

Abstract
Time-resolved fluorescence dynamics are investigated in two mutants of a thermophilic alcohol dehydrogenase (ht-ADH): Y25A (at the dimer interface) and V260A (at the cofactor-binding domain). These residues, ca. 32 Å apart, are shown to exhibit opposing low-temperature effects on the hydride tunneling step. Using single-tryptophan constructs at the active site (Trp87) and a remote, surface-exposed site (Trp167), time-dependent Stokes shifts and collisional quenching data allow an analysis of intra-protein dynamical communication. A double mutant, Y25A:V260A, was also inserted into each single-Trp construct and analyzed accordingly. None of the mutations affect fluorescence lifetimes, Stokes shift relaxation rates, and quenching data for the surface-exposed Trp167 to an appreciable extent. By contrast, fluorescent probes of the active-site tryptophan 87 reveal distinctive forms of dynamical communication. Stokes shifts show that the distal Y25A increases active-site flexibility, V260A introduces a temperature-dependent equilibration process not previously reported by such measurements, and the double mutant (Y25A:V260A) eliminates the temperature-dependent transition sensed by the active-site tryptophan in the presence of V260A. Collisional quenching data at Trp87 further show a structural change in the active-site environment/solvation for V260A. In the aggregate, the temperature dependencies of the fluorescence data are distinct from the breaks in behavior previously reported for catalysis and hydrogen/deuterium exchange, attributed to time scales for the interconversion of protein conformational substates that are slower and more global than the local motions monitored within. An extended network of dynamical communication between the protein dimer surface and substrate- and cofactor-binding domains emerges from the flourescent data.
Introduction
Rationalizing the catalytic impact of protein side chains that are not directly involved in enzyme–substrate binding, bond cleavage events, or networks of allostery can be a very challenging endeavor. Studies of hydrogen-transfer systems,1−4 phosphotriesterase,5 superoxide dismutase,6 purine nucleoside phosphorylase,7,8 and proline isomerase9−11 have attempted to address this issue. The effects of remote mutations on hydrogen transfer are of particular interest because of the importance of quantum mechanical hydrogen tunneling and its dependence on protein motions that occur on reaction coordinates distinct from the hydrogenic wave function overlap itself.12 The heavy atom motions that accompany active-site reorganization are expected to be affected by sampling of protein substates that alter amino acid side chain packing, subunit interactions, and overall electrostatic stabilization. Given the enormous landscape available within a protein’s conformational space, many ideas have emerged that aim to simplify the degrees of freedom required to describe the catalytically relevant microstates within an enzyme.13−16
A thermophilic alcohol dehydrogenase isolated from Bacillus stearothermophilus (ht-ADH) has emerged as a model system for interrogating the relationship between a protein’s conformational landscape and the properties of hydrogen tunneling.17 The temperature dependence of the hydride-transfer step that reduces nicotinamide adenine dinucleotide (NAD+) to NADH reveals an Arrhenius break at 30 °C that separates a catalytically impaired regime at low temperatures (EA = 21 kcal/mol) from a catalytically optimized regime at high temperatures (EA = 15 kcal/mol).18 This difference of ca. 6 kcal/mol in wild-type (WT) ht-ADH is believed to originate from an inherently large thermal barrier associated with the recovery to catalytically competent protein microstates at low temperature.17,19,20 Use of hydrogen/deuterium (H/D) exchange linked to mass spectrometry (HDX-MS) as a biophysical probe of changes in apo-protein also displays a marked transition in protein dynamics at 30 °C,21 implicating similar temperature dependent changes in protein flexibility within free and substrate bound forms of enzyme. The time scale of the HDX-MS measurements (seconds to hours) reflects an aggregate property that is distinct from real-time motions at localized protein positions.22
In this work, we focus on understanding the underlying behavior of two protein side chains, Tyr25 and Val260, which drastically alter the temperature dependence of the hydride-transfer properties in ht-ADH. In the case of Tyr25, X-ray characterization indicates a π-stacked residue that is connected over a distance of ca. 17 Å to the substrate-binding pocket by a series of intervening β-sheets23,24 (Figure 1). These β-sheets correspond to the region of ht-ADH that undergoes an abrupt increase in HDX above 30 °C.21 Kinetic analyses of Y25A in ht-ADH further reveal a complete absence of a break in the Arrhenius plot of kcat that is characterized by an enthalpy of activation similar to that of WT in the high-temperature regime.24 The failure to observe a protein “rigidification” below 30 °C in Y25A is attributed to a change in flexibility within the network of β-sheets connecting the dimer interface to the substrate-binding pocket.
Figure 1.
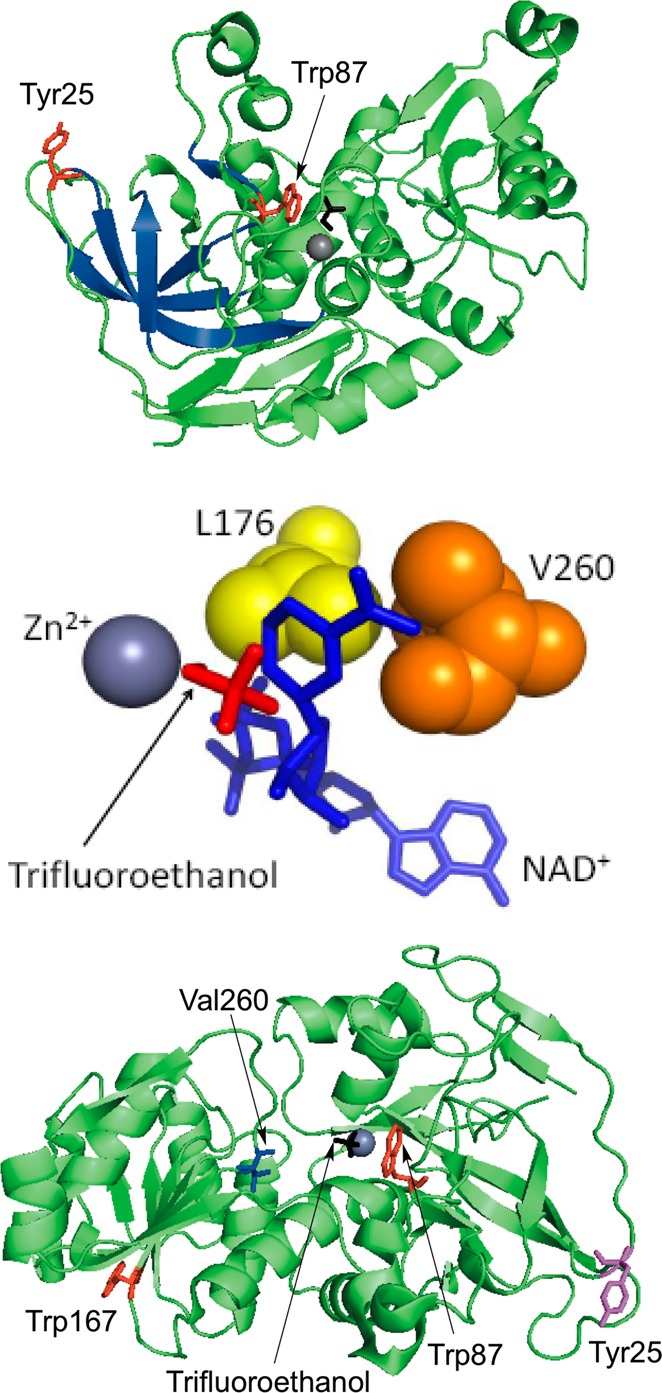
Structures of ht-ADH (PDB: 1RJW). Top panel: The structure is rotated to show the network of β-sheets (blue) connecting Tyr25 (red) at the dimer interface to the substrate-binding site Trp87 (red). In the crystal structure, Trp87 lies within van der Waals contact of the substrate analogue trifluorethanol (black) which coordinates to the catalytic Zn2+ (gray). Middle panel: Close-up of the cofactor-binding site and residues Leu176 (yellow) and Val260 (orange). Both residues lie within van der Waals of NAD+ (blue) in the docked structure. Image reproduced with permission from ref (37). Bottom panel: Monomeric structure of ht-ADH showing the relative positioning of both residues of interest, Tyr25 (magenta) and Val260 (blue), and the tryptophans used as probes (red).
