

Abstract
Opposite phenotypic and behavioural traits associated with copy number variation and disruptions to imprinted genes with parent-of-origin effects have led to the hypothesis that autism and schizophrenia share molecular risk factors and pathogenic mechanisms, but a direct phenotypic comparison of how their risks covary has not been attempted. Here, we use health registry data collected on Denmark's roughly 5 million residents between 1978 and 2009 to detect opposing risks of autism and schizophrenia depending on normal variation (mean ± 1 s.d.) in adjusted birth size, which we use as a proxy for diametric gene-dosage variation in utero. Above-average-sized babies (weight, 3691–4090 g; length, 52.8–54.3 cm) had significantly higher risk for autism spectrum (AS) and significantly lower risk for schizophrenia spectrum (SS) disorders. By contrast, below-average-sized babies (2891–3290 g; 49.7–51.2 cm) had significantly lower risk for AS and significantly higher risk for SS disorders. This is the first study directly comparing autism and schizophrenia risks in the same population, and provides the first large-scale empirical support for the hypothesis that diametric gene-dosage effects contribute to these disorders. Only the kinship theory of genomic imprinting predicts the opposing risk patterns that we discovered, suggesting that molecular research on mental disease risk would benefit from considering evolutionary theory.
Keywords: autism, schizophrenia, birth size, parent-of-origin, genomic imprinting, copy number
1. Introduction
Autism and schizophrenia are complex disorders that remain difficult to explain with currently identified risk factors, which include genetic, developmental and environmental influences (for recent reviews, see [1–4]). Heritability estimates place the familial component between 50 and 80% for autism and schizophrenia [5–8], but only a fraction of the contributing genetic variants have been identified [9–11], and such estimates are often inflated [12]. Recent work has begun to uncover the potential importance of imprinted genes with parent-of-origin effects on autism, schizophrenia and other neuropsychiatric disorders [13–15]. In 2008, Badcock and Crespi [16,17] hypothesized that autism and schizophrenia are diametric opposites joined by a spectrum of less severe disorders and normal cognition (figure 1).
Figure 1.
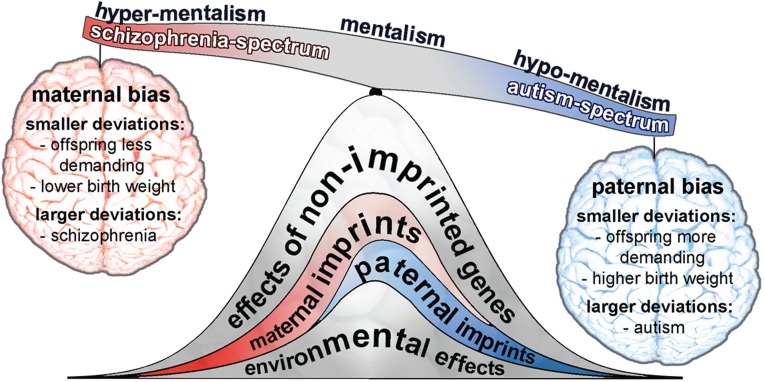
The imprinted brain theory of autism and schizophrenia. This theory suggests that autism and schizophrenia are diametric opposites balanced by normal cognition (mentalism), in part caused by small-to-large genome-wide imbalances in imprinted genes or CNVs that have effects on neurodevelopment. Deviations can be either maternally or paternally biased (see Ubeda & Gardner [18,19] for further discussion of these effects), with smaller deviations influencing resource-demanding traits such as birth weight or suckling behaviour in offspring, whereas larger deviations lead to schizophrenia or autism, respectively (see [16,17]). Other causal factors (non-imprinted genes and environment) also contribute to the risk of these disorders.
Genomic imprinting, also known as parent-of-origin imprinting, normally involves the expression of only one copy of a bi-parentally inherited gene because it is tagged for expression by one parent and silenced by the other parent during early development [18–21]. Genetic mechanisms for achieving parent-of-origin expression are deletion, loss of imprint and obtaining two rather than one copy from one of the parents (uniparental disomy) [18,19]. For example, insulin-like growth factor (IGF2) is a mammalian gene with diametric gene-dosage effects. When perturbed, IGF2 can cause opposite outcomes in fetal development, birth size and subsequent behavioural syndromes [22,23] because the maternal copy is normally silenced and the paternal copy expressed. When both copies are expressed (paternal bias), the result is birth weight up to 50% above normal (with other overgrowth defects) and the behavioural disorder Beckwith–Wiedemann syndrome (BWS) [24]; when both copies are silenced (maternal bias), the result is the undergrowth disorder Silver–Russell syndrome (SRS) [25].
Badcock and Crespi [16,17] proposed that smaller deviations in imprinted gene expression with paternally biased gene-dosage effects lead to larger-born and behaviourally more demanding children, while larger deviations should increase the risk of autism. Conversely, smaller deviations in imprinted gene expression with maternally biased gene-dosage effects should lead to smaller-born and behaviourally less demanding children, while larger deviations should increase the risk of schizophrenia [16,17]. Several studies have provided circumstantial support for this idea, including opposite phenotypic and behavioural outcomes associated with deviations in genomic copy number variants (CNVs) [26], imprinted gene expression alterations [16,27], and transgenic and knockout mouse models [28]. Evidence from extreme and pathological parent-of-origin gene-dosage differences is also consistent with the Badcock and Crespi hypothesis [16,17]: IGF2 expression is higher in individuals with autism [29], and individuals with BWS have a higher risk of autism [30]. Angelman syndrome (ANS) and Prader-Willi syndrome (PWS), caused by expression versus silencing defects of imprinted genes on a fragment of chromosome 15, are also often associated with severe forms of autism and schizophrenia, respectively [31].
