

Abstract
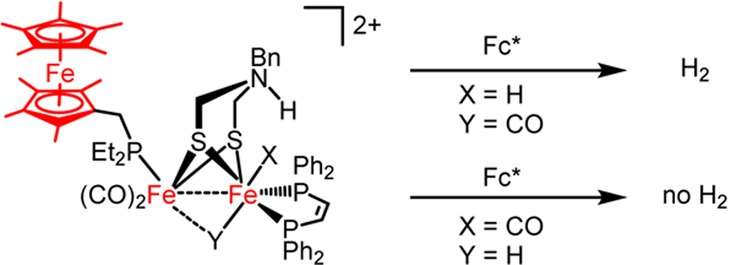
Active site mimics of [FeFe]-hydrogenase are shown to be bidirectional catalysts, producing H2 upon treatment with protons and reducing equivalents. This reactivity complements the previously reported oxidation of H2 by these same catalysts in the presence of oxidants. The complex Fe2(adtBn)(CO)3(dppv)(PFc*Et2) ([1]0; adtBn = (SCH2)2NBn, dppv = cis-1,2-bis(diphenylphosphino)ethylene, PFc*Et2 = Et2PCH2C5Me4FeCp*) reacts with excess [H(OEt2)2]BArF4 (BArF4– = B(C6H3-3,5-(CF3)2)4–) to give ∼0.5 equiv of H2 and [Fe2(adtBnH)(CO)3(dppv)(PFc*Et2)]2+ ([1H]2+). The species [1H]2+ consists of a ferrocenium ligand, an N-protonated amine, and an FeIFeI core. In the presence of additional reducing equivalents in the form of decamethylferrocene (Fc*), hydrogen evolution is catalytic, albeit slow. The related catalyst Fe2(adtBn)(CO)3(dppv)(PMe3) (3) behaves similarly in the presence of Fc*, except that in the absence of excess reducing agent it converts to the catalytically inactive μ-hydride derivative [μ-H3]+. Replacement of the adt in [1]0 with propanedithiolate (pdt) results in a catalytically inactive complex. In the course of synthesizing [FeFe]-hydrogenase mimics, new routes to ferrocenylphosphine ligands and nonamethylferrocene were developed.
Introduction
In recent years models for the active sites of the hydrogenase (H2ase) enzymes have been developed with respect to structural and, to a lesser extent, functional fidelity. This progress is a result of our deepening biophysical knowledge of the enzymes1−3 and innovations in synthetic organometallic chemistry.4−6 Early work showed that complexes of the type Fe2(dithiolate)(CO)6 are electrocatalysts for the hydrogen evolution reaction (HER).7 Catalysis proceeds via initial reduction of the FeFe core followed by protonation. The mechanism for HER is quite different for FeFe dithiolates substituted with multiple phosphine or cyanide ligands. Catalysis by such electron-rich complexes begins with protonation followed by reduction of the intermediate diferrous μ-hydride.4,5 These early designs have been superseded, at least with respect to HER, by diferrous complexes with biomimetic stereochemistry featuring a terminal hydride adjacent to the aminodithiolate (adt) cofactor (Figure 1).8 The essential nature of this cofactor was recently confirmed by reconstitution of the apoenzyme with [Fe2[(SCH2)2X](CN)2(CO)4]2–.9,10 Efficient hydrogen evolution was only observed in the case of X = NH, even though the protein also assembled for derivatives where X = O, CH2.
Figure 1.
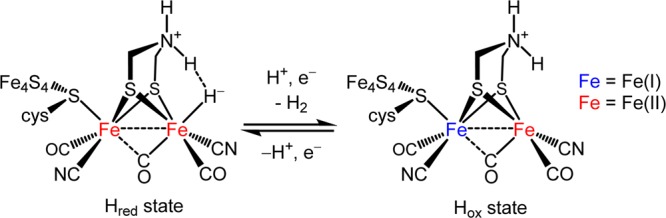
Active site of [FeFe]-H2ase in two catalytically significant states. The location and presence of some H atoms remain speculative.
Almost all active site models for the [FeFe]- and [NiFe]-H2ases are electrocatalysts for the HER. The enzymes are however bidirectional, catalyzing both the oxidation of H2and the reduction of protons. For both enzyme classes, the relative rates of these two reactions can differ by an order of magnitude, but both rates are rather fast and proceed at low overpotentials.1,11 Described here is the first example of bidirectional catalysis by a biomimetic [FeFe]-hydrogenase model.
In a recent report, the model complex Fe2(adtBn)(CO)3(dppv)(PFc*Et2) ([1]0) was shown to catalyze the oxidation of H2 in the presence of ferrocenium (Fc+ = FeCp2+) and base. Although the rates are modest compared to those for the enzyme, multiple turnovers were achieved,12 and this methodology has been expanded.13 Catalysis by this compound requires a redox agent covalently attached to the FeFe center, as oxidation of hydrogen by Fe2(adtBn)(CO)3(dppv)(PMe3) (3) is stoichiometric in the presence of external oxidant.14 That work was guided by knowledge that the H2/2H+ interconversion requires two redox equivalents and only one of these equivalents is provided by the FeIFeI/FeIFeII couple. This 1e−-couple ranges from −0.5 to −1.2 V (all potentials in this paper are referenced to Fc+/0) for complexes of the type Fe2(SR)2(CO)3(PR3)3 and Fe2(SR)2(CO)2(PR3)4. In our model systems, the second redox equivalent is supplied by ferrocenium reagents, whereas in the enzyme the appended [4Fe-4S] cluster supplies the second redox equivalent. The same principles apply to HER: the single reducing equivalent derived from the diiron(I) core must be supplemented.
Thermodynamic considerations show that, in MeCN solution, HER is favorable at potentials more negative than −0.026 V, depending on the acid’s strength.15,16 With a potential of ∼−0.5 V, the Fc*+/0 couple should be sufficient to simulate the role of the 4Fe-4S cluster. Thus, inclusion of an appropriate ferrocenylphosphine ligand into [FeFe]-H2ase models may enable HER in addition to the previously established H2 oxidation reaction. Several FeFe models were studied in the course of this work, and their structures are shown in Figure 2.
Figure 2.
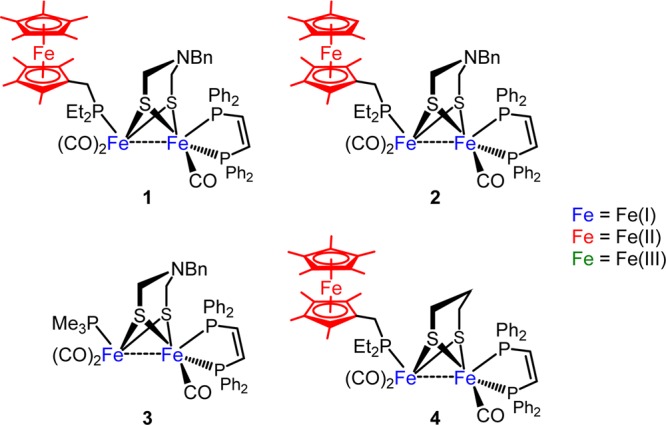
Model FeFe complexes studied in the course of this work. Oxidation states of Fe atoms are denoted by color.
Results
Synthesis and Acid−Base Properties of Redox-Active Phosphine Ligands
A new synthesis of ferrocenylphosphines was developed that allows for variation of the substituents on both the phosphine and the cyclopentadienyl groups (Scheme 1). The new methods improve upon the synthesis of Fe(C5Me5)(C5Me4CH2PEt2) (PFc*Et2),12 which suffers from the need to generate the unstable intermediate [Cp*Fe(μ-Cl)]2.
Scheme 1. Synthesis of PFc*R2 Ligands.

The new method builds on the versatile chemistry of formyl ferrocenes. With octamethylferrocene (Fc#, Fe(C5Me4H)2) as the starting material, a modified Vilsmeier reaction afforded the aldehyde Fe(C5Me4H)(C5Me4CHO)(FcMe8CHO). Reduction of this aldehyde with either LiAlH4 or LiBEt3H efficiently generated the alcohol FcMe8CH2OH.17
This work uncovered an improved route to nonamethylferrocene,18 which is otherwise difficult to prepare. Experiments revealed that borane–tetrahydrofuran cleanly converts FcMe8CHO (as well as FcMe8CH2OH)) into Fe(C5Me4H)(C5Me5)(FcMe9).19,20 The electrochemical potential of FcMe9 was investigated. While it is assumed that each additional methyl group attached to the ferrocene rings lowers the oxidation potential by roughly 50 mV, mainly symmetrically methylated ferrocenes have been studied.21 At −490 mV in CH2Cl2/[Bu4N]PF6, the [FcMe9]+/0 couple was found to lie almost exactly between the [FcMe8]+/0 (−440 mV) and [FcMe10]+/0 (−550 mV) couples.