The second residue of interest, Val260, resides in the cofactor-binding domain. Based on the impact of a similar side chain in horse liver ADH,25 this residue was predicted to influence active-site geometry in ht-ADH. Kinetic analysis has shown that V260A amplifies the low-temperature impairment of WT, increasing the enthalpy of activation for hydride transfer to over 32 kcal/mol and inflating the Arrhenius prefactor to nearly 1024 s–1. Such results have been explained in the context of a low-temperature rigidification of protein that generates high enthalpic barriers to the recovery of catalytically viable microstates.17
Given the strong inference of opposing dynamical behaviors in Y25A and V260A of ht-ADH, time-resolved fluorescence methodologies have been implemented to compare the temperature-dependent behaviors of mutant constructs to their WT counterpart. Previous work in this regard characterized the temperature dependence of the time-dependent Stokes shift and the collisional Stern–Volmer quenching behavior of two single-tryptophan variants in ht-ADH.26 Trp87, a residue located fortuitously in the active site within van der Waals contact of substrate, exhibits a sizable temperature-dependent behavior on the local Stokes shift relaxation, whereas the surface-exposed Trp167 shows no temperature dependence for the same process. In contrast, collisional quenching data indicate that both residues are equally accessible to solvent with thermal barriers comparable to that measured for quenching of free N-acetyltryptophanamide.27 In the present work, we have inserted Y25A and V260A into each single-Trp variant, and compared the temperature dependence of the Stokes shift relaxation and the collisional quenching behavior to the parent single-Trp counterpart (designated W87in and W167in). In addition, a double mutant, Y25A:V260A, has been generated and characterized. The data demonstrate an interplay between Tyr25 and Val260, extending the previously described dynamical network24 from the dimer interface to the substrate-binding domain across the active site to include the cofactor-binding domain.
Experimental Section
Site-Directed Mutagenesis and Protein Purification
The previously generated single-Trp plasmids (W87in containing W49F:W167Y; W167in containing W49F:W87F)26 were used to create W87in:Y25A, W167in:Y25A, W87in:V260A, W167in:V260A, W87in:Y25A:V260A, and W167in:Y25A:V260A. Changes to the pET-24b(+) expression vector were incorporated using the following primers (Operon) and their reverse complements (not shown):
-
Y25A:
5′-GTAGAAAAACCAACCATTTCAGCTGGAGAAGTATTAGTCCGC-3
-
V260A:
5′-CTTGTGTGCTTGCCGGATTGCCACC-3′
Both W87in:Y25A:V260A and W167in:Y25A:V260A were constructed by inserting Y25A into each respective single-Trp plasmid initially containing V260A. All DNA sequencing was performed at the UC Berkeley DNA Sequencing Facility. Subsequent cell harvest, lysis, and protein purification was previously optimized and described elsewhere.21,26
Steady-State Kinetics
Multi-substrate enzyme kinetics were analyzed for all six mutants by simultaneously varying the concentrations of NAD+ (Sigma-Aldrich) and either all protio-benzyl alcohol (h-BnOH, Sigma-Aldrich) or α,α-d2-benzyl alcohol (d-BnOH) (CDN Isotopes), in a manner previously described with minor alterations.26 Because of varying KM values for substrate and cofactor in each mutant, five appropriate concentrations of BnOH or d-BnOH between 0.5 and 18 mM and six appropriate concentrations between 0.05 and 20 mM of NAD+ were assayed to extract enzyme kinetic parameters for all mutants. Kinetic traces were collected for at least 5 min. Assays were performed at least in triplicate over a temperature range from 10 to 50 °C in 5 °C increments.
Time-Dependent Inactivation
Enzyme stability was also assessed for the mutants as previously described.26 Because of the differing KM values, the concentration of substrate and cofactor used were at 15 mM d-BnOH and 20 mM NAD+, respectively, for both single-Trp V260A and Y25A:V260A mutants. For W87in:Y25A, inactivation was monitored at 8 mM d-BnOH and 5 mM NAD+; for W167in:Y25A, inactivation was monitored at 16 mM d-BnOH and 3 mM NAD+. Where applicable, fresh samples were introduced to the fluorimeter cell at the time thar enzyme had lost more than 25% of its original activity. This threshold is plotted as a dotted line within the time- and temperature-dependent inactivation profiles shown in Figure S1 (Supporting Information).
Gel Filtration Chromatography
The oligomeric distribution of all single-Trp mutants were assessed at 4 °C by size-exclusion chromatography by methods previously described.17 All single-Trp variants eluted entirely as an active tetramer at 4 °C except for W87in:Y25A and W87in:Y25A:V260A (Figure S2, Supporting Information). While there is a modest amount of dimer and monomer present in elution profiles for these constructs, their behavior is in line with that previously observed in the WT counterpart, Y25A.24 Hence, we conclude that the removal of Trp49 and Trp167 has no additional effect on the inherent equilibrium distribution of oligomeric states normally observed at low temperatures following the insertion of Y25A.
Steady-State and Time-Resolved Fluorescence Measurements
All techniques, instrument specifications, and subsequent downstream analyses of time-resolved emission spectra (TRES), Stokes shifts, Stern–Volmer quenching data, and fluorescence lifetimes were executed as previously described.26 These studies have been restricted to the apo-form of enzyme, analogous to HDX experiments.21 As discussed in our previous study, fluorescence characterization of enzyme complexes with either NADH, NAD+, or substrate was precluded by a variety of complications in each instance.26
Results
Steady-State Kinetics
In general, single-tryptophan variants of ht-ADH, introduced into Y25A or V260A, produce only small differences in kinetic parameters at 30 °C (Table 1). Arrhenius plots (Figure S3, Supporting Information) also show similar temperature dependencies for both W87in and W167in in the presence of V260A, Y25A, or Y25A:V260A. The most noteworthy kinetic differences between the mutated single-Trp mutants lie with the relative KM values and the isotope effects on kcat and kcat/KM. The KM(h-BnOH) for W167in:Y25A (10.9 ± 2.0 mM) is roughly 1 order of magnitude greater than that found for W87in:Y25A (0.8 ± 0.1 mM) and closer to that for Y25A alone. Analysis of the isotope effect data on first- and second-order kinetics for the Y25A series further reveals that while hydride transfer is at least partially rate-determining at substrate saturation (Dkcat > 1 in all cases), the ratio of Dkcat to D(kcat/KM(BnOH)) is <1. Fully rate-limiting hydride transfer, under all conditions, necessitates a Dkcat/D(kcat/KM) that is equal to unity. The most likely explanation is that a second step (other than hydride transfer) contributes to kcat, reducing its deuterium isotope effect below that for kcat/KM(BnOH). This effect appears to be greatest for W167in:Y25A which shows a Dkcat ≃ 2 and a reduction in Dkcat/D(kcat/KM(NAD+)) as well. Despite these modest differences, the most important kinetic features of Y25A are observed in all constructs; these include the size of the enthalpy of activation and the absence of a break at 30 °C (Table 1, footnote h). We have concluded that single-site tryptophan constructs within Y25A are suitable variants of ht-ADH for the interrogation of time-resolved fluorescence spectroscopy.
Table 1. Comparative Kinetic Parameters of the Single-Trp Mutants Following Insertion of Y25A and V260A, 30 °C.
|
KM, mM |
Dkcat/D(kcat/KM)b |
||||||
|---|---|---|---|---|---|---|---|
| mutant | kcat, s–1 | NAD+ | h-BnOH | Dkcata | BnOH | NAD+ | ΔΔH⧧, kcal/molc |
| WTd | 8.0 (1.0) | 1.1 (0.1) | 6.8 (0.5) | 3.1 (0.2) | 0.8 (0.2) | 1.0 (0.1) | 6.7 (0.9) |
| Y25Ae | 5.1(0.5) | 1.0 (0.5) | 16.5 (2.1) | 2.7 (0.1) | 0.4 (0.1) | 1.0 (0.3) | N.A.h |
| V260Af | 1.9 (0.2) | 10.0 (1.6) | 4.2 (0.8) | 4.1 (0.7) | 1.0 (0.1) | 1.0 (0.2) | 18.7 (1.6) |
| W87ing | 3.4 (0.2) | 1.8 (0.2) | 3.1 (0.4) | 2.5 (0.1) | 1.0 (0.2) | 1.0 (0.2) | 5.4 (0.4) |
| W87in:Y25A | 2.5 (0.3) | 0.5 (0.1) | 0.8 (0.1) | 3.5 (0.3) | 0.5 (0.1) | 1.0 (0.1) | N.A.h |
| W87in:V260A | 1.4 (0.2) | 8.3 (1.1) | 8.2 (1.6) | 4.2 (0.2) | 1.0 (0.1) | 1.0 (0.1) | 15.2 (1.2) |
| W87in:Y25A:V260A | 1.9 (0.2) | 9.7 (1.5) | 5.4 (1.3) | 3.5 (0.6) | 1.1 (0.4) | 1.4 (0.5) | 17.8 (1.3) |
| W167ing | 3.0 (0.1) | 1.3 (0.3) | 6.0 (0.2) | 2.2 (0.1) | 0.9 (0.1) | 0.8 (0.1) | 5.7 (0.4) |
| W167in:Y25A | 6.3 (0.3) | 0.9 (0.2) | 10.9 (2.0) | 1.8 (0.1) | 0.4 (0.1) | 0.3 (0.1) | N.A.h |
| W167in:V260A | 1.0 (0.1) | 10.2 (0.8) | 12.9 (1.5) | 2.8 (0.3) | 1.0 (0.1) | 0.9 (0.2) | 12.2 (1.3) |
| W167in:Y25A:V260A | 2.5 (0.1) | 14.8 (0.6) | 10.1 (0.7) | 3.5 (0.5) | 0.9 (0.2) | 0.9 (0.2) | 14.5 (2.5) |
Computed as kcat(h-BnOH)/kcat(d-BnOH).
