich) was started on day 3, and continued throughout differentiation. On day 10, the cells were treated with adipogenesis induction media (basal media supplemented with rosiglitazone 1 μM [Cayman Chemicals], insulin 25 mg/mL [Sigma Aldrich], dexamethasone 1 nM [Sigma Aldrich], IBMX 0.5mM [Sigma Aldrich]) for 48 hours followed by adipogenesis maintenance media (basal media supplemented with rosiglitazone 1 μM, insulin 25 mg/mL) for 5 days, in the presence of either DMSO or Rald. Oil Red O (ORO) staining (41) and the Adipored assay (Lonza) were performed to quantitate intracellular lipid accumulation.
In vitro Osteoclastogenesis Cultures
Bone marrow cells were isolated from femurs and tibias of matched 12-week old female WT and Aldh1a1−/− mice as described above. Bone marrow cells (4.5×105 cells/well) were then plated in a 96-well dish in differentiation medium consisting of αMEM supplemented with 10% fetal calf serum (VWR), 1% Penicillin-Streptomycin 1000U (Invitrogen), 30 ng/mL macrophage colony-stimulating factor (m-CSF) and 100 ng/mL soluble receptor activator of nuclear factor-κB ligand (sRANKL) (PeproTech, Inc.). When indicated, cells were treated with either 1μM of Rosiglitazone (Cayman Chemicals), Rald, (Sigma Aldrich), or the combination of rosiglitazone and Rald. The culture medium and treatments were changed at day 3 and day 6. At day 7, the culture was terminated; cells were then fixed with 2.5% glutaraldehyde (Electron Microscopy Sciences) and stained for tartrate-resistant acid phosphatase activity (TRAP5b) using the Sigma Leucocyte acid phosphatase (TRAP) kit. Osteoclasts were identified by TRAP5b positive multinucleated cells (more than four nuclei) using light microscopy.
ST2 Marrow Stromal Cell Culture
ST2 cells were purchased from Riken (Japan) and maintained in 1640 RPMI media supplemented with 10% FBS and 1% penicillin-streptomycin (Life Technologies). ST2 cells were differentiated into osteoblasts using osteogenic media consisting of basal growth media basal media supplemented with AA at 50 μg/mL and BGP at 10 mM) in the presence of DMSO, rosiglitazone (1 μm), Rald (1 μm), AGN 193109 (Sigma), or HX531 (a kind gift from Dr. H. Kagechika, University of Tokyo) for 7 days. Alkaline phosphatase (ALP) staining (Sigma 86R-KT) was then performed. For the siRNA studies, either Co siRNA, RARγ siRNA or RXRα siRNA was transfected into ST2 cells using the DeliverX™ siRNA transfection system (Affymetrics/Panomics) as per the manufacturer’s protocol. Following transfection, the ST2 cells were then treated with Rald (1 μm) for 24 hours prior to staining for ALP.
Bone resorption assay
Bone marrow cells were cultured and treated as described above, and the resorption assay was performed as before (36). Briefly, cells were plated in the Corning Osteo Assay Surface 96-well plates (VWR). The culture medium and treatments were changed every 3 days until day 13. Cells were then washed with PBS, incubated with 10% sodium hypochlorite for 5 minutes at room temperature. The wells were washed twice with water and stained with a modified von Kossa stain [5% (w/v) aqueous silver nitrate solution] for 30 minutes in the dark at room temperature. Following staining, wells were then soaked in water for 5 minutes and treated with 100μl of 5% sodium carbonate (w/v in 10% formalin) for 5 minutes. Pit formation was quantified and analyzed using Image Pro Software.
Reverse Transcriptase (RT) PCR and Quantitative Real Time PCR
RNA was harvested using Trizol (Invitrogen) and RT-PCR was performed using 0.5–1 μg of RNA using the High Capacity cDNA synthesis kit (Applied Biosystems). Gene expression analysis was performed using an iQ quantitative real time thermal cycler system (Bio-Rad). One μL of diluted cDNA (diluted 1:5) generated from RT-PCR was then used as template for qPCR amplification using 2X iQ SYBR green mastermix (Bio-Rad) in a total reaction volume of 25 μL. Gene expression was normalized using 36B4.
Statistical Analysis
Unpaired student t-tests were employed in the statistical analysis of WT and Aldh1a1−/− body weights, fat depot weights, MRI fat spectral peaks, and in vitro assays (ORO, Adipored, gene expression). For the WT osteoclastogenesis assays, the ANOVA statistical method was used in analyzing differences in TRAP positive multinucleated cells and gene expression between rosiglitazone, Rald, or combined rosiglitazone and Rald treatments.
RESULTS
Aldh1a1 deficiency protects mice against rosiglitazone-induced bone loss and marrow adiposity
To investigate whether Aldh1a1 modulates PPARγ-RXR skeletal responses in vivo, matched, standard chow-fed 18 week-old female WT and Aldh1a1−/− mice were treated with the PPARγ agonist rosiglitazone (20 mg/kg/day in chow) for 12 weeks. At the conclusion of these studies, total body weight (TBW) was similar in WT and Aldh1a1−/− mice (Fig. 1A) (27.8 ± 1.2 g versus 25.5 ± 2.8 g, p= 0.17). To determine body fat composition of the treated animals, perigonadal visceral white fat and interscapular brown fat (BAT) pads were dissected and weighed individually. Aldh1a1−/− mice had less visceral white adipose tissue (VWAT) than WT mice as a percentage of TBW (0.80 ± 0.19% versus 1.95 ± 0.23% of TBW, p<0.05) when treated with rosiglitazone (Fig. 1B). Compared to WT controls, there was also a trend toward less interscapular brown fat (0.64 ± 0.07 versus 0.80 ± 0.18% of TBW, p= 0.08) in Aldh1a1−/− mice treated with rosiglitazone, which is known to increase murine BAT depot size (34) (Fig. 1B). Liver weight and femur length were not significantly different in the two groups (data not shown). When we examined bone density in WT and Aldh1a1−/− mice treated with rosiglitazone, dual energy x-ray absorptiometry (DEXA) scanning (Fig. 1C) and micro computed tomography (μCT) demonstrated significantly higher bone mass in the Aldh1a1−/− group. Both total femoral mineral content (8.58 ± 0.153 μg vs 1.02 ± 0.286 μg, p< 0.001) and bone density (0.0722 ± 0.00150 versus 0.0607 ± 0.00133 g/cm2, p< 0.001) were significantly higher in the Aldh1a1−/− mice, as measured by PIXImus densitometry.
Figure 1. Aldh1a1 deficiency protects against rosiglitazone-induced bone loss.