Like Fc#, FcMe9 is amenable to functionalization via the Vilsmeier reaction to give FcMe9CHO. The reported oxidation of Fc* to the formyl derivative proceeded inefficiently in our hands.22 In near-quantitative yields, the Vilsmeier route produced FcMe9CHO, which can be efficiently reduced to FcMe9CH2OH.
Protonation of the ferrocenyl alcohols generates the corresponding cationic fulvene complexes,17 which readily add secondary phosphines to give the targeted redox-active tertiary phosphines. In addition to PFc*Et2, related ligands were prepared, including PFc#Et2 from FcMe8CH2OH and PFc*Cy2 and PFc*Ph2 from FcMe9CH2OH. Crystallographic analyses of PFc#Et2, PFc*Et2, and [PFc*Et2]BF4 confirmed the close similarity of the neutral and oxidized ligands (Figures 3–5), which highlights the small reorganization energies associated with oxidation of ferrocenes.
Figure 3.
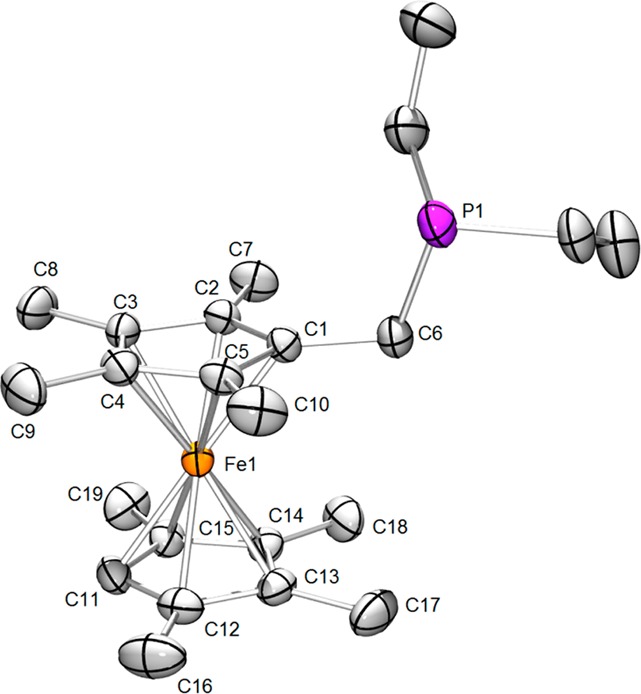
Structure of PFc#Et2. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at the 50% probability level. Selected distances (Å): Fe1–C1, 2.057(2); Fe1–C2, 2.054(2); Fe1–C3, 2.052(2); Fe1–C4, 2.049(2); Fe1–C5, 2.053(2); Fe1–C11, 2.048(2); Fe–C12, 2.056(2); Fe1–C13, 2.060(2); Fe1–C14, 2.055(2); Fe1–C15, 2.057(2); Fe1–centroid (C1–C5), 1.654(2); Fe1–centroid (C11–C15), 1.658(2); C1–C6, 1.502(3); C6–P1, 1.865(2).
Figure 5.
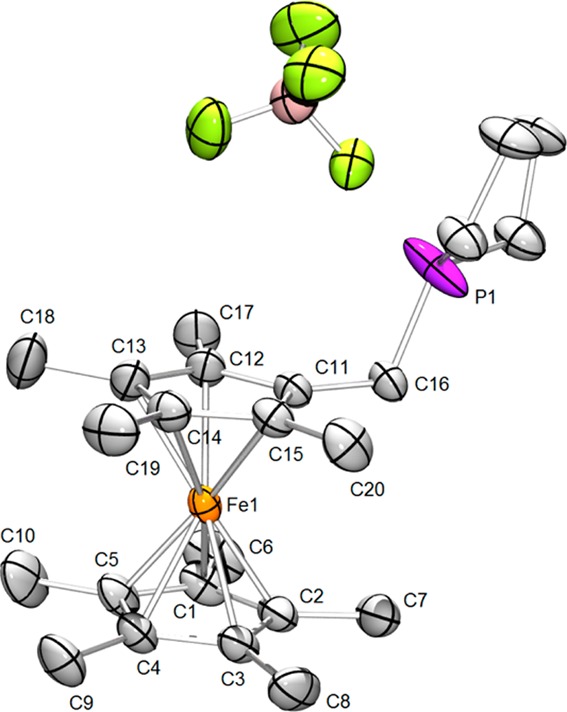
Structure of [PFc*Et2]BF4. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at the 50% probability level. Selected distances (Å): Fe1–C1, 2.062(2); Fe1–C2, 2.058(2); Fe1–C3, 2.057(2); Fe1–C4, 2.0567(18); Fe1–C5, 2.0553(19); Fe1–C11, 2.0419(18); Fe–C12, 2.0507(18); Fe1–C13, 2.061(2); Fe1–C14, 2.072(2); Fe1–C15, 2.0605(18); Fe1–centroid (C1–C5), 1.661(2); Fe1–centroid (C11–C15), 1.657(2); C11–C16, 1.508(2); C16–P1, 1.768(5).
Figure 4.
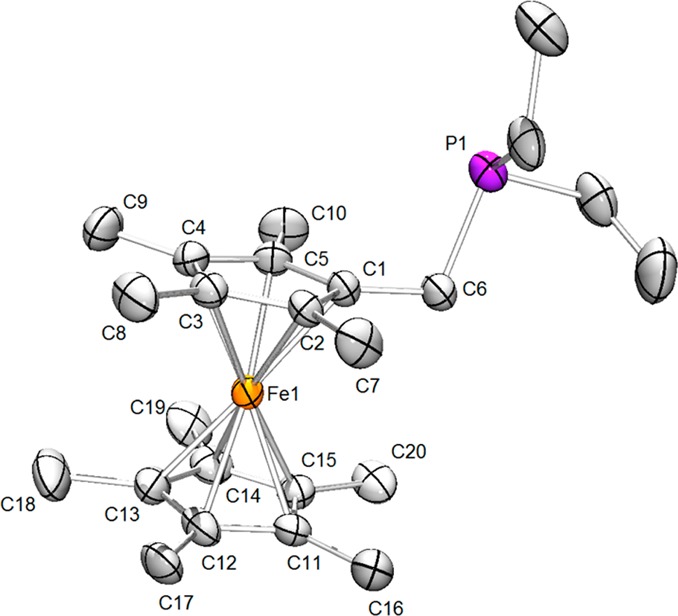
Structure of PFc*Et2. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at the 50% probability level. Selected distances (Å): Fe1–C1, 2.055(2); Fe1–C2, 2.049(2); Fe1–C3, 2.055(2); Fe1–C4, 2.058(2); Fe1–C5, 2.053(2); Fe1–C11, 2.048(2); Fe–C12, 2.042(2); Fe1–C13, 2.052(2); Fe1–C14, 2.065(2); Fe1–C15, 2.062(2); Fe1–centroid (C1–C5), 1.656(2); Fe1–centroid (C11–C15), 1.661(2); C1–C6, 1.496(3); C6–P1, 1.858(3).
The redox properties of the four ferrocenyl ligands were investigated by cyclic voltammetry (Table 1). Removal or addition of a methyl group of the cyclopentadienyl rings shifts the redox potential by 50 and 55 mV in [Bu4N]PF6 and [Bu4N]BArF4,23 respectively. The substituents on phosphorus only weakly affect the redox couples.
Table 1. Redox Potentials of Ferrocenes and Phosphine Derivativesa.
| ferrocene | E1/2, mV vs Fc0/+, [Bu4N]PF6 (ΔEp, mV) | ipa/ipc | E1/2, mV vs Fc0/+, [Bu4N]BArF4 (ΔEp, mV) | ipa/ipc |
|---|---|---|---|---|
| Fc# | –440 (86) | 0.96 | –500 (61) | 0.95 |
| FcMe9 | –490 (74) | 0.88 | –556 (60) | 0.95 |
| PFc#Et2 | –475 (76) | 1.0 | –536 (63) | 0.98 |
| PFc*Et2 | –524 (74) | 0.97 | –591 (61) | 0.9512 |
| PFc*Cy2 | –539 (82) | 0.96 | –602 (63) | 0.96 |
| PFc*Ph2 | –501 (61) | 0.98 | –572 (50) | 0.95 |
| Fc* | –550 (59) | 1.0 | –61023 | n/a |
Conditions: 1 mM analyte, CH2Cl2 solvent, 0.1 M [Bu4N]PF6 or 0.025 M [Bu4N]BArF4.