kcat /KM for substrate and cofactor are extracted from fits of the individual trials of h-BnOH or d-BnOH.
Computed as the difference of ΔH⧧ <30 °C and ΔH⧧ >30 °C.
From ref (18).
From ref (24).
From ref (17).
From ref (26).
N.A., not available, due to a lack of a break in the Arrhenius plots. ΔH⧧ = 12.1 (0.5) kcal/mol for Y25A24 and 13.9 (0.5) and 13.8 (0.5) kcal/mol for W87in:Y25A and W167in:Y25A, respectively.
The constructs of W87in and W167in also preserve the general Arrhenius features exhibited by V260A. One of these features includes an elevation of the temperature at which the break in kinetic behavior occurs, from 30 °C for WT to 40 °C for V260A; the W87in:V260A break is well-modeled at 40 °C and W167in:V260A shows a break at 35 °C (Figure S3). Moreover, both W87in:V260A and W167in:V260A exhibit identical low temperature enthalpies of activation, measured at 28.9 ± 0.5 and 29.1 ± 1.1 kcal/mol, respectively. These values are significantly greater than reported for WT (21.2 ± 1.0 kcal/mol) and are in line with the ΔH⧧ = 32.3 ± 1.3 kcal/mol for V260A.17,18 The Dkcat for W87in:V260A is within error of V260A (4.2 ± 0.2 vs 4.1 ± 0.7, respectively), while W167in:V260A reveals a slightly depressed kinetic isotope effect (KIE) on hydride transfer of 2.8 ± 0.3. In contrast to W167in:Y25A, W167in:V260A shows values for Dkcat/(Dkcat /KM) for both substrate and cofactor within error of unity (Table 1), in support of a hydride-transfer step that is largely rate-determining under both first- and second-order kinetic conditions.
Though the WT-version of Y25A:V260A mutant has not been kinetically characterized for comparison, the kinetic analyses of this work and previous studies20 show that generation of W87in or W167in within mutant variants does not greatly affect the inherent Arrhenius behavior.26 Studies here are focused on the single-Trp variants that additionally contain Y25A:V260A. W87in:Y25A:V260A and W167in:Y25A:V260A exhibit a distinct break at the increased temperature of 35 °C, aligning their behavior with V260A (Figure S3). Both variants mirror the low-temperature Arrhenius behavior of V260A, having a ΔH⧧ of 30.6 ± 0.9 and 27.0 ± 2.5 kcal/mol, respectively. The KIE on hydride transfer of 3.5 ± 0.5 together with Dkcat/D(kcat/KM) ≃ 1 further indicates that binding of cofactor and substrate remain fast, relative to a rate-limiting hydride transfer. Finally, of relevance to spectroscopic characterizations below, the addition of Y25A to V260A is unable to reverse the low-temperature protein rigidification generated by Val260 alone (Figure S3).
Steady-State Fluorescence
Steady-state emission spectra for the six newly characterized single-Trp mutants are shown in Figure S4 (Supporting Information). Peak emission wavelengths are located between 337.0 and 339.0 ± 0.5 nm for all mutants. Moreover, none of the mutants has a detectable break in wavelength or intensity as a function of temperature (Figure S5, Supporting Information). There is, thus, no evidence for unfolding or a major conformational change occurring as a function of temperature, implicating other causes for the observed breaks in kinetic behavior.17,26
Picosecond Fluorescence Lifetime Decays
Figure 2 shows the raw fluorescence counts, curve fitting, and residuals associated with the exponential fits analyzed at their respective peak wavelengths for the three mutations within W87in. The mutant that has the least change in fluorescence behavior relative to W87in is W87in:Y25A, with regard to the number of exponents (three) needed to properly fit the raw fluorescence decay curves and the amplitudes and lifetimes of each component (Table 2). In contrast to W87in, however, the temperature dependencies of the three lifetime components are linear (Figure S6, Supporting Information), whereas W87in indicated a distinctive break in τ1.26
Figure 2.

Fluorescence transients of W87in:Y25A (left), W87in:V260A (middle), and W87in:Y25A:V260A (right) at each mutant’s steady-state peak emission wavelength at 30 °C. Each decay panel contains the instrument response function (black), the raw fluorescence counts (green) and the fit (red). The residuals associated with the number of exponentials used to fit the data are shown under each decay panel. The reduced χ2 values were 12.9, 1.4, and 1.1 for one, two, and three exponentials used to fit W87in:Y25A, respectively. For W87in:V260A, the values were 9.8 and 1.0 for one and two exponentials; for W87in:Y25A:V260A, these respective values were 9.7 and 1.0.
Table 2. Comparison of Fluorescence Decay Parameters for Each Single-Tryptophan Construct and Its Respective Mutant Series at 30 °C.
| mutanta | α1b | τ1c | α2 | τ2 | α3 | τ3 |
|---|---|---|---|---|---|---|
| W87ind | 0.08 | 0.67 | 0.42 | 2.17 | 0.50 | 4.64 |
| W87in:Y25A | 0.07 | 0.65 | 0.50 | 2.24 | 0.43 | 5.02 |
| W87in:V260A | 0.48 | 2.00 | 0.52 | 4.56 | ||
| W87in:Y25A:V260A | 0.46 | 1.92 | 0.54 | 4.54 | ||
| W167ind | 0.08 | 3.10 | 0.92 | 6.36 | ||
| W167in:Y25A | 0.09 | 3.03 | 0.91 | 6.18 | ||
| W167in:V260A | 0.08 | 2.91 | 0.92 | 6.18 | ||
| W167in:Y25A:V260A | 0.06 | 2.36 | 0.94 | 6.16 |
All parameters were collected at the respective steady state peak emission wavelength: W87in, 337.0 nm; W87in:Y25A:V260A,W167in:V260A, W167in:Y25A:V260A, 337.5 nm; W87in:Y25A, W167in, W167in:Y25A, 338.0 nm; W87in:V260A, 339.0 nm.
Relative amplitudes for each decay process. Typical errors are ±3%.
Decay times are presented in ns. Typical errors are ±10% for τ1, and ±3% for τ2 and τ3.
From ref (26).
A global analysis of W87in:Y25A’s fluorescence parameters as a function of wavelength reveals further significant differences in the absolute amplitudes and lifetimes at all temperatures. Focusing on illustrative changes at 30 °C (Table 3), α1, the fluorescence component previously implicated as an excited-state Trp-Zn2+ interaction for W87in, has the greatest weight at blue wavelengths and systematically decreases as the wavelength increases. Its Y25A counterpart, however, shows an opposing trend in amplitudes and is characterized by a τ1 that increases almost an entire order of magnitude ranging from ca. 190 to 1500 ps between 310 and 370 nm. We propose that either the angular orientation of the excited state Trp87 dipole points farther away from the catalytic Zn2+ or the Trp-Zn2+ distance has increased with time in the case of W87in:Y25A. It is also clear that the amplitude decrease in α3 is larger for W87in:Y25A than W87in, translating into an increased contribution to the fastest transient (τ1). The two shorter lifetime components comprise 74% of the total fluorescence at 370 nm as opposed to 61% for W87in.
