

Abstract
Major climatic and geological events but also population history (secondary contacts) have generated cycles of population isolation and connection of long and short periods. Recent empirical and theoretical studies suggest that fast evolutionary processes might be triggered by such events, as commonly illustrated in ecology by the adaptive radiation of cichlid fishes (isolation and reconnection of lakes and watersheds) and in epidemiology by the fast adaptation of the influenza virus (isolation and reconnection in hosts). We test whether cyclic population isolation and connection provide the raw material (standing genetic variation) for species evolution and diversification. Our analytical results demonstrate that population isolation and connection can provide, to populations, a high excess of genetic diversity compared with what is expected at equilibrium. This excess is either cyclic (high allele turnover) or cumulates with time depending on the duration of the isolation and the connection periods and the mutation rate. We show that diversification rates of animal clades are associated with specific periods of climatic cycles in the Quaternary. We finally discuss the importance of our results for macroevolutionary patterns and for the inference of population history from genomic data.
Keywords: migration, population subdivision, secondary contact, diversification
1. Introduction
Successive environmental changes have often modified species habitat in the past, repeatedly isolating and connecting populations, successively suppressing and allowing migration between them. During the climatic cycles of the Quaternary period, many species experienced repeated long periods of isolation into refugia followed by population reconnection [1]. At least five wet–dry periods occurred within the African continent in which water-level fluctuations have successively isolated and connected ecosystems [2]. Such dynamics are often associated with rapid species and population diversification, for example, allopatric differentiations of African buffalo populations [3], giraffe [4] and their associated predators (e.g. lions [5]). Similarly, repeated hybridizations within and between watersheds are expected to have played an important role for Cichlid adaptive radiation [6,7]. In Europe and North America, past climatic cycles led to isolation and connection events in many land animals such as mammals [1], birds [8], insects [9], and also in marine, coastal and freshwater species [10,11]. It is thus now widely recognized that climatic and geological events impact population genetics and speciation patterns at different temporal and spatial scales [12].
Isolation and connection events are common features of virus history. Their dependency on the host isolate viral populations in between transmission events. Events of reconnection of populations occur through superinfections (co-infections) [13,14]. Such isolation and connection events have been widely observed and constitute a major cause of pandemics and failures in virus control. For example, a mix of swine, avian and human influenza was responsible for the 2009 swine flu outbreak [15].
Previous studies showed that population genetic diversity is slightly impacted by migration fluctuation and that its influence can be approximated with an effective migration rate [16,17]. By contrast, high transient values of genetic diversity can be observed following a single [18] or multiple [19] connection events. Those results raised several questions. In particular, would these high transient values of genetic diversity cumulate, be maintained or decrease under cycles of isolation and connection? Answers to this question are not only of theoretical interest, but also have strong implications for the understanding of the mechanisms of adaptation and diversification. Adaptation from standing genetic variation might be more common than from de novo mutations as alleles from stranding genetic variation are present at higher frequency, can switch selective value and are compatible with the background genome [20–23]. Transient excesses of genetic diversity could then provide the raw material for adaptation from standing genetic variation. Similarly, it could increase diversification by promoting speciation [24] and decreasing extinction rate (preventing inbreeding depression [25], but see outbreeding depression [26]). This view is supported by theoretical and empirical works on speciation and adaptive radiation, which suggest that a high level of pre-existing neutral genetic diversity in founder populations is determinant to have a high rate of speciation [21,27–30]. Finally, simulation studies have shown that cycles of isolation and connection of populations can trigger species diversification [31].
While a single [18] or multiple [19] events of populations connection can generate a large excess of neutral genetic diversity within populations, the conditions of existence and maintenance of such genetic diversity excesses remain to be determined. To investigate these questions, we analytically describe the dynamics of genetic diversity under successive periods of isolation and connection of populations (migration cyclically takes a value of 0 and m) of different durations. Assuming that all alleles are selectively neutral, we disentangle the relative importance of the length of the isolation and connection periods in regard to the mutation rate, the migration rate, the number of populations and the initial population genetic diversity. We characterize equilibrium trajectories and transient values of genetic diversity towards equilibrium. We determine four domains in which genetic diversity has a determined behaviour in regards to genetic diversity accumulation and turnover. Then, using clades from the main animal orders, we assess the relationship between diversification rate and length of the isolation and connection periods during the climatic cycles of the Quaternary. Finally, we discuss the implication of our results for species adaptation to novel environments, macroevolution and the inference of population history.