The acid–base properties of PFc*Et2 were examined. This ligand undergoes protonation upon treatment with [NPh2H2]BArF4 (pKaMeCN = 5.97),24 as observed by 31P{1H} and 1H NMR spectroscopy. Solutions of Fc* are reported to evolve hydrogen in the presence of strong acids,25−27 but a solution of PFc*Et2, Fc* (5 equiv) and HBF4·Et2O (10 equiv) does not produce any observable hydrogen at −15 °C.
Structure of [1]0
The structure of [1]0 was confirmed by X-ray crystallography (Figure 6). Like related derivatives of the type Fe2(dithiolate)(CO)3(chel)(PR3),28,29 the monophosphine (PFc*Et2) occupies an apical position on the Fe(CO)2 site, while the dppv ligand spans apical and basal sites. The ferrocenyl substituent is oriented away from the FeFe center.
Figure 6.
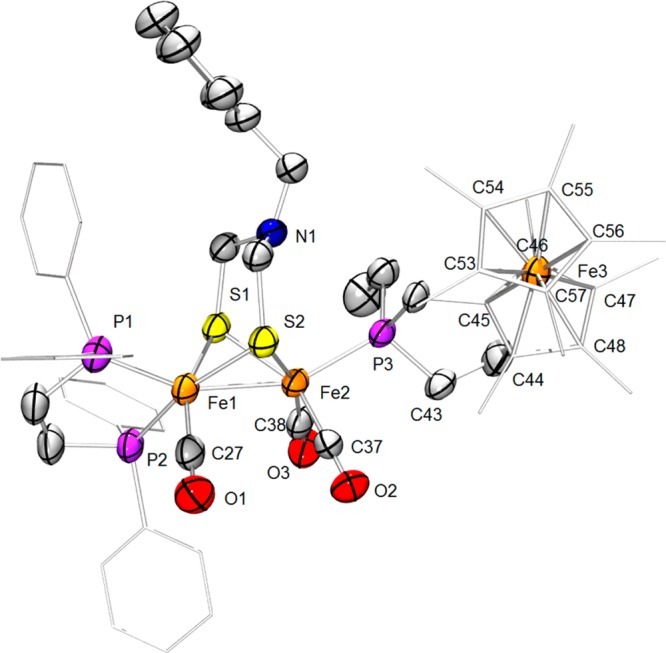
Structure of 1. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at the 50% probability level. Phenyl groups and ferrocenyl backbone have been simplified for clarity. Selected distances (Å): Fe1–Fe2, 2.5379(7); Fe1–S1, 2.251(1); Fe1–S2, 2.269(1); Fe2–S1, 2.286(1); Fe2–S2, 2.270(1); Fe1–P1, 2.179(1); Fe1–P2, 2.200(2); Fe1–C27, 1.749(4); C27–O1, 1.157(4); Fe2–C37, 1.746(3); Fe2–C38, 1.751(6); C37–O2, 1.159(4); C38–O3, 1.154(7); Fe2–N1, 3.343(7); Fe2–P3, 2.229(1); Fe3–C44, 2.07(3); Fe3–C45, 2.01(1); Fe3- C46, 2.02(1); Fe3–C47, 2.04 (1); Fe3–C48, 2.05 (1); Fe3–C53, 2.028(5); Fe3–C54, 2.023(7); Fe3–C55, 2.024(8); Fe3–C56, 2.05(1); Fe3–C57, 2.064(6); Fe3–centroid (C53–C57), 1.646(7); Fe3–centroid (C44–C48), 1.64(1); C43–C44, 1.508(2); C43–P1, 1.768(5).
Protonation of Fe2(adtBn)(CO)3(dppv)(PFc*Et2): High and Low Acid Scenarios
A noteworthy feature of [1]0 is its ability to reduce protons to H2 directly: i.e., without the addition of reductant. The yields of the HER are sensitive to several factors: order of addition of reagents, stoichiometry, and the presence of reducing equivalents. Treatment of a CH2Cl2 solution of [1]0 at −15 °C with 1 equiv of [H(OEt2)2]BArF4 afforded the bridging hydride complex [μ-H1]+ over the course of several hours. When this reaction was monitored by IR or NMR spectroscopy, rapid formation of the ammonium derivative [1H]+ was observed. Over the course of 30 min at room temperature, [1H]+ isomerizes to [μ-H1]+ via a first-order pathway with a rate constant of 2.2(2) × 10–4 s–1 at 0 °C (Supporting Information). All subsequent protonation experiments were undertaken at −15 °C in order to minimize the isomerization of [1H]+ to [μ-H1]+.
Although treatment of [1]0 with 1 equiv of [H(OEt2)2]BArF4 gave no H2, different results were obtained upon addition of excess acid to [1]0. With the addition of ≥2 equiv of [H(OEt2)2]BArF4, H2 yields reached 0.5 equiv (Table 2). Similarly, H2 is also produced when acid is added to a solution of [1H]+ (before isomerization to [μ-H1]+). The bridging hydride complex [μ-H1]+ does not yield H2 upon further treatment with [H(OEt2)2]BArF4, even in the presence of Fc* (Figure 7).
Table 2. Yield of H2 from the Reaction of [1]0 (4.2 mM in CH2Cl2) with Various Amounts of [H(OEt2)2]BArF4a.
| amt of H+, equiv | amt of H2, equiv |
|---|---|
| 2.0 | 0.30 ± 0.09 |
| 5.0 | 0.45 ± 0.08 |
| 10.0 | 0.56 ± 0.09 |
| 20.0 | 0.47 ± 0.07 |
GC analyses were performed 30 min after addition of components. Yields were obtained in triplicate unless otherwise noted (see the Experimental Section).
Figure 7.
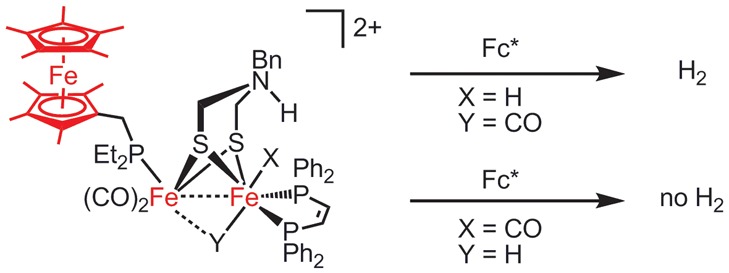
Fc*-triggered catalytic hydrogen evolution occurring from the terminal hydride [term-H1H]2+, not from the isomeric bridging hydride [μ-H1H]2+.
Mixed-Valence Species [Fe2(adtBnH)(CO)3(dppv)(PFc*Et2)]2+ [1H]2+
A single organometallic product results from the rapid addition of excess [H(OEt2)2]BArF4 to [1]0. The IR spectrum of the resulting solution (νCO 1957, 1912 cm–1; Figure 8, bottom) is consistent with [Fe2(adtBnH)(CO)3(dppv)(PFc*Et2)]2+ ([1H]2+). This species features a ferrocenium ligand, a tertiary ammonium center, and an FeIFeI core. Complex [1H]2+ was independently generated by two additional methods: (i) protonation of [1]0 followed by oxidation and (ii) oxidation of [1]0 followed by protonation, using [H(OEt2)2]BArF4 and[Fc]BArF4 as the acid and oxidant, respectively (Figure 8). The IR spectrum of [1H]2+ generated by these methods matched that for product of treatment of [1]0 with excess acid.
Figure 8.
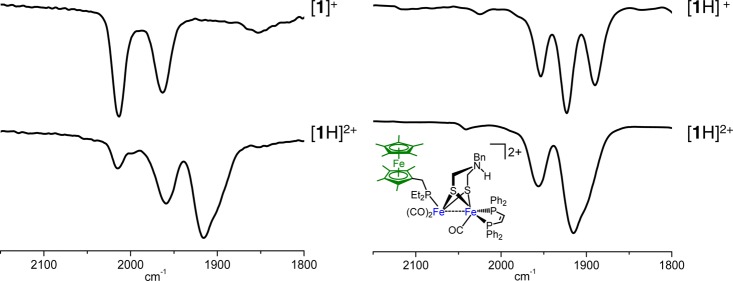
IR spectra for the generation of [1H]2+: (left) by protonation of [1]+ with 1 equiv of [H(OEt2)2]BArF4; (right) by oxidation of [1H]+ with 1 equiv of [Fc]BArF4. Inset: structure of [1H]2+.