Table 3. Comparative Global Emission Wavelength Analysis of Fluorescence Decay Parameters for W87in and Additionally Inserted Mutations at 30 °Ca–d.
| W87in |
W87in:Y25A |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wavelength, nm | α1 | α2 | α3 | τ1 | τ2 | τ3 | α1 | α2 | α3 | τ1 | τ2 | τ3 |
| 310 | 0.20 | 0.33 | 0.47 | 0.36 | 1.55 | 4.15 | 0.10 | 0.39 | 0.51 | 0.19 | 1.38 | 4.27 |
| 320 | 0.16 | 0.36 | 0.48 | 0.45 | 1.79 | 4.26 | 0.07 | 0.42 | 0.51 | 0.32 | 1.66 | 4.50 |
| 330 | 0.14 | 0.43 | 0.44 | 0.56 | 2.16 | 4.57 | 0.08 | 0.48 | 0.44 | 0.56 | 2.06 | 4.92 |
| 340 | 0.12 | 0.44 | 0.44 | 0.63 | 2.28 | 4.67 | 0.10 | 0.51 | 0.39 | 0.80 | 2.44 | 5.27 |
| 350 | 0.11 | 0.49 | 0.40 | 0.69 | 2.47 | 4.91 | 0.11 | 0.53 | 0.37 | 0.94 | 2.56 | 5.52 |
| 360 | 0.11 | 0.48 | 0.41 | 0.76 | 2.50 | 4.92 | 0.16 | 0.52 | 0.32 | 1.21 | 2.86 | 5.84 |
| 370 | 0.09 | 0.52 | 0.39 | 0.70 | 2.61 | 5.25 | 0.23 | 0.51 | 0.26 | 1.51 | 3.25 | 6.23 |
| W87in:V260A |
W87in:Y25A:V260A |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wavelength, nm | α1 | α2 | α3 | τ1 | τ2 | τ3 | α1 | α2 | α3 | τ1 | τ2 | τ3 |
| 310 | 0.42 | 0.58 | 1.30 | 4.17 | 0.43 | 0.57 | 1.32 | 4.11 | ||||
| 320 | 0.42 | 0.58 | 1.53 | 4.34 | 0.42 | 0.58 | 1.55 | 4.23 | ||||
| 330 | 0.43 | 0.57 | 1.78 | 4.54 | 0.47 | 0.53 | 1.85 | 4.44 | ||||
| 340 | 0.45 | 0.55 | 1.96 | 4.67 | 0.49 | 0.51 | 1.99 | 4.56 | ||||
| 350 | 0.47 | 0.53 | 2.09 | 4.79 | 0.52 | 0.48 | 2.07 | 4.64 | ||||
| 360 | 0.48 | 0.52 | 2.15 | 4.86 | 0.54 | 0.46 | 2.16 | 4.72 | ||||
| 370 | 0.49 | 0.51 | 2.20 | 4.93 | 0.54 | 0.46 | 2.21 | 4.81 | ||||
The decay times are presented in ns.
The wavelengths summarized here are distinct from the peak steady-state wavelength emissions shown in Table 2.
Typical errors are ±10% for τ1 and ±3% for τ2 and τ3.
Amplitudes are reported such that ∑I αi = 1, where i equals the number of exponentials used to model the decay. Typical error for αi, ±3%.
W87in:V260A and W87in:Y25A:V260A differ most distinctly from W87in due to the absence of a short lifetime component in their multi-exponential fits (Figure 2 and Table 2). An interesting observation is that inserting Y25A into W87in:V260A has virtually no effect on the fluorescence lifetimes, appearing almost identical to W87in:V260A at all wavelengths and temperatures. Another noteworthy observation is that the emission wavelength-dependent change in the distribution of amplitudes for W87in:V260A and W87in:Y25A:V260A generally mirrors those of the two longer components of W87in. As the emission wavelength is increased, α2 systematically increases while α3 systematically decreases. The magnitude of this redistribution is not quite as large in the mutants as for W87in. However, there are small differences in the τ2 values which, together with the loss of τ1, indicates a redistribution in the population of protein conformers.
In contrast to the differences observed at Trp87 in the presence of Y25A and V260A, either alone or in combination, the observations at Trp167 indicate virtually no change in the fluorescence properties regardless of mutation. Figure 3 shows the raw fluorescence data and fitting parameters for W167in:Y25A, W167in:V260A, and W167in:Y25A:V260A. The relative amplitudes and lifetimes are similarly unaffected by any mutation (Table 2). All constructs within W167in can be described by a biexponential process having a 92 ± 2% component with a fluorescence lifetime of 6.2 ± 0.1 ns. Also, unlike the significant changes in the global wavelength behavior seen for the W87in mutant series in Table 3, there is no significant variation in the relative amplitudes and lifetimes across mutants at any given wavelength (data not shown). Though it appears that W167in:Y25A:V260A shortens τ2 relative to the other W167in constructs (Table 2), this difference is effectively negligible when calculating amplitude-weighted or average lifetimes as they affect TRES or collisional quenching analyses.28
Figure 3.

Fluorescence transients of W167in:Y25A (left), W167in:V260A (middle), and W167in:Y25A:V260A (right) at each mutant’s respective peak emission wavelength at 30 °C. The color scheme is the same as that used in Figure 2. The data for all mutants could be fit to two exponentials. The reduced χ2 values were found to be 1.7 and 1.1 for W167in:Y25A, 1.5 and 1.2 for W167in:V260A, and 1.8 and 1.1 for W87in:Y25A:V260A for one and two exponentials, respectively.
Temperature Dependence of the Stokes Shifts
The time-dependent Stokes shift probes the ability of local side chain interactions in proteins to rearrange during the lifetime of an excited state tryptophan. Electronic excitation of tryptophan induces a distorted electric field within its local environment, and protein motions such as side chain rotations, backbone fluctuations, and side chain translations are expected to relax on the time scales of these experiments to accommodate the new electrostatic environment.26,29,30 Select timeslices showing the incremental spectral evolution of each mutant’s red shift were constructed (Figures S7 and S8, Supporting Information). Figure 4 shows the temperature-dependence of the time-resolved red shift for W87in, W167in, and their respective mutant series, expressed in terms of the solvation correlation function, c(t). The decay times and the total spectral shift associated with each c(t) are expressed in Table 4. Arrhenius plots (Figure 5) lead to the activation parameters summarized in Table 5.
Figure 4.

Temperature-dependent solvation correlation decays, c(t), of the time-dependent Stokes shift for all eight mutants studied. The W87in mutant series is shown in the left column and the W167in mutant series is shown in the right column. The temperatures shown are at 10 °C (black), 20 °C (blue), 30 °C (green), 40 °C (orange), and 50 °C (red). All fits are to a single exponential except for W167in. W87in and W167in are reproduced with permission from ref (26).
Table 4. Total Stokes Shifts and Relaxation Rates of the W87in and W167in Mutant Series.
| decay
timesa,b |
total
red shiftc,d |
|||||||
|---|---|---|---|---|---|---|---|---|
| T, °C | W87ine | +Y25A | +V260A | +Y25A: V260A | W87ine | +Y25A | +V260A | +Y25A: V260A |
| 10 | 1640(80) | 943 (15) | 1580(50) | 710 (30) | 483 | 334 | 255 | 152 |
| 15 | 1070(60) | 743 (2) | 1580(70) | 670 (50) | 490 | 326 | 261 | 156 |
| 20 | 1010(40) | 665 (14) | 1520(10) | 640 (30) | 402 | 322 | 252 | 148 |
| 25 | 750(20) | 490 (18) | 1530(50) | 620 (40) | 365 | 258 | 252 | 101 |
| 30 | 460(20) | 409 (15) | 1900(20) | 610 (30) | 255 | 229 | 248 | 154 |
| 35 | 400(20) | 307 (15) | 2670(50) | 670 (80) | 234 | 235 | 257 | 116 |
| 40 | 220(30) | 217 (35) | 3220(270) | 600 (50) | 146 | 99 | 281 | 101 |
| 45 | 270(30) | >100f | 3090(80) | 750 (40) | 187 | 10f | 259 | 151 |
| 50 | 150(30) | N.D.g | 3170(270) | 600 (40) | 100 | N.D.g | 239 | 88 |
| decay
timesa,b |
total
red shiftc,d |
|||||||
|---|---|---|---|---|---|---|---|---|
| T, °C | W167ine | +Y25A | +V260A | +Y25A: V260A | W167ine | +Y25A | +V260A | +Y25A: V260A |
| 10 | 1110(30) | 1670(90) | 1030 (10) | 830 (60) | 100 | 85 | 105 | 57 |
| 15 | 1150(90) | 1780(60) | 1110 (20) | 790 (50) | 120 | 99 | 113 | 52 |
| 20 | 920(120) | 1890(40) | 1250 (20) | 610 (30) | 114 | 85 | 118 | 57 |
| 25 | 1140(180) | 1640(70) | 1270 (20) | 830 (40) | 99 | 85 | 118 | 58 |
| 30 | 1010(50) | 1530(70) | 1240 (20) | 1000 (40) | 111 | 105 | 127 | 69 |
| 35 | 1060(140) | 1540(50) | 1250 (20) | 980 (80) | 109 | 93 | 130 | 71 |
| 40 | 1110(30) | 1310(60) | 1250 (10) | 970 (50) | 96 | 92 | 129 | 76 |
| 45 | 770(50) | 1680(90) | 1220 (20) | 870 (40) | 122 | 98 | 128 | 78 |
| 50 | N.D.g | N.D.g | 1250 (20) | 860 (40) | N.D.g | N.D.g | 129 | 75 |
Reported in ps.