Badcock and Crespi's hypothesis was built on genomic imprinting theory developed by Haig [21,32–34] as an extension of Hamiltonian inclusive fitness theory [35,36]. Ubeda & Gardner [18,19] later extended the theory, emphasizing that the phenotypic effects of imprinting apply not only to maternal resource allocation during pregnancy and infant provisioning, but can also be expected to affect the mental health of adults whose life in the womb and infancy was affected by imprinting imbalances.
We used the population-wide health registries of Denmark, containing three decades of data for roughly 5 million nationals and residents, to test whether autism and schizophrenia are diametrically associated with phenotypic proxies of diametric gene-dosage effects in utero that affect birth size. We tested for opposite risks for autism and schizophrenia as a function of birth size variation, which itself has been shown to correlate with underlying deviations in diametric gene-dosage effects (see the electronic supplementary material, Material and Methods, for details). While previous smaller-scale epidemiological studies have examined the association between birth size and risk of autistic [37,38] or schizophrenic disorders [39–41], none have attempted to test for diametric autism and schizophrenia risks in a single study of the same population. Neither have previous studies attempted to exclude the pathological neurodevelopmental effects associated with being born extremely small or large, which may confound inferred associations between phenotypic markers of prenatal disrupted gene-dosages and later psychiatric disorders.
To test Badcock and Crespi's theory that autism and schizophrenia are diametrically connected, we asked four questions: (1) Is there an opposite pattern of risk present between autism and schizophrenia within ±1 s.d. of mean birth size (to avoid confounding pathological effects at distribution extremes)? That is, does above-average birth weight/length predict increased autism (but not schizophrenia) risk, and does below-average birth weight/length predict increased schizophrenia (but not autism) risk? (2) If this diametric risk pattern exists, does it increase as average birth size deviates beyond ±1 s.d., as expected if these disorders are linked by a spectrum of small-to-large diametric gene-dosage effects? (3) Are these patterns supported across the range of autistic versus schizophrenic disorders? (4) Do other known risk factors affect the hypothesized axis between autism and schizophrenia?
2. Material and methods
To answer questions (1) and (2), we focused on comparing risk patterns between autism spectrum (AS) and schizophrenia spectrum (SS) disorders based on deviations from average birth weight and length. Other autistic, schizophrenic and related disorders were included largely to answer question (3). We used Cox regression (proportional hazards) to model the risk of autism and schizophrenia as a function of birth weight and length. Covariates included to control for known familial and environmental effects on these disorders were also used to answer question (4).
(a). Study population
We identified all singleton live births (n = 1 787 447) between January 1978 and January 2009 in the Danish Fertility Database, which contains identification of parents and other birth-related information. We used unique personal identification numbers (de-identified from central-person register numbers) to link information about individuals between different registries to near completion, including the Danish National Patient Registry (which holds nationwide data on all admissions to any Danish hospital since 1977), the Danish Psychiatric Central Register (with recorded diagnoses for all psychiatric inpatient admissions since 1969), the Danish Civil Registration System and the Danish Cause of Death Registry (which contains records of changes in vital status, date of death, migration and socioeconomic status). Of the initial 1 787 447 singleton births, 29 677 (1.6%) were removed due to missing data and removal of outliers (to ensure normality), reducing the sample size to 1 757 770. Of these, 95 345 (5.42%) had an autistic or schizophrenic (or related) disorder (see study sample characteristics, electronic supplementary material, table S1).
(b). Birth size as a phenotypic marker of gene-dosage variation in utero
We assumed that normal population-level variation in birth size should (after accounting for other factors that influence birth size) contain a detectable signal of underlying deviations from average gene-dosage variation in utero. This assumption is based on the demonstrated effects of disrupted imprinted genes on birth size and subsequent postnatal behaviour (see the electronic supplementary material, Material and Methods, for details). We explored the validity of this assumption in the Danish population by examining variation in birth size for individuals with BWS, SRS, PWS and ANS (figure 2). This showed that adjusted birth sizes differ significantly depending on whether the imprinting disorder was maternally or paternally biased, strongly so for BWS, SRS and PWS, and less pronounced for ANS. This confirms that population-wide variation in birth size has the potential to capture perturbations to imprinted genes and copy number variation, and supported the use of birth size as a phenotypic marker when investigating whether autism and schizophrenia might be linked to putative diametric gene-dosage deviations in utero.
Figure 2.