The X-band EPR spectrum of [1H]2+ is also consistent with it being a ferrocenium derivative. The spectrum consisted of a very broad signal at 77 K, typical for a ferrocenium derivative.30,31 In contrast, the EPR spectrum of [1]+ and related FeFe-centered radicals feature axial spectra with significant (ca. 50 MHz) hyperfine coupling to equivalent phosphorus ligands.12
Hydrogen Evolution Catalyzed by Fe2(adtBn)(CO)3(dppv)(PFc*Et2)
The reduction of protons by [1]0 becomes catalytic in the presence of multiple equivalents of Fc* (Table 3). Furthermore, the stoichiometry of HER approaches one H2 per two Fc*. Thus, when a solution of [1]0 and 5 equiv of Fc* was added to 10 equiv of [H(OEt2)2]BArF4, 3.3(±0.3) equiv of H2 was obtained after 30 min. (theoretical yield: 3.0 equiv, 0.5 equiv from 1, and an additional 2.5 equiv from added Fc*). Catalysis by [1]0 was further probed by serial addition of acids (Figure 9). A mixture of solid [1]0 and 20 equiv of [H(OEt2)2]BArF4 was treated with a CH2Cl2 solution of 5 equiv of Fc*. Gas chromatographic (GC) analysis after 30 min revealed the formation of 2.3 equiv of H2 (theoretical yield: 3.0 equiv). Addition of a further 5 equiv of Fc* gave an additional 2.5 equiv of H2 (theory: 2.5 equiv). Repeating this procedure yielded a further 1.6 equiv of H2 before the catalyst became inactive. Under these conditions, the yield of hydrogen was ∼80% (6.4 equiv of H2 from 16 equiv of Fc*). The inefficiency of the system is attributed to catalyst degradation to [μ-H1]+ and its N-protonated derivative [μ-H1H]2+, as observed by IR spectroscopy (Supporting Information). HER from [1H]+ and acids can also be driven by the addition of octamethylferrocene (Fc#), although the reaction is slower than with Fc* (Table 4).
Table 3. Yield of H2 by Treatment of [1]0 (4.2 mM in CH2Cl2) with [H(OEt2)2]BArF4 and Fc* at −15 °Ca.
| amt of 1, equiv | amt of H+, equiv | amt of Fc*, equiv | solvent | amt of H2, equiv |
|---|---|---|---|---|
| 1 | 5 | 0 | CH2Cl2 | 0.45 ± 0.08 |
| 1 | 5 | 1 | CH2Cl2 | 1.1 ± 0.2 |
| 1 | 5 | 4 | CH2Cl2 | 1.5 ± 0.3 |
| 1 | 5 | 10 | CH2Cl2 | 1.5 ± 0.3 |
| 1 | 10 | 5 | CH2Cl2 | 3.3 ± 0.3 |
| 1 | 10 | 5 | MeCN | 0.28 ± 0.03 |
| 0 | 10 | 5 | CH2Cl2 | 0.01 ± 0.01 |
GC analysis was performed 30 min after addition of components.
Figure 9.

Results of serial addition of Fc* to [1]0. The solution began with 5 equiv of Fc* and 20 equiv of [H(OEt2)2]BArF4. Asterisks mark the time that the headspace was analyzed and then evacuated. To the resulting solid was added another solution of 5 equiv of Fc*, and the headspace was reanalyzed after 30 min and then evacuated.
Table 4. Yield of H2 by Treatment of [1]0 (4.2 mM in CH2Cl2) with [H(OEt2)2]BArF4 and Reducing Agent.
| amt of H+, equiv | amt of Fc*, equiv | amt of Fc#, equiv | amt of H2, equiv | time,a h |
|---|---|---|---|---|
| 10 | 5 | 0 | 3.3 ± 0.3 | 0.5 |
| 10 | 0 | 5 | 3.3 ± 0.4b | 3.0 |
Approximate period to give the maximum yield of H2.
Experiment was repeated twice.
Yields of hydrogen are strongly affected by acid strength. Using 10 equiv of [NPh2H2]BArF4 (pKa, MeCN = 5.97)24 in place of [H(OEt2)2]BArF4 (together with 5 equiv of Fc* and 1) yielded only 0.30 equiv of hydrogen even after an extended period of time. IR analysis of the reaction mixture showed the amine-protonated species [1H]+ as the dominant organometallic species in solution.
Oxidation States of Protonated Models
Protonation of the amine affects the site of oxidation in the triiron ensembles. The first oxidation of [1]0 occurs at the FeFe core, the second oxidation being localized at the ferrocenyl ligand. In [1H]+, the sequence is reversed: initial oxidation occurs at the ferrocenyl ligand followed by oxidation at the FeFe center. The localization of oxidation is reflected by differences in νCO(av) upon oxidation. Thus, the [1]0/+ and [1H]+/2+ couples are associated with ΔνCO(av) values of 60 and 5 cm–1, respectively (Figure 10). Small shifts of νCO on the order of 5–15 cm–1 are expected for the oxidation of ferrocenyl ligands.12,32,33 Furthermore, the addition of acid to [1]+ results in redox tautomerization in such a way that the electron hole has moved from the FeFe core to the appended ligand (Figure 11). While not directly measured, the [1H]2+/3+ couple is estimated to be positive of 0 mV.34 Chemical oxidation of [1H]2+ with acetylferrocenium (E1/2 = 270 mV)35 shifts νCO by >60 cm–1 (Supporting Information).
Figure 10.

IR spectra of [1]0 (left) and [1H]+ (right) before (top) and after (bottom) oxidation with [Fc]BArF4.
Figure 11.
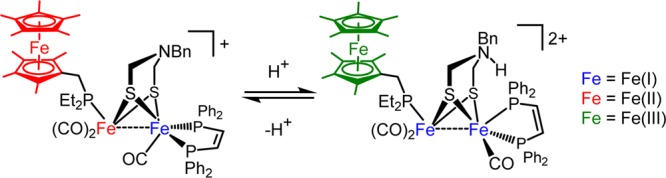
Redox tautomerization induced by protonation of [1]+.
Variations of the Design of 1
Upon the discovery that [1]0 catalyzes HER, variations of the catalyst structure were examined. The complex Fe2(adtBn)(CO)3(dppv)(PFc#Et2) (2) has one fewer methyl group on the ferrocenyl ligand. Correspondingly, the [2]+/2+ couple, which is ferrocenyl ligand localized, is 78 mV more positive than the [1]+/2+ couple, although the [1]0/+ and [2]0/+ couples are almost identical (Table 5). Apparently reflecting its diminished reduction potential, 2 is a slower, less efficient catalyst than [1]0 (Table 6). The organometallic product obtained by treatment of 2 with excess acid is spectroscopically similar to [1H]2+ (Supporting Information).
Table 5. Electrochemical Potentials of Pertinent Ligands and Their Diiron Complexesa.
| compd | PFcx couple | ipa/ipc | FeIFeI/FeIFeII couple | ipa/ipc |
|---|---|---|---|---|
| FcP*Et2 | –591 | 0.95 | n/a | n/a |
| FcP#Et2 | –536 | 1.0 | n/a | n/a |
| Fe2(adtBn)(CO)3(dppv)(PFc*Et2) ([1]0) | –393 | 0.90 | –700 | 0.8512 |
| Fe2(adtBn)(CO)3(dppv)(PFc#Et2) ([2]0) | –315 | 0.86 | –713 | 0.77 |
| Fe2(adtBn)(CO)3(dppv)(PMe3) ([3]0) | n/a | n/a | –715 | 0.934 |
| Fe2(pdt)(CO)3(dppv)(PFc*Et2) ([4]0) | –382 | 0.97 | –675 | 0.73 |
All potentials were measured in CH2Cl2 with [Bu4N]BArF4 electrolyte and are given in mV. All potentials are either reversible or quasi-reversible.
Table 6. Yield of H2 by Treatment of 2 (4.2 mM in CH2Cl2) with [H(OEt2)2]BArF4.
| amt of H+, equiv | amt of Fc*, equiv | amt of H2, equiv | time,a h |
|---|---|---|---|
| 5 | 0 | 0.26 ± 0.03 | 0.5 |
| 10 | 5 | 3.3 ± 0.3 | 2.0 |
Approximate period for maximum yield of H2.
