

Abstract
The pathology of type 2 diabetes is complex, with multiple stages culminating in a functional β‐cell mass that is insufficient to meet the body's needs. Although the broad outlines of the disease etiology are known, many critical questions remain to be answered before next‐generation therapeutics can be developed. In order to further elucidate the pathobiology of this disease, animal models mimicking the pathology of human type 2 diabetes are of great value. One example of a type 2 diabetes animal model is the high‐fat diet‐fed, streptozotocin (HFD/STZ)‐treated rat model. The present review first summarizes the current understanding of the metabolic profile and pathology involved in the different stages of the type 2 diabetes disease progression in humans. Second, the known characteristics of the HFD/STZ rat model are reviewed and compared with the pathophysiology of human type 2 diabetes. Next, the suitability of the HFD/STZ model as a model of type 2 diabetes with a focus on identifying critical caveats and unanswered questions about the model is discussed. The improved understanding of refined animal models will hopefully lead to more relevant preclinical studies and development of improved therapeutics for diabetes. Depending on the amount of residual functional β‐cells mass, the HFD/STZ rat model might be a suitable animal model of the final stage of type 2 diabetes.
Keywords: High‐fat diet, Streptozotocin, Type 2 diabetes
Introduction
Type 2 diabetes is increasing in prevalence worldwide1, and it is strongly associated with obesity and insulin resistance3, as well as defects in pancreatic β‐cell function and mass5. These metabolic disorders impede the critical regulatory influence of insulin on glucose, lipid and protein metabolism, thus precipitating a disease characterized by impairments in these physiological processes. However, it takes years to develop frank diabetes. Patients developing type 2 diabetes have often gone through a state of obesity associated with reduced insulin sensitivity along with an activated β‐cell compensatory mechanism, such as excess basal insulin secretion and hyperproinsulinemia, as a part of their metabolic profile7. These pathological conditions occur early in the disease progression of type 2 diabetes8, and before the β‐cells severely fail in late stage (insulin‐dependent) type 2 diabetes8.
To combat type 2 diabetes, there is an urgent need for more effective treatments and therapeutic regimens. Thoroughly characterized and clinically relevant type 2 diabetes animal models are required to achieve this aim of testing new and better therapeutics. Both genetic spontaneous diabetes models and experimentally‐induced non‐spontaneous diabetes models exist. An example of an experimentally‐induced animal model of diabetes is the high‐fat diet/streptozotocin treated (HFD/STZ) rat model. This model involves a combination of a diet high in fat, and in some cases sugar, to bring about hyperinsulinemia, insulin resistance and/or glucose intolerance followed by treatment with the β‐cell toxin STZ, which results in a severe reduction in functional β‐cell mass10. Together, these two stressors are designed to mimic the pathology of type 2 diabetes, though on a shorter timescale than found in the human condition.
The aim of the present review is to clarify and discuss critical caveats and unanswered questions regarding the HFD/STZ rat model, which have not been discussed in the literature. First, the impact of and differences between the diet regimens in relation to obesity and type 2 diabetes will be discussed. Second, the effect of the various STZ treatments, as well as the importance of age, with respect to type 1 and type 2 diabetes, will be focused on. Finally, whether the HFD/STZ rat model mimics the early or late stages of type 2 diabetes are discussed. This disease stage classification is based on comparison of circulating metabolic measures provided in studies using the HF/STZ rat model. Classification of type 2 diabetes is an important consideration when choosing the best therapeutic intervention in patients.
In order to discuss these topics at the end of the present review, the human metabolic profile of the different stages in the disease progression of type 2 diabetes will be summarized first. Next, the history of the development of the HFD/STZ rat model will be recounted. It is beyond the scope of this review to present an overview of all existing diabetes rodent models, as many of the other models have been reviewed recently12. To summarize, the aim of the present review is to provide a guide of the factors to take into account when modeling and working with the HFD/STZ rat model.
Metabolic Profile of Healthy and Prediabetes Humans
Before discussing the HFD/STZ rat model, it is important to review the stages and transitions in the progression of type 2 diabetes, which the HFD and STZ treatments are meant to emulate. The first transition is the shift from a healthy state to a prediabetes state. In prediabetes, patients have either impaired fasting glucose, impaired glucose tolerance, or both, and is often associated with insulin resistance. In healthy individuals, the adipose tissue functions as a safe storage site for lipids during a positive caloric balance16. Likewise, excess circulating glucose is accommodated by the liver and muscle tissue in the form of glycogen. In the context of fully occupied glycogen stores, high glucose levels might also bring about de novo lipogenesis, occurring mainly in the liver and, to a lesser extent, in the adipose tissue17. De novo lipogenesis helps maintain normal blood glucose levels by sequestering away excess glucose from the circulation. Normoglycemia in healthy individuals is maintained by the unique interplay between the almost opposing hormones, insulin and glucagon. The dialogue between these two hormones becomes perturbed with the disease progression of type 2 diabetes20. The transition from a metabolically healthy state to prediabetes often includes an obese state characterized by hyperinsulinemia, insulin resistance, and dyslipidemia8. However, it should be stressed that both metabolically healthy obese individuals, as well as metabolically unhealthy lean individuals, can be found in the general population25. This implies that obesity might not automatically or immediately result in the development of type 2 diabetes, and highlights that type 2 diabetes is a highly polygenic and heterogenous disease25. The nutritional overload, which in the long term leads to obesity, can quickly induce insulin resistance in skeletal muscle as well as in the liver (Figure 1)28. Insulin resistance in skeletal muscle might reduce the occurrence of lipotoxic effects in muscle by redirecting the excess energy to the adipose tissue stores23, and can thus be seen as a normal physiological function in healthy individuals.
Figure 1.
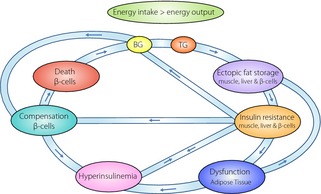
Simplified overview of the interactions between multiple tissues in type 2 diabetes. When energy input exceeds output, both blood glucose (BG) and blood triglycerides (TG) will increase, which eventually lead to ectopic fat accumulation in muscle and the liver. The consequence is insulin resistance, thus directing lipids to the adipose tissue. When the adipocytes become dysfunctional, extra ectopic fat accumulation including fat accumulation in the β‐cells occurs. Whether insulin resistance brings about hyperinsulinemia or vice versa is a highly debated topic. An increase in BG and insulin resistance both lead to induction of β‐cell compensatory mechanisms including β‐cell hypertrophy and increased insulin secretion, further contributing to hyperinsulinemia. This is a vicious cycle of first physiological events, then pathological events, and finally β‐cell death leading to a severe BG increase and full‐blown diabetes.
Severe expansion of the adipose tissue is tightly associated with adipose inflammation and a distorted adipokine profile, marked by high leptin and low adiponectin levels30 representing dysfunctional adipocytes. Dysfunctional adipose tissue leads to ectopic fat accumulation in non‐adipose tissue, such as muscle, liver, and β‐cells (Figure 1)31. Intramyocellular lipid accumulation is associated with insulin resistance33. Insulin‐resistant muscles have lower glycogen synthesis and redirect glucose to the liver, where it contributes to hepatic lipid accumulation through de novo lipogenesis (Figure 1)35. Hepatic fat accumulation can induce hepatic insulin resistance (Figure 1)36, with decreased glycogen synthesis and increased gluconeogenesis36. This impaired insulin‐induced suppression of hepatic glucose output may contribute to hyperglycemia (Figure 1). Further inflammation of the abdominal adipose tissue may worsen the dysfunctional state of the adipocytes30, leading to more ectopic fat accumulation, insulin resistance and hyperinsulinemia, in a negative feedback loop (Figure 1). However, beneficial aspects of inflammation, such as proliferation of certain classes of macrophages in the adipose tissue, has been illustrated39.