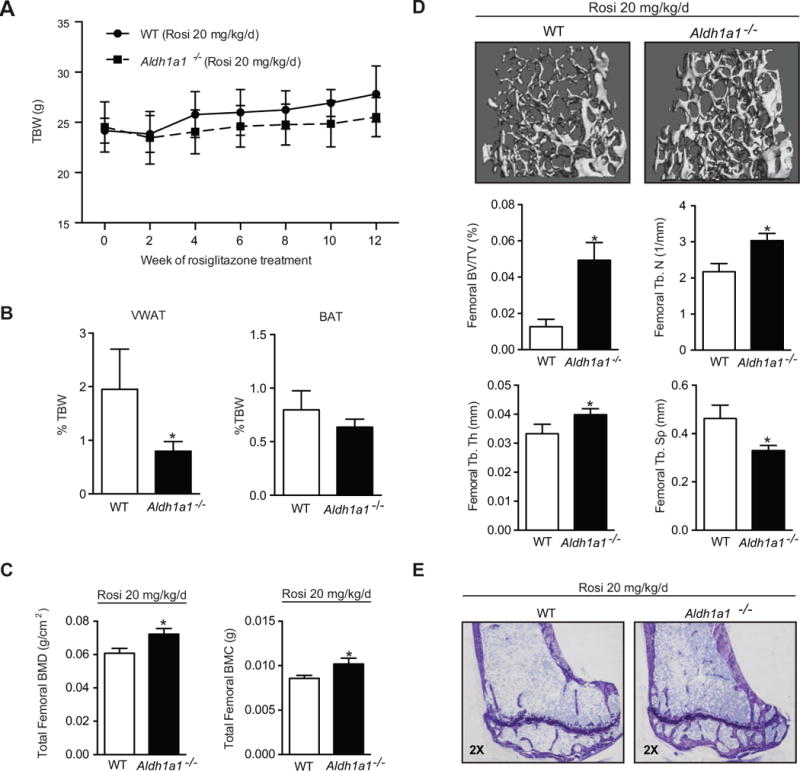
A. Weekly weights of matched 30-week old wild type (WT, solid line) and Aldh1a1−/− (dashed line) female mice treated with the PPARγ agonist rosiglitazone (n=5/genotype, 20 mg/kg/day) for 12 weeks (age at start of treatment was 18 weeks age and age at the time of harvest and analysis was 30 weeks). B. Fat depot composition of matched 30-week old WT and Aldh1a1−/− female mice treated with the PPARγ agonist rosiglitazone. C. PIXImus of 30-week old WT and Aldh1a1−/− female mice treated with rosiglitazone. D. Micro CT of 30-week old WT and Aldh1a1−/− female mice treated with rosiglitazone. E. Toludine blue sections of tibiae from rosiglitazone-treated WT and Aldh1a1−/− mice. VWAT – perigonadal visceral white adipose tissue; BAT- interscapular brown adipose tissue; TBW- total body weight; Rosi-rosiglitazone. *p<0.05
Micro CT (μCT) demonstrated significant microarchitectural differences between WT and Aldh1a1−/− mice exposed to rosiglitazone (Figure 1D). Femoral bone volume/total volume (BV/TV) was significantly lower in WT mice as compared to the Aldh1a1−/− group (0.0126 ± 0.00185 % versus 0.0493 ± 0.00435 %; p <0.0001). Aldh1a1−/− mice on a standard chow diet have higher femoral BV/TV at multiple developmental time points (24); however, rosiglitazone treatment led to a greater decrease in bone mass in the WT cohort. WT mice treated with rosiglitazone demonstrated a larger relative decline in femoral BV/TV on μCT as compared to the Aldh1a1−/− group (−66% vs −35%) as determined by comparison to relevant historical controls (18 week old female WT and Aldh1a1−/− on a standard chow diet; supplementary data, Fig. S1). The higher femoral BV/TV seen in rosiglitazone-treated Aldh1a1−/− mice was also accompanied by greater trabecular number (TbN) (3.035 ± 0.200 versus 2.175 ± 0.226 /mm, p<0.005) and trabecular thickness (TbTh) (0.0399 ± 0.00210 versus 0.0333 ± 0.00330 μm, p<0.005), and lower trabecular spacing (TbS) (0.330 ± 0.0210 versus 0.4621 ± 0.0556, p<0.005). Notably, histomorphometry (Table 1 and Fig 1E) demonstrated significantly lower number of osteoclasts per total area (N.Oc/T.Ar) and bone perimeter (N.Oc/B.Pm) in the Aldh1a1−/− group as compared to WT mice (p< 0.05). The osteoclast surface to bone surface ratio (Oc.S/BS) was also lower in the Aldh1a1−/− mice (3.96 ± 1.18 versus. 8.55 ± 1.82, p< 0.01) compared to WT mice. In addition, there were trends toward higher osteoid volume to total volume (OV/TV), osteoid surface to bone surface (OS/BS), number of osteoblasts per total area (N.Ob/T.Ar) and TbN in Aldh1a1−/− mice as compared to WT controls.
Table 1.
Histomorphometry of WT and Aldh1a1−/− female mice after 12 weeks of rosiglitazone treatment
| Parameter | WT (n=5) | Aldh1a1−/− (n=5) | p value |
|---|---|---|---|
| BV/TV (%) | 9.48±2.16 | 10.54±3.21 | 0.56 |
| OV/TV (%) | 0.40±0.15 | 0.61±0.24 | 0.15 |
| OV/BV (%) | 4.27±1.49 | 5.87±2.39 | 0.24 |
| BS/BV (%) | 50.28±6.36 | 54.83±7.47 | 0.33 |
| OS/BS (%) | 28.31±5.17 | 35.39±9.31 | 0.18 |
| Tb.Th (μm) | 40.29±5.07 | 37.02±5.04 | 0.34 |
| Tb.N (/mm) | 2.35±0.36 | 2.83±0.64 | 0.18 |
| Tb.Sp (μm) | 393.08±63.82 | 332.19±94.73 | 0.27 |
| Ob S/OS (%) | 47.22±6.31 | 46.61±9.98 | 0.91 |
| Ob.S/BS (%) | 13.34±2.84 | 17.05±6.98 | 0.30 |
| N.Ob/T.Ar (/mm2) | 45.84±16.54 | 72.08±33.03 | 0.15 |
| N.Ob/B.Pm (/mm) | 12.15±2.89 | 15.72±5.95 | 0.26 |
| N.Ob/O.Pm (/mm) | 90.75±8.31 | 94.34±9.67 | 0.55 |
| O.Th (μm) | 2.89±0.29 | 2.92±0.18 | 0.18 |
| Oc.S/BS (%) | 8.55±1.82 | 3.96±1.18 | 0.0014 |
| N.Oc/T.Ar (/mm2) | 8.30±2.20 | 5.12±1.93 | 0.040 |
| N.Oc/B.Pm (/mm) | 2.23±0.35 | 1.17±0.41 | 0.0022 |
Given the well-characterized effects of PPARγ-RXR activation on marrow adiposity (34), we then examined marrow fat accumulation in WT and Aldh1a1−/− mice treated with rosiglitazone. Although Aldh1a1−/− mice on a standard chow diet have higher marrow adipocyte content by histological analysis at baseline (24), after 12 weeks of rosiglitazone treatment, MRI spectroscopy of the proximal and mid shaft of the tibiae demonstrated a significantly lower fat peak in the Aldh1a1−/− group as compared to control mice (Fig 2A). In addition, T1-weighted MRI imaging of tibiae revealed greater fat accumulation (hyperintense signal) in WT vs Aldh1a1−/− mice (Fig 2A); quantitative MRI spectral analysis of fat peaks showed significantly less marrow adiposity in Aldh1a1−/− mice as compared to WT controls (2.38 × 109 versus 4.1 × 109 A.U, p<0.05; Fig 2A). On histological analysis, Aldh1a1−/− mice treated with rosiglitazone had significantly less marrow adipocytes (18.6 ± 3.5 versus 24.7 ± 1.8 adipocytes/mm2, p<0.05) than WT controls, as seen by quantification of adipocyte ghosts adjacent to the growth plate of the tibia (Fig. 2B).
Figure 2. Aldh1a1 deficiency protects against rosiglitazone-induced marrow adiposity.
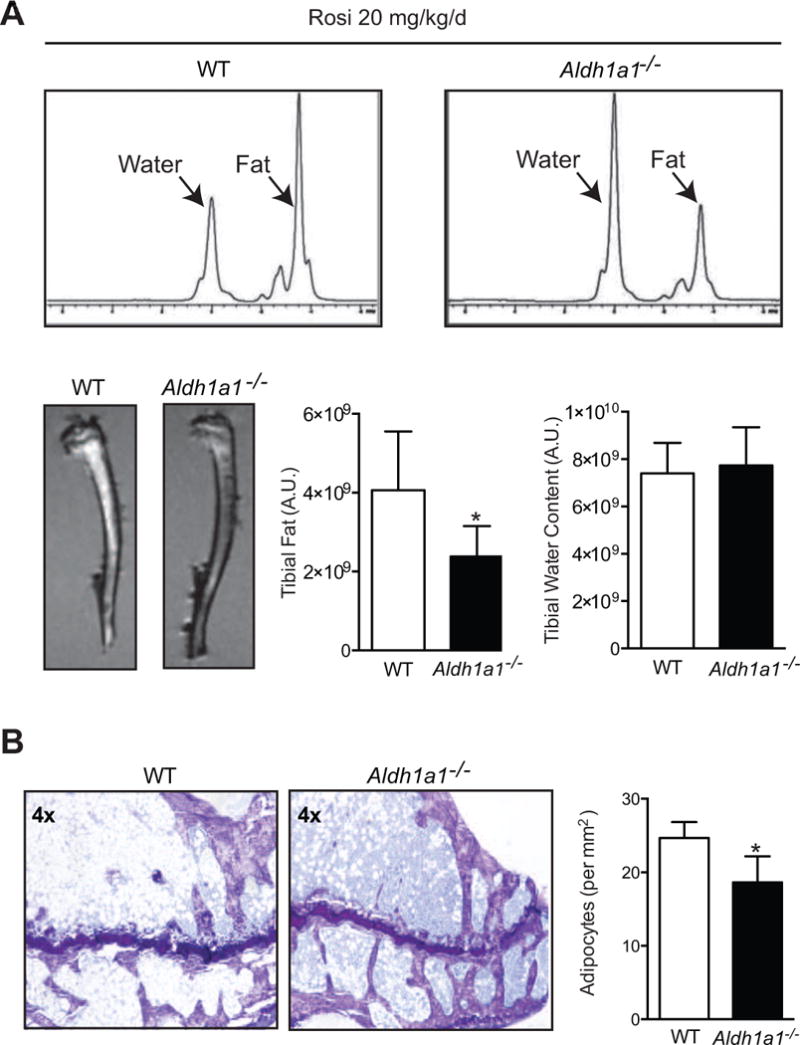
A. MRI spectral analysis of marrow fat of WT and Aldh1a1−/− female mice treated with rosiglitazone (n=5/genotype, 20 mg/kg/day) for 12 weeks (age at start of treatment was 18 weeks and age at the time of harvest and analysis was 30 weeks). T1-weighted MRI (lower left panels) and quantitative MRI fat spectral peak analysis (upper and lower right panels) of tibiae of rosiglitazone-treated WT and Aldh1a1−/− mice. B. Histological assessment of fat content of tibiae of rosiglitazone-treated WT and Aldh1a1−/− mice. Representative toludine blue staining of WT and Aldh1a1−/− mice treated with rosiglitazone. * p<0.05.