Catalysis by Fe2(adtBn)(CO)3(dppv)(PMe3)
In contrast to the case for [1]0, the reference compound Fe2(adtBn)(CO)3(dppv)(PMe3) ([3]0) does not generate hydrogen upon treatment with 5 equiv of [H(OEt2)2]BArF4. The result is significant because the [1]0/+ and [3]0/+ couples are nearly identical. As indicated by the IR signatures (two νCO bands in unprotonated to three νCO bands in N-protonated), addition of acid to [3]0 immediately produces [Fe2(adtBnH)(CO)3(dppv)(PMe3)]+ ([3H]+). 31P{1H} NMR analysis confirms the formation of [3H]+ (δ 92.8 (dppv) and 33.0 (PMe3)). Over the course of hours at −15 °C in the presence of excess [H(OEt2)2]BArF4, [3H]+ converts to the ammonium hydride [μ-HFe2(adtBnH)(CO)3(dppv)(PMe3)]2+ ([μ-H3H]2+; Figure 12).
Figure 12.

IR spectrum (CH2Cl2 solution) of 3 in the presence of 5 equiv of [H(OEt2)2]BArF4 at −15 °C: (top) after 40 min; (bottom) after 150 min. Peaks marked with asterisks are assigned as [3H]+.
Although 3 will not reduce protons to H2, it does so in the presence of Fc*. Thus, treatment of 3 with 5 equiv of [H(OEt2)2]BArF4 and 1 equiv of Fc* produced 0.94 ± 0.18 equiv of H2. The immediate organometallic product is the ammonium complex [3H]+, as indicated by IR spectroscopy. The reaction is catalytic in the presence of 10 equiv of acid and 5 equiv of reductant (Table 7). Over the period of several hours, solutions of [3H]+ decay to [μ-H3]+, which is inactive.
Table 7. Yield of H2 from the Reaction of 3 (4.2 mM in CH2Cl2) with [H(OEt2)2]BArF4 and Varying Amounts of Fc*.
| amt of H+, equiv | amt of Fc*, equiv | amt of H2, equiv | time,a h |
|---|---|---|---|
| 5 | 0 | 0 | 0.5 |
| 5 | 1 | 0.9 ± 0.2 | 0.5b |
| 10 | 5 | 2.7 ± 0.5 | 1.5 |
Approximate period for maximum yield of H2.
The concentration was 5.8 mM.
Attempted Catalysis by Fe2(pdt)(CO)3(dppv)(PFc*Et2)
Although HER is possible both with and without attachment of a reducing agent, the amine is critical to catalysis. Catalysis was attempted with a propanedithiolate (pdt) analogue of [1]0, Fe2(pdt)(CO)3(dppv)(PFc*Et2) (4). Like [1]0, [4]0 undergoes two reversible, one-electron oxidations, one centered on the FeFe core and another being ligand-centered (Table 5). These couples are very similar to those for [1]0/+ and [1]+/2+. Treatment of [4]0 with 5 equiv of [H(OEt2)2]BArF4 gave <0.01 equiv of H2, as the FeFe precursor converted to the bridging hydride species [(μ-H)Fe2(pdt)(CO)3(dppv)(PFc*Et2)]BArF4 ([μ-H4]+). Treatment of Fe2(pdt)(CO)3(dppv)(PFc*Et2) with 10 equiv of [H(OEt2)2]BArF4 and 5 equiv of Fc* produced only traces of hydrogen (<0.05 equiv) even after 2 h. The main product, on the basis of IR spectroscopy, was [μ-H4]+. The effects of changes to the redox-active ligand in this FeFe system are summarized in Table 8.
Table 8. Yields of H2 by Treatment of Various Catalysts (4.2 mM FeFe Complex in CH2Cl2) with [H(OEt2)2]BArF4.
| FeFe complex | amt of H+, equiv | amt of Fc*, equiv | amt of H2, equiv | time,a h |
|---|---|---|---|---|
| 1 | 10 | 5 | 3.3 ± 0.3 | 0.5 |
| 2 | 10 | 5 | 3.3 ± 0.3 | 2.0 |
| 3 | 10 | 5 | 2.7 ± 0.5 | 1.5 |
| 4 | 10 | 5 | 0.04 ± 0.01 | 0.5 |
Approximate period for maximum yield of H2.
Discussion
Mechanism of Hydrogen Evolution
A proposed mechanism for the reaction of [1]0 with protons to produce H2 is shown in Figure 13. Generation of H2 is proposed to proceed via the following steps. Compound [1]0 initially protonates at the amine to give [1H]+, which we can observe. Compound [1H]+ then undergoes protonation at iron to give a terminal hydride, the ammonium center not serving as a proton relay. Possibly concomitant with this second protonation is electron transfer from PFc*Et2, inducing elimination of H2 from the nascent ammonium hydride, producing [1]2+. Aspects of the catalysis are discussed in the following sections.
Figure 13.

Proposed hydrogen evolution mechanism for [1]0 (and [2]0, where R = H) in the presence of excess acid and reducing agent.
Comproportionation
The formation of only 0.5 equiv of H2 from the reaction of [1]0 with excess H+ results from comproportionation (Figure 13, center arrows). Comproportionation arises because the immediate product of HER, [1]2+, is reduced by [1]0, yielding [1]+. Analogous processes are favorable for the redox between [1]2+ and [1H]+. The comproportionation of [1]0 and [1]2+ is favored by 307 mV, as the potentials for [1]0/+ and [1]+/2+ are at −700 and −393 mV, respectively. Although comproportionation complicates analysis for the organometallic complexes, the stoichiometry of catalysis is unaffected. In the protein, redox reactions between H clusters would be precluded.
Role of Azadithiolate
Hydrogen generation in these systems requires the azadithiolate.8 The amine is the kinetic, but not thermodynamic, site of protonation. In the present case, however, the amine cofactor serves two roles: as a proton donor and as a regulator of the reducing power of the FeFe subunit.
The proton-relay function of the azadithiolate is unusual in the present systems. In contrast to other biomimetic models, HER by [1]0 and [2]0 requires strong acids: the likely rate-determining step is protonation of [1H]+/[2H]+ at the weakly basic Fe center. In these cases, the ammonium center does not relay protons. In fact, N-protonation interferes with hydride formation, since it decreases the basicity of the Fe(I)Fe(I) center. Subsequent to the second protonation (to give [H1H]2+/[H2H]2+), intramolecular electron transfer is proposed to occur. In the resulting mixed-valence species [H1H]+/[H2H]+, the ammonium proton couples to the terminal hydride.
N-protonation of Fe2(adt)(CO)6–x(L)x complexes affects the redox properties of the diiron core. N-protonation shifts the FeIFeI/FeIFeII couple about 0.5 V.34,36,37 Because of this shift, the [1]0/+ couple (−700 mV) is localized on the diiron center, whereas the [1H]+/2+ couple (estimated at −390 mV) is ferrocene-based.
H2 Elimination
Previous work showed that diferrous ammonium hydrides [HFe2(adtBnH)(CO)2L4]2+ do not eliminate dihydrogen.8,38 Elimination of H2 would afford the 32e dications, which are high-energy species, as confirmed by electrochemical measurements.28,34 Instead, H2 release is triggered by reduction, which we propose is localized on the proximal (non-hydride-bearing) iron center.39 In this way, hydrogenogenesis (and the reverse reaction, hydrogen oxidation) is regulated by the redox potential of the catalyst’s environment. The present work does not distinguish a mixed-valence ammonium hydride intermediate from a concerted PCET pathway. We do know that reduction-induced HER from the ammonium hydride is very fast, since otherwise terminal hydrides rapidly isomerize to the catalytically incompetent μ-hydride species (see below).
Terminal vs Bridging Iron Hydrides
A recurring challenge to biomimetic HER is the tendency of terminal hydrides of FeFe dithiolates to isomerize to μ-hydrido derivatives. This isomerization is of great interest, since the [FeFe]-H2ases operate via terminal hydrides and synthetic models are also faster for terminal hydrides relative to the isomeric bridging hydrides.8 The terminal to bridging hydride isomerization is slow with bulky terminal hydrides, e.g., [HFe2(xdt)(CO)2(PMe3)4]+ and [HFe2(xdt)(CO)2(dppv)2]+ (xdt = pdt, adt), with half-lives of minutes at room temperature.8,38 For less bulky complexes, e.g., [HFe2(xdt)(CO)3(PMe3)(dppv)]+ and the complexes discussed in this work, the isomerization proceeds is rapid even at −90 °C.40 For catalytic HER to occur with 1, reduction of the ammonium hydride must be faster than the unimolecular isomerization to bridging hydrides.