Error reported in parentheses is the standard error in ps.
Reported in cm–1.
Typical error: ±20 cm–1.
From ref (26).
Rate constant faster than the time resolution of the measurement.
N.D. indicates not determined.
Figure 5.

Arrhenius plots of the temperature-dependent Stokes shift relaxation rates extracted from Figure 4 and Table 4. Each panel contains a comparison between the W87in (red) and the W167in (blue) parent construct (upper left panel); the additional insertions are noted at the top of the panel. The data for the parent constructs are from ref (26).
Table 5. Arrhenius Parameters of the Stokes Shift Relaxation Decay Rates.
| mutant | ΔH⧧, kcal/mol | TΔS⧧, kcal/molb |
|---|---|---|
| W87ina | 9.4 (0.3) | 34.2 (1.3) |
| W87in:Y25A | 7.0 (0.3) | 29.2 (0.5) |
| W87in:V260A | equilibrium | equilibrium |
| W87in:Y25A:V260A | –0.4 (1.0) | 11.4 (0.4) |
| W167ina | 1.3 (0.7) | 13.1 (1.0) |
| W167in:Y25A | 1.8 (0.6) | 13.9 (0.6) |
| W167in:V260A | –1.4 (0.6) | 9.6 (0.2) |
| W167in:Y25A:V260A | –1.6 (1.2) | 8.4 (0.6) |
From ref (26).
At 30 °C.
Of considerable interest is the obvious diversity of temperature-dependent behavior shown among the W87in, W87in:Y25A, W87in:V260A, and W87in:Y25A:V260A series. Paralleling the observed fluorescence lifetime behavior (Table 2), W87in:Y25A demonstrates the greatest similarities to W87in regarding the temperature dependence of its Stokes shift, exhibiting a monotonic decrease in decay times with temperature (Figure 5). However, its spectral shift is lower in magnitude than W87in and its decay rates are generally faster. This behavior leads to a relative decrease in the enthalpy of activation for the Stokes shifts to 7.0 ± 0.3 kcal/mol. Correspondingly, TΔS⧧ at 30 °C drops from 34.2 ± 1.3 kcal/mol for W87in to 29.2 ± 0.5 kcal/mol (Table 5).
Among the W87in mutant series, W87in:V260A exhibits the most unusual behavior regarding its temperature-dependent red shift (Figure 5). The magnitude of the total shift generally does not change with temperature between 10 and 50 °C (Table 4). The decay times between 10 and 25 °C show an average value of approximately 1550 ± 40 ps. Between 25 and 40 °C, the decay time gradually decreases, converging at an approximately 2-fold longer process than that observed at low temperatures (3150 ± 100 ps). The temperature-dependent titration in W87in:V260A reveals an apparent equilibrium between two distinct states with a midpoint at approximately 30 °C.
The temperature dependence of the red shift behavior in W87in:Y25A:V260A exhibits a feature quite different from either single mutation. The relaxation data are interpreted as effectively temperature-independent (ΔH⧧ ≈ −0.4 ± 1.0 kcal/mol) with an average decay time of 650 ± 55 ps at all temperatures. The spectral shifts incorporate features of both W87in:V260A and W87in:Y25A. Like W87in:V260A, the magnitude of the Stokes shift in the double mutant changes very little as a function of temperature. The double mutant’s resemblance to W87in:Y25A, however, originates from faster decay times relative to W87in (Table 4); additionally, there is no break in behavior with temperature (Figure 5).
The temperature-dependent behavior for time-dependent Stokes shifts in W167in, W167in:Y25A, W167in:V260A, and W167in:Y25A:V260A displays a very low barrier process with enthalpies of activation ranging from −1.6 to 1.8 kcal/mol. The only possible exception to these trends is the low temperature data in W167in:Y25A:V260A. At 10, 15, and 20 °C the Arrhenius behavior may suggest a more negative enthalpy of activation. However, the large relative error associated with rate constants for this variant, a function of the quite small magnitude of its Stokes shifts (Table 4), is more likely a consequence of poor signal-to-noise. Neglecting any low-temperature trend, the behavior of W167in:Y25A:V260A parallels that of the other W167in variants (Table 5). One potentially important observation to be noted from the W167in series derives from the absolute decay times. With the exception of 20 and 45 °C with W167in, it appears that W167in and W167in:V260A have very comparable decay times ranging from 1010 to 1270 ps. However, the two mutants additionally containing Y25A affect the decay times in opposite directions. In the double mutant, the average decay time decreases slightly to ca. 960 ps, while in W167in:Y25A it increases to ca. 1630 ps. While Val260 and Trp167 are located within the cofactor-binding domain, Tyr25, a residue in the substrate-binding domain ca. 44 Å away from Trp167 in the same subunit, has the greater effect on the lifetime of its Stokes shift.
Stern–Volmer Collisional Quenching
The temperature dependence of the collisional process between an excited state tryptophan and acrylamide was measured in all six single-Trp mutants and compared to W87in and W167in (Figure 6). A summary of the activation parameters associated with this process is given in Table 6. The temperature-dependent Stern–Volmer plots from which the bimolecular quenching constant, kq, is derived, are available in Figures S9 and S10 (Supporting Information). The rate constants for collisional quenching are found to be on the order of 108 M–1 s–1 at 30 °C for all eight variants, suggesting little change in accessibility of acrylamide to tryptophan under this condition.
Figure 6.

Arrhenius plots of the temperature-dependent Stern–Volmer collisional quenching rate constants extracted from Figures S9 and S10 in the Supporting Information. Each panel contains the W87in (red) and W167in (blue) parent construct (upper left panel), with the additional mutation noted at the top of the remaining panels.
Table 6. Temperature-Dependent Activation Parameters on kq Derived from Collisional Stern–Volmer Data.
| mutant | ΔH⧧a | TΔS⧧, 30 °Ca | kq × 10–8, 30 °Cb |
|---|---|---|---|
| W87inc | 3.4 (0.1) | 18.1 (0.2) | 3.2 (0.1) |
| W87in:Y25A | 7.8 (0.3) | 28.8 (0.5) | 4.7 (0.2) |
| W87in:V260A | 1.4 (0.1) | 13.6 (0.1) | 3.4 (0.1) |
| W87in:Y25A:V260A | 1.8 (0.1) | 14.7 (0.1) | 4.2 (0.2) |
| W167inc | 4.7 (0.1) | 21.8 (0.1) | 5.7 (0.1) |
| W167in:Y25A | 5.2 (0.1) | 23.2 (0.2) | 6.8 (0.2) |
| W167in:V260A | 4.2 (0.2) | 20.9 (0.4) | 6.5 (0.2) |
| W167in:Y25A:V260A | 4.2 (0.1) | 21.0 (0.2) | 7.1 (0.1) |
On the other hand, the temperature dependencies controlling quencher penetration to each tryptophan differs considerably. W167in and its mutant series exhibit enthalpies of activation ranging from 4.2 to 5.2 kcal/mol, all slightly elevated relative to acrylamide quenching of free N-acetyltryptophanamide of 3.7 kcal/mol.27 Though the slightly elevated values can be attributed to the lack of bulk solvent surrounding Trp167 due to shielding of this fluorescent probe by the opposite dimer, it is clear that any additional mutation associated with affecting catalysis has little effect on the accessibility of quencher to the surface tryptophan.