Aldh1a1−/− mice challenged with a HFD have higher bone density and develop significantly less marrow adiposity than WT controls
Since Aldh1a1−/− mice were protected from rosiglitazone-mediated bone loss and marrow adiposity, we were interested in determining whether the effect of Aldh1a1 deficiency in offsetting the skeletal effects of rosiglitazone extended to other models in which PPARγ activation is thought to cause skeletal remodeling, namely HFD feeding (42–45). We studied bone density and marrow adiposity in matched 12-week-old WT and Aldh1a1−/− female mice maintained on a HFD (60% fat kCal) for 18 weeks. Consistent with our prior studies (28), despite having similar baseline weights, Aldh1a1−/− mice weighed less than WT controls (Fig. 3A) after 18 weeks of HFD (22.6 ± 2.0 g versus 38.5 ± 6.7 g, p < 0.0005). High fat-fed Aldh1a1−/− mice also had less VWAT than WT mice (1.9 ± 2.7% versus 10.7 ± 1.1%, p<1.0 × 10−6), with no significant difference in BAT content (Fig. 3A). In terms of bone, Aldh1a1−/− mice had significantly higher femoral BV/TV (0.063 ± 0.018 % vs. 0.018 ± 0.012 % p< 0.001) and cortical thickness (0.278 ± 0.022 μm vs. 0.232 ± 0.0079 μm, p<0.001) in response to HFD as compared to WT mice (Fig. 3B). In addition, T1-weighted MRI imaging of tibiae of WT and Aldh1a1−/− mice maintained on a HFD demonstrated significantly greater bone marrow fat accumulation in the WT group (Fig. 3C). Quantitative MRI spectroscopy of the proximal and mid shaft of the tibia demonstrated a significantly lower fat peak in the Aldh1a1−/− group (1.9 × 107 versus 2.92 × 108 A.U., p< 0.05, Fig. 3C).
Figure 3. Aldh1a1 deficiency inhibits high fat diet (HFD)-induced one loss and marrow adiposity.
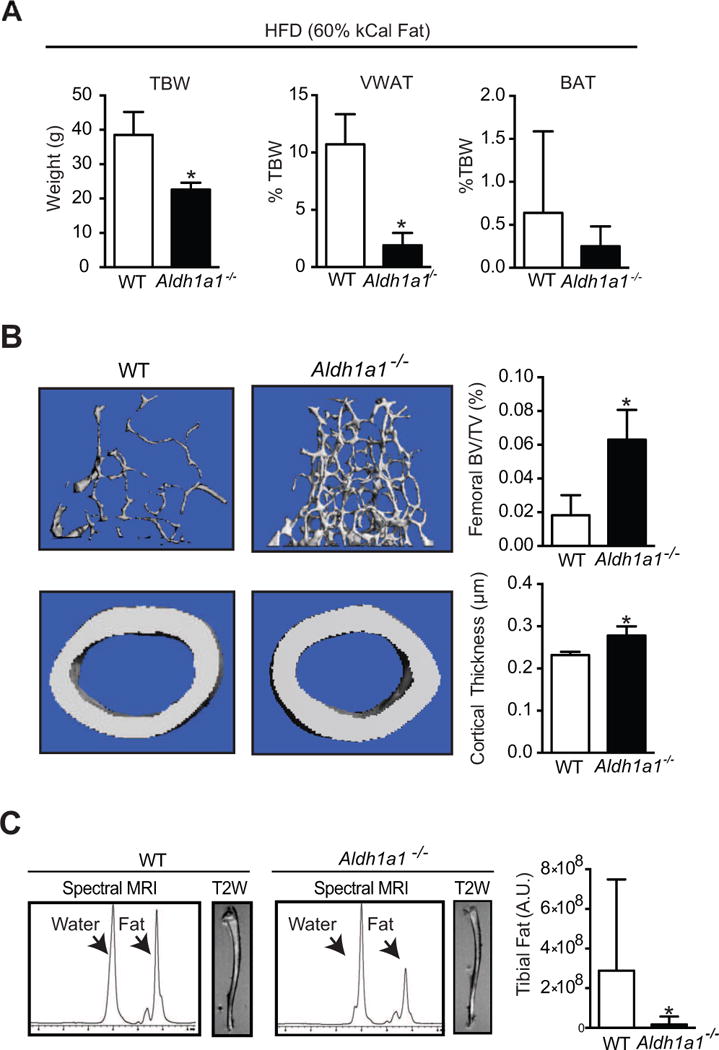
A. Total body weights and fat depot composition (VWAT and BAT) of matched 12 week-old WT and Aldh1a1−/− female mice fed a HFD (60% kcal% fat) for 18 weeks. B. Micro CT of WT and Aldh1a1−/− female mice maintained on HFD for 18 weeks C. T1-weighted (T1W) MRI and MRI spectral analysis of fat content of tibiae of WT and Aldh1a1−/− mice on a HFD. * p<0.05.
Rald and Aldh1a1 deficiency modulate PPARγ-RXR effects on MSC lineage determination
Given these results, we were interested in exploring mechanisms contributing to the protection seen in Aldh1a1 deficiency against TZD and HFD-induced skeletal remodeling. Activation of the PPARγ-RXR transcriptional complex inhibits in vitro osteoblastogenesis by multiple mechanisms including the inhibition of expression and activity of key osteogenic transcription factors such as runt-related transcription factor 2 (Runx2) (34, 46, 47). Simultaneously, PPARγ agonists such as rosiglitazone promote in vitro MSC adipogenesis, thereby skewing MSC allocation toward an adipocyte lineage (34). Given the impact of Aldh1a1 deficiency on PPARγ-RXR effects on bone density and marrow adiposity in vivo, next we examined how Rald and Aldh1a1 modulate PPARγ-RXR-mediated transcriptional responses during MSC osteoblastogenesis and adipogenesis. In both primary WT and Aldh1a1−/− murine marrow stromal (MSC) cultures induced to differentiate into osteoblasts with ascorbic acid (AA) and beta-glycerol phosphate (BGP), rosiglitazone (1 μM) decreased expression of alkaline phosphatase (ALP), a marker of early osteoblasts; however, this inhibitory rosiglitazone effect was significantly diminished in Aldh1a1−/− versus WT MSCs (Fig 4A). Aldh1a1−/− MSCs exposed to rosiglitazone during osteoblastogenesis expressed significantly higher levels of ALP as compared to WT control cultures (Fig. 4A). To investigate if these pro-osteoblastic effects in Aldh1a1 deficiency were due to Rald itself, we repeated experiments under osteoblastic conditions in the murine bone marrow stromal cell line ST2 (48). Although rosiglitazone completely blocked ALP expression in ST2 cells treated with AA and BGP, this inhibitory effect of rosiglitazone was completely abrogated in the presence of Rald (1 μM) (Fig. 4B). Moreover, Rald directly induced ALP expression in ST2 cells even in the absence of AA and BGP (Fig. 4B). To test if the action of Rald on inducing osteoblastic markers were due to changes in retinoid receptor responses, the experiments in ST2 cells were repeated in the presence of chemical inhibitors or validated siRNA-mediated knockdown of RAR and RXR. Indeed, in the presence of either the RAR inhibitor AGN193109 and the RXR inhibitor HX531, or siRNA directed against RAR or RXR, the Rald-mediated induction of ALP expression was significantly diminished (Fig. 4B).
Figure 4. Rald and Aldh1a1 deficiency inhibits PPARγ-RXR effects on MSC allocation in vitro.