Role of Appended Fc* Group
The mechanism for HER by catalysts 1–3 is the same. In all cases, protonation at the amine is followed by protonation at iron and then electron transfer from a ferrocene group. In the absence of Fc* or PFc*R2 isomerization of terminal hydrides to the catalytically inactive bridging hydrido complexes occurs. Additionally, with [1]0 and [2]0, unique species are observed ([1H]2+and [2H]2+), which display enhanced stability with respect to formation of bridging hydrides in comparison to the respective ammonium counterparts, e.g., [3H]+.
Overpotential
The overpotentials for the HER are estimated on the assumption that EMeCN ≈ ECH2Cl2. In MeCN solution, HER from fully dissociated acid occurs at −0.026 V.16 With EMeCN(Fc*0/+,[Bu4N]BArF4) = −0.61 V, the overpotential for HER by [1]0 is 0.54 V, on the basis of the [Fc*]+/0 couple. Using Fc# (ECH2Cl2, [Bu4N]BArF4 = −0.50 V) for catalysis (Table 4), the overpotential drops to 0.43 V, although the rate of hydrogen evolution also slows relative to Fc* (for Fc*, 6.6 TO/h; Fc#, 1.1 TO/h).
Conclusions
Several [FeFe]-H2ase models have been found to catalyze the reduction of protons to H2 in the presence of acid and soluble reductant. The complexes [1]0 and [2]0 react with acid to yield H2, even without additional reducing agents, which is unprecedented in H2ase models. The new results underlines the critical role of the 4Fe-4S cluster in catalysis.41 In the absence of additional Fc* or FcP*, catalysis does not occur; rather, bridging hydride species are generated. The catalytic reaction can be summarized according to the equation
In MeCN solution, HER from fully dissociated acid is calculated to occur at −0.026 V;16 thus, HER is thermodynamically favorable by 580 mV for Fc*. In living systems, [4Fe-4S] clusters (ca. −1.4 V) serve as donors.42 In both living and synthetic systems, the diiron–adt–carbonyl catalyst is required for HER, although the redox cofactors ([4Fe-4S] clusters, Fc*) provide the thermodynamic driving force.
Other redox-active ligands have been incorporated into hydrogenase mimics without enhancing catalysis.33,43−45 These catalyst candidates, however, lack the adt functionality and contain ferrocenes with very mild reduction potentials. The catalysts presented in this work show enhanced reactivity due to the combined effect of three factors: (i) the adt cofactor, (ii) a sufficiently basic FeFe core to enable formation of terminal hydrides, and (iii) the presence of a redox-active ligand with sufficient driving force. The complete FeFe model provides a location to bring a hydride and a proton together.
Further work on FeFe-H2ase modeling could focus on catalysts that are more robust and operate faster at lower overpotentials. Both goals would be met by bulkier, more basic diiron centers. The Fe2(adtR)(CO)2(dppv)2 system meets some of these criteria, as the terminal hydride is stable for minutes at room temperature and the basicities of the amine and the diiron(I) center are matched. The [HFe2(Hadt)(CO)2(dppv)2]2+/+ couple (−1.4 V) requires strong reductants that do not react directly with proton donors. In living systems, [4Fe-4S] clusters (ca. −1.4 V) serve as donors.42
Experimental Section
Unless otherwise noted, reactions were performed using standard Schlenk and glovebox techniques. Most reagents were purchased from either Strem or Sigma-Aldrich. Solvents were HPLC grade or better and were dried and deoxygenated by passage through activated alumina and sparging with Ar or by distillation under nitrogen. The compounds Fe(C5Me4H)(C5Me4CHO) and Fe(C5Me4H)(C5Me4CH2OH),17 [H(OEt2)2]BArF4,46 [Bu4N]BArF4,47 Fe2(adtBn)(CO)3(dppv)(PFc*Et2) ([1]0),12 and Fe2(adtBn)(CO)3(dppv)(PMe3) ([3]0)48 were prepared according to literature procedures. [Bu4N]PF6 was recrystallized from ethanol. 1H NMR spectra (500 MHz) are referenced to residual solvent referenced to TMS. 31P{1H} NMR spectra (202 MHz) are referenced to external 85% H3PO4. FT-IR spectra were recorded on a Perkin-Elmer 100 FT-IR spectrometer, focusing primarily on the ν(CO) region. ESI-MS data were recorded of dilute CH2Cl2 solutions on a Waters Micromass Quattro II spectrometer. Chromatography was performed on silica gel (40–63 μm, 230–400 mesh). Gas chromatography was performed using an Agilent 7820A instrument equipped with a thermal conductivity detector and a 5 Å molecular sieve (80–100 mesh) column. The response factor for H2/CH4 was 3.8 under our conditions, as established by calibrations of standard H2 and CH4. Irradiation reactions were undertaken using Pyrex Schlenk flasks using a light-emitting diode array from Opto Technology with a light output of 365 nm. CV measurements were recorded on a CHI Model 630D instrument, using Pt working and counter electrodes. An Ag bar was used as a pseudo reference electrode. After each CV measurement, Fc was added as an internal standard. Unless indicated otherwise, the analyte concentration was 1 mM, the [Bu4N]PF6 concentration was 0.1 M, and the [Bu4N]BArF4 concentration was 0.025 M, with a sweep rate of 100 mV/s. An iR compensation was undertaken prior to all measurements.
Synthesis of FcMe9
A 100 mL Schlenk flask was charged with 2.5 g of Fe(C5Me4H)(C5Me4CHO) and 30 mL of CH2Cl2 to produce a red solution. A 23.0 mL amount of BH3·THF (1.0 M) was added via gastight syringe, resulting in an immediate color change from deep red to orange. The solution was stirred for 17 h, after which it was slowly quenched with 20 mL of aqueous saturated NH4Cl. At this point, the product can be manipulated in air for short periods. The mixture was transferred into a separatory funnel, and the aqueous layer was discarded. The organic layer was washed twice with 20 mL of water and once with 20 mL of brine. The organic layer was dried over MgSO4 and stripped of solvent. The residue was passed through a column of silica gel, with a 9/1 mixture of hexane/Et2O as eluent. Removal of solvent produced an orange-yellow solid. Yield: 2.29 g (96%). Analytically pure samples were obtained by vacuum sublimation (0.01 Torr) overnight at 120 °C. 1H NMR (CD2Cl2): δ 3.16 (s, 1H), 1.72 (s), 1.71 (s, overlapping, total to 21H), 1.65 (s, 6H). 13C NMR (CD2Cl2): δ 80.27, 79.94, 79.24, 71.27, 11.46, 10.17, 9.56. ESI-MS: m/z 312.3 [M]+. Anal. Calcd for C19H28Fe (found): C, 73.07 (73.21); H, 9.04 (9.19).
Synthesis of PFc#Et2
A 200 mL Schlenk flask was charged with 1.22 g (3.7 mmol) of Fe(C5Me4H)(C5Me4CH2OH) and 40 mL of Et2O. Once the solution was homogeneous, 625 μL (6.6 mmol) of Ac2O was added, and the flask was cooled to −78 °C (some solid precipitate appeared). The cold solution was then treated in one portion with 550 μL of HBF4·Et2O (4.04 mmol), resulting in the immediate formation of a pale red precipitate. After it was stirred for 30 min, the cold slurry was treated with 50 mL of pentane to enhance precipitation of the product. The solution was filtered at low temperature, and the solid was washed with an additional 100 mL of Et2O and dried briefly under vacuum. While the temperature was maintained at −78 °C, a red slurry was formed by the addition of 30 mL of Et2O. A solution of 450 μL of HPEt2 (3.91 mmol) in 20 mL of Et2O was transferred into the red slurry. The slurry was stirred at low temperatures for 10 min, after which 40 mL of CH2Cl2 was added, resulting in a color change to yellow. The reaction mixture was maintained at low temperatures for 1 h before it was warmed to room temperature. Excess K2CO3 and MgSO4 were added under argon pressure. The following morning, all of the volatiles were removed under vacuum, and the solid was extracted with pentane. The pentane solution was filtered through a pad of Celite. Evaporation of solvent under vacuum gave Fe(C5Me4H)(C5Me4CH2PEt2) as an air-sensitive orange-yellow solid. Yield: 1.22 g (78% based on PEt2H). Crystals were grown from a concentrated solution of pentane at −30 °C. The compound can be further purified by filtering a pentane extract through a plug of silica, upon which the compound was retained. After the silica plug was washed with pentane, the compound was extracted by eluting with Et2O. Removal of solvent resulted in the orange-yellow solid. Mp: 37–38 °C dec. 1H NMR (CD2Cl2): δ 3.15 (s, 1H), 2.34, (s, 2H), 1.75 (s, 6H), 1.73 (s, 6H), 1.70 (s, 6H), 1.64 (s, 6H), 1.33 (m, 4H), 1.02 (m, 6H). 31P{1H} NMR (CD2Cl2): δ −17.4. ESI-MS: m/z 400.4 [M]+. Anal. Calcd for C23H37FeP (found): C, 69.00 (68.94); H, 9.31 (9.79).