2. Dynamics of genetic diversity under cycles of connection and isolation
To study the dynamics of genetic diversity, we consider n populations of size N (diploid individuals) that are cyclically connected and isolated. Connection and isolation events are of regular periods of length P generations each. Under this model, we determine the recursion equations describing the dynamics of genetic diversities within and across isolation and connection periods and the long-term equilibrium trajectory of genetic diversity. We assume a finite island model with migration that varies between 0 (isolation) and (n − 1)/n (panmixia [32,33]), non-overlapping generations (Wright–Fisher model [34,35]) and consider that mutations (rate μ) follow the infinite allele model [36].
(a). The dynamics of genetic diversities within connection and isolation periods
The dynamics of genes in populations under the finite island model can be described by transitions in gene identities between generations [33]. Using the relationship between gene identity, F, and genetic diversity, H, H = 1 − F, the genetic diversity H(t) at generation t can be decomposed into the within-population genetic diversity, hs(t), and the between-population genetic diversity, hb(t) [33]:
| 2.1 |
where hs(t) and hb(t) correspond to the probability that two genes, randomly sampled from the same and from different populations, respectively, are different [37].
The dynamics of genetic diversity (equation (2.1)) can be described solving the equations of [33] (electronic supplementary material, S1). The change in genetic diversities after P generations during a connection period and during an isolation period, Hc(P) and Hi(P), respectively, become
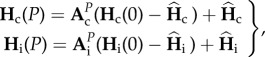 |
2.2 |
where Hc(0) and Hi(0) are the initial genetic diversities during the connection and the isolation periods and 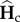 and
and  are their expected value at equilibrium [36,38]. Matrices Ac and Ai are transition matrices which determine the probability of identity of two genes in the same or in different populations, given their previous identity in the same or in different populations, they follow [33]
are their expected value at equilibrium [36,38]. Matrices Ac and Ai are transition matrices which determine the probability of identity of two genes in the same or in different populations, given their previous identity in the same or in different populations, they follow [33]
| 2.3a |
with parameters
| 2.3b |
| 2.3c |
| 2.3d |
| 2.4 |
In equation (2.3a), a (resp. b) corresponds to the probability that two genes sampled in the same population (resp. different populations) were in the same population in the previous generation, and c corresponds to the probability that two genes sampled in the same population are copies of the same gene (from [39]); (1 − μ)2 corresponds to the probability that neither of the two genes mutated. Thus, (1 − μ)2a(1 − c) (resp. (1 − μ)2b(1 − c)) is the probability that two genes that were identical and in the same population (resp. different populations) at a given generation are still identical and in the same population at the next generation; (1 − μ)2(1 − a) (resp. (1 − μ)2(1 − b)) is the probability that two genes that were identical and in the same population (resp. different populations) at a given generation are still identical and in different populations at the next generation.
(b). The dynamics of genetic diversities across cycles of connection and isolation
From equation (2.2), we derive the recurrence equations describing the dynamics of genetic diversity across multiple cycles k of connection and isolation. We obtain the genetic diversities, 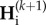 , at the end of any cycle k + 1 (end of isolation period), given genetic diversities,
, at the end of any cycle k + 1 (end of isolation period), given genetic diversities, 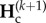 , at the end of the connection period of the same cycle k + 1. Similarly,
, at the end of the connection period of the same cycle k + 1. Similarly, 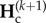 depends on
depends on 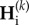 , the genetic diversities at the end of the previous cycle k, thus we have
, the genetic diversities at the end of the previous cycle k, thus we have
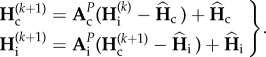 |
2.5 |
These equations provide a full description of the transient dynamics of genetic diversities across any number of cycles k of duration P. Equation (2.5) can be solved analytically. To do so, we first substitute the expression of 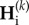 in the equation of
in the equation of 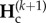 (equation (2.5)), which leads to an inhomogeneous linear recurrence equation:
(equation (2.5)), which leads to an inhomogeneous linear recurrence equation:
| 2.6a |
where
| 2.6b |
and C is a constant vector which depends on the values of 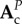 ,
, 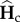 and
and 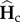 .
.
As matrix I − Γc is invertible, equation (2.6) has an equilibrium value, 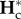 , and its solution is
, and its solution is
| 2.7 |
Consequently, the dynamics of genetic diversities at the end of each cycle k are determined by matrix Γc. Similarly, the matrix 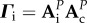 determines the dynamics of
determines the dynamics of 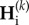 . As Γi and Γc have the same eigenvalues, the behaviour of genetic diversity studied here is independent of the order of the sequence of connection and isolation events.
. As Γi and Γc have the same eigenvalues, the behaviour of genetic diversity studied here is independent of the order of the sequence of connection and isolation events.