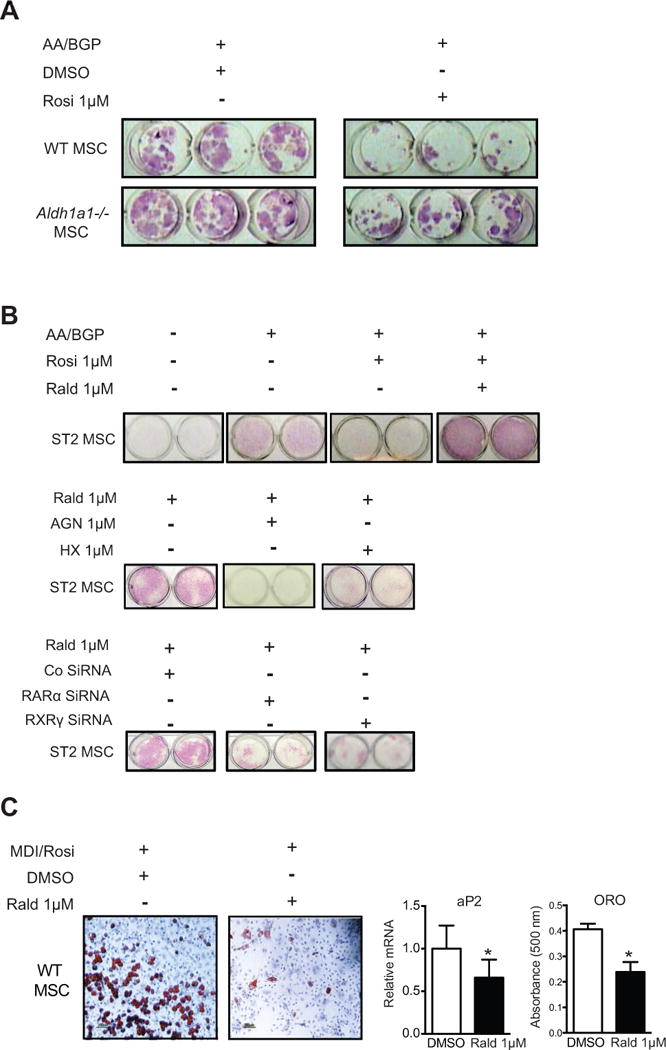
A. Alkaline phosphatase (ALP) staining of in vitro osteoblastogenesis cultures of primary WT and Aldh1a1−/− MSCs in the absence and presence of rosiglitazone. B. ALP staining of ST2 MSC osteoblastogenesis cultures. Rald blunted rosiglitazone-mediated inhibition of ALP expression during osteoblastogenesis in ST2 MSCs (top panel). In addition, Rald directly induced ALP even in the absence of AA and BGP (top panel). Chemical inhibition of RAR (using AGN) and RXR (using HX531) attenuated Rald-mediated induction of ALP (middle panel). Similar effects were observed with siRNA-mediated knockdown of RAR and RXR (bottom panel). C. In vitro adipogenesis cultures of primary WT MSC cultures in the absence and presence of Rald. * p<0.05.
These results suggested fundamental switches in osteoblast versus adipogenic MSC differentiation in the presence of Rald. To further evaluate this, we induced WT MSCs to undergo adipogenesis by stimulation with the PPARγ agonist rosiglitazone (1 μM) and a standard pro-adipogenic cocktail consisting of 3-isobutyl-1-methylxanthine, dexamethasone, and insulin (MDI). As expected, rosiglitazone enhanced adipogenesis of primary WT MSC cultures, as evident on oil red O (ORO) staining and expression of the adipogenic marker aP2. In marked contrast to the stimulatory effect on osteoblastogenesis demonstrated above, Rald potently inhibited adipogenesis in primary WT MSCs (Fig. 4C).
Rald blocks PGC-1β induction and inhibits in vitro osteoclastogenesis
PPARγ-RXR signaling plays a central role in osteoclastogenesis (12, 35, 36), suggesting another mechanism through which Rald may preserve bone mass in the setting of TZD treatment and HFD. Given the significantly lower number of osteoclasts observed on histomorphometry in Aldh1a1−/− mice after treatment with rosiglitazone (Table 2), we studied the impact of Rald and Aldh1a1 deficiency on in vitro osteoclastogenesis in primary WT and Aldh1a1−/− bone marrow cultures differentiated with m-CSF and RANKL (Fig 5). Under these conditions, primary Aldh1a1−/− bone marrow cultures manifested fewer multinucleated osteoclasts on tartrate-resistant acid phosphatase (TRAP) staining when compared with WT cultures (Fig 5A). In considering potential drivers of this response, we found that the mRNA expression of peroxisome proliferator-activated receptor gamma coactivator-1 beta (PGC-1β), a transcriptional co-activator that is potently induced by both RANKL and PPARγ during osteoclastogenesis (12, 51), was significantly lower in Aldh1a1−/− osteoclast cultures compared to WT controls (Fig 5B). Recent evidence indicates that PGC-1β is necessary for osteoclastogenesis in vitro and in vivo (51), and is required for PPARγ and TZD-mediated stimulation of osteoclastogenesis and bone resorption (12). Furthermore, the expression of c-Fos, a key upstream regulator of osteoclastogenesis that is a direct transcriptional target of PPARγ, was also lower in Aldh1a1−/− osteoclast cultures (0.20 ± 0.081 versus 1.0 ± 0.24, p< 0.05). Consistent with these findings, expression of Trap5b, a marker of mature osteoclasts, was lower in Aldh1a1−/− osteoclast cultures (0.39 ± 0.066 versus 1.0 ± 0.33, p= 0.051). Despite trending lower in Aldh1a1−/− osteoclasts, cathepsin K (CatK) expression did not differ significantly between WT and Aldh1a1−/− osteoclast cultures (1.0 ± 0.086 versus 1.27 ± 0.39, p=0.31). In addition, Aldh1a1−/− osteoclasts demonstrated corresponding decreases in functional osteoclast activity with evidence of attenuated pit formation over a synthetic inorganic bone surface (Fig 5C). To determine whether Rald, the metabolic intermediary that accumulates in vivo in Aldh1a1 deficiency (27, 28), mediated these inhibitory effects on osteoclastogenesis through modulation of PPARγ signaling, osteoclast differentiation assays were repeated using WT marrow cultures in the presence of rosiglitazone and/or Rald (Figs 5D and 5E). As expected, rosiglitazone increased the formation of multinucleated osteoclasts and pits over a synthetic inorganic bone surface compared to mCSF/RANKL alone (Fig 5D). Consistent with the findings of reduced osteoclastogenesis in Aldh1a1−/− cultures, Rald potently blocked osteoclastogenesis by TRAP staining and pit formation in WT marrow cultures treated with mCSF/RANKL (Fig 5D). Furthermore, Rald completely blocked the stimulatory effects of rosiglitazone in WT osteoclast cultures in these assays (Fig 5D). These findings were accompanied by corresponding shifts in gene expression patterns (Fig 5E). Rosiglitazone increased PGC-1β, Trap5b, and CatK expression in WT marrow cultures stimulated with mCSF and RANKL. As expected, Rald blocked these rosiglitazone transcriptional effects. In addition, Rald decreased PGC-1β, Trap5b, and CatK expression in WT marrow cultures treated with mCSF and RANKL (Fig 5E).
Figure 5. Rald and Aldh1a1 deficiency inhibit osteoclastogenesis in vitro.