Synthesis of PFc*Et2
The following procedure is an improvement over the literature method.12 The compound PFc*Et2 was prepared from FcMe9CH2OH following the method for PFc#Et2. Crystals were grown from a concentrated solution of pentane at −30 °C. Yield: 56%. Mp: 84 °C dec. 1H NMR (CD2Cl2): δ 2.29 (s, 2H), 1.71 (s, 6H), 1.69 (s, 6H), 1.67 (s, 15H), 1.33 (m, 4H), 1.03 (m, 6H). 31P{1H} NMR (CD2Cl2): δ −17.4. ESI-MS: m/z 414.4 [M]+. Anal. Calcd for C24H39FeP (found): C, 69.56 (69.79); H, 9.49 (9.51).
Synthesis of [PFc*Et2]BF4
A mixture of PFc*Et2 (41.4 mg, 100 μmol) and FcBF4 (24.6 mg, 90 μmol, 0.9 equiv) was dissolved in CH2Cl2 (1 mL). After 1 min, pentane (15 mL) was added and the mixture was allowed to stand for 1 h. Decanting the solvent allowed for isolation of an oily solid, which was dissolved in CH2Cl2 (1 mL) and precipitated by addition of pentane (15 mL). The solids were isolated by filtration, washed with pentane (5 mL), and dried briefly to afford the title compound as a green microcrystalline powder (35.2 mg, 78%). Green prismatic single crystals were grown by layering a concentrated CH2Cl2 solution with pentane and allowing the mixture to stand at −30 °C. ESI-MS: m/z 415.5 [M – BF4–]+. UV–vis: 796 (ε = 180 M–1 cm–1).
((Dicyclohexylphosphino)methyl)octamethylferrocene (PFc*Cy2)
The compound PFc*Cy2 was prepared from FcMe9CH2OH following the method for PFc*Et2, but using PCy2H. Yield: 55%. Mp: 127–128 °C dec. 1H NMR (CD2Cl2): δ 2.35 (s, 1H), 1.72 (s, 6H), 1.67 (s, 6H), 1.47–1.12 (m, 22H). 31P{1H} NMR (CD2Cl2): δ −4.07. ESI-MS: m/z 523.3 [M]+. Anal. Calcd for C32H51FeP (found): C, 73.55 (73.09); H, 9.84 (10.02).
((Diphenylphosphino)methyl)octamethylferrocene (PFc*Ph2)
The compound PFc*Ph2 was prepared from FcMe9CH2OH following the method for PFc*Et2, but using PPh2H. Yield: 36%. Mp: 147 °C dec. 1H NMR (CD2Cl2): δ 7.37–7.29 (broad, m, 10H), 2.97 (s, 2H), 1.66 (s, 15H), 1.64 (s, 6H), 1.13 (s, 6H). 31P{1H} NMR (CD2Cl2): δ −18.8. ESI-MS m/z 510.4 [M]+. Anal. Calcd for C32H39FeP (found): C, 75.29 (75.35); H, 7.70 (7.83).
Protonation of Ferrocenylphosphines
A J. Young tube was charged with 5 mg of PFc*Et2 (12 μmol) and 12.5 mg of [NH2Ph2]BArF4 (12 μmol). Approximately 500 μL of CD2Cl2 was distilled onto the solids, forming a yellow solution. The signal in the 31P{1H} NMR spectrum shifts from δ −17.4 for the phosphine (PFc*Et2) to δ +15.5 for the phosphonium derivative. In the 1H NMR spectrum, the signals for the methyl groups on the ferrocene do not change drastically upon protonation, but a pair of multiplet signals is observed centered at δ 4.98 and 5.90 (JP–H = 186 Hz), assigned to PH. The 1H NMR signal for the PH center in HPEt3+ is reported at δ 5.97.49 The spectrum of the phosphonium species remained unchanged over a period of 2 days at room temperature. Addition of strong acids (even 1 equiv) to PFc*Et2 caused the 31P{1H} NMR signal to disappear, an effect we attribute to the generation of a small amount of [PFc*Et2]+, a paramagnetic species in rapid exchange with the parent ferrocene.
Synthesis of Fe2(adtBn)(CO)3(dppv)(PFc#Et2)
This compound was prepared in a fashion analogous to that for compound 1, using PFc#Et2 in place of PFc*Et2. Yield: 70%. 1H NMR (CD2Cl2): δ 8.06–7.95, 7.43–7.14 (broad, m, 27H), 6.77 (d, 2H), 3.14 (s, 1H), 3.10 (d, 2H), 3.00 (s, 2H), 2.78 (d, 2H), 1.87 (s, 6H), 1.77 (s, 6H), 1.70 (m, 4H, overlapping), 1.69 (s, 6H), 1.64 (s, 6H), 1.06 (m, 6H). 31P{1H} NMR (CD2Cl2): δ 93.96 (s), 58.43 (s). IR (CH2Cl2): 1955, 1900 cm–1. Anal. Calcd for C61H70Fe3NO3P3S2 (found): C, 61.58 (61.75); H, 5.93 (6.05); N, 1.18 (1.68).
Synthesis of Fe2(pdt)(CO)3(dppv)(PFc*Et2)
A 300 mL Schlenk flask was loaded with 255 mg of Fe2(pdt)(CO)4(dppv) (0.35 mmol) and 162 mg of PFc*Et2 (0.39 mmol). The solids were dissolved in 150 mL of dry PhMe, and the solution was photolyzed at 365 nm while the flask was flushed with Ar to remove CO. The reaction was monitored by IR, and upon completion (no further decrease in the carbonyl band at ∼2020 cm–1), the solvent was removed under vacuum. The residue was chromatographed inside a glovebox on a column of silica gel. Elution with 5% Et2O in pentane yielded a fast-moving orange-yellow band (excess ligand), followed by a slower-moving brown-red band. The beginning of the brown-red band contained the desired product; however, as the band continued to elute, contamination of unreacted Fe2(pdt)(CO)4(dppv) with the product was observed. Removal of solvent from the fractions containing only Fe2(pdt)(CO)3(dppv)(PFc*Et2) gave a red-brown solid. Yield: 125 mg (32%). 1H NMR (CD2Cl2): δ 8.07–7.29 (broad, m, 20H), 3.07 (d, 2H), 1.82 (s, 6H), 1.74 (m, overlapping, 4H), 1.71 (s, 6H), 1.65 (s, 15H), 1.12 (m, 6H). 31P{1H} NMR (CD2Cl2): δ 93.95 (s), 57.85 (s). IR (CH2Cl2): 1955, 1900. Anal. Calcd for C56H67Fe3O3P3S2 (found): C, 60.45 (60.14); H, 6.07 (5.94)
Hydrogen Evolution Experiments
Within a nitrogen-filled glovebox, a 7.5 mL GC vial was charged with 4.2 μmol of FeFe compound, followed by the appropriate mass of [H(OEt2)2]BArF4 and reductant and a triangular stir bar. A septum was affixed and wired down with copper wire, and the vial was brought out of the box and cooled to −15 ± 2.5 °C. Simultaneously, 1.0 mL of dry CH2Cl2 and 60 μL of methane (internal standard) were added, and grease iswas applied at the needle puncture site. After the appropriate amount of time (30 min, unless otherwise specified), grease was removed, 500 μL of headspace was withdrawn, and grease was reapplied. In the event that multiple samples of headspace were removed and tested, the hydrogen output and methane standard were recalculated to account for losses during the previous GC analysis.
Acknowledgments
We thank Dr. David Schilter for crystals of [PFc*Et2]BF4. This work was supported by the National Institutes of Health (Grant GM61153).