In the early state of type 2 diabetes progression, β‐cell compensatory mechanisms have typically adapted to preserve normoglycemia8. The compensatory mechanisms might include increased β‐cell mass, augmented β‐cell function, or a combination of both (Figure 1)6. β‐Cell function in this metabolic state seems to be improved through elevated insulin biosynthesis, altered glucose and lipid metabolism, as well as through enhanced incretin sensitivity and parasympathetic nervous system activity ensuing normoglycemia and hyperinsulinemia9. Despite the tight association between obesity and hyperinsulinemia and/or insulin resistance, the exact causal relationship between these phenomena is still being elucidated. Some investigators have proposed that elevated circulating insulin levels found very early in the disease progression might play a causal role in obesity and/or insulin resistance in at least some individuals40. To summarize, a simplified version of the transition from a metabolically healthy state to an obese and prediabetic state involves a vicious cycle comprising hyperinsulinemia, insulin resistance, dyslipidemia, inflamed and dysfunctional adipose tissue, ectopic fat deposition in liver and muscle, and failure of β‐cells (see Figure 1).
Metabolic Profile of Human Type 2 Diabetes
Genetic susceptibility increases the odds that an individual will progress from prediabetes to frank diabetes26, which is frequently defined as fasting blood glucose above 7 mmol/L (126 mg/dL)43. Interestingly, the majority of the genes discovered in genome‐wide association studies are thought to play a primary role in the pancreatic β‐cells, while having only minor roles in so‐called ‘classical’ insulin target tissues26. In clinical studies, β‐cell failure typically manifests as a loss of first phase insulin secretion, loss of pulsatile insulin oscillations45 and an increased circulating pro‐insulin‐to‐insulin ratio46. As in the case of prediabetes, type 2 diabetes is also intimately related to dyslipidemia33 and hepatic steatosis47, as well as with hypoadiponectinemia48 and increased levels of the liver damage marker alanine aminotransferase24. However, essentially, the transition from the prediabetes state to frank type 2 diabetes requires the loss of a significant portion of the functional β‐cell mass6.
Functional β‐cell mass is the product of physical β‐cell mass and β‐cell function, which includes an appropriate level of glucose‐stimulated pulsatile insulin release and the appropriate suppression of basal insulin secretion. Failure of these β‐cell functions can to some extent been explained by the twin cycle hypothesis50. The idea behind this hypothesis is that hepatic lipid accumulation leads to β‐cell lipid uptake51, worsening insulin resistance, and promoting β‐cell failure and death9. The factors and mechanisms involved in programmed β‐cell death have recently been reviewed52. Importantly, the amount of physical β‐cell mass left in an individual with type 2 diabetes seems to be dependent of the duration of the disease. Consequently, the metabolic profile in type 2 diabetes patients will depend on the duration of their disease (an early vs late stage of type 2 diabetes). The choice of therapeutics will differ between patients being in either an early or late state of type 2 diabetes. The type of therapeutic intervention, lifestyle vs pharmacological therapeutics, such as insulin therapy, will further affect the metabolic profiles.
Finally, it is important to note that the cell types involved in the pathogenesis of type 2 diabetes are not solely limited to adipocytes, myocytes, hepatocytes and β‐cells. In the 2009 Banting lecture, Dr Ralph Defronzo stressed the pivotal role of all members of the ‘ominous octet’ in the development of glucose intolerance, which includes the brain, kidneys, alpha cells, and the gastrointestinal tract, besides the four cell types already mentioned.53.
History of the Development of the HFD/STZ Rat Model
In the beginning of the new millennium, Reed et al.54 reported a new rat model of type 2 diabetes. This model is today known as the HFD/STZ rat, as well as by other names (e.g. high energy/STZ rat). Recently, the model is most often referred to simply as a type 2 diabetes model. The aim of the study by Reed et al. was to develop a rat model simulating the natural diabetes pathology progression, from prediabetes and/or insulin resistance to a state of type 2 diabetes and hypoinsulinemia, in a condensed timeline. Reed et al.54 fed 7‐week‐old Sprague–Dawley rats a diet with 40% kcal fat for 2 weeks. The presence of insulin resistance was indicated through the observation of equal glucose clearance profiles in fat and lean rats, respectively, with an increase in glucose‐induced insulin responses in the fat‐fed rats54. Subsequently, overnight‐fasted animals were dosed (i.v.) once with STZ (50 mg/kg). A total of 3 days after STZ treatment, rats that had reached an elevated blood glucose plateau were included in the study and their response to metformin was tested54. The metformin‐induced lowering of blood glucose further established the HFD/STZ model to be a rat model of type 2 diabetes relevant to the human condition. Later, another HFD/STZ rat model was generated by using a low dose of STZ55. The HFD/STZ model was then further modified by Zhang et al.56, wherein the STZ‐treatment comprised multiple low doses of STZ instead of a single dose. This approach was inspired by the type 1 diabetes animal model involving multiple low doses of STZ. This approach has been reported to induce an inflammation‐mediated destruction of the β‐cells instead of the fast induction of the β‐cell death induced by a single dose of STZ56. After these three key publications, several versions of the HFD/STZ rat have appeared in the literature.
Discussion of the HFD/STZ Rat Model: Disease Modeling
Impact of the Diet Regimen in Relation to Obesity and Type 2 Diabetes
In the HFD/STZ rat models, the state of obesity, insulin resistance and/or glucose intolerance in prediabetes is simulated by a period of a high‐fat or ‘Western’ diet. Whether the rats actually reach a state of true overweight or obesity within this time period seems to depend on the duration of the fat feeding, which tends to be either relatively long (≥ 3 months)57 or relatively short (2–4 weeks; Table 1). Furthermore, the classification of overweight and obesity in humans is based on body mass index and can also be defined as abnormal or excessive fat accumulation that might impair health (www.who.int). However, it is often unclear how one can apply this definition to rodents. Unfortunately, no standardized definition of rodent obesity exists. Hence, the actual presence of obesity in the various HFD/STZ rats should always be taken into consideration when working with this model. Besides the duration of the HFD feeding timeframe, the composition of the diet seems to greatly affect the weight gain and fat distribution58. The diets used in the HFD/STZ model vary in both nutritional composition and source of the nutrients (Table 1). Some studies utilized a diet high in carbohydrates to produce a ‘high energy’ feeding regimen. However, the most commonly used approach is to feed rats with a diet high in fat, but with normal levels of sugar.