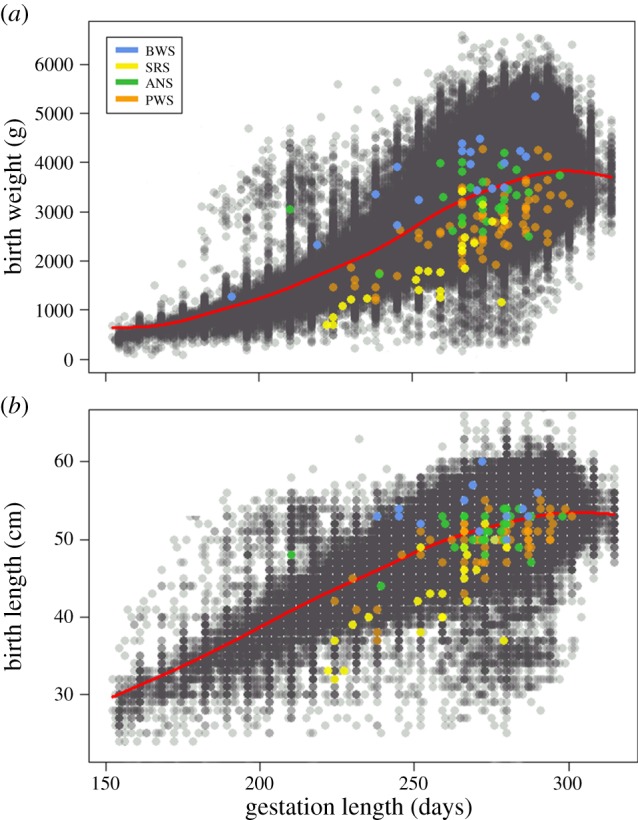
Birth weight (a) and birth length (b) as a function of gestation length for Danish individuals with (coloured dots) or without (grey dots) four known imprinted gene disorders (BWS, Beckwith–Wiedemann syndrome; SRS, Silver–Russell Syndrome; ANS, Angelman syndrome; PWS, Prader–Willi syndrome). Individuals with BWS (n = 18, paternally biased over-expression of IGF2, chromosome 11) had significantly higher birth weight (t = 9.35, p < 0.001) and length (t = 6.50, p < 0.001) than did those with SRS (n = 23, maternally biased under-expression of IGF2). Individuals with ANS (n = 29, paternally biased inactivation of certain genes on the maternally inherited chromosome 15) had significantly higher birth weight (t = 3.79, p < 0.001) and length (t = 1.69, p = 0.046) than did individuals with PWS (n = 68, maternally biased through loss of certain paternally inherited genes on chromosome 15). To remove the effect of gestation length on birth weight and length prior to running t-tests, residuals were taken from a generalized additive model (red line in plots).
(c). Removing confounding effects from birth size
We removed the effect of gestation length on birth weight and length by taking residuals from generalized additive models of these traits against gestation length (figure 2) with the global mean added to convert each residual back to its original metric. Both traits were then broken into seven categorical groups (including a mean reference group), each coded as binary, that indicated whether birth weights or lengths fell below (groups 1 to 3) or above (groups 4 to 6) the mean (figure 3; electronic supplementary material, table S1): group 1 (1850–2490 g or 45–48.0 cm); group 2 (2491–2890 g or 48.1–49.6 cm); group 3 (2891–3290 g or 49.7–51.2 cm); central group (3291–3690 g or 51.3–52.7 cm); group 4 (3691–4090 g or 52.8–54.3 cm); group 5 (4091–4490 g or 54.4–55.9 cm); group 6 (4491–5400 g or 56.0–59 cm). This categorical grouping allowed for statistical comparisons within approximately ±1 s.d. of the mean (approximated by the outer limits of groups 3 and 4) to test for presence/absence of diametric risk patterns between AS and SS while avoiding potential confounding effects associated with extreme birth sizes [42,43]. It also allowed for statistical comparisons of birth size groups between the mean and distribution edges to test for cumulative increases in risk related to hypothesized smaller–larger gene-dosage deviations.
Figure 3.

Risk of AS and SS disorders by birth weight (a) and birth length (b). Points show relative risks (RRs) obtained from Cox regression for each disorder (see the electronic supplementary material, table S4, for corresponding risk ratios, confidence intervals and p-values). Risk for other disorders related to AS and SS are also shown. Grey histogram bars represent the majority of birth data within approximately ±1 s.d. of the mean, including groups 3 (2891–3290 g or 49.7–51.2 cm), central (3291–3690 g or 51.3–52.7 cm) and 4 (3691–4090 g or 52.8–54.3 cm). Smaller babies beyond ±1 s.d. included groups 1 (1850–2490 g or 45–48.0 cm) and 2 (2491–2890 g or 48.1–49.6 cm). Larger babies included groups 5 (4091–4490 g or 54.4–55.9 cm) and 6 (4491–5400 g or 56.0–59 cm). Histograms represent birth weight and length distributions by frequency (×1000) on the right y-axis.
(d). Defining autistic and schizophrenia disorder groups
Our two main disorder groups were AS disorders and a broad SS disorder group. AS included autism (infantile and atypical), Asperger's syndrome and pervasive developmental disorder not otherwise specified. SS included schizophrenia, bipolar disorder and major depression (see ICD codes and structure, electronic supplementary material, table S2 and figure S1). Both spectra have been described before [44,45], have overlapping genetic risk loci [46], and were chosen specifically to test questions (1) and (2) because they cover the range of autistic and schizophrenic behaviours well while avoiding smaller sample sizes associated with more specific diagnoses.
The use of spectra is becoming increasingly accepted due to overlapping aetiology and behaviours, but we recognized that their definitions continue to be debated and revised [47]. We therefore also did separate analyses that used narrower and broader disorder groups related to both AS and SS (electronic supplementary material, table S2 and figure S1) to address question (3). Narrower groups derived from AS included infantile autism and infantile and atypical autism. Narrower groups derived from SS included bipolar disorder, major depression and schizophrenia. Broader groups related to AS included disorders of psychological development (as both AS and autism are derivatives from this broader group; electronic supplementary material, figure S1) and behavioural and emotional disorders with onset in childhood and adolescence. This group was chosen for the typical early onset and self-oriented behaviours, which ultimately make these individuals demanding on their carers (usually the mother) and thus functionally similar to autistic behaviours associated with the paternally biased gene-dosage effects described by Badcock & Crespi [16,17]. Broader groups related to SS included schizophrenia, plus schizotypal and delusional disorders, chosen because schizophrenia is derived from this broader group (electronic supplementary material, figure S1). For details of data validation, see the electronic supplementary material, Material and Methods.