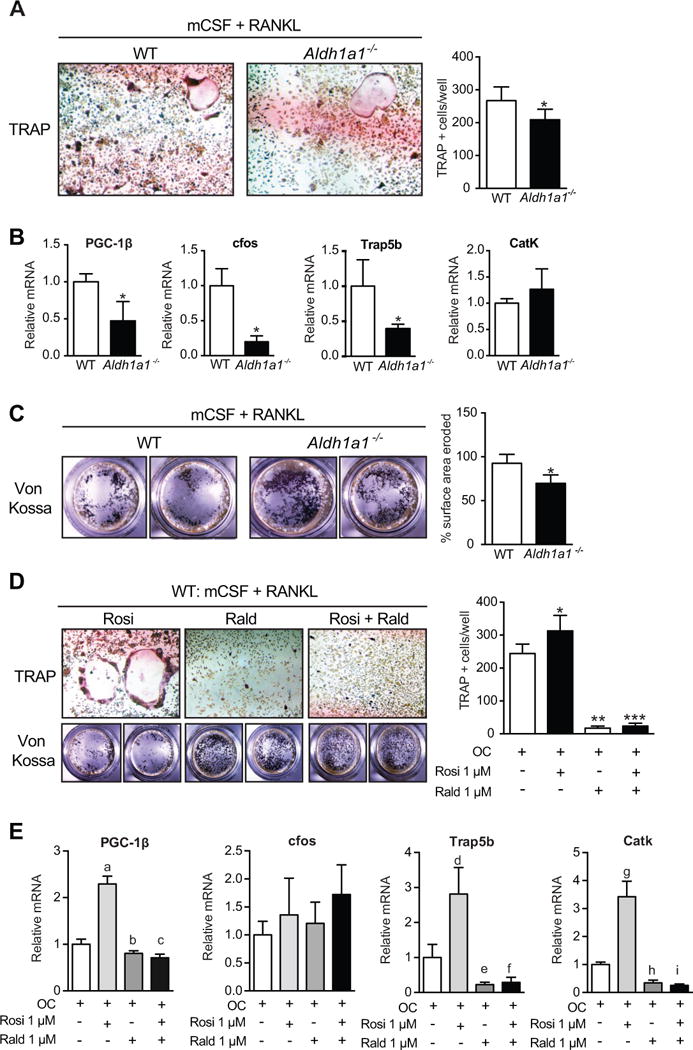
A. TRAP staining of in vitro osteoclastogenesis cultures of primary WT and Aldh1a1−/− marrow cells. B. Gene expression patterns during osteoclastogenesis in primary WT and Aldh1a1−/− marrow cells. C. Pit formation assay of primary WT and Aldh1a1−/− in vitro osteoclastogenesis cultures. D. TRAP staining and pit formation assays in primary WT marrow cells induced to differentiate into osteoclasts with mCSF and RANKL, in the presence of rosiglitazone, Rald, or both. Effects of Rald and rosiglitazone on osteoclastogenesis in WT marrow cultures. *p<0.05; **p<0.05 compared to OC alone (mCSF/RANKL) and OC + rosiglitazone; ***p<0.05 compared to OC alone (mCSF/RANKL) and OC + rosiglitazone; a- p<0.05 compared to OC; b- p<0.05 compared to OC and OC + rosiglitazone; c- p<0.05 compared to OC and OC + rosiglitazone; d- p<0.05 compared to OC; e- p<0.05 compared to OC and OC+ rosiglitazone; f- p<0.05 compared to OC and OC + rosiglitazone; g- p<0.05 compared to OC; h- p<0.05 compared to OC and OC + rosiglitazone; and i- p<0.05 compared to OC and OC + rosiglitazone.
DISCUSSION
In this report, we present evidence establishing Aldh1a1 and its substrate Rald as novel determinants of PPARγ-RXR responses in the skeleton. The absence of Aldh1a1 in mice and the known associated accumulation of Rald in vivo (27, 28) strongly protect against the deleterious skeletal effects of PPARγ transactivation. Aldh1a1−/− mice demonstrate higher bone density (by μCT) and less marrow fat (by MRI spectral analysis) than WT controls when challenged with either rosiglitazone (Fig 1, 2) or a HFD (Fig 3). Importantly, both rosiglitazone and HFD potently activate PPARγ-RXR and have been linked to bone loss, marrow adiposity, and fractures in humans (13, 14, 16, 17, 19). To investigate the basis for this protection, we systematically examined how Rald and Aldh1a1 deficiency modulate key PPARγ-regulated MSC and HSC differentiation events. Consistent with our in vivo data, Aldh1a1−/− MSCs manifest distinct stem cell lineage determination events when compared to WT MSCs (Fig 4, 5). Although rosiglitazone decreased osteoblastogenesis in both WT and Aldh1a1−/− MSCs, Aldh1a1−/− osteogenic cultures demonstrated significantly higher ALP expression (Fig 4A). In ST2 marrow stromal cells, Rald blocks the inhibitory effect of rosiglitazone on osteoblastogenesis and directly induces ALP expression in an RAR and RXR-dependent manner (Fig 4B). Collectively, these data suggest Rald and Aldh1a1 deficiency limit PPARγ-mediated inhibition of osteoblastogenesis. In contrast, Rald potently blocks PPARγ-dependent adipogenic responses in MSC models (Fig 4) while limiting osteoclastogenesis in HSCs in vitro (Fig 5) and in response to rosiglitazone in vivo (Fig 1, Table 2). Collectively, these findings argue that specific retinoids and retinoid metabolizing enzymes exert coordinated effects on PPARγ control of MSC and HSC differentiation in vitro and in vivo.
The nuclear receptor PPARγ is a master regulator of adipogenesis that controls the expression of multiple genes within complex transcriptional networks (52). Increasing evidence argues for a similar integrative role for PPARγ in bone remodeling. Recent studies link age-dependent increases in skeletal PPARγ expression to dysregulated bone homeostasis and disorders of low bone mineral density like osteoporosis (53). On balance, PPARγ activation promotes bone resorption while decreasing bone formation by coordinating stem cell differentiation and lineage commitment events in the marrow niche (8). PPARγ-deficient murine embryonic stem cells fail to differentiate into adipocytes and spontaneously form bone nodules, while PPARγ haploinsufficiency increases bone mass and promotes osteoblastogenesis in vivo (9). PPARγ-RXR transactivation also promotes osteoblast apoptosis (54) and inhibits MSC osteoblastogenesis through multiple mechanisms, including Runx2 repression (55, 56). At the same time, PPARγ agonists promote MSC adipogenesis in vitro and in vivo (8, 34, 47). In further support of this coordinating role for PPARγ in bone, recent work indicates that PPARγ also controls osteoclastogenesis in HSCs through PGC1β-dependent induction of c-Fos (12, 49). Indeed, PPARγ may help support normal remodeling since genetic deletion of PPARγ in HSCs results in osteopetrosis (49).
Recent clinical evidence reinforces this emerging importance of PPARγ in skeletal remodeling as well. TZDs, which induce insulin sensitization by activating PPARγ, were found unexpectedly to promote bone loss, marrow adiposity, and fractures in patients with diabetes (13–15). In addition, high fat diets, which are know to increase PPARγ levels, have been linked with decreased skeletal integrity. A multivariate analysis from NHANES III reported that saturated fat intake was negatively associated with bone mineral density in a large cohort of women (16). Saturated fat intake has also been linked to hip fracture risk in postmenopausal women (17). Pre-clinical mouse models further support the notion that a diet enriched in saturated fats induce bone loss and promote marrow adiposity (44, 61–63).
The data establishing PPARγ as a critical player in bone homeostasis must be coupled to RXR, the obligate, heterodimeric partner required for PPARγ transcriptional effects. Although expanding links between retinoid metabolism and PPARγ transcriptional activity exist (3, 24, 28, 31), very little is known about how the metabolism of retinoids, the ligands of RXR, influence PPARγ-RXR action in the skeleton. PPARγ-RXR responses in bone remains poorly understood and underexplored. To the best of our knowledge, no prior studies have specifically examined how retinoid metabolism influences PPARγ-RXR transactivation within the marrow niche and its broader implications for bone homeostasis. Considerable evidence is provided here that Rald and Aldh1a1 serve as novel proximal regulators of PPARγ-RXR in bone that modulate marrow stem cell lineage determination events through effects on PPARγ transactivation. Genetic deletion of Aldh1a1 results in a unique in vivo phenotype characterized by protection against PPARγ-mediated bone loss and accumulation of marrow adiposity. Notably, our data on the effects of Rald and Aldh1a1 deficiency closely track prior reports regarding the broad actions of PPARγ in bone. Rald and Aldh1a1 deficiency protect against TZD and HFD induced skeletal changes by coordinately promoting osteoblastogenesis, decreasing osteoclastogenesis, and limiting bone marrow adipogenesis. Moreover, Rald and Aldh1a1 deficiency modulate key regulators of bone, including alkaline phosphatase, PGC-1β, and c-Fos.
Global Aldh1a1 deficiency has been shown to increase Rald levels, which is consistent with a model in which increased Rald concentrations in bone in Aldh1a1−/− mice results in impaired exogenous and endogenous PPARγ transactivation in the marrow microenvironment. Our previous studies indicate that Rald directly binds the PPARγ-RXR complex and inhibits the activation of a transfected PPAR response element (PPRE) after agonist stimulation with rosiglitazone (28). Further studies are required to elucidate the exact molecular mechanisms by which Rald inhibits PPARγ-RXR activation. RXR-selective ligands can independently modulate the PPARγ-RXR transcriptional complex, allowing for the possibility of RXR dependent effects (22, 23). Rald action on bone may depend on its interaction with RXR, changes in RA levels, or alterations in retinoid receptor activity. Rald may also modulate PPARγ-RXR through posttranslation modifications (PTMs) of PPARγ. Interestingly, phosphorylation of serine 112 on PPARγ by MAP kinase represses PPARγ transcriptional activity (64). Given that retinoids influence MAP and p38 kinase pathways (65, 66), Rald may also induce repressive PTMs of PPARγ, thereby impairing its transcriptional activity. Finally, our data on the inhibitory effect of Rald and Aldh1a1 deficiency on RANKL-mediated induction of PGC-1β specifically raise the possibility that Rald may regulate PPARγ transcriptional responses through transcriptional coregulators. Such possibilities are intriguing prospects for future work based on our findings here.