Table 1. Summary of high‐fat diet‐fed, streptozotocin rat studies.
| References | STZ* | Diet†,‡ | Initial age/BW | Strain§ | Metabolic measures¶ | T2D stage (**,††) |
|---|---|---|---|---|---|---|
| Hu et al.83 | 1 × 30–35 | 12W, 26K, 15.2P, 58FL, % | 10–12 weeks | SD | PG = 22 (H), PI = 191 (H), HOMA‐IRς = (H) | Early (T2D) |
| Abo‐Elmatty et al.78 | 1 × 35 | 2W, 17C, 25P, 58FL, % | – | A | FBG = 18 (H), SI = 86 (S), HbA1c = 10 (H), TG = 1.5 (H), LDL = 3.2 (H), HDL = 0.6 (L) | Late (T2D) |
| Gandhi et al.79 | 1 × 40 | 2W, 73ND, 25FCN, 2CO | 180 ± 10 g | W | FBG = 17 (H), PI = 198 (H), TG = 1.7 (H), TC = 2.5 (H) | Early (T2D) |
| Khan et al.84 | 1 × 35 | 2W, 20CS, 10FL, 2.5CO, 1O | 230 ± 20 g | SD | FG = 14 (H), FI = 111 (L), TG = 1.4 (H), C = 5.7 (H), HDL = 0.5 (L), LDL = 1.5 (H), HOMA‐IR = (H), HOMA‐B = (L) | Late (T2D) |
| Ren et al.85 | 1 × 30 | 6W, 67ND, 20CS, 10FL, 2CO, 1O | 8 weeks/180–220 g | SD | – | NA (T2D) |
| Hou et al.86 | 1 × 25 | 20C, 20P, 59F | 200–220 g | SD | TG = 18 (H) | NA (T2D) |
| Guo et al.60 | 1 × 30 | 4W, 67ND, 20CS, 10FL, 1O | 140–180 g | W | ISI = (L) | NA (T2D) |
| Mahmoud et al.87 | 1 × 35 | 2W, 41C, 18P, 40F | 190 ± 10 g | RN | G = 16 (H), HbA1c = 9 (H), I = 108 (L), HOMA‐IR = (H) | Late (T2D) |
| Si et al.69 | 1 × 50 i.v. | 2W, 41C, 18P, 40F | 7 weeks, 200 g | SD | BG = (H) | NA |
| Guo et al.88 | 1 × 30 | 4W, 67ND, 20CS, 10FL, 1O | 140–180 g | W | PG = 18 (H), PI = 206 (H), TG = 1.7 (H), TC = 3.5 (H), IRI = (H) | Early (T2D) |
| Hussein et al.89 | 1 × 35 i.v. | 2W, 3CS, ST, 74PSB, MP, 23FVO, 1O | 15–21 weeks | SD | FG = 13, FI = 107, TG = 2.1, TC = 5.2, HD = 1.0, LDL = 3.7, ¶¶ | NA (T2D) |
| Guo et al.90 | 1 × 30 | 4W, 67ND, 20CS, 10FL, 1O | 140–180 g | W | PG = 17 (H), PI = 299 (H), TG = 1.5 (H), TC = 2.8 (H), IRI = (L) | Early (T2D) |
| Sharma et al.80 | 1 × 40 | 1.5W, 73ND, 25FCN, 2CO | 170–200 g | W | BG = 17 (H), I = 123 (H), APQ = (H), TG = (H), HDL = (L), LDL (H), HOMA‐IR = (H), HOMA‐B = (L) | Early (T2D) |
| Albersen et al.59 | 2 × 20 | 2W, ND added 10FL, 2CO | 12 weeks | SD | BG>17, SI = (L), TG = (H), TC = (H), LDL = H), HDL = (H) | NA (T2D) |
| Lu et al.65 | 1 × 30 | 8W, 30C, 22P, 12F, 3O | 8 weeks, 250 ± 20 g | W | BG>11, HbA1C = 8, TG = 1.4, C = 2.0, HDL = 1.5, LDL = 0.2, ¶¶ | NA (T2D) |
| Parveen et al.91 | 1 × 40 | 2W, 41C, 18P, 40F, % | 160–200 g | W | HbA1c = 11, ¶¶ | NA (DS) |
| Zou et al. 201092 | 1 × 25 | 8W, 25.6C, 16.4P, 58F, % | 220–250 g | SD | FBG = 20, SI = 86, HbA1c = 7, ¶¶ | NA (T2D) |
| Xing et al.93 | 1 × 30 | 6W, 66ND, 20CS, 10FL, 2.5CO, 1O | 170–200 g | SD§§§ | FBG (H), ISI (L), ¶¶ | NA (D) |
| Zhang et al.94 | 1 × 25 | 8W, 60 FL, % | 180–200 g | SD | FPG = 21 (H), FPI = 190 (H), FTG = 2.7 (H), PTC = 2.1 (H) | Early (DS) |
| Islam et al.95 | 1 × 40 | 2W, 47CS, ST, 20PCA, 20FL, 10O | 5 weeks, 120–140 g | SD | FBG = 181.7 ± 60.1 (mg/dL), FBI = 61.8 ± 27.3 (pmol/L), HbA1C% = 7.8¶¶ | NA (D) |
| Zhang et al.56 | 2 × 30 | 4W, 48C, 20P, 22F | 200–250 g | W | FBG = 14 (H), FI = 64 (S), TG = 1.7 (H), TC = 3.0 (H) | Late (DS) |
| Gao et al. 200796 | 1 × 25 | 4W, 30CS, 15FL | 210–220 g | SD | BG = (H), TG = 0.9 (H), C = 2.6 (S) | NA (DS) |
| Sahin et al.61 | 1 × 40 | 2W, 30CS, S, 20PCA, 40FAF, 10O | 8 weeks, 200–250 g | SD | G = 26 (H), I = 161 (L), TG = 4.4 (H), TC = 6.5 (H) | Late ‐ NA |
| Danda et al.97 | 1 × 35 i.v. | 5W, 60FAF, % | 175–200 g | SD | BG = (H), Hba1c = 6 (H), C = 3.1 (H), TG = 3.3 (H), ¶¶¶SI = 84.3 | NA (T2D) |
| Srinivasan et al.55 | 1 × 35 | 2W, 17K, 25P, 58F, % | 160–180 g | SD | PGL = 23 (H), PI = 217 (S), PTG = 2.0 (H), PTC = 4.6 (H) | Late (DS) |
| Zhou et al.82 | 1 × 40 | 4W, 54C, 13P, 20FL, 5O | 4 weeks, 83 ± 5 g | SD | FBG = 14 (H), FSI = 60 (S), TG = 3.7 (H), C = 2.6 (H) | Late (DS) |
| Wu et al. 200498 | 1 × 30 i.v. | 2W, 41K, 18P, 41F, % | 8 weeks | SD | FBG = 7 (H), I = 77 (S) | Late (DS) |
| Zhang et al.81 | 1 × 15 i.v. | 8W, 50C, 13P, 30F | 8 weeks | SD | FBG = 17 (H), FSI = 120 (S), TG = 3.8 (H), C = 2.4 (H) | Late (DS) |
| Yang et al. 200371 | 1 × 15 i.v. | 8W, 40C, 13P, 40F, 7O | 8 weeks | SD | FBG = (H), TG = (H), C = (H) | NA (DS) |
| Reed et al.54 | 1 × 50 | 2W, 41C, 18P, 40F | 7 weeks, 200 g | SD | BG = 21 (H), I = 186 (H), TG = 7.5 (H) | Early (DS) |
*STZ treatment: Number of doses × dose (mg/kg) of Streptozotocin. The route of administration was intraperitoneally unless otherwise indicated; intraveneously (i.v.). †Diet (duration): Duration of diet regimen in weeks (W) before STZ treatment. ‡Diet (nutritional content): dietary Carbohydrate percentage (C): Starch (ST), Sucrose (s); dietary Fat percentage (F): Animal Fat (AF), Lard (L), Coconut Oil (CN), Vegetable Oil (VO): dietary Protein percentage (P): Casein (CA), Milk Powder (MP), Soy Bean (SB); dietary Cholesterol percentage (CO); dietary percentage of Other components than C, F, P and CO (O); Normal diet (ND); %kca is specified with ‘%’. §Strain of male rats: Sprague Dawly (SD); Wistar (W); Albino (A); **Female. §§Metabolic measures: The metabolic measure was either Higher (H) or Lower (L) in the HFD/STZ rat than in lean controls, or the same (S). All data have been converted to SI units. The mM Unit was used for: Glucose (G), Blood Glucose (BG), Plasma Glucose (PG), Fasting Glucose (FG), Fasting Blood Glucose (FBG), Triglycerides (TG), Fasting Triglycerides (FTG), Total Cholesterol (TC), Plasma Triglycerides (PTG), Plasma Total Cholesterol (PTC), High‐Density Lipoprotein (HDL), Low‐Density Lipoprotein (LDL); The pM Unit was used for: Insulin (I), Plasma Insulin (PI), Fasting Blood Insulin (FBI), Fasting Serum Insulin (FSI); The mg/mL Unit was used for serum adiponectin (APQ); Homeostasis Model of Assessment of β‐cell function (HOMA‐B); Homeostasis Model of Assessment of Insulin Resistance (HOMA‐IR); Insulin Sensitivity Index (ISI), Insulin Resistance Index (IRI); ¶¶Statistics against controls were not provided, ¶¶¶No lean controls were included. **Stage of type 2 diabetes (T2D): Whether the animal model mimics the Early versus the Late stage of type 2 diabetes was based on the levels of insulin and glucose provided in the study. The model was concluded be mimic the early stage if glucose and insulin levels were higher than in controls, whereas the model was concluded to mimic the late stage if insulin levels were lower than or the same as in controls. ††Nomenclature for the HFD/STZ rat model: Type 2 diabetes (T2D); diabetes (D); according to the Diet and STZ treatments (DS); lack of meatabolic parameters to classify the model or lack of nomenclature (NA).