The change in the temperature-dependent quenching behavior occurring at the active-site tryptophan with inserted mutations is of greater interest. While W87in reveals a quenching behavior similar to that of free tryptophan, the presence of either Y25A or V260A demonstrates opposing effects relative to W87in. The barrier for quenching in W87in:Y25A was measured at nearly 8 kcal/mol, more than double than that observed for W87in. On the other hand, the enthalpy of activation for W87in:V260A was measured at only 1.4 ± 0.1 kcal/mol, lowering this barrier by more than 2-fold, relative to W87in. With an enthalpy of activation of 1.8 ± 0.1 kcal/mol, the quenching behavior of W87in:Y25A:V260A is found to be extremely similar to that observed in W87in:V260A. In sum, there appears to be an inverse correlation between the large enthalpies of activation in kinetic traces of V260A (Figure S1) and the temperature dependence of the Stern–Volmer quenching behavior at W87in.
Discussion
While much work has been performed on various protein systems using flourescence upconversion techniques on the fs-ps timescale,31−36 we have focused on studying flourescence lifetimes, time-dependent Stokes shifts, and collisional quenching data on the ns-ps timescale. These results show that while some noteworthy change can occur at Trp167 in the presence of catalysis-altering mutations, the aggregate data (representing the temperature dependence of all three fluorescence properties) rule out a significant disruption in the environment surrounding a remotely positioned tryptophan. Trp167 serves, thus, as an excellent control for the changes detected in the environment surrounding the active-site Trp87. Below, we highlight the major impact of mutation at Tyr25 and Val260 on the behavior of Trp87, discussing possible physical interpretations from the observed fluorescence properties. The data that form the focus of this discussion are summarized in Table 7.
Table 7. Summary of Time-Resolved Fluorescence Properties for the Trp87in Mutant Series.
| lifetimes |
Stokes
shifts |
collisional
quenching |
||||
|---|---|---|---|---|---|---|
| construct | observation of τ1 | observation of temperature transition | ΔH⧧, kcal/mol | observation of temperature transition | ΔH⧧, kcal/mol | observation of temperature transition |
| W87in | yes | yes | 9.4 | no | 3.4 | no |
| W87in:Y25A | yes | no | 7.0 | no | 7.8 | no |
| W87in:V260A | no | no | – | yes | 1.4 | no |
| W87in:Y25A:V260A | no | no | –0.4 | no | 1.8 | no |
Fluorescence Lifetimes
The fluorescence lifetime data indicate two modes of behavior for ht-ADH: the triexponential behavior observed in W87in and W87in:Y25A, and the biexponential behavior in the two mutants containing V260A. At the steady-state λmax for emission, the short lifetime component in W87in:Y25A, previously attributed to a Zn2+-Trp interaction, maintains a relative amplitude and lifetime similar to that observed in W87in.26 However, the clear-cut temperature break seen in W87in for τ1 is absent from W87in:Y25A (Figure S8). More insight regarding τ1 in W87:Y25A emerges when the lifetime is examined as a function of wavelength (Table 3). At 310 nm, the amplitude and lifetime measured for τ1 in W87in:Y25A are decreased relative to that observed for W87in alone, whereas at 370 nm both the amplitude and lifetime of the fast transient have increased for W87:Y25A. This trend suggests a time dependent alteration in the active-site configuration that is unique to W87in:Y25A, and results in a redistribution of the total fluorescence lifetimes. Together with the loss of a temperature break in τ1, these behaviors support the earlier proposal that Y25A alters active-site flexibility through an intervening network of β-sheets (Figure 1).24
After V260A has been inserted into W87in, the aggregate data indicate that the short, sub-ns lifetime component no longer contributes to the fluorescence properties. The remaining radiative processes observed in both W87in:V260A and W87in:Y25A:V260A appear to be tightly controlled by the presence of V260A because the amplitude and lifetimes measured are within error for both variants (Table 2). On top of the loss of τ1, the comparative lifetimes for τ2 and τ3 at all wavelengths suggest that packing defects within the cofactor-binding domain relay subtle information to the Trp87 at the substrate-binding domain that alters the distribution of its fluorescence processes. Moreover, in the presence of V260A, Y25A clearly has less control over the observed radiative processes at Trp87, as the double mutant more closely resembles W87in:V260A.
Temperature-Dependent Stokes Shifts
Some of the most compelling data gleaned from this study lie with the unique changes in Stokes shifts that Y25A, V260A, and Y25A:V260A relay to the active site. The Arrhenius behavior of W87in:Y25A exhibits a decreased enthalpic barrier for excited state reorganization of 7.0 kcal/mol (Table 7). This decrease, while modest relative to W87in, is consistent with previous implications of increased active-site flexibility facilitated by Y25A.24 In the case of W87in:V260A, Stokes shift data bear no resemblance to the patterns exhibited by W87in and W87in:Y25A. Its Arrhenius behavior, shown in Figure 5, indicates a temperature-dependent transition between two averaged conformational landscapes having inherently temperature-independent relaxation times of approximately 1550 ps at low temperature and 3150 ps at high temperature. In the region between 25 and 40 °C, a weighted average time constant reflects the shift between the two populations. This behavior can be directly related to the temperature-dependent equilibrium process that alters kinetic parameters above and below 30 °C, demonstrating a sensitivity at Trp87 to the alternate conformational space that determines the catalytic properties for both WT18 and the V260A variant.17 There is the question of why the enthalpy of activation is essentially zero for the relaxations observed in W87in:V260A, either above or below the transition temperatures. This behavior is closest to W167in and its mutated forms, suggesting that the structural disruption introduced at the cofactor-binding site has created an environment for Trp87 that is likely to be more solvent-exposed (see Collisional Stern–Volmer Quenching below). This structural deformation in the apo-enzyme must be sensitive to the presence of cofactor and substrate, since the kinetic behavior of V260A resembles WT ht-ADH at elevated temperatures.17
The Stokes shift data for the W87in:Y25A:V260A construct are equally puzzling. Once again, its Arrhenius behavior appears to resemble that observed in the W167in mutant series, containing the hallmark of small Stokes shifts and a temperature-independent relaxation barrier. However, the average relaxation time of 650 ps is considerably shorter than either of the relaxation times observed for W87in:V260A and there is no temperature break. Both of these features are consistent with the growing body of evidence for increased flexibility at the substrate-binding site in Y25A.24 While Y25A communicates increased active-site flexibility and V260A introduces active-site impairment, the combination yields a variant in which the active-site fluorescence at Trp87 now closely resembles the surface behavior of Trp167 (Table 5). The observed sensitivity of Trp87 to the remote mutational sites, either individually or in tandem, demonstrates a “fluid” native protein structure with an active site that can be easily perturbed into alternate conformational substates while simultaneously leaving Trp167 at the protein surface relatively unaffected.
Collisional Stern–Volmer Quenching
In the case of the W87in construct, Y25A and V260A exhibit opposing temperature-dependent trends. That is, W87in:Y25A possesses a barrier for acrylamide quenching (7.8 kcal/mol) that is more than double that observed for W87in (3.4 kcal/mol). By contrast, W87in:V260A has a barrier that is less than half that of W87in for the same process (1.4 kcal/mol). The latter behavior is fully consistent with the temperature independence of Stokes shifts at W87in in the presence of V260A, suggesting a marked change in the environment surrounding Trp87. It would appear that conformations for the constructs containing V260A lead to Trp87 being further from charged residues and in closer proximity to hydrophobic side chains, while Y25A retains the WT-dipolar interactions between the excited state chromophore and active-site side chains within a more dynamical environment. The latter may provide access of Trp87 to positions with increased hydrogen-bonding and/or polar interactions that would decrease access to acrylamide and increase the thermal activation for quenching. With regard to catalytic function in the temperature range of 5 to 50 °C, the introduction of Y25A has been concluded to introduce sufficient flexibility that the protein can avoid being trapped into the reduced activity configurations observed for native enzyme below 30 °C.24 In the case of V260A, the change in environment within the active site detected by fluorescence measurements, when coupled with reduced temperatures (<30 °C), appears too severe to be reversed by cofactor and substrate-binding, leading to the characteristic signature of greatly elevated values for the enthalpy and entropy of activation that control hydride transfer.17,37
Further indication that collisional quenching data with acrylamide can mirror catalytic features comes from W87in:Y25A:V260A. This variant exhibits a signature break at 35 °C with a low temperature enthalpy of activation of 31 kcal/mol, mirroring that of W87in:V260A (Figure S3). At the same time, it exhibits a temperature-dependent fluorescent quenching barrier of 1.8 kcal/mol, a value almost within error of that measured in W87in:V260A.