(e). Statistical analyses and comparisons of risks
We used Cox regression in R v. 15 to estimate relative risks (RRs) of AS, SS and related disorders in offspring depending on birth weight or length grouped as defined above. Other influences were controlled for by adding them as covariates in Cox regressions; they included known familial effects, pre-existing conditions, pregnancy- and birth-related factors, and environmental and socioeconomic effects (for details on covariates, see the electronic supplementary material, Material and Methods, and table S3). To directly test for diametric risks between AS and SS disorders within ±1 s.d. of the mean, we compared Cox regression coefficients and standard errors within groups 3 or 4 with Student's t-tests (for dependent samples, survcomp package in R). Specifically, we tested whether AS risk was significantly higher than SS risk for larger babies (group 4) and vice versa for smaller babies (group 3). To test for broader cumulative risk patterns within AS or SS, we used survcomp to compare Cox regression coefficients and standard errors, specifically asking whether AS risk was significantly lower in group 4 than 5 and lower in group 5 than 6, and whether SS risk was significantly lower in group 3 than 2 and lower in group 2 than 1.
3. Results
(a). Autism spectrum and schizophrenia spectrum disorders
(i). Relative risk patterns within ±1 s.d. of mean birth size
For larger-born babies (group 4 in figure 3), AS risk was significantly higher (6% for birth weight and 18% for birth length) and SS risk was significantly lower (6% for both weight and length at birth) relative to the central group (figure 3; electronic supplementary material, table S4). Risk for AS for these above-average babies was significantly higher than risk for SS disorders (birth weight, t = 20.03, p < 0.001; birth length, t = 41.6, p < 0.001). For smaller-born babies (group 3 in figure 3), AS risk was significantly lower (8% for birth weight and 11% for birth length) and SS risk was significantly higher (8% for birth weight and 12% for birth length) relative to the central group (figure 3). For these below-average babies, SS risk was significantly higher than AS risk (birth weight, t = 19.3, p < 0.001; birth length, t = 26.4, p < 0.001).
(ii). Relative risk patterns across all birth sizes
Considering the full birth size range (groups 1 to 6 in figure 3; electronic supplementary material, table S4) an opposing pattern of risks between AS and SS persisted for all groups when considering birth length and for all groups except group 1 when considering birth weight. The magnitude of AS risks increased cumulatively across groups 4–5–6 (weight, 6–23–53%; length 18–31–56%; figure 3) as babies were born increasingly larger, with each risk significantly larger than the previous for weight (group 4 < 5, t = 3.12, p < 0.001; group 5 < 6, t = 3.03, p = 0.001) and length (group 4 < 5, t = 2.21, p < 0.013; group 5 < 6, t = 2.71, p = 0.003). The magnitude of SS risks across groups 4–5–6 remained below average with a significant monotonic decrease with increasing birth length (figure 3). Across groups 3–2–1 (figure 3), the magnitude of SS risks increased as babies were born increasingly smaller (weight, 8–21–25%; length, 12–24–34%), with all risks significantly larger than the previous for length (group 3 < 2, t = 2.94, p = 0.001; 2 < 1, t = 1.73, p = 0.041) and between one of the two group comparisons for weight (group 3 < 2, t = 3.35, p < 0.001; group 2 < 1, t = 0.59, p = 0.277).
(b). Other disorders related to the autism and schizophrenia spectra
Within ±1 s.d. of mean birth size (groups 3 and 4 in figure 3; electronic supplementary material, table S4), patterns of RR for narrower and broader disorder groups displayed a similar pattern of risk to those found for AS and SS disorders. Larger-born babies (group 4) were at increased risk for most disorders related to AS (birth weight, 3 out of 4; birth length, 4 of 4) and at decreased risk for 2 of 4 disorders related to SS for both birth size predictor variables (figure 3). Smaller-born babies (group 3) were at increased risk for all disorders related to SS (both predictor variables, 4 out of 4) and decreased risk for most disorders related to AS (both predictor variables, 3 of 4).
Considering the entire birth size range (groups 1 to 6; figure 3), RR patterns for the narrower and broader disorder groups were likewise largely consistent with those found for AS and SS. For larger-born babies (groups 4–5–6), risks were higher for autistic-related relative to schizophrenic-related disorders, with autistic risks tending to increase in magnitude when further from the mean (figure 3). For smaller-born babies (groups 3–2–1), risks tended to be larger for schizophrenic-related disorders relative to autistic-related disorders between groups 3 to 2. However, the clarity of this separation disappeared at the smallest birth size in group 1, indicating that only considering the smallest babies does not provide encompassing insight in the overall trends that underlie risk for mental disorders across all birth sizes.
(c). Other risk factors
Patterns of RR related to covariates (figure 4; electronic supplementary material, tables S5–S14) showed that autistic disorders tended to be higher in male offspring and schizophrenic disorders more prevalent in female offspring, except for two groups (schizophrenia, schizophrenia—schizotypal–delusional disorders) that were higher in males. This may be due to differences in severity [16] and age at detection [48,49] between the sexes, where schizophrenia is more often severe in males (and perhaps obvious earlier in life) with onset delayed in females, including a second peak for women aged 45–79 [49]. The 30 years of follow-up available for this study (electronic supplementary material, figure S2) may thus not have allowed sufficient time to capture all cases of these disorders in females, especially for those born closer to 2009.
Figure 4.

Risks patterns for autistic (left) and schizophrenic (right) disorders based on covariates. Red shades indicate magnitudes of increased risk and blue shades decreased risks (see the electronic supplementary material, tables S5–S14, for detailed values). Blank panels represent coefficients that could not be calculated due to lack of variation. White dots (centre of square) indicate significance (p < 0.05).