In our experience, 5 weeks of high‐fat/high‐sucrose (HF/HS) feeding induced a higher body fat percentage and impaired glucose tolerance along with hyperinsulinemia (S. Skovsø, unpublished data). In contrast, induction of insulin resistance, by even shorter high‐fat diet regimens, has been reported.54 Furthermore, short periods of HFD feeding have been reported to simulate insulin resistance in lean patients, which is different from ‘true’ obese state that might take a much longer time to replicate in rats57. Thus, one should carefully consider the length and nutritional composition of the diet regimen, depending on what state one wants to mimic. Short diet regimens (2 weeks) tend to just induce insulin resistance and/or glucose intolerance, whereas relatively longer diet periods (5 weeks) can also induce a higher body fat percentage. Hence, this relatively short diet feeding seems to mimic the human situation of prediabetes, including obesity and hyperinsulinemia, more appropriately than the short diet regimens. Even longer feeding timeframes (> 3 months) are preferred when aiming for ‘true’ obesity including significant bodyweight increases.
HFD/STZ rats are often reported to be dyslipidemic, similar to the metabolic profile of type 2 diabetes in humans. Whether this is a direct consequence of the diet regimen alone is rarely reported in the literature. Data comprising the presence of hyperinsulinemia, obesity and impaired glucose tolerance, all representing the prediabetes state, are also rarely reported in the literature at the time‐point before initiation of STZ treatment. Such data, before establishment of severe hyperglycemia with STZ, would be required to underscore similarities with the progression of human type 2 diabetes, with respect to the order of the main pathological events. In unpublished studies, we have found that it is possible to maintain normal fasting glucose levels while significantly increasing the levels of total body fat (magnetic resonance imaging scanning), the liver fat (computed tomography scanning), plasma C‐peptide and triglyceride, as a consequence of a 5‐week HFD diet regimen consisting of 4 kcal% fat (lard), 35% carbohydrate (corn starch, sucrose, and maltodextrin) and 20% protein (casein), before initiation of STZ treatment (S. Skovsø, unpublished data). Furthermore, similar to the findings of other groups54, we have observed glucose intolerance in HFD‐fed rats. After STZ treatment of HFD‐fed rats, we and others have observed profound hyperglycemia, low levels of circulating adiponectin60, and high levels of plasma alanine aminotransferase61 (S. Skovsø, unpublished data). In summary, the diet regimen is one of the most important factors to consider in the HFD/STZ rat model.
Does STZ Treatment Make the Model a Type 1 or a Type 2 Diabetes Animal Model?
The final event involved in the development of type 2 diabetes is β‐cell failure/death5. This is also the case in type 1 diabetes62. Hence, the β‐cell toxin, STZ, has been used in both type 1 and type 2 diabetes animal models55. The STZ dose will greatly affect the β‐cell mass remaining in the rats. Likewise, variation in the amount of β‐cell mass left in both type 1 and type 2 diabetes exists in humans6. Despite the lack of non‐invasive measurement techniques, it has been suggested that 60–80% of the functional β‐cell mass is lost by the time of diagnosis of type 1 diabetes63. In contrast, only a 24% reduction has been observed in patients with a < 5 years’ history of type 2 diabetes compared to controls64. However, another study has reported a 54% reduction in β‐cell mass 15 years after diagnosis of type 2 diabetes64. Collectively, these data suggest a similarity in β‐cell mass, when comparing early type 1 and late stage type 2 diabetes.
This observation suggests that the HFD/STZ rat model could mimic the case of early type 1 diabetes coexisting with obesity. However, obesity is more often seen in patients after their type 1 diabetes diagnosis, whereas obesity is often seen decades before the diagnosis of type 2 diabetes. Thus, the order of the pathological events, obesity followed by β‐cell failure, seen in HFD/STZ rats favors a mimicking of type 2 diabetes rather than type 1 diabetes, despite the observed similarity between early type 1 diabetes and late type 2 diabetes. Furthermore, the loss of β‐cell mass in the pathogenesis of type 1 diabetes occurs mainly as a result of an autoimmune reaction63, which is not the case in HFD/STZ rats. In contrast, the events leading to β‐cell compensatory mechanisms and subsequent β‐cell failure in type 2 diabetes involve lipotoxicity and/or glucolipotoxicity, insulin resistance, hyperinsulinemia, and stress, with a modest contribution from low‐level inflammation9. In other words, the different causalities that induce β‐cell death in type 1 and type 2 diabetes cannot be mimicked to perfection by STZ treatment in animal models.
Despite the lack of the autoimmune component, HFD‐fed rats treated with just a single high dose of STZ show clear features of type 1 diabetes, such as hyperglycemia, insulin deficiency, drastic weight loss and resistance towards insulin‐sensitizing therapeutics55. Furthermore, STZ treatment of both lean‐STZ (a frequently used model of type 1 diabetes55; 3 × 30 mg STZ/kg bodyweight, once daily for 3 days) and HFD/STZ rats (3 × 20 mg STZ/kg bodyweight, once daily for 3 days) are often associated with an initial weight loss (S. Skovsø, unpublished data), whereas both models respond with a weight gain after 3 weeks of insulin therapy (S. Skovsø, unpublished data). An insulin‐induced weight gain is commonly seen during insulin therapy in type 1 and type 2 diabetes patients.67 In contrast to type 1 diabetes, a clear and sudden weight loss is not observed on diagnosis of type 2 diabetes. However, patients with undiagnosed and/or non‐insulin‐treated overt type 2 diabetes would inevitably also face a significant weight loss with time. Thus, the STZ‐induced weight loss and gain in weight on insulin therapy, which we have observed in the HFD/STZ rat model, does not make the model a better model of type 1 diabetes than of type 2 diabetes, as they are phenomena potentially occurring in both diseases.