The skeletal effects of limiting PPARγ-RXR-mediated responses through Rald and Aldh1a1 inhibition may point to new therapeutic options for minimizing the untoward effects of PPARγ-RXR transactivation in bone across a broad spectrum of clinical conditions. Although the use of TZDs in the treatment of diabetes will likely remain limited, the potent insulin sensitizing effects seen with PPARγ activation continues to draw attention for other therapeutic strategies (67, 68). Aldh1a1 inhibition may offer a way to offset possible bone issues linked to PPARγ activation. Rald and Aldh1a1 inhibition also limit the negative skeletal effects of a HFD and obesity. Interestingly, obesity may magnify the bone effects of aging although this data is controversial (69). It remains unknown how aging or obesity alter levels of Rald or other specific retinoids, retinoid binding proteins, or the activity or levels of enzymes involved with retinoid formation. The action of Rald and Aldh1a1 deficiency on bone as reported here provides a strong rationale for such studies. In osteoporosis and aging, bone loss correlates with decreased bone formation, increased osteoclast activity, and increased marrow adiposity (70–72). Several studies have linked these changes to increased skeletal levels of PPARγ2 and putative fatty acids ligands (53, 73–75). The effects of Rald and Aldh1a1 deficiency on MSC allocation and osteoclastogenesis may mitigate PPARγ-mediated changes in bone formation and resorption seen during aging. Consistent with this notion, Aldh1a1−/− mice acquire higher peak bone mineral density and maintain a higher bone mass up to 9 months of age (24) compared to matched controls. Taken together, the negative impact of commonly encountered clinical issues like obesity and anti-diabetic therapies on bone homeostasis, along with the protection against these issues seen with Rald and Aldh1a1 deficiency, argue for the need to explore further how retinoid metabolism modulates PPARγ and bone remodeling.
Supplementary Material
Compared to historical controls (18 week old female WT and Aldh1a1−/− mice on a regular chow diet), Aldh1a1−/− mice treated with rosiglitazone (20 mg/kg/d × 12 weeks) lost a smaller percentage of their femoral BV/TV and Tb.N on micro CT compared to WT controls.
HIGHLIGHTS.
Delineating novel pathways that modulate PPARγ action in bone holds important implications for disorders of low bone mineral density
PPARγ requires its heterodimeric partner RXR, the receptor for retinoids (vitamin A derivatives), for transcriptional activity
Mice lacking retinaldehyde dehydrogenase 1, which converts retinaldehyde into retinoic acid, are protected against PPARγ-mediated bone loss and marrow adiposity
Retinaldehyde (Rald), which accumulates in vivo in Aldh1a1−/− mice, protects against rosiglitazone-mediated inhibition of osteoblastogenesis in vitro
Rald potently inhibits in vitro adipogenesis and osteoclastogenesis in WT mesenchymal stem cells (MSCs) and hematopoietic stem cells (HSCs) respectively
Acknowledgments
We thank Sheila Bornstein for technical assistance with marrow cultures, and Marilena Preda and Ilka Pinz for conducting MRI studies.
S.N. received supported from the NIH Training Grant 5T32HL007208-34.
Abbreviations
- Aldh1a1
retinaldehyde dehydrogenase 1
- AA
Ascorbic acid
- ALP
alkaline phosphatase
- AR
alizarin red
- BGP
beta glycerol phosphate
- BMP2
bone morphogenetic protein 2
- CSF
colony stimulating factor
- HFD
high fat diet
- HSC
hematopoietic stem cell
- MSC
mesenchymal stem cell
- NR
nuclear receptor
- OC
osteoclast
- PPARγ
peroxisome proliferator-activated receptor-gamma
- PGC-1β
peroxisome proliferator-activated receptor gamma coactivator-1 beta
- RA
all trans retinoic acid
- Rald
retinaldehyde
- 9cRA
9-cis retinoic acid
- RANKL
receptor activator of nuclear factor-κB ligand
- RAR
retinoic acid receptor
- RXR
retinoid X receptor
- Rosi
rosiglitazone
- TZD
thiazolidinediones
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Napoli JL. Biochemical pathways of retinoid transport, metabolism, and signal transduction. Clin Immunol Immunopathol. 1996;80:S52–62. doi: 10.1006/clin.1996.0142. [DOI] [PubMed] [Google Scholar]
- 2.Napoli JL. Interactions of retinoid binding proteins and enzymes in retinoid metabolism. Biochim Biophys Acta. 1999;1440:139–162. doi: 10.1016/s1388-1981(99)00117-1. [DOI] [PubMed] [Google Scholar]
- 3.Ziouzenkova O, Plutzky J. Retinoid metabolism and nuclear receptor responses: New insights into coordinated regulation of the PPAR-RXR complex. FEBS Lett. 2008;582:32–38. doi: 10.1016/j.febslet.2007.11.081. [DOI] [PubMed] [Google Scholar]
- 4.Rosen ED, Spiegelman BM. PPARgamma : a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem. 2001;276:37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- 5.Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature. 1992;358:771–774. doi: 10.1038/358771a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 7.Kawai M, Rosen CJ. PPARgamma: a circadian transcription factor in adipogenesis and osteogenesis. Nat Rev Endocrinol. 2010;6:629–636. doi: 10.1038/nrendo.2010.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wan Y. PPARgamma in bone homeostasis. Trends Endocrinol Metab. 2010;21:722–728. doi: 10.1016/j.tem.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 9.Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung UI, Kubota N, Terauchi Y, Harada Y, Azuma Y, Nakamura K, Kadowaki T, Kawaguchi H. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113:846–855. doi: 10.1172/JCI19900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, Lecka-Czernik B. PPARgamma2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. J Cell Biochem. 2009;106:232–246. doi: 10.1002/jcb.21994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wan Y, Chong LW, Evans RM. PPAR-gamma regulates osteoclastogenesis in mice. Nat Med. 2007;13:1496–1503. doi: 10.1038/nm1672. [DOI] [PubMed] [Google Scholar]
- 12.Wei W, Wang X, Yang M, Smith LC, Dechow PC, Sonoda J, Evans RM, Wan Y. PGC1beta mediates PPARgamma activation of osteoclastogenesis and rosiglitazone-induced bone loss. Cell Metab. 2010;11:503–516. doi: 10.1016/j.cmet.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bilik D, McEwen LN, Brown MB, Pomeroy NE, Kim C, Asao K, Crosson JC, Duru OK, Ferrara A, Hsiao VC, Karter AJ, Lee PG, Marrero DG, Selby JV, Subramanian U, Herman WH. Thiazolidinediones and fractures: evidence from translating research into action for diabetes. J Clin Endocrinol Metab. 2010;95:4560–4565. doi: 10.1210/jc.2009-2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loke YK, Singh S, Furberg CD. Long-term use of thiazolidinediones and fractures in type 2 diabetes: a meta-analysis. CMAJ. 2009;180:32–39. doi: 10.1503/cmaj.080486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwartz AV, Sellmeyer DE, Vittinghoff E, Palermo L, Lecka-Czernik B, Feingold KR, Strotmeyer ES, Resnick HE, Carbone L, Beamer BA, Park SW, Lane NE, Harris TB, Cummings SR. Thiazolidinedione use and bone loss in older diabetic adults. J Clin Endocrinol Metab. 2006;91:3349–3354. doi: 10.1210/jc.2005-2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corwin RL, Hartman TJ, Maczuga SA, Graubard BI. Dietary saturated fat intake is inversely associated with bone density in humans: analysis of NHANES III. J Nutr. 2006;136:159–165. doi: 10.1093/jn/136.1.159. [DOI] [PubMed] [Google Scholar]