Offspring born from older fathers had increased risks for all disorders, with risks for 8 of 10 disorders significantly increased. These risks tended to be larger for autistic than schizophrenic disorders. By contrast, increasing maternal age decreased risk for four out of five schizophrenic-related disorders and one autistic-related group, but increased risk for three autistic-related groups. Compared with first-born children, second- and later-born children had relatively lower risk for autistic disorders but higher risk for schizophrenic disorders (figure 4).
4. Discussion
(a). Opposite risk patterns present for autism and schizophrenia
The results of our study consistently support the Badcock and Crespi hypothesis [16,17] that autism and schizophrenia are opposite extremes of a single gradient of mental disorders. This is remarkable, for the inclusive fitness theory that produced these predictions [18,19,21,32,34–36] was initially blind to any clinical applications and remains at present the only theory that predicts the diametrically opposed risk patterns that our study uncovered. The evolutionary logic and our results for normal birth sizes within ±1 s.d. (which answered the first question that we posed) underscore that, in an evolutionary sense, our physical and mental health is always at risk of being compromised by deep kinship conflicts that we share with most other animals [21,32–34,50].
Our results lend credibility to Badcock & Crespi's [16,17] idea that the opposed risk factors will at least partly be due to diametric gene-dosage deviations caused by disrupted genomic imprinting or copy number variants. However, this support is indirect. We still lack most of the genetic evidence needed to understand which genes became subject to parent-of-origin imprinting and why other genes escaped that fate, but the evidence available so far seems encouraging. For example, reciprocal deletions and duplications at two loci 1q21.1 and 16p11.2 appear to be linked with diametric phenotypes in the autistic–schizophrenic spectrum [28,51]. Duplications of 1q21.1 and deletions of 16p11.2 are correlated with larger head size in childhood and increased risk of autism, whereas deletions of 1q21.1 and duplications of 16p11.2 are associated with smaller head size and higher risk of schizophrenia [52,53]. A recent analysis of CNVs found four chromosomal regions where reciprocal deletions and duplications contributed to the diametric association between autism and schizophrenia. However, CNV variation in three other regions affected autism and schizophrenia similarly rather than diametrically [26].
It is important to note that our results are consistent with previous work examining risk of autism or schizophrenia depending on birth size. Using a sample of 1.49 million singleton births between 1973 and 1986 from Denmark and Sweden, Abel et al. [39] found an overall significant negative trend in risk for schizophrenia and affective disorders based on eight birth weight groups, indicating higher and lower risks for smaller and larger babies, respectively. Using a sample of 589 114 children aged 0–17 who resided in Stockholm County from 2001 to 2007, Abel et al. [37] later found a significant nonlinear trend in risk for AS disorders based on nine birth weight z-score groups, indicating higher risks in very small and large babies relative to those born of average size. Both studies support the risk patterns found across the entire birth size range examined here, but they examined autism and schizophrenia independently, and their aim was not to test for presence or absence of a diametric association between both disorders in the same population and for normal birth sizes. Other epidemiological studies [38,40,41] have also examined risk of either autism or schizophrenia based on birth size, but most were limited by very small sample sizes and were likewise not testing for opposite patterns of risk between autism and schizophrenia.
(b). Larger deviations in birth size increase risks for most disorders
The second question that we addressed concerned Badcock and Crespi's prediction [16,17] that disturbances in paternal and maternal gene-dosage deviations should directionally increase or decrease risks of specific psychiatric disorders outwards from the centre of average birth size and normal cognition. Our results testing the full birth size range mostly supported this prediction. AS risk (and other related behaviours) continued to increase with increasing birth weight and length, and SS risk (and related behaviours) continued to increase with decreasing birth weight and length. This pattern was consistent across the entire birth length range, but at extremely low birth weight (group 1, figure 3), the separation between AS and SS became less clear. This is consistent with earlier findings that abnormal gestations resulting in very low birth sizes may be harmful for brain development in general, increasing the risks for many psychiatric disorders [37,39] irrespective of imprinting or copy number predispositions that may bias risk in a specific direction when birth size is closer to the population mean.
The third question we asked was whether risk patterns would consistently support the Badcock and Crespi hypothesis whether diagnoses were grouped broadly or narrowly. As figure 3 illustrates, this appeared to be the case, with remarkably few exceptions. This level of coherence also suggests that the results that we obtained are robust in spite of ongoing discussions among clinicians on how best to categorize and refine diagnoses of mental disease [47].
(c). Effects of parental age, maternal hypertension and parity
We found a consistent effect of paternal age on risk of both autistic- and schizophrenic-related disorders, which relates to predictions from previous studies suggesting that age-related increases in the mutational load of male sperm may negatively impact offspring psychiatric health [1,54]. Errors in sperm production could increase risk for either autism or schizophrenia, for example through duplication of paternally expressed genes or expression of normally paternally silenced genes with effects on neurodevelopment. Such age effects are not expected for maternal gametes as they are mostly produced while the female is still in her mother's womb.
In light of this, the effects of maternal age in our analyses are interesting, for they appear to be directional, with largely increasing risks of autistic and decreasing risks of schizophrenic disorders in offspring. However, our finding that first-born children have an elevated risk of autistic diagnoses later in life suggests that the relationship between risk of psychiatric disorders and maternal age may be U-shaped. Maternal hypertension during pregnancy significantly increased risk for all autistic (but not schizophrenic) disorders, consistent with autistic-type disorders being related to paternally induced higher demands on fetal provisioning that require higher maternal metabolism during pregnancy [32,55].