Confusion has been added into the literature by the fact that some high‐fat fed rat models, treated with the same high dose of STZ used when modeling type 1 diabetes, have also been referred to as models of type 2 diabetes in other studies54. However, when the STZ dose is changed from a single high dose to a single low dose or multiple lower doses of STZ, researchers tend to agree on the HFD/STZ rat as a suitable model of type 2 diabetes55 (Table 1). Thus, the dose of STZ in itself obviously has a significant impact on the phenotype of HFD‐fed rats. STZ treatment induces robust (but not absolute) β‐cell ablation in a manner that depends on the dose, the number of doses, the time interval between doses, the route of administration, the fed/fasted state upon STZ administration, and the rat strain/vendor. There are great variations in the STZ treatments, which affect the level of β‐cell depletion. Variations among these parameters also exist in studies working with the HFD/STZ rat model (Table 1).
The same STZ treatment has been applied to rats on different diet regimens, and thus rats with potentially different body compositions might also result in different phenotypes. Data supporting this concept are found in studies comparing lean STZ rats with HFD/STZ rats treated with the same amount of STZ. These rats do not show the same phenotype in respect to blood glucose levels.54 This might be related to the fact that STZ has been shown not to interact with lipids70. Another possibility could be varying levels of glucose transporter 2, required for STZ entry into β‐cells, in the two models. Another caveat to remember when treating HFD fed animals with STZ is that diabetes induced by STZ treatment can lead to increased insulin sensitivity when compared with controls71. In contrast, type 2 diabetes in humans is characterized by insulin resistance9. Finally, when discussing the affect of STZ treatment with respect to the HFD/STZ model, it should be stressed that the STZ treatment leads to a transition from an insulin‐resistant state to a state of type 2 diabetes in a very fast and unnatural way. This means that the time aspect of the disease progression/transition is not mimicked ideally in this animal model. In summary, the design of the STZ treatment superimposed on the choice of the diet regimen will greatly affect the phenotype of the HFD/STZ rat model. No absolute agreement of the STZ treatment approach exists in the literature when it comes to modeling of type 2 diabetes in the HFD/STZ rat model, though some tendencies appear (Table 1).
Impact of Age in HFD/STZ Models When Deciding on the Type of Diabetes Model
Type 2 diabetes remains mainly a disease of older humans72. Thus, another important factor in modeling the HFD/STZ diabetes rat model is age. The vast majority of HFD/STZ rats used in the literature are young rats (< 6 months; Table 1). The young age of the rats make them a potential disease model for diabetes present in young human individuals. It is arguable that young HFD/STZ rats with a massive loss of functional β‐cell mass mimic type 1 diabetes in children who are obese, but without the autoimmune component hallmarking of type 1 diabetes73. In contrast, young HFD/STZ rats bearing a somewhat lower depletion of β‐cell mass mimic obese children with type 2 diabetes. Notably, the prevalence of type 2 diabetes in young children and adolescents has increased with a rapid pace74. Importantly, the pathogenesis of type 2 diabetes in young vs older individuals has indirectly been shown to be different from one another75; where young type 2 diabetes patients have a tendency to be insulin deficient, the elderly have a tendency to be more insulin resistant. This fits with knowledge from genome‐wide association studies pointing to genetic defects in β‐cell function, and the general concept that earlier diagnosis would be associated with a greater genetic contribution. This point also favors the young HFD/STZ rats to be a model for type 2 diabetes in young individuals, as STZ treatment brings about insulin secretion deficiency rather than insulin resistance. It is important to mention that young rodents, like young people, have the capacity to increase β‐cell mass76. Older rodents (aged >1 year) and older people (aged >30 years) do not seem to have this capacity76. This partly explains why it can be so challenging to administer the correct dose of STZ to induce the state of diabetes intended for the investigation. Collectively, these observations stress the importance of choosing the age of the rats when modeling type 2 diabetes in the HFD/STZ rat model.
Early vs Late Stage Type 2 Diabetes: STZ Treatment and β‐Cell Functionality in High‐Fat Fed Rats
So far, different ways of modeling type 2 diabetes, in the HFD/STZ rat model, have been discussed in the present review. However, another important question centers around whether the HFD/STZ rat mimics an early or late stage of type 2 diabetes. This is an important issue because of the principal metabolic differences present in subjects having had type 2 diabetes for either a shorter or longer period, including the level of remnant functional β‐cell mass6. This emphasizes the importance of characterizing the amount of, and more importantly, the function of the remaining β‐cells and the level of insulin resistance when working with the HFD/STZ rats. This is not reported consistently in preclinical studies. In contrast, clinical studies have stressed the importance of dividing type 2 diabetes patients into subgroups, based on their disease duration and severity, as well as on the length, intensity and choice of77. Feyter et al.77 investigated muscle mitochondrial dysfunction and divided their patients into four different subgroups: (i) long‐standing insulin‐treated type 2 diabetes patients; (ii) patients with impaired fasting glucose; (iii) impaired glucose tolerance and/or recently diagnosed type 2 diabetes; and (iv) healthy, normoglycemic controls. Such clinical studies raise the awareness of the importance to also divide the type 2 diabetes animal models in respect to the state of the disease. It can be argued as to whether classifications should be the same in animal models. It should be possible to make a ‘rough’ division of type 2 diabetes patients based on the duration of the disease alone, dividing them into an early phase and late phase cohort. Both early and late stage type 2 diabetes patients have hyperglycemia, but individuals in the early phase might still have relatively high levels of insulin. In contrast, late phase patients have similar or lower levels of insulin compared with weight‐matched controls.
Likewise, one can find examples of HFD/STZ rat models with elevated blood glucose levels coexisting with either higher, lower or the same levels of insulin when compared with lean controls (Table 1). Despite the demonstration of these data in some studies, the categorization (early vs late) is rarely reported in studies using the HFD/STZ rat model. Such a grouping is especially important when testing therapeutics for type 2 diabetes, aiming towards either the early or the late stage of type 2 diabetes.
In this review, the HFD/STZ rat models used in the various studies have been classified into models representing either the early or the late stage of type 2 diabetes (Table 1). The classification is based on the levels of glucose and insulin, and has thus only been possible for the studies providing these parameters for both the HFD/STZ rats and their HFD controls. Only few studies have more thoroughly investigated the stage of type 2 diabetes by examining the pathological state of the pancreas through investigation of oxidative stress markers, anti‐oxidative markers, islets area, insulin positive cells and/or cell death in the endocrine pancreas61. Naturally, without this information, it is not possible to fully understand whether the HFD/STZ rat model corresponds to an early or a late stage of type 2 diabetes. Researchers working with the HFD/STZ rat animal model, as well as researchers working with other type 2 diabetes models, are encouraged to provide data (at least glucose and insulin/c‐peptide levels), which can be used for identifying the stage of type 2 diabetes mimicked in the specific HFD/STZ rat model.