(c). Equilibrium trajectory of genetic diversities under cycles of connection and isolation
The equilibrium trajectory of genetic diversities is reached when two consecutive isolation events, with a connection event in between, have the same genetic diversity values, i.e. when
| 2.8 |
Denoting 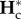 and
and 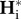 the equilibrium values of
the equilibrium values of 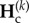 and
and 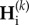 , respectively, equations (2.5) and (2.8) lead to
, respectively, equations (2.5) and (2.8) lead to
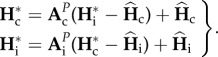 |
2.9 |
By solving equation (2.9), we have
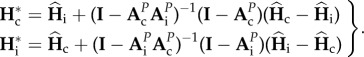 |
2.10 |
From equation (2.10), it can be seen that the equilibrium trajectory depends only on the parameters of the model (M, θ, n, N and P), and are independent from the initial value of genetic diversities.
(d). The duration of the transient dynamics and the behaviour of genetic diversities
Here, we determine the duration of the transient dynamics (relaxation time or time to equilibrium values) for the within- and between-population genetic diversities during connection and isolation periods—respectively PC, PT, PI and PS—and we show how they influence the behaviour of genetic diversities across and within cycles. We define the relaxation time τ as the time required to converge to equilibrium value, such that λτ = δ and thus τ = ln(δ)/ln(λ) with δ a small value (0 < δ < 1) and λ the corresponding eigenvalue in the transition matrix. The relaxation times for hs and hb within a connection (PC and PT) and an isolation event (PI and PS) are thus determined by the two eigenvalues of matrix Ac and the two eigenvalues of matrix Ai (from highest to lowest). From equations (2.3a) and (2.4), and assuming small migration and mutation rates (i.e. 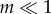 and
and 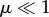 ) and large population sizes (i.e.
) and large population sizes (i.e. 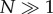 ), and as ln(1 − X) = −X + o(X), we have
), and as ln(1 − X) = −X + o(X), we have
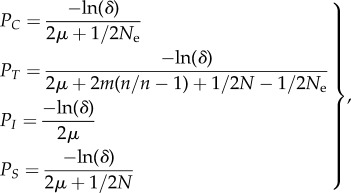 |
2.11 |
where 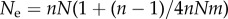 corresponds to the effective population size under the finite island model [40].
corresponds to the effective population size under the finite island model [40].
The expressions of PC, PI and PS in equation (2.11) have also an interpretation in the coalescence framework (the ‘coalescent with killings’ [41]) as the denominator in expressions corresponds to the log probability that lineages did not mutate or coalesce within a generation.
Given the relaxation times and the period length P, we can determine whether genetic diversities remain in transient states. Five situations can be described.
First, both within- and between-population genetic diversities reach successively their equilibrium values during isolation and connection periods within a timeframe shorter than P (i.e. the highest relaxation time from equation (2.11), PI, is smaller than P; figure 1a). Therefore, after each event, 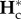 and
and 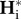 tend to their respective equilibrium,
tend to their respective equilibrium, 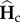 and
and  .
.
Figure 1.

Trajectories of within- (hs) and between-population (hb) genetic diversities under cycles of isolation and connection: (a) in the long-period domain P > PI and (b) in the short-period domain P < PW. The dashed and dotted lines represent the expected equilibrium value when populations are connected and isolated, respectively. In (a), both hs and hb reach their expected equilibrium value at the end of each connection and isolation period. In (b), both hs and hb tend to their equilibrium value of connection (dashed line) with very small fluctuations. Parameters are M = 40, n = 10, N = 2 000, μ = 2.5 × 10−5. (a) P = 60 000, (b) P = 150.
Second, genetic diversities reach their equilibrium value during the connection periods, but not during isolation periods, i.e. PI and PC (from equation (2.11)) are larger and smaller than P, respectively. 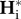 does not reach the isolation equilibrium value within P, whereas
does not reach the isolation equilibrium value within P, whereas 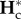 tends to its equilibrium value within P. Genetic diversities at the beginning of the connection period have transient values corresponding to the one observed at the end of the isolation period.
tends to its equilibrium value within P. Genetic diversities at the beginning of the connection period have transient values corresponding to the one observed at the end of the isolation period.
Third, genetic diversities do not reach their equilibrium value, neither during isolation nor during connection periods. This implies that all relaxation times during connection and isolation are larger than P. The trajectory of the successive peaks of hs (i.e. the successive maximum values of hs in each cycle) monotonously tends to an equilibrium value (arrows in figure 2, and electronic supplementary material S2 for panmixia). However, the trajectory of genetic diversities to their equilibrium values presents strong fluctuations (figure 2), caused by the succession of peaks of genetic diversity generated by connection events. Their direction, increasing or decreasing, depends on the initial value of between-population genetic diversity, hb(0). When hb(0) is below its equilibrium value, successive connection events generate peaks of genetic diversity of increasing values (figure 2a). When hb(0) is above its equilibrium value, successive connection events generate peaks of genetic diversity with decreasing values (figure 2b).