(d). Caveats and perspectives
Two main caveats should be noted. First, the diametric pattern predicted by Badcock and Crespi (supported by figure 3) is only one, albeit a very coherent, factor contributing to the causation and prevalence of autism and schizophrenia. The finding that many other variables we included also contributed significant risk to both disorders (figure 4; electronic supplementary material, tables S5–S14) echoes this. Second, the statistical trends we uncovered represent relatively modest changes in absolute disease risk, which were 0.65% (or 10 890 of 1 673 315) for AS disorders and 1.23% (or 20 672 of 1 683 097) for SS disorders. Thus, for example, the 53% enhanced AS relative risk of group 6 babies (birth weights 4491–5400 g) increased their absolute risk to 1% and the 25% increased SS relative risk of group 1 babies (1850–2490 g) increased their absolute risk to 1.54%.
These reservations notwithstanding, the shifts that we found in RR of being diagnosed with autism or schizophrenia (or related) disorders were remarkably consistent. This robustness of correlative evidence suggests that it will be worthwhile to extend large-scale genomic studies [5,11] with epigenetic studies that could clarify which genes are involved in parent-of-origin expression, and how their imprints and copy numbers are established. Collecting enough brain samples to make meaningful connections between disorders and differential imprinting or copy numbers of genes that affect fetal provisioning and the behaviour of children and young adolescents will be challenging. Our results make clear, however, that such studies deserve priority given the enormous significance of neuropsychiatric illnesses for the human condition.
Supplementary Material
Supplementary Material
Acknowledgements
Statistics Denmark provided access to all de-identified data hosted on a secure computer server. We thank Bernard Crespi and Christopher Badcock for comments on an earlier version of the manuscript, Birgitte Hollegaard for assistance in gaining access to data and discussion, Charlotte Nielsen and Morten Lindboe at Statistics Denmark for facilitating data access and technical support, and the referees for their constructive suggestions.
Ethics statement
Approval for our study was obtained from the Danish Data Protection Agency, the Danish National Board of Health, the Danish Psychiatric Central Research Registry and Statistics Denmark.
Data accessibility
Data for this study are regulated by public authorities in accordance with The Danish Act on Processing of Personal Data (Act No. 429 of 31 May 2000). Data have been deposited under terms of a contract at Statistics Denmark (www.dst.dk) and cannot leave the servers at Statistics Denmark. Access to the data used in this study can be granted to other researchers through an affiliation with Centre for Social Evolution, University of Copenhagen, if approved by Statistics Denmark. For further information please contact Professor Jacobus J Boomsma, Centre for Social Evolution (JJBoomsma@bio.ku.dk) and the Head of Division for Research Services, Ivan Thaulow (ITH@DST.dk), Statistics Denmark.
Funding statement
The Centre for Social Evolution and its Evolutionary Medicine programme are funded by the Danish National Research Foundation (DNRF57). S.G.B. was also funded by a Marie Curie International Incoming Fellowship FP7-PEOPLE-2010-IIF-276565.
References
- 1.Geschwind DH. 2009. Advances in autism. Annu. Rev. Med. 60, 367–380. ( 10.1146/annurev.med.60.053107.121225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geschwind DH. 2011. Genetics of autism spectrum disorders. Trends Cogn. Sci. 15, 409–416. ( 10.1016/j.tics.2011.07.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Svrakic DM, Zorumski CF, Svrakic NM, Zwir I, Cloninger CR. 2013. Risk architecture of schizophrenia: the role of epigenetics. Curr. Opin. Psychiatry 26, 188–195. ( 10.1097/YCO.0b013e32835d8329) [DOI] [PubMed] [Google Scholar]
- 4.van Os J, Kapur S. 2009. Schizophrenia. Lancet 374, 635–645. ( 10.1016/S0140-6736(09)60995-8) [DOI] [PubMed] [Google Scholar]
- 5.Gaugler T, et al. 2014. Most genetic risk for autism resides with common variation. Nat. Genet. 46, 881–887. ( 10.1038/ng.3039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lichtenstein P, Carlstrom E, Rastam M, Gillberg C, Anckarsater H. 2010. The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am. J. Psychiatry 167, 1357–1363. ( 10.1176/appi.ajp.2010.10020223) [DOI] [PubMed] [Google Scholar]
- 7.Lichtenstein P, Yip BH, Bjork C, Pawitan Y, Cannon TD, Sullivan PF, Hultman CM. 2009. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet 373, 234–239. ( 10.1016/S0140-6736(09)60072-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sullivan PF, Kendler KS, Neale MC. 2003. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 60, 1187–1192. ( 10.1001/archpsyc.60.12.1187) [DOI] [PubMed] [Google Scholar]