Conclusions
In the present review, the metabolic profile of the different stages of type 2 diabetes progression in both humans and HFD/STZ rats are reviewed, and some similarities and differences are highlighted. The evolution of this model is reviewed in the context of efforts to more accurately model human type 2 diabetes. The specific variations in the dietary regimen, STZ treatment and age used in HFD/STZ rat models are discussed thoroughly. Finally, the importance of considering whether a specific HFD/STZ rat model mimics an early or late stage of human type 2 diabetes is considered. It is clear that more basic characterization needs to be carried out on this model, and this review has provided some guidance on how to proceed. Finally and most importantly, it is the opinion of this author that, despite its limitations and the wide variety of both the high‐fat fed regimen and the STZ treatment, the HFD/STZ is a reasonable animal model of type 2 diabetes mainly representing the later stage of the disease depending on the amount of residual β‐cell mass.
Acknowledgments
The author gratefully thanks Jim Johnson, University of British Columbia, for helpful and critical comments during the writing of this manuscript. Furthermore, Xenia Asbæk Wolf and Bidda Rolin from the department of Translational Pharmacology, Novo Nordisk, Måløv, Denmark, are thanked for assistance and encouragement in the writing of this review. The author declares that there is no conflict of interest associated with this work.
J Diabetes Invest 2014; 5: 349–358
References
- 1.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 2010; 87: 4–14 [DOI] [PubMed] [Google Scholar]
- 2.Unwin N, Gan D, Whiting D. The IDF Diabetes Atlas: providing evidence, raising awareness and promoting action. Diabetes Res Clin Pract 2010; 87: 2–3 [DOI] [PubMed] [Google Scholar]
- 3.Guilherme A, Virbasius J, Vishwajeet P, et al. Adipocyte dysfunction linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 2008; 9: 367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoon KH, Lee JH, Kim JW, et al. Epidemic obesity and type 2 diabetes in Asia. Lancet 2006; 368: 1681–1688 [DOI] [PubMed] [Google Scholar]
- 5.Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in beta‐cell function. Nature 2001; 414: 788–791 [DOI] [PubMed] [Google Scholar]
- 6.Butler AE, Janson J, Bonner‐Weir S, et al. Beta‐cell deficit and increased beta‐cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52: 102–110 [DOI] [PubMed] [Google Scholar]
- 7.Kahn SE, Halban PA. Release of incompletely processed proinsulin is the cause of the disproportionate proinsulinemia of NIDDM. Diabetes 1997; 46: 1725–1732 [DOI] [PubMed] [Google Scholar]
- 8.Tabak AG, Jokela M, Akbaraly TN, et al. Trajectories of glycemia, insulin sensitivity and insulin secretion preceeding the diagnosis of type 2 diabetes: The Whitehall II Study. Lancet 2009; 373: 2215–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest 2006; 116: 1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lenzen S. The mechanisms of alloxan‐ and streptozotocin‐induced diabetes. Diabetologia 2008; 51: 216–226 [DOI] [PubMed] [Google Scholar]
- 11.Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res 2001; 50: 537–546 [PubMed] [Google Scholar]
- 12.Wang YW, Sun GD, Sun J, et al. Spontaneous type 2 diabetic rodent models. J Diabetes Res 2013; 2013: 401723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Islam MS and Loots DT. Experimental rodent models of type 2 diabetes: a review. Methods Find Exp Clin Pharmacol 2009; 31: 249–261 [DOI] [PubMed] [Google Scholar]
- 14.King AJ. The use of animal models in diabetes research. Br J Pharmacol 2012; 166: 877–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shafrir E. Contribution of animal models to the research of the causes of diabetes. World J Diabetes 2010; 1: 137–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frayn KN. Adipose tissue as a buffer for daily lipid flux. Diabetologia 2002; 45: 1201–1210 [DOI] [PubMed] [Google Scholar]
- 17.Acheson KJ, Schutz Y, Bessard T, et al. Glycogen storage capacity and de novo lipogenesis during massive carbohydrate overfeeding in man. Am J Clin Nutr 1988; 48: 240–247 [DOI] [PubMed] [Google Scholar]
- 18.Diraison F, Yankah V, Letexier D, et al. Differences in the regulation of adipose tissue and liver lipogenesis by carbohydrates in humans. J Lipid Res 2003; 44: 846–853 [DOI] [PubMed] [Google Scholar]
- 19.Donnelly KL, Smith CI, Schwarzenberg SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005; 115: 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherrington AD, Stevenson RW, Steiner KE, et al. Insulin, glucagon, and glucose as regulators of hepatic glucose uptake and production in vivo. Diabetes Metab Rev 1987; 3: 307–332 [DOI] [PubMed] [Google Scholar]
- 21.Reaven GM, Chen YD, Golay A, et al. Documentation of hyperglucagonemia throughout the day in nonobese and obese patients with noninsulin‐dependent diabetes mellitus. J Clin Endocrinol Metab 1987; 64: 106–110 [DOI] [PubMed] [Google Scholar]
- 22.Rohrer S, Menge BA, Gruber L, et al. Impaired crosstalk between pulsatile insulin and glucagon secretion in prediabetic individuals. J Clin Endocrinol Metab 2012; 97: E791–E795 [DOI] [PubMed] [Google Scholar]
- 23.Nolan CJ, Damm P, Prentki M. Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet 2011; 378: 169–181 [DOI] [PubMed] [Google Scholar]
- 24.Sattar N, McConnachie A, Ford I, et al. Serial metabolic measurements and conversion to type 2 diabetes in the west of Scotland coronary prevention study: specific elevations in alanine aminotransferase and triglycerides suggest hepatic fat accumulation as a potential contributing factor. Diabetes 2007; 56: 984–991 [DOI] [PubMed] [Google Scholar]
- 25.Bluher M. The distinction of metabolically ‘healthy’ from ‘unhealthy’ obese individuals. Curr Opin Lipidol 2010; 21: 38–43 [DOI] [PubMed] [Google Scholar]
- 26.Permutt MA, Wasson J, Cox N. Genetic epidemiology of diabetes. J Clin Invest 2005; 115: 1431–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saxena R, Voight BF, Lyssenko V, et al. Genome‐wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007; 316: 1331–1336 [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Obici S, Morgan K, et al. Overfeeding rapidly induces leptin and insulin resistance. Diabetes 2001; 50: 2786–2791 [DOI] [PubMed] [Google Scholar]
- 29.Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest 2008; 118: 2992–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han JM, Levings MK. Immune regulation in obesity‐associated adipose inflammation. J Immunol 2013; 191: 527–532 [DOI] [PubMed] [Google Scholar]
- 31.Ravussin E, Smith SR. Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus. Ann N Y Acad Sci 2002; 967: 363–378 [DOI] [PubMed] [Google Scholar]