Progress Toward Understanding Networks of Protein Motion in ht-ADH
One of the significant findings from this study is that a packing defect introduced at Val260 affects the local fluorescence phenomenon of Trp87 over 11 Å away and across separate enzyme domains. Though not as highly structured as the β-sheet network of residues connecting Tyr25 at the subunit interface and Trp87, there is a plausible network of residues linking Val260 to Trp87 through a combination of van der Waals and hydrogen bonding interactions (Figure 7, top). Ternary complex structures of a prokaryotic ADH show the equivalent of Val260 and Leu176 engaging in side chain packing interactions against the amide group and nicotinamide ring of NAD+,38 while kinetic studies of ht-ADH indicate a role for both Val260 and Leu176 in controlling the low-temperature enthalpy of activation. The network illustrated in Figure 7 for ht-ADH suggests that two additional residues, Val286, ca. 4 Å from Trp87, or Thr152, which resides on an α-helix that topologically divides the substrate and cofactor catalytic domains, may be critical in conveying structural/dynamical information between the substrate and cofactor domains. It is proposed that packing defects arising from mutation at Val260 (or putatively Leu176) disrupt this network, and move it away from the native interactions sampled by W87in. This change in communication between domains at the intersection of the active site is likely to be a major factor in producing the exaggerated enthalpies of activation observed below 30 °C for V260A and Leu176A.
Figure 7.
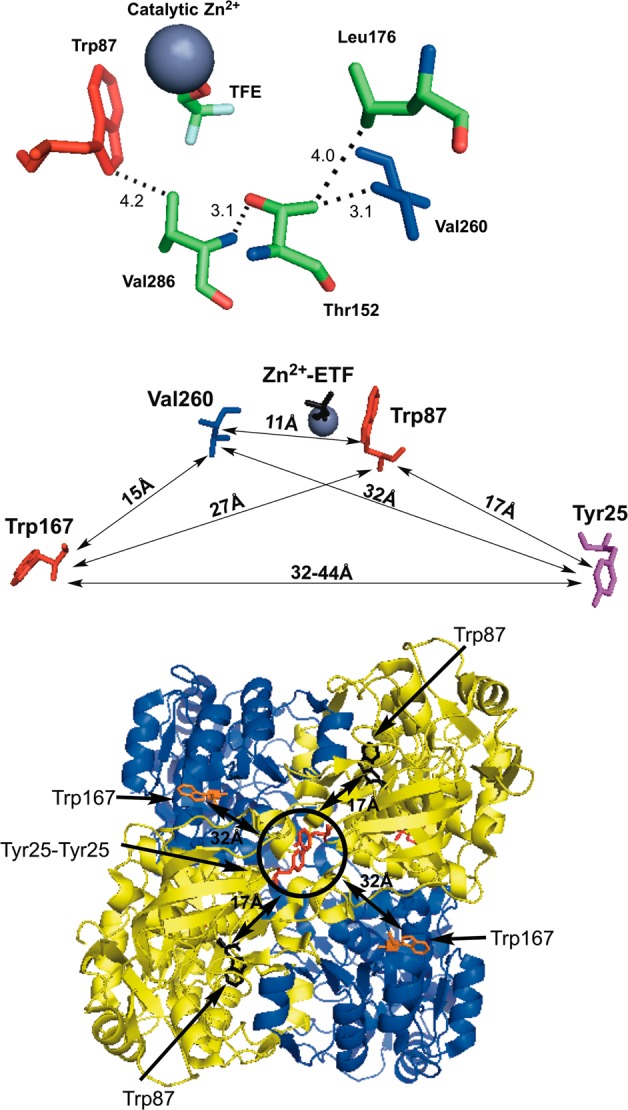
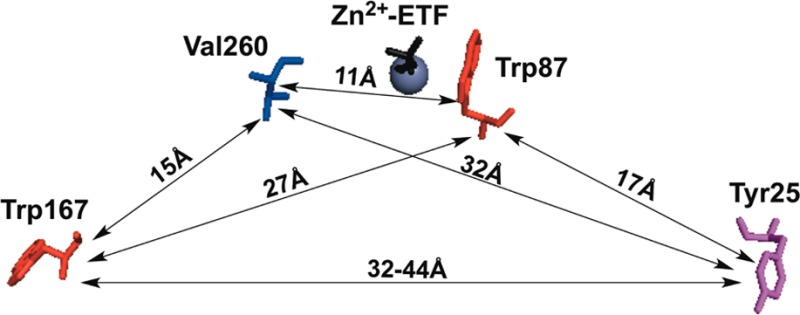
Residues within ht-ADH shown to be dynamically linked. (Top) Close-up of the network connecting Val260 (blue) in the cofactor-binding domain to Trp87 (red) in the substrate-binding domain. The numbers indicate the distance in angstroms between atoms connected to their respective residues. The modeled substrate, trifluorethanol, is abbreviated TFE. (Middle) Map of intra-subunit protein residues showing the relative distances among the single-Trp probes (red) at positions 87 and 167 relative to the catalytically relevant residues of Tyr25 (plum) and Val260 (blue). (Bottom) Birds-eye view of the tetrameric ht-ADH structure looking down on the crystal structure’s axis of symmetry. The circled Tyr25 π-stacking interaction (red sticks) belongs to both gold monomer subunits. Though intrasubunit distances between Tyr 25 and Trp167 are measured at ca. 44 Å (cf. Middle), the distance to Trp167 (orange sticks) on the opposite blue monomers is roughly 32 Å. For reference, the Trp87 (black sticks) is shown at a distance of 17 Å to Tyr25.
With the data presented herein, we now have three independent probes of the temperature-dependent transition in the properties of ht-ADH: the time-dependent fluorescence of the present study (apo-enzyme), HDX (apo-enzyme)21,38 and catalysis (ternary complex).17,37 While the limits of the experimental measurements of HDX39 and tryptophan fluorescence26 have precluded in-depth studies of enzyme cofactor complexes, the catalytically relevant transition of ca. 30 °C can be detected within apoenzyme in both instances. The previously measured patterns of HDX provided spatial resolution that identified a series of β-sheets regulating communication between the dimer interface and substrate-binding domain.21,24 Until the completion of this work, HDX was the only structural evidence that related networks of amino acid residues in ht-ADH to the distribution of conformational microstates as a function of temperature.
Our ability to examine the sensitivity of spatially distinctive fluorescent probes at the solvent interface (W167in) and the active site (W87in) to catalysis-altering mutations has both confirmed long distance communication from the dimer interface to the substrate binding site24 and uncovered a number of previously uncharacterized networks of communication throughout the enzyme. Though Tyr25 is ca. 17 Å from the active-site tryptophan (Figure 7, middle), insertion of Y25A at the dimer interface (Figure 7, bottom) introduces notable changes to the observed fluorescent lifetimes and their respective weighted amplitude for Trp87 (Table 3). The temperature dependence of the Stokes shift measurements also indicates a decreased barrier for excited-state reorganization around the active-site tryptophan in W87in:Y25A (Table 5), mainly arising from accelerated relaxation times (Table 4). Such data corroborate previous HDX21 and kinetic24 data that suggested a long-range interplay between Tyr25 and Trp87 mediated by a network of β-sheets connecting the dimer interface to the active site. In addition to the accelerated relaxation times observed at Trp87, it appears that Y25A invokes some mode of communication that somewhat retards the relaxation rates around the surface tryptophan at position 167 (Table 4). Such a feature may be better rationalized from the tetrameric structure of ht-ADH, where Tyr25 and Trp167 share a more proximal mode of communication between dimers (ca. 32 Å apart) than within each monomer (ca. 45 Å apart).