- 9.Sullivan PF, Daly MJ, O'Donovan M. 2012. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat. Rev. Genet. 13, 537–551. ( 10.1038/nrg3240) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Dongen J, Boomsma DI. 2013. The evolutionary paradox and the missing heritability of schizophrenia. Am. J. Med. Genet. B 162, 122–136. ( 10.1002/ajmg.b.32135) [DOI] [PubMed] [Google Scholar]
- 11.Schizophrenia Working Group of the Psychiatric Genomics. 2014. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. ( 10.1038/nature13595) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuk O, Hechter E, Sunyaev SR, Lander ES. 2012. The mystery of missing heritability: genetic interactions create phantom heritability. Proc. Natl Acad. Sci. USA 109, 1193–1198. ( 10.1073/pnas.1119675109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abel KM. 2004. Foetal origins of schizophrenia: testable hypotheses of genetic and environmental influences. Br. J. Psychiatry 184, 383–385. ( 10.1192/bjp.184.5.383) [DOI] [PubMed] [Google Scholar]
- 14.Davies W, Isles AR, Wilkinson LS. 2005. Imprinted gene expression in the brain. Neurosci. Biobehav. Rev. 29, 421–430. ( 10.1016/j.neubiorev.2004.11.007) [DOI] [PubMed] [Google Scholar]
- 15.Kopsida E, Mikaelsson MA, Davies W. 2011. The role of imprinted genes in mediating susceptibility to neuropsychiatric disorders. Horm. Behav. 59, 375–382. ( 10.1016/j.yhbeh.2010.04.005) [DOI] [PubMed] [Google Scholar]
- 16.Badcock C, Crespi B. 2008. Battle of the sexes may set the brain. Nature 454, 1054–1055. ( 10.1038/4541054a) [DOI] [PubMed] [Google Scholar]
- 17.Crespi B, Badcock C. 2008. Psychosis and autism as diametrical disorders of the social brain. Behav. Brain Sci. 31, 241. [DOI] [PubMed] [Google Scholar]
- 18.Ubeda F, Gardner A. 2010. A model for genomic imprinting in the social brain: juveniles. Evolution 64, 2587–2600. ( 10.1111/j.1558-5646.2010.01015.x) [DOI] [PubMed] [Google Scholar]
- 19.Ubeda F, Gardner A. 2011. A model for genomic imprinting in the social brain: adults. Evolution 65, 462–475. ( 10.1111/j.1558-5646.2010.01115.x) [DOI] [PubMed] [Google Scholar]
- 20.Ferguson-Smith AC. 2011. Genomic imprinting: the emergence of an epigenetic paradigm. Nat. Rev. Genet. 12, 565–575. ( 10.1038/nrg3032) [DOI] [PubMed] [Google Scholar]
- 21.Haig D. 2004. Genomic imprinting and kinship: how good is the evidence? Annu. Rev. Genet. 38, 553–585. ( 10.1146/annurev.genet.37.110801.142741) [DOI] [PubMed] [Google Scholar]
- 22.Constancia M, et al. 2002. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 417, 945–948. ( 10.1038/nature00819) [DOI] [PubMed] [Google Scholar]
- 23.Eggermann T, Eggermann K, Schonherr N. 2008. Growth retardation versus overgrowth: Silver–Russell syndrome is genetically opposite to Beckwith–Wiedemann syndrome. Trends Genet. 24, 195–204. ( 10.1016/j.tig.2008.01.003) [DOI] [PubMed] [Google Scholar]
- 24.Weksberg R, Shen DR, Fei YL, Song QL, Squire J. 1993. Disruption of insulin-like growth factor 2 imprinting in Beckwith–Wiedemann syndrome. Nat. Genet. 5, 143–150. ( 10.1038/ng1093-143) [DOI] [PubMed] [Google Scholar]
- 25.Gicquel C, et al. 2005. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver–Russell syndrome. Nat. Genet. 37, 1003–1007. ( 10.1038/ng1629) [DOI] [PubMed] [Google Scholar]
- 26.Crespi B, Stead P, Elliot M. 2010. Comparative genomics of autism and schizophrenia. Proc. Natl Acad. Sci. USA 107(Suppl. 1), 1736–1741. ( 10.1073/pnas.0906080106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isles AR, Davies W, Wilkinson LS. 2006. Genomic imprinting and the social brain. Phil. Trans. R. Soc. B 361, 2229–2237. ( 10.1098/rstb.2006.1942) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crespi B. 2013. Diametric gene-dosage effects as windows into neurogenetic architecture. Curr. Opin. Neurobiol. 23, 143–151. ( 10.1016/j.conb.2012.08.005) [DOI] [PubMed] [Google Scholar]
- 29.Mills JL, Hediger ML, Molloy CA, Chrousos GP, Manning-Courtney P, Yu KF, Brasington M, England LJ. 2007. Elevated levels of growth-related hormones in autism and autism spectrum disorder. Clin. Endocrinol. 67, 230–237. ( 10.1111/j.1365-2265.2007.02868.x) [DOI] [PubMed] [Google Scholar]
- 30.Kent L, Bowdin S, Kirby GA, Cooper WN, Maher ER. 2008. Beckwith Weidemann syndrome: a behavioral phenotype–genotype study. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 1295–1297. ( 10.1002/ajmg.b.30729) [DOI] [PubMed] [Google Scholar]
- 31.Nicholls RD, Saitoh S, Horsthemke B. 1998. Imprinting in Prader–Willi and Angelman syndromes. Trends Genet. 14, 194–200. ( 10.1016/S0168-9525(98)01432-2) [DOI] [PubMed] [Google Scholar]
- 32.Haig D. 1993. Genetic conflicts in human-pregnancy. Q. Rev. Biol. 68, 495–532. ( 10.1086/418300) [DOI] [PubMed] [Google Scholar]
- 33.Haig D. 2010. Colloquium papers: transfers and transitions: parent–offspring conflict, genomic imprinting, and the evolution of human life history. Proc. Natl Acad. Sci. USA 107(Suppl. 1), 1731–1735. ( 10.1073/pnas.0904111106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore T, Haig D. 1991. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 7, 45–49. ( 10.1016/0168-9525(91)90230-N) [DOI] [PubMed] [Google Scholar]