- 32.Snel M, Jonker JT, Schoones J, et al. Ectopic fat and insulin resistance: pathophysiology and effect of diet and lifestyle interventions. Int J Endocrinol 2012; 2012: 983814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002; 51: 7–18 [DOI] [PubMed] [Google Scholar]
- 34.Roden M, Price TB, Perseghin G, et al. Mechanism of free fatty acid‐induced insulin resistance in humans. J Clin Invest 1996; 97: 2859–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petersen KF, Dufour S, Savage DB, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci USA 2007; 104: 12587–12594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 2012; 148: 852–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galbo T, Shulman GI. Lipid‐induced hepatic insulin resistance. Aging (Albany NY) 2013; 5: 582–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pettersson US, Walden TB, Carlsson PO, et al. Female mice are protected against high‐fat diet induced metabolic syndrome and increase the regulatory T cell population in adipose tissue. PLoS ONE 2012; 7: e46057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amano SU, Cohen JL, Vangala P, et al. Local proliferation of macrophages contributes to obesity‐associated adipose tissue inflammation. Cell Metab 2014; 19: 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corkey BE. Banting lecture 2011: hyperinsulinemia: cause or consequence? Diabetes 2012; 61: 4–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dankner R, Chetrit A, Shanik MH, et al. Basal state hyperinsulinemia in healthy normoglycemic adults heralds dysglycemia after more than two decades of follow up. Diabetes Metab Res Rev 2012; 28: 618–624 [DOI] [PubMed] [Google Scholar]
- 42.Mehran AE, Templeman NM, Brigidi GS, et al. Hyperinsulinemia drives diet‐induced obesity independently of brain insulin production. Cell Metab 2012; 16: 723–737 [DOI] [PubMed] [Google Scholar]
- 43.Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med 1998; 15: 539–553 [DOI] [PubMed] [Google Scholar]
- 44.Cornelis MC, Hu FB. Gene‐environment interactions in the development of type 2 diabetes: recent progress and continuing challenges. Annu Rev Nutr 2012; 32: 245–259 [DOI] [PubMed] [Google Scholar]
- 45.Hollingdal M, Juhl CB, Pincus SM, et al. Failure of physiological plasma glucose excursions to entrain high‐frequency pulsatile insulin secretion in type 2 diabetes. Diabetes 2000; 49: 1334–1340 [DOI] [PubMed] [Google Scholar]
- 46.Gorden P, Hendricks CM, Roth J. Circulating proinsulin‐like component in man: increased proportion in hypoinsulinemic states. Diabetologia 1974; 10: 469–474 [DOI] [PubMed] [Google Scholar]
- 47.Kotronen A, Juurinen L, Tiikkainen M, et al. Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology 2008; 135: 122–130 [DOI] [PubMed] [Google Scholar]
- 48.Sheng T, Yang K. Adiponectin and its association with insulin resistance and type 2 diabetes. J Genet Genomics 2008; 35: 321–326 [DOI] [PubMed] [Google Scholar]
- 49.Johnson JD, Alejandro EU. Control of pancreatic beta‐cell fate by insulin signaling: the sweet spot hypothesis. Cell Cycle 2008; 7: 1343–1347 [DOI] [PubMed] [Google Scholar]
- 50.Taylor R. Banting Memorial lecture 2012: reversing the twin cycles of type 2 diabetes. Diabet Med 2013; 30: 267–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taylor R. Pathogenesis of type 2 diabetes: tracing the reverse route from cure to cause. Diabetologia 2008; 51: 1781–1789 [DOI] [PubMed] [Google Scholar]
- 52.Johnson JD, Luciani DS. Mechanisms of pancreatic beta‐cell apoptosis in diabetes and its therapies. Adv Exp Med Biol 2010; 654: 447–462 [DOI] [PubMed] [Google Scholar]
- 53.Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009; 58: 773–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reed MJ, Meszaros K, Entes LJ, et al. A new rat model of type 2 diabetes: the fat‐fed, streptozotocin‐treated rat. Metabolism 2000; 49: 1390–1394 [DOI] [PubMed] [Google Scholar]
- 55.Srinivasan K, Viswanad B, Asrat L, et al. Combination of high‐fat diet‐fed and low‐dose streptozotocin‐treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol Res 2005; 52: 313–320 [DOI] [PubMed] [Google Scholar]
- 56.Zhang M, Lv XY, Li J, et al. The characterization of high‐fat diet and multiple low‐dose streptozotocin induced type 2 diabetes rat model. Exp Diabetes Res 2008; 2008: 704045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Browning KN, Fortna SR, Hajnal A. Roux‐en‐Y gastric bypass reverses the effects of diet‐induced obesity to inhibit the responsiveness of central vagal motoneurones. J Physiol 2013; 591(Pt 9): 2357–2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buettner R, Parhofer KG, Woenckhaus M, et al. Defining high‐fat‐diet rat models: metabolic and molecular effects of different fat types. J Mol Endocrinol 2006; 36: 485–501 [DOI] [PubMed] [Google Scholar]
- 59.Albersen M, Lin G, Fandel TM, et al. Functional, metabolic, and morphologic characteristics of a novel rat model of type 2 diabetes‐associated erectile dysfunction. Urology 2011; 78: 476–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo Z, Zhang R, Li J, et al. Effect of telmisartan on the expression of adiponectin receptors and nicotinamide adenine dinucleotide phosphate oxidase in the heart and aorta in type 2 diabetic rats. Cardiovasc Diabetol 2012; 11: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sahin K, Onderci M, Tuzcu M, et al. Effect of chromium on carbohydrate and lipid metabolism in a rat model of type 2 diabetes mellitus: the fat‐fed, streptozotocin‐treated rat. Metabolism 2007; 56: 1233–1240 [DOI] [PubMed] [Google Scholar]
- 62.Tisch R, McDevitt H. Insulin‐dependent diabetes mellitus. Cell 1996; 85: 291–297 [DOI] [PubMed] [Google Scholar]
- 63.Di Gialleonardo V, de Vries EF, Di Girolamo M, et al. Imaging of beta‐cell mass and insulitis in insulin‐dependent (Type 1) diabetes mellitus. Endocr Rev 2012; 33: 892–919 [DOI] [PubMed] [Google Scholar]
- 64.Rahier J, Guiot Y, Goebbels RM, et al. Pancreatic beta‐cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 2008; 10(Suppl 4): 32–42 [DOI] [PubMed] [Google Scholar]
- 65.Lu HE, Jian CH, Chen SF, et al. Hypoglycaemic effects of fermented mycelium of Paecilomyces farinosus (G30801) on high‐fat fed rats with streptozotocin‐induced diabetes. Indian J Med Res 2010; 131: 696–701 [PubMed] [Google Scholar]
- 66.Kim E, Sohn S, Lee M, et al. Differential responses of the growth hormone axis in two rat models of streptozotocin‐induced insulinopenic diabetes. J Endocrinol 2006; 188: 263–270 [DOI] [PubMed] [Google Scholar]
- 67.Intensive blood‐glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352: 837–853 [PubMed] [Google Scholar]