An additional and significantly different pattern of communication exists between Val260 and Trp87, specifically a temperature dependence of the Stokes shift relaxation which results in an active-site titration between two distinct forms of enzyme with a midpoint at ca. 30 °C (Table 4 and Figure 5). To our knowledge, such a phenomenon has not been previously detected by Stokes shift measurements of proteins and is the most direct spectroscopic evidence for a temperature-dependent equilibration between multiple forms of enzyme in ht-ADH. Moreover, this behavior unveils a mode of communication that connects the substrate and cofactor-binding domains, revealing new possibilities for how the equilibrium between active and inactive conformers is mediated at low temperature. That Tyr25 and Val260, 17 and 11 Å from Trp87, respectively (Figure 7 middle), can communicate to the active site and modulate relaxation phenomena raises a multitude of questions regarding the modes of protein motion sampled and the associated ensemble of microstates that are perturbed at low temperature and/or upon mutation in ht-ADH. Such questions are germane to a plethora of biochemical phenomena, that include protein–protein interactions,40−42 allostery,43−45 ligand binding,46−48 and proteins in crowded cellular environments.49,50
Conclusion
The data from this work extend our previous efforts to uncover the dynamical and structural changes in ht-ADH that alter the properties of hydrogen tunneling properties above and below a breakpoint of 30 °C.17,18,21,37 The relatively slow time scales of earlier studies, millisecond for catalysis and up to hours for HDX reflect the probability of achieving the subset of protein conformational states amenable to the measured parameter. By contrast, the picosecond-resolution fluorescence measurements presented herein, by virtue of their rapidity, are expected to be fairly local51 and too rapid to allow protein re-equilibration on the time scale of the measurements. Curiously, in no instance to date has the actual transition between the low and high temperature regimes of distinctive protein conformational landscapes in ht-ADH been measured in real time. Nonetheless, the available measurements are extremely informative, as shown by their ability to detect and extend a pattern of long-range interactions. Perhaps the most important conclusion to emerge from the present studies is the exquisite sensitivity of fluorescent probes to mutations up to 30–40 Å away (Figure 7, middle), thereby providing a roadmap for the long-range structural networks that determine active-site catalytic efficiency.
Acknowledgments
This work was funded by the National Institutes of Health (GM025765 and GM025765-32S1 to J.P.K.).
Supporting Information Available
Steady-state fluorescence spectra, fluorescence lifetime data for W87in:Y25A, time-resolved emission spectra, Stern–Volmer plots, enzyme kinetics, and gel-filtration data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Meyer M. P.; Tomchick D. R.; Klinman J. P. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Goodey N. M.; Benkovic S. J.; Kohen A. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 15753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Abeysinghe T.; Finer-Moore J. S.; Stroud R. M.; Kohen A. J. Am. Chem. Soc. 2012, 134, 17722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rod T. H.; Radkiewicz J. L.; Brooks C. L. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson C. J.; Foo J. L.; Tokuriki N.; Afriat L.; Carr P. D.; Kim H. K.; Schenk G.; Tawfik D. S.; Ollis D. L. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 21631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards R. A.; Whittaker M. M.; Whittaker J. W.; Baker E. N.; Jameson G. B. Biochemistry 2001, 40, 4622. [DOI] [PubMed] [Google Scholar]
- Ghanem M.; Li L.; Wing C.; Schramm V. L. Biochemistry 2008, 47, 2559. [DOI] [PubMed] [Google Scholar]
- Saen-Oon S.; Ghanem M.; Schramm V. L.; Schwartz S. D. Biophys. J. 2008, 94, 4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser J. S.; Clarkson M. W.; Degnan S. C.; Erion R.; Kern D.; Alber T. Nature 2009, 462, 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P. K.; Geist A.; Gorin A. Biochemistry 2004, 43, 10605. [DOI] [PubMed] [Google Scholar]
- Eisenmesser E. Z.; Millet O.; Labeikovsky W.; Korzhnev D. M.; Wolf-Watz M.; Bosco D. A.; Skalicky J. J.; Kay L. E.; Kern D. Nature 2005, 438, 117. [DOI] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Acc. Chem. Res. 2006, 39, 93. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K. A.; Thai V.; Lei M.; Ott M.; Wolf-Watz M.; Fenn T.; Pozharski E.; Wilson M. A.; Petsko G. A.; Karplus M.; Hubner C. G.; Kern D. Nature 2007, 450, 838. [DOI] [PubMed] [Google Scholar]
- Baldwin A. J.; Kay L. E. Nat. Chem. Biol. 2009, 5, 808. [DOI] [PubMed] [Google Scholar]
- Min W.; Xie X. S.; Bagchi B. J. Phys. Chem. B 2008, 112, 454. [DOI] [PubMed] [Google Scholar]
- Schrank T. P.; Bolen D. W.; Hilser V. J. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 16984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel Z. D.; Dong M.; Bahnson B. J.; Klinman J. P. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohen A.; Cannio R.; Bartolucci S.; Klinman J. P. Nature 1999, 399, 496. [DOI] [PubMed] [Google Scholar]
- Limbach H. H.; Lopez J. M.; Kohen A. Philos. Trans. R. Soc. B-Biol. Sci. 2006, 361, 1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truhlar D. G.; Kohen A. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z.-X.; Lee T.; Resing K. A.; Ahn N. G.; Klinman J. P. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 9556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoofnagle A. N.; Resing K. A.; Ahn N. G. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 1. [DOI] [PubMed] [Google Scholar]
- Ceccarelli C.; Liang Z.-X.; Strickler M.; Prehna G.; Goldstein B. M.; Klinman J. P.; Bahnson B. J. Biochemistry 2004, 43, 5266. [DOI] [PubMed] [Google Scholar]
- Nagel Z. D.; Cun S.; Klinman J. P. J. Biol. Chem. 2013, 288, 14087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahnson B.; Colby T.; Chin J.; Goldstein B.; Klinman J. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows C. W.; Ou R.; Klinman J. P. J. Phys. Chem. B 2014, 6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftink M. R.; Ghiron C. A. Biochemistry 1977, 16, 5546. [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R.Principles of fluorescence spectroscopy, 3rd ed.; Springer: New York, 2006. [Google Scholar]
- Horng M. L.; Gardecki J. A.; Maroncelli M. J. Phys. Chem. A 1997, 101, 1030. [Google Scholar]
- Badea M. G.; Brand L. Methods Enzymol. 1979, 61, 378. [DOI] [PubMed] [Google Scholar]
- Pal S. K.; Peon J.; Bagchi B.; Zewail A. H. J. Phys. Chem. B 2002, 106, 12376. [Google Scholar]
- Qiu W.; Zhang L.; Okobiah O.; Yang Y.; Wang L.; Zhong D.; Zewail A. H. J. Phys. Chem. B 2006, 110, 10540. [DOI] [PubMed] [Google Scholar]
- Qiu W.; Wang L.; Lu W.; Boechler A.; Sanders D. A.; Zhong D. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Zhang L.; Wang L.; Zhong D. J. Am. Chem. Soc. 2012, 134, 16460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon O. H.; Yoo T. H.; Othon C. M.; Van Deventer J. A.; Tirrell D. A.; Zewail A. H. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 17101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. W.; He T. F.; Guo L.; Stevens J. A.; Li T.; Wang L.; Zhong D. J. Am. Chem. Soc. 2010, 132, 12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel Z. D.; Meadows C. W.; Dong M.; Bahnson B. J.; Klinman J. P. Biochemistry 2012, 51, 4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z.-X.; Tsigos I.; Lee T.; Bouriotis V.; Resing K. A.; Ahn N. G.; Klinman J. P. Biochemistry 2004, 43, 14676. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Q.; Wietgrefe S. W.; Li Q.; Shore M. D.; Duan L.; Reilly C.; Lifson J. D.; Haase A. T. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S.; Thornton J. M. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 13.8552589 [Google Scholar]
- Maslov S.; Sneppen K. Science 2002, 296, 910. [DOI] [PubMed] [Google Scholar]
- Hilser V. J.; Dowdy D.; Oas T. G.; Freire E. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern D.; Zuiderweg E. R. P. Curr. Opin. Struct. Biol. 2003, 13, 748. [DOI] [PubMed] [Google Scholar]
- Gunasekaran K.; Ma B. Y.; Nussinov R. Proteins: Struct., Funct. Bioinf. 2004, 57, 433. [DOI] [PubMed] [Google Scholar]
- Popovych N.; Sun S. J.; Ebright R. H.; Kalodimos C. G. Nat. Struct. Mol. Biol. 2006, 13, 831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.-J.; Lai C.-T.; Pan P.; Yu W.; Liu N.; Bommineni G. R.; Garcia-Diaz M.; Simmerling C.; Tonge P. J. ACS Chem. Biol. 2014, 9, 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr D. D.; Nussinov R.; Wright P. E. Nat. Chem. Biol. 2009, 5, 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobi D.; Bahar I. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 18908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar A.; Samiotakis A.; Ebbinghaus S.; Nienhaus L.; Homouz D.; Gruebele M.; Cheung M. S. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 17586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cino E. A.; Karttunen M.; Choy W. Y. PLoS One 2012, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The distance dependence between the light-induced dipole and regions of charge within the protein can be estimated: r–6 for induced dipole–induced dipole, r–6 for induced dipole–dipole, and r–4 for induced dipole–fixed charge interactions.Anslyn E. V.; Dougherty D. A.. Modern Physical Organic Chemistry, 1st ed.; University Science Books: Sausalito, CA, 2006, p 188. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.