Figure 2.

Trajectories of within- (hs) and between-population (hb) genetic diversities under cycles of isolation and connection in the domain PW < P < PI when initial genetic diversity hb(0) is (a) low and (b) high. In (a), when hb(0) is low, the successive peaks of within-population genetic diversity (grey arrows) increase in size, while in (b) they decrease in size. In both (a) and (b), the equilibrium trajectories of genetic diversity (framed by a rectangle) are the same. Parameters are M = 400, θ = 0.1 n = 4, N = 10 000, P = 50 000 generations.
Fourth, periods of isolation P are so short that they do not impact values of genetic diversity (negligible genetic differentiation during isolation). In this situation, the value of genetic diversities at the end of the isolation period is close to the one at the end of the connection period of the same cycle, k, and we have 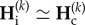 . From equation (2.5), this implies that the smallest eigenvalue, λ, of matrix Ai at the power P is close to 1,
. From equation (2.5), this implies that the smallest eigenvalue, λ, of matrix Ai at the power P is close to 1, 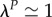 . Consequently, we can define the maximum number of generations, PW, for which genetic diversities are not impacted by isolation periods. PW can be obtained from
. Consequently, we can define the maximum number of generations, PW, for which genetic diversities are not impacted by isolation periods. PW can be obtained from 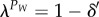 , where δ′ is a small number. Thus, we have
, where δ′ is a small number. Thus, we have
| 2.12 |
The dynamics of genetic diversities when the period is smaller than PW are illustrated in figure 1a: the first event of connection generates a large peak of genetic diversity, owing to the quick distribution among populations of mutations accumulated during isolation. Following this major change in genetic diversity, the successive isolation and connection events generate only very small fluctuations of genetic diversity values in their trajectories to their equilibrium. The time to reach their equilibrium value is the same as under constant migration (without cycles; electronic supplementary material, S2).
Fifth, genetic diversity values are very high owing to a very high mutation rate and are weakly impacted by connection and isolation events. The limit of the domain can be derived with a threshold value of the scaled mutation rate, θsat (the scaled mutation rate is 4Nμ) for which genetic diversities remain high and approximately constant. This situation occurs when 4Nμ/(1 + 4Nμ) > 1 − α, where α is a small number, and implies that 4Nμ > θsat = (1 − α)/α. Note that the saturation of genetic diversity values is a consequence of having an infinite allele model.
In summary, we have found five situations where cycles of isolation and connection have a different impact on the trajectories of genetic diversity. Interestingly, the limit between these situations depends mainly on the mutation rate μ, the population size N and the number of generations (duration) of the connection and isolation periods P (see equation (2.11)).
Those results are robust to stochasticity. Indeed, considering that the length of the isolation and connection periods P is a random variable leads to qualitatively similar results, as long as the variance of P is smaller than a determined threshold (electronic supplementary material, S3).
(e). The dynamics of the excess of genetic diversity within populations
We now describe the dynamics of the excess of within-population genetic diversities under cycles of connection and isolation, relative to their expected equilibrium values (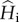 and
and 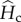 ), as a function of the period length P and model parameters. We focus on two measures that are representative of two important processes: genetic diversity turnover and accumulation. Genetic diversity turnover is measured as the amplitude of the variations of the excess of within-population genetic diversity within an isolation and connection cycle (i.e. the difference between maximum and minimum value of hs). This quantity provides information on whether the excess of hs is fluctuating or stationary. Genetic diversity accumulation is measured as the mean excess of within-population genetic diversity during the cycles. This quantity provides information on the overall excess of hs produced by the cycles of isolation and connection. These values are computed numerically from equations (2.2) and (2.5) and represented in figure 3.
), as a function of the period length P and model parameters. We focus on two measures that are representative of two important processes: genetic diversity turnover and accumulation. Genetic diversity turnover is measured as the amplitude of the variations of the excess of within-population genetic diversity within an isolation and connection cycle (i.e. the difference between maximum and minimum value of hs). This quantity provides information on whether the excess of hs is fluctuating or stationary. Genetic diversity accumulation is measured as the mean excess of within-population genetic diversity during the cycles. This quantity provides information on the overall excess of hs produced by the cycles of isolation and connection. These values are computed numerically from equations (2.2) and (2.5) and represented in figure 3.