- 68.Purnell JQ, Zinman B, Brunzell JD. The effect of excess weight gain with intensive diabetes mellitus treatment on cardiovascular disease risk factors and atherosclerosis in type 1 diabetes mellitus: results from the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study (DCCT/EDIC) study. Circulation 2013; 127: 180–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Si Y, Zhao Y, Hao H, et al. Infusion of mesenchymal stem cells ameliorates hyperglycemia in type 2 diabetic rats: identification of a novel role in improving insulin sensitivity. Diabetes 2012; 61: 1616–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Masiello P, Karunanayake EH, Bergamini E, et al. Streptozotocin: its distribution and interaction with nucleic acids and proteins. Biochem Pharmacol 1981; 30: 1907–1913 [DOI] [PubMed] [Google Scholar]
- 71.Yang J, Li G, Zhang F, et al. Identification of variations of gene expression of visceral adipose and renal tissue in type 2 diabetic rats using cDNA representational difference analysis. Chin Med J (Engl) 2003; 116: 529–533 [PubMed] [Google Scholar]
- 72.Koopman RJ, Mainous AG III, Diaz VA, et al. Changes in age at diagnosis of type 2 diabetes mellitus in the United States, 1988 to 2000. Ann Fam Med 2005; 3: 60–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010; 464: 1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Demmer RT, Zuk AM, Rosenbaum M, et al. Prevalence of diagnosed and undiagnosed type 2 diabetes mellitus among US adolescents: results from the continuous NHANES, 1999–2010. Am J Epidemiol 2013; 178: 1106–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pani LN, Nathan DM, Grant RW. Clinical predictors of disease progression and medication initiation in untreated patients with type 2 diabetes and A1C less than 7%. Diabetes Care 2008; 31: 386–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kushner JA. The role of aging upon beta cell turnover. J Clin Invest 2013; 123: 990–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.De Feyter HM, van den Broek NM, Praet SF, et al. Early or advanced stage type 2 diabetes is not accompanied by in vivo skeletal muscle mitochondrial dysfunction. Eur J Endocrinol 2008; 158: 643–653 [DOI] [PubMed] [Google Scholar]
- 78.Abo‐elmatty DM, Essawy SS, Badr JM, et al. Antioxidant and anti‐inflammatory effects of Urtica pilulifera extracts in type 2 diabetic rats. J Ethnopharmacol 2013; 145: 269–277 [DOI] [PubMed] [Google Scholar]
- 79.Gandhi GR, Stalin A, Balakrishna K, et al. Insulin sensitization via partial agonism of PPARgamma and glucose uptake through translocation and activation of GLUT4 in PI3K/p‐Akt signaling pathway by embelin in type 2 diabetic rats. Biochim Biophys Acta 2013; 1830: 2243–2255 [DOI] [PubMed] [Google Scholar]
- 80.Sharma AK, Bharti S, Ojha S, et al. Up‐regulation of PPARgamma, heat shock protein‐27 and ‐72 by naringin attenuates insulin resistance, beta‐cell dysfunction, hepatic steatosis and kidney damage in a rat model of type 2 diabetes. Br J Nutr 2011; 106: 1713–1723 [DOI] [PubMed] [Google Scholar]
- 81.Zhang F, Ye C, Li G, et al. The rat model of type 2 diabetic mellitus and its glycometabolism characters. Exp Anim 2003; 52: 401–407 [DOI] [PubMed] [Google Scholar]
- 82.Zhou YS, Gao Y, Guo XH, et al. Effects of timely insulin treatment on protection of beta cells in a rat model of type 2 diabetes mellitus. Chin Med J (Engl) 2004; 117: 1523–1529 [PubMed] [Google Scholar]
- 83.Hu SH, Jiang T, Yang SS, et al. Pioglitazone ameliorates intracerebral insulin resistance and tau‐protein hyperphosphorylation in rats with type 2 diabetes. Exp Clin Endocrinol Diabetes 2013; 121: 220–224 [DOI] [PubMed] [Google Scholar]
- 84.Khan HB, Vinayagam KS, Moorthy BT, et al. Anti‐inflammatory and anti‐hyperlipidemic effect of Semecarpus anacardium in a high fat diet: STZ‐induced type 2 diabetic rat model. Inflammopharmacology 2013; 21: 37–46 [DOI] [PubMed] [Google Scholar]
- 85.Ren Z, Li W, Zhao Q, et al. The impact of 1,25‐dihydroxy vitamin D3 on the expressions of vascular endothelial growth factor and transforming growth factor‐beta(1) in the retinas of rats with diabetes. Diabetes Res Clin Pract 2012; 98: 474–480 [DOI] [PubMed] [Google Scholar]
- 86.Hou L, Lian K, Yao M, et al. Reduction of n‐3 PUFAs, specifically DHA and EPA, and enhancement of peroxisomal beta‐oxidation in type 2 diabetic rat heart. Cardiovasc Diabetol 2012; 11: 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mahmoud AM, Ashour MB, Abdel‐Moneim A, et al. Hesperidin and naringin attenuate hyperglycemia‐mediated oxidative stress and proinflammatory cytokine production in high fat fed/streptozotocin‐induced type 2 diabetic rats. J Diabetes Complications 2012; 26: 483–490 [DOI] [PubMed] [Google Scholar]
- 88.Guo Z, Qin Z, Zhang R, et al. Effect of rosiglitazone on the expression of cardiac adiponectin receptors and NADPH oxidase in type 2 diabetic rats. Eur J Pharmacol 2012; 685: 116–125 [DOI] [PubMed] [Google Scholar]
- 89.Hussein AA, Abdel‐Aziz A, Gabr M, et al. Myocardial and metabolic dysfunction in type 2 diabetic rats: impact of ghrelin. Can J Physiol Pharmacol 2012; 90: 99–111 [DOI] [PubMed] [Google Scholar]
- 90.Guo Z, Zheng C, Qin Z, et al. Effect of telmisartan on the expression of cardiac adiponectin and its receptor 1 in type 2 diabetic rats. J Pharm Pharmacol 2011; 63: 87–94 [DOI] [PubMed] [Google Scholar]
- 91.Parveen K, Khan MR, Mujeeb M, et al. Protective effects of Pycnogenol on hyperglycemia‐induced oxidative damage in the liver of type 2 diabetic rats. Chem Biol Interact 2010; 186: 219–227 [DOI] [PubMed] [Google Scholar]
- 92.Zou F, Mao XQ, Wang N, et al. Astragalus polysaccharides alleviates glucose toxicity and restores glucose homeostasis in diabetic states via activation of AMPK. Acta Pharmacol Sin 2009; 30: 1607–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xing XH, Zhang ZM, Hu XZ, et al. Antidiabetic effects of Artemisia sphaerocephala Krasch. gum, a novel food additive in China, on streptozotocin‐induced type 2 diabetic rats. J Ethnopharmacol 2009; 125: 410–416 [DOI] [PubMed] [Google Scholar]
- 94.Zhang T, Pan BS, Zhao B, et al. Exacerbation of poststroke dementia by type 2 diabetes is associated with synergistic increases of beta‐secretase activation and beta‐amyloid generation in rat brains. Neuroscience 2009; 161: 1045–1056 [DOI] [PubMed] [Google Scholar]
- 95.Islam MS, Choi H, Loots DT. Effects of dietary onion (Allium cepa L.) in a high‐fat diet streptozotocin‐induced diabetes rodent model. Ann Nutr Metab 2008; 53: 6–12 [DOI] [PubMed] [Google Scholar]
- 96.Gao L, Niu Y, Liu W, et al. The antilipolytic action of bis(alpha‐furancarboxylato)oxovanadium(IV) in adipocytes. Clin Chim Acta 2008; 388: 89–94 [DOI] [PubMed] [Google Scholar]
- 97.Danda RS, Habiba NM, Rincon‐Choles H, et al. Kidney involvement in a nongenetic rat model of type 2 diabetes. Kidney Int 2005; 68: 2562–2571 [DOI] [PubMed] [Google Scholar]
- 98.Wu Y, Ouyang JP, Zhou YF, et al. Mechanism of improving effect of losartan on insulin sensitivity of non‐insulin‐dependent diabetes mellitus rats. Acta Physiologica Sinica 2004; 56: 539–549 [PubMed] [Google Scholar]