- 17.Orchard TS, Cauley JA, Frank GC, Neuhouser ML, Robinson JG, Snetselaar L, Tylavsky F, Wactawski-Wende J, Young AM, Lu B, Jackson RD. Fatty acid consumption and risk of fracture in the Women’s Health Initiative. Am J Clin Nutr. 2010;92:1452–1460. doi: 10.3945/ajcn.2010.29955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loke YK, Singh S, Furberg CD. Long-term use of thiazolidinediones and fractures in type 2 diabetes: a meta-analysis. CMAJ. 2009;180:32–39. doi: 10.1503/cmaj.080486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwartz AV, Sellmeyer DE, Vittinghoff E, Palermo L, Lecka-Czernik B, Feingold KR, Strotmeyer ES, Resnick HE, Carbone L, Beamer BA, Park SW, Lane NE, Harris TB, Cummings SR. Thiazolidinedione use and bone loss in older diabetic adults. J Clin Endocrinol Metab. 2006;91:3349–3354. doi: 10.1210/jc.2005-2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kane MA, Folias AE, Pingitore A, Perri M, Obrochta KM, Krois CR, Cione E, Ryu JY, Napoli JL. Identification of 9-cis-retinoic acid as a pancreas-specific autacoid that attenuates glucose-stimulated insulin secretion. Proc Natl Acad Sci U S A. 2010;107:21884–21889. doi: 10.1073/pnas.1008859107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mukherjee R, Davies PJ, Crombie DL, Bischoff ED, Cesario RM, Jow L, Hamann LG, Boehm MF, Mondon CE, Nadzan AM, Paterniti JR, Jr, Heyman RA. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature. 1997;386:407–410. doi: 10.1038/386407a0. [DOI] [PubMed] [Google Scholar]
- 22.Lenhard JM, Lancaster ME, Paulik MA, Weiel JE, Binz JG, Sundseth SS, Gaskill BA, Lightfoot RM, Brown HR. The RXR agonist LG100268 causes hepatomegaly, improves glycaemic control and decreases cardiovascular risk and cachexia in diabetic mice suffering from pancreatic beta-cell dysfunction. Diabetologia. 1999;42:545–554. doi: 10.1007/s001250051193. [DOI] [PubMed] [Google Scholar]
- 23.Claudel T, Leibowitz MD, Fievet C, Tailleux A, Wagner B, Repa JJ, Torpier G, Lobaccaro JM, Paterniti JR, Mangelsdorf DJ, Heyman RA, Auwerx J. Reduction of atherosclerosis in apolipoprotein E knockout mice by activation of the retinoid X receptor. Proc Natl Acad Sci U S A. 2001;98:2610–2615. doi: 10.1073/pnas.041609298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nallamshetty S, Wang H, Rhee EJ, Kiefer FW, Brown JD, Lotinun S, Le P, Baron R, Rosen CJ, Plutzky J. Deficiency of retinaldehyde dehydrogenase 1 induces BMP2 and increases bone mass in vivo. PLoS One. 2013;8:e71307. doi: 10.1371/journal.pone.0071307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dupe V, Matt N, Garnier JM, Chambon P, Mark M, Ghyselinck NB. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc Natl Acad Sci U S A. 2003;100:14036–14041. doi: 10.1073/pnas.2336223100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mic FA, Haselbeck RJ, Cuenca AE, Duester G. Novel retinoic acid generating activities in the neural tube and heart identified by conditional rescue of Raldh2 null mutant mice. Development. 2002;129:2271–2282. doi: 10.1242/dev.129.9.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molotkov A, Duester G. Genetic evidence that retinaldehyde dehydrogenase Raldh1 (Aldh1a1) functions downstream of alcohol dehydrogenase Adh1 in metabolism of retinol to retinoic acid. J Biol Chem. 2003;278:36085–36090. doi: 10.1074/jbc.M303709200. [DOI] [PubMed] [Google Scholar]
- 28.Ziouzenkova O, Orasanu G, Sharlach M, Akiyama TE, Berger JP, Viereck J, Hamilton JA, Tang G, Dolnikowski GG, Vogel S, Duester G, Plutzky J. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat Med. 2007;13:695–702. doi: 10.1038/nm1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amann PM, Schadendorf D, Owen RW, Korn B, Eichmuller SB, Bazhin AV. Retinal and retinol are potential regulators of gene expression in the keratinocyte cell line HaCaT. Exp Dermatol. 2011;20:373–375. doi: 10.1111/j.1600-0625.2010.01127.x. [DOI] [PubMed] [Google Scholar]
- 30.Kiefer FW, Orasanu G, Nallamshetty S, Brown JD, Wang H, Luger P, Qi NR, Burant CF, Duester G, Plutzky J. Retinaldehyde dehydrogenase 1 coordinates hepatic gluconeogenesis and lipid metabolism. Endocrinology. 2012;153:3089–3099. doi: 10.1210/en.2011-2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiefer FW, Vernochet C, O’Brien P, Spoerl S, Brown JD, Nallamshetty S, Zeyda M, Stulnig TM, Cohen DE, Kahn CR, Plutzky J. Retinaldehyde dehydrogenase 1 regulates a thermogenic program in white adipose tissue. Nat Med. 2012;18:918–925. doi: 10.1038/nm.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Zhang Y, Li R, Chen W, Howell M, Zhang R, Chen G. The hepatic Raldh1 expression is elevated in Zucker fatty rats and its over-expression introduced the retinal-induced Srebp-1c expression in INS-1 cells. PLoS One. 2012;7:e45210. doi: 10.1371/journal.pone.0045210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeon EJ, Yoon BY, Lim JY, Oh HJ, Park HS, Park MJ, Lim MA, Park MK, Kim KW, Cho ML, Cho SG. Adoptive transfer of all-trans-retinal-induced regulatory T cells ameliorates experimental autoimmune arthritis in an interferon-gamma knockout model. Autoimmunity. 2012;45:460–469. doi: 10.3109/08916934.2012.682666. [DOI] [PubMed] [Google Scholar]
- 34.Ali AA, Weinstein RS, Stewart SA, Parfitt AM, Manolagas SC, Jilka RL. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology. 2005;146:1226–1235. doi: 10.1210/en.2004-0735. [DOI] [PubMed] [Google Scholar]
- 35.Chen JR, Lazarenko OP, Wu X, Tong Y, Blackburn ML, Shankar K, Badger TM, Ronis MJ. Obesity reduces bone density associated with activation of PPARgamma and suppression of Wnt/beta-catenin in rapidly growing male rats. PLoS One. 2010;5:e13704. doi: 10.1371/journal.pone.0013704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeMambro VE, Clemmons DR, Horton LG, Bouxsein ML, Wood TL, Beamer WG, Canalis E, Rosen CJ. Gender-specific changes in bone turnover and skeletal architecture in igfbp-2-null mice. Endocrinology. 2008;149:2051–2061. doi: 10.1210/en.2007-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hesslein DG, Fretz JA, Xi Y, Nelson T, Zhou S, Lorenzo JA, Schatz DG, Horowitz MC. Ebf1-dependent control of the osteoblast and adipocyte lineages. Bone. 2009;44:537–546. doi: 10.1016/j.bone.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tong J, Li W, Vidal C, Yeo LS, Fatkin D, Duque G. Lamin A/C deficiency is associated with fat infiltration of muscle and bone. Mech Ageing Dev. 2011;132:552–559. doi: 10.1016/j.mad.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 39.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28:2–17. doi: 10.1002/jbmr.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urs S, Venkatesh D, Tang Y, Henderson T, Yang X, Friesel RE, Rosen CJ, Liaw L. Sprouty1 is a critical regulatory switch of mesenchymal stem cell lineage allocation. FASEB J. 2010;24:3264–3273. doi: 10.1096/fj.10-155127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramirez-Zacarias JL, Castro-Munozledo F, Kuri-Harcuch W. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil red O. Histochemistry. 1992;97:493–497. doi: 10.1007/BF00316069. [DOI] [PubMed] [Google Scholar]
- 42.Xiao Y, Cui J, Li YX, Shi YH, Le GW. Expression of genes associated with bone resorption is increased and bone formation is decreased in mice fed a high-fat diet. Lipids. 2010;45:345–355. doi: 10.1007/s11745-010-3397-0. [DOI] [PubMed] [Google Scholar]
- 43.Xiao Y, Cui J, Li YX, Shi YH, Wang B, Le GW, Wang ZP. Dyslipidemic high-fat diet affects adversely bone metabolism in mice associated with impaired antioxidant capacity. Nutrition. 2011;27:214–220. doi: 10.1016/j.nut.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 44.Lu XM, Zhao H, Wang EH. A high-fat diet induces obesity and impairs bone acquisition in young male mice. Mol Med Rep. 2013;7:1203–1208. doi: 10.3892/mmr.2013.1297. [DOI] [PubMed] [Google Scholar]