Supporting Information Available
Figures and CIF files giving spectroscopic and cyclic voltammetric data for all new compounds, kinetic plots, and crystallographic data for PFc#Et2, PFc*Et2, [PFc*Et2]BF4, and 1. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Department of Chemistry, Yeshiva University, 500 W. 185th St, New York, NY 10033.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fontecilla-Camps J. C.; Volbeda A.; Cavazza C.; Nicolet Y. Chem. Rev. 2007, 107, 4273. [DOI] [PubMed] [Google Scholar]
- Lubitz W.; Ogata H.; Rüdiger O.; Reijerse E. Chem. Rev. 2014, 114, 4081. [DOI] [PubMed] [Google Scholar]
- Silakov A.; Wenk B.; Reijerse E.; Lubitz W. Phys. Chem. Chem. Phys. 2009, 11, 6592. [DOI] [PubMed] [Google Scholar]
- Gloaguen F.; Rauchfuss T. B. Chem. Soc. Rev. 2009, 38, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tard C. d.; Pickett C. J. Chem. Rev. 2009, 109, 2245. [DOI] [PubMed] [Google Scholar]
- Bullock R. M.; Appel A. M.; Helm M. L. Chem. Commun. 2014, 50, 3125. [DOI] [PubMed] [Google Scholar]
- Gloaguen F.; Lawrence J. D.; Rauchfuss T. B. J. Am. Chem. Soc. 2001, 123, 9476. [DOI] [PubMed] [Google Scholar]
- Carroll M. E.; Barton B. E.; Rauchfuss T. B.; Carroll P. J. J. Am. Chem. Soc. 2012, 134, 18843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berggren G.; Adamska A.; Lambertz C.; Simmons T. R.; Esselborn J.; Atta M.; Gambarelli S.; Mouesca J. M.; Reijerse E.; Lubitz W.; Happe T.; Artero V.; Fontecave M. Nature 2013, 499, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esselborn J.; Lambertz C.; Adamska-Venkatesh A.; Simmons T.; Berggren G.; Noth J.; Siebel J.; Hemschemeier A.; Artero V.; Reijerse E.; Fontecave M.; Lubitz W.; Happe T. Nat. Chem. Biol. 2013, 9, 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cracknell J. A.; Vincent K. A.; Armstrong F. A. Chem. Rev. 2008, 108, 2439. [DOI] [PubMed] [Google Scholar]
- Camara J. M.; Rauchfuss T. B. Nat. Chem. 2012, 4, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N.; Wang M.; Wang Y.; Zheng D.; Han H.; Ahlquist M. S. G.; Sun L. J. Am. Chem. Soc. 2013, 135, 13688. [DOI] [PubMed] [Google Scholar]
- Camara J. M.; Rauchfuss T. B. J. Am. Chem. Soc. 2011, 133, 8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felton G. A. N.; Glass R. S.; Lichtenberger D. L.; Evans D. H. Inorg. Chem. 2006, 45, 9181. [DOI] [PubMed] [Google Scholar]
- Roberts J. A. S.; Bullock R. M. Inorg. Chem. 2013, 52, 3823. [DOI] [PubMed] [Google Scholar]
- Zou C.; Wrighton M. S. J. Am. Chem. Soc. 1990, 112, 7578. [Google Scholar]
- Herber R. H.; Nowik I.; Schottenberger H.; Wurst K.; Schuler N.; Mueller A. G. J. Organomet. Chem. 2003, 682, 163. [Google Scholar]
- Routaboul L.; Chiffre J.; Balavoine G. G. A.; Daran J.-C.; Manoury E. J. Organomet. Chem. 2001, 637–639, 364.19594449 [Google Scholar]
- Kim D.-H.; Ryu E.-S.; Cho C. S.; Shim S. C.; Kim H.-S.; Kim T.-J. Organometallics 2000, 19, 5784. [Google Scholar]
- Trojánek A.; Langmaier J.; Samec Z. Electrochim. Acta 2012, 82, 457. [Google Scholar]
- Kelly A. M.; Katif N.; James T. D.; Marken F. New J. Chem. 2010, 34, 1261. [Google Scholar]
- Barrière F.; Kirss R. U.; Geiger W. E. Organometallics 2004, 24, 48. [Google Scholar]
- Kaljurand I.; Kütt A.; Sooväli L.; Rodima T.; Mäemets V.; Leito I.; Koppel I. A. J. Org. Chem. 2005, 70, 1019. [DOI] [PubMed] [Google Scholar]
- Su B.; Hatay I.; Ge P. Y.; Mendez M.; Corminboeuf C.; Samec Z.; Ersoz M.; Girault H. H. Chem. Commun. 2010, 46, 2918. [DOI] [PubMed] [Google Scholar]
- Hatay I.; Ge P. Y.; Vrubel H.; Hu X.; Girault H. H. Energy Environ. Sci. 2011, 4, 4246. [Google Scholar]
- Scanlon M. D.; Bian X.; Vrubel H.; Amstutz V.; Schenk K.; Hu X.; Liu B.; Girault H. H. Phys. Chem. Chem. Phys. 2013, 15, 2847. [DOI] [PubMed] [Google Scholar]
- Justice A. K.; Zampella G.; De Gioia L.; Rauchfuss T. B.; van der Vlugt J. I.; Wilson S. R. Inorg. Chem. 2007, 46, 1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orain P.-Y.; Capon J.-F.; Gloaguen F.; Schollhammer P.; Talarmin J. Int. J. Hydrogen Energy 2010, 35, 10797. [Google Scholar]
- Prins R.; Reinders F. J. J. Am. Chem. Soc. 1969, 91, 4929. [Google Scholar]
- Prins R. Mol. Phys. 1970, 19, 603. [Google Scholar]
- Miller T. M.; Ahmed K. J.; Wrighton M. S. Inorg. Chem. 1989, 28, 2347. [Google Scholar]
- Liu Y.-C.; Lee C.-H.; Lee G.-H.; Chiang M.-H. Eur. J. Inorg. Chem. 2011, 2011, 1155. [Google Scholar]
- Olsen M. T.; Rauchfuss T. B.; Wilson S. R. J. Am. Chem. Soc. 2010, 132, 17733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly N. G.; Geiger W. E. Chem. Rev. 1996, 96, 877. [DOI] [PubMed] [Google Scholar]
- Eilers G.; Schwartz L.; Stein M.; Zampella G.; de Gioia L.; Ott S.; Lomoth R. Chem. Eur. J. 2007, 13, 7075. [DOI] [PubMed] [Google Scholar]
- Ezzaher S.; Orain P.-Y.; Capon J.-F.; Gloaguen F.; Petillon F. Y.; Roisnel T.; Schollhammer P.; Talarmin J. Chem. Commun. 2008, 2547. [DOI] [PubMed] [Google Scholar]
- Zaffaroni R.; Rauchfuss T. B.; Gray D. L.; De Gioia L.; Zampella G. J. Am. Chem. Soc. 2012, 134, 19260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Nilges M. J.; Rauchfuss T. B.; Stein M. J. Am. Chem. Soc. 2013, 135, 3633. [DOI] [PubMed] [Google Scholar]
- Barton B. E.; Zampella G.; Justice A. K.; De Gioia L.; Rauchfuss T. B.; Wilson S. R. Dalton Trans. 2010, 39, 3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder D. W.; Ratzloff M. W.; Shepard E. M.; Byer A. S.; Noone S. M.; Peters J. W.; Broderick J. B.; King P. W. J. Am. Chem. Soc. 2013, 135, 6921. [DOI] [PubMed] [Google Scholar]
- Tard C.; Liu X.; Ibrahim S. K.; Bruschi M.; Gioia L. D.; Davies S. C.; Yang X.; Wang L.-S.; Sawers G.; Pickett C. J. Nature 2005, 433, 610. [DOI] [PubMed] [Google Scholar]
- Gimbert-Suriñach C.; Bhadbhade M.; Colbran S. B. Organometallics 2012, 31, 3480. [Google Scholar]
- Roy S.; Groy T. L.; Jones A. K. Dalton Trans. 2013, 42, 3843. [DOI] [PubMed] [Google Scholar]
- Ghosh S.; Hogarth G.; Hollingsworth N.; Holt K. B.; Kabir S. E.; Sanchez B. E. Chem. Commun. 2014, 50, 945. [DOI] [PubMed] [Google Scholar]
- Brookhart M.; Grant B.; Volpe A. F. Organometallics 1992, 11, 3920. [Google Scholar]
- LeSuer R. J.; Buttolph C.; Geiger W. E. Anal. Chem. 2004, 76, 6395. [DOI] [PubMed] [Google Scholar]
- Olsen M. T.; Barton B. E.; Rauchfuss T. B. Inorg. Chem. 2009, 48, 7507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah G. A.; McFarland C. W. J. Org. Chem. 1969, 34, 1832. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.