- 35.Abbot P, et al. 2011. Inclusive fitness theory and eusociality. Nature 471, E1–E4; author reply E9–10 ( 10.1038/nature09831) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamilton WD. 1972. Altruism and related phenomena mainly in the social insects. Annu. Rev. Ecol. Syst. 3, 193–232. ( 10.1146/annurev.es.03.110172.001205) [DOI] [Google Scholar]
- 37.Abel KM, Dalman C, Svensson AC, Susser E, Dal H, Idring S, Webb RT, Rai D, Magnusson C. 2013. Deviance in fetal growth and risk of autism spectrum disorder. Am. J. Psychiatry 170, 391–398. ( 10.1176/appi.ajp.2012.12040543) [DOI] [PubMed] [Google Scholar]
- 38.Losh M, Esserman D, Anckarsater H, Sullivan PF, Lichtenstein P. 2012. Lower birth weight indicates higher risk of autistic traits in discordant twin pairs. Psychol. Med. 42, 1091–1102. ( 10.1017/S0033291711002339) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abel KM, Wicks S, Susser ES, Dalman C, Pedersen MG, Mortensen PB, Webb RT. 2010. Birth weight, schizophrenia, and adult mental disorder: is risk confined to the smallest babies? Arch. Gen. Psychiatry 67, 923–930. ( 10.1001/archgenpsychiatry.2010.100) [DOI] [PubMed] [Google Scholar]
- 40.Byrne M, Agerbo E, Bennedsen B, Eaton WW, Mortensen PB. 2007. Obstetric conditions and risk of first admission with schizophrenia: a Danish national register based study. Schizophr. Res. 97, 51–59. ( 10.1016/j.schres.2007.07.018) [DOI] [PubMed] [Google Scholar]
- 41.Eaton WW, Mortensen PB, Frydenberg M. 2000. Obstetric factors, urbanization and psychosis. Schizophr. Res. 43, 117–123. ( 10.1016/S0920-9964(99)00152-8) [DOI] [PubMed] [Google Scholar]
- 42.Curhan GC, Willett WC, Rimm EB, Spiegelman D, Ascherio AL, Stampfer MJ. 1996. Birth weight and adult hypertension, diabetes mellitus, and obesity in US men. Circulation 94, 3246–3250. ( 10.1161/01.CIR.94.12.3246) [DOI] [PubMed] [Google Scholar]
- 43.Gluckman PD, Hanson MA, Beedle AS. 2007. Early life events and their consequences for later disease: a life history and evolutionary perspective. Am. J. Hum. Biol. 19, 1–19. ( 10.1002/Ajhb.20590) [DOI] [PubMed] [Google Scholar]
- 44.Newschaffer CJ, et al. 2007. The epidemiology of autism spectrum disorders. Annu. Rev. Public Health 28, 235–258. ( 10.1146/annurev.publhealth.28.021406.144007) [DOI] [PubMed] [Google Scholar]
- 45.Tienari P, Wynne LC, Laksy K, Moring J, Nieminen P, Sorri A, Lahti I, Wahlberg KE. 2003. Genetic boundaries of the schizophrenia spectrum: evidence from the Finnish adoptive family study of schizophrenia. Am. J. Psychiatry 160, 1587–1594. ( 10.1176/appi.ajp.160.9.1587) [DOI] [PubMed] [Google Scholar]
- 46.Cross-Disorder Group of the Psychiatric Genomics et al. 2013. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 45, 984–994. ( 10.1038/ng.2711) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adam D. 2013. Mental health: on the spectrum. Nature 496, 416–418. ( 10.1038/496416a) [DOI] [PubMed] [Google Scholar]
- 48.Castle D, Sham P, Murray R. 1998. Differences in distribution of ages of onset in males and females with schizophrenia. Schizophr. Res. 33, 179–183. ( 10.1016/S0920-9964(98)00070-X) [DOI] [PubMed] [Google Scholar]
- 49.Hafner H, Maurer K, Loffler W, Riecher-Rossler A. 1993. The influence of age and sex on the onset and early course of schizophrenia. Br. J. Psychiatry 162, 80–86. ( 10.1192/bjp.162.1.80) [DOI] [PubMed] [Google Scholar]
- 50.Hamilton WD. 1996. Narrow roads of gene land: the collected papers of W.D. Hamilton. Oxford, UK: WH Freeman/Spektrum. [Google Scholar]
- 51.Crespi BJ, Crofts HJ. 2012. Association testing of copy number variants in schizophrenia and autism spectrum disorders. J. Neurodev. Disord. 4, 15 ( 10.1186/1866-1955-4-15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, Barnes CC, Pierce K. 2011. Neuron number and size in prefrontal cortex of children with autism. JAMA 306, 2001–2010. ( 10.1001/jama.2011.1638) [DOI] [PubMed] [Google Scholar]
- 53.Rees E, et al. 2014. Evidence that duplications of 22q11.2 protect against schizophrenia. Mol. Psychiatry 19, 37–40. ( 10.1038/Mp.2013.156) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kong A, et al. 2012. Rate of de novo mutations and the importance of father's age to disease risk. Nature 488, 471–475. ( 10.1038/nature11396) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hollegaard B, Byars SG, Lykke J, Boomsma JJ. 2013. Parent–offspring conflict and the persistence of pregnancy-induced hypertension in modern humans. PLoS ONE 8, e56821 ( 10.1371/journal.pone.0056821) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data for this study are regulated by public authorities in accordance with The Danish Act on Processing of Personal Data (Act No. 429 of 31 May 2000). Data have been deposited under terms of a contract at Statistics Denmark (www.dst.dk) and cannot leave the servers at Statistics Denmark. Access to the data used in this study can be granted to other researchers through an affiliation with Centre for Social Evolution, University of Copenhagen, if approved by Statistics Denmark. For further information please contact Professor Jacobus J Boomsma, Centre for Social Evolution (JJBoomsma@bio.ku.dk) and the Head of Division for Research Services, Ivan Thaulow (ITH@DST.dk), Statistics Denmark.