- 45.Gautam J, Choudhary D, Khedgikar V, Kushwaha P, Singh RS, Singh D, Tiwari S, Trivedi R. Micro-architectural changes in cancellous bone differ in female and male C57BL/6 mice with high-fat diet-induced low bone mineral density. Br J Nutr. 2014:1–11. doi: 10.1017/S0007114514000051. [DOI] [PubMed] [Google Scholar]
- 46.Jeon MJ, Kim JA, Kwon SH, Kim SW, Park KS, Park SW, Kim SY, Shin CS. Activation of peroxisome proliferator-activated receptor-gamma inhibits the Runx2-mediated transcription of osteocalcin in osteoblasts. J Biol Chem. 2003;278:23270–23277. doi: 10.1074/jbc.M211610200. [DOI] [PubMed] [Google Scholar]
- 47.Benvenuti S, Cellai I, Luciani P, Deledda C, Baglioni S, Giuliani C, Saccardi R, Mazzanti B, Dal Pozzo S, Mannucci E, Peri A, Serio M. Rosiglitazone stimulates adipogenesis and decreases osteoblastogenesis in human mesenchymal stem cells. J Endocrinol Invest. 2007;30:RC26–30. doi: 10.1007/BF03350807. [DOI] [PubMed] [Google Scholar]
- 48.Kang S, Bennett CN, Gerin I, Rapp LA, Hankenson KD, Macdougald OA. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma. J Biol Chem. 2007;282:14515–14524. doi: 10.1074/jbc.M700030200. [DOI] [PubMed] [Google Scholar]
- 49.Wan Y, Chong LW, Evans RM. PPAR-gamma regulates osteoclastogenesis in mice. Nat Med. 2007;13:1496–1503. doi: 10.1038/nm1672. [DOI] [PubMed] [Google Scholar]
- 50.Chan BY, Gartland A, Wilson PJ, Buckley KA, Dillon JP, Fraser WD, Gallagher JA. PPAR agonists modulate human osteoclast formation and activity in vitro. Bone. 2007;40:149–159. doi: 10.1016/j.bone.2006.07.029. [DOI] [PubMed] [Google Scholar]
- 51.Ishii KA, Fumoto T, Iwai K, Takeshita S, Ito M, Shimohata N, Aburatani H, Taketani S, Lelliott CJ, Vidal-Puig A, Ikeda K. Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat Med. 2009;15:259–266. doi: 10.1038/nm.1910. [DOI] [PubMed] [Google Scholar]
- 52.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7:885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 53.Moerman EJ, Teng K, Lipschitz DA, Lecka-Czernik B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell. 2004;3:379–389. doi: 10.1111/j.1474-9728.2004.00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bruedigam C, Eijken M, Koedam M, van de Peppel J, Drabek K, Chiba H, van Leeuwen JP. A new concept underlying stem cell lineage skewing that explains the detrimental effects of thiazolidinediones on bone. Stem Cells. 2010;28:916–927. doi: 10.1002/stem.405. [DOI] [PubMed] [Google Scholar]
- 55.Lecka-Czernik B, Gubrij I, Moerman EJ, Kajkenova O, Lipschitz DA, Manolagas SC, Jilka RL. Inhibition of Osf2/Cbfa1 expression and terminal osteoblast differentiation by PPARgamma2. J Cell Biochem. 1999;74:357–371. [PubMed] [Google Scholar]
- 56.Jeon MJ, Kim JA, Kwon SH, Kim SW, Park KS, Park SW, Kim SY, Shin CS. Activation of peroxisome proliferator-activated receptor-gamma inhibits the Runx2-mediated transcription of osteocalcin in osteoblasts. J Biol Chem. 2003;278:23270–23277. doi: 10.1074/jbc.M211610200. [DOI] [PubMed] [Google Scholar]
- 57.Berry DC, DeSantis D, Soltanian H, Croniger CM, Noy N. Retinoic acid upregulates preadipocyte genes to block adipogenesis and suppress diet-induced obesity. Diabetes. 2012;61:1112–1121. doi: 10.2337/db11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.D’Ambrosio DN, Clugston RD, Blaner WS. Vitamin A metabolism: an update. Nutrients. 2011;3:63–103. doi: 10.3390/nu3010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwarz EJ, Reginato MJ, Shao D, Krakow SL, Lazar MA. Retinoic acid blocks adipogenesis by inhibiting C/EBPbeta-mediated transcription. Mol Cell Biol. 1997;17:1552–1561. doi: 10.1128/mcb.17.3.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Waltner-Law M, Duong DT, Daniels MC, Herzog B, Wang XL, Prasad R, Granner DK. Elements of the glucocorticoid and retinoic acid response units are involved in cAMP-mediated expression of the PEPCK gene. J Biol Chem. 2003;278:10427–10435. doi: 10.1074/jbc.M211846200. [DOI] [PubMed] [Google Scholar]
- 61.Cao JJ, Gregoire BR, Gao H. High-fat diet decreases cancellous bone mass but has no effect on cortical bone mass in the tibia in mice. Bone. 2009;44:1097–1104. doi: 10.1016/j.bone.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 62.Chen JR, Lazarenko OP, Wu X, Tong Y, Blackburn ML, Shankar K, Badger TM, Ronis MJ. Obesity reduces bone density associated with activation of PPARgamma and suppression of Wnt/beta-catenin in rapidly growing male rats. PLoS One. 2010;5:e13704. doi: 10.1371/journal.pone.0013704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gautam J, Choudhary D, Khedgikar V, Kushwaha P, Singh RS, Singh D, Tiwari S, Trivedi R. Micro-architectural changes in cancellous bone differ in female and male C57BL/6 mice with high-fat diet-induced low bone mineral density. Br J Nutr. 2014:1–11. doi: 10.1017/S0007114514000051. [DOI] [PubMed] [Google Scholar]
- 64.van Beekum O, Fleskens V, Kalkhoven E. Posttranslational modifications of PPAR-gamma: fine-tuning the metabolic master regulator. Obesity (Silver Spring) 2009;17:213–219. doi: 10.1038/oby.2008.473. [DOI] [PubMed] [Google Scholar]
- 65.Yen A, Roberson MS, Varvayanis S, Lee AT. Retinoic acid induced mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) kinase-dependent MAP kinase activation needed to elicit HL-60 cell differentiation and growth arrest. Cancer Res. 1998;58:3163–3172. [PubMed] [Google Scholar]
- 66.De Genaro P, Simon MV, Rotstein NP, Politi LE. Retinoic acid promotes apoptosis and differentiation in photoreceptors by activating the P38 MAP kinase pathway. Invest Ophthalmol Vis Sci. 2013;54:3143–3156. doi: 10.1167/iovs.12-11049. [DOI] [PubMed] [Google Scholar]
- 67.Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, Griffin PR, Spiegelman BM. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS, Shin Y, He Y, Bruning JB, Marciano DP, Cameron MD, Laznik D, Jurczak MJ, Schurer SC, Vidovic D, Shulman GI, Spiegelman BM, Griffin PR. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477:477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cao JJ. Effects of obesity on bone metabolism. J Orthop Surg Res. 2011;6 doi: 10.1186/1749-799X-6-30. 30-799X-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chan GK, Duque G. Age-related bone loss: old bone, new facts. Gerontology. 2002;48:62–71. doi: 10.1159/000048929. [DOI] [PubMed] [Google Scholar]
- 71.Jilka RL. Biology of the basic multicellular unit and the pathophysiology of osteoporosis. Med Pediatr Oncol. 2003;41:182–185. doi: 10.1002/mpo.10334. [DOI] [PubMed] [Google Scholar]
- 72.Rosen CJ, Bouxsein ML. Mechanisms of disease: is osteoporosis the obesity of bone? Nat Clin Pract Rheumatol. 2006;2:35–43. doi: 10.1038/ncprheum0070. [DOI] [PubMed] [Google Scholar]
- 73.Maurin AC, Chavassieux PM, Vericel E, Meunier PJ. Role of polyunsaturated fatty acids in the inhibitory effect of human adipocytes on osteoblastic proliferation. Bone. 2002;31:260–266. doi: 10.1016/s8756-3282(02)00805-0. [DOI] [PubMed] [Google Scholar]
- 74.Maurin AC, Chavassieux PM, Meunier PJ. Expression of PPARgamma and beta/delta in human primary osteoblastic cells: influence of polyunsaturated fatty acids. Calcif Tissue Int. 2005;76:385–392. doi: 10.1007/s00223-004-0108-y. [DOI] [PubMed] [Google Scholar]
- 75.Spiteller G. The important role of lipid peroxidation processes in aging and age dependent diseases. Mol Biotechnol. 2007;37:5–12. doi: 10.1007/s12033-007-0057-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Compared to historical controls (18 week old female WT and Aldh1a1−/− mice on a regular chow diet), Aldh1a1−/− mice treated with rosiglitazone (20 mg/kg/d × 12 weeks) lost a smaller percentage of their femoral BV/TV and Tb.N on micro CT compared to WT controls.