Figure 3.

(a) Turnover and (b) accumulation of within-population genetic diversity hs during cycles of isolation and connection, as a function of the period P and the scaled mutation rate 4Nμ, (c) domains of turnover and accumulation of hs, (d) relationship between P and the net diversification rate of clades from the major animal orders. Turnover of hs is measured as the amplitude of the variations of hs within a cycle. Accumulation of hs is measured as the mean excess of hs per generation compared to its expected equilibrium value. In (c), Domain A presents large turnover of hs. Domain B presents both turnover and accumulation of hs. Domain C presents small turnover and large accumulation of hs. In domain D, hs saturates and all hs values are larger than 0.9 during the cycles. Parameters in (a–c) are n = 10 populations, m = 0.01 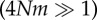 , 2N = 1000 for each population, 200 000 generations. (d) P and net diversification rate for species representative of the main animal orders: mammals (Muridae, Ursidae, Bovidae, Cervidae, Hystricidae, Cercopithecidae, Cebidae, Cricetidae [42]), birds (passerines, [43], Darwin's finches, [44]), reptiles (collubrid snakes, [42], annoline lizards, [45]), bony fishes (Ictalurus, Cyprinidae, [42], cichlids, [46]), insects (Tipulidae, Formicinae [42]), bivalves (Petricolidae, Mesodematidae, Semelidae [42]), barnacles [42] and echinoids (Mellitidae, Clypeasteridae, Laganidae [42]). P corresponds to the period of climatic cycles in generations (100 000 years cycles divided by the generation time of each species). The shaded area represents the range of diversification rates, computed in sliding windows of 0.2 log10(years) (see alternative window sizes in the electronic supplementary material, figure S1). The dashed and dotted lines represent the values of PI for a gene of 1 and 10 kb (haplotype blocks in Eukaryotes range from 10 kb to hundreds of kb [47]), respectively, and a mutation rate of 10−8 bp−1 (mutation rates in animals range from 10−9 bp−1 to 5.10−8 bp−1 [48]).
, 2N = 1000 for each population, 200 000 generations. (d) P and net diversification rate for species representative of the main animal orders: mammals (Muridae, Ursidae, Bovidae, Cervidae, Hystricidae, Cercopithecidae, Cebidae, Cricetidae [42]), birds (passerines, [43], Darwin's finches, [44]), reptiles (collubrid snakes, [42], annoline lizards, [45]), bony fishes (Ictalurus, Cyprinidae, [42], cichlids, [46]), insects (Tipulidae, Formicinae [42]), bivalves (Petricolidae, Mesodematidae, Semelidae [42]), barnacles [42] and echinoids (Mellitidae, Clypeasteridae, Laganidae [42]). P corresponds to the period of climatic cycles in generations (100 000 years cycles divided by the generation time of each species). The shaded area represents the range of diversification rates, computed in sliding windows of 0.2 log10(years) (see alternative window sizes in the electronic supplementary material, figure S1). The dashed and dotted lines represent the values of PI for a gene of 1 and 10 kb (haplotype blocks in Eukaryotes range from 10 kb to hundreds of kb [47]), respectively, and a mutation rate of 10−8 bp−1 (mutation rates in animals range from 10−9 bp−1 to 5.10−8 bp−1 [48]).
The highest turnover of genetic diversity is observed when the period lengths are large (P > 2N; figure 3a) and when the mutation rate is moderate (4Nμ < θsat = 9). The lowest turnover of genetic diversity is observed when the mutation rate is high (4Nμ > θsat = 9) or when 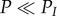 . When 4Nμ > θsat, genetic diversity is saturated by mutations: isolation and connection events do not impact the value of genetic diversity in populations as the genetic diversity is at its maximum value (close to 1).
. When 4Nμ > θsat, genetic diversity is saturated by mutations: isolation and connection events do not impact the value of genetic diversity in populations as the genetic diversity is at its maximum value (close to 1).
The highest amount of cumulated genetic diversity is observed when isolation and connection periods are smaller than 2N. In this case, relaxation times are larger than the periods. Consequently, genetic diversities remain in a transient state and can cumulate over time. The lowest cumulated genetic diversity is observed when the mutation rate is high and genetic diversity is saturated by mutations (4Nμ > θsat), or when P > PI.
From the estimation of the value of genetic turnover and accumulation, we can define four domains in which genetic diversities have a specific behaviour in regard to turnover and accumulation (figure 3c). In domain A, genetic diversity turnover is observed and no accumulation of genetic diversity occurs. Domain A is defined by P > PI and 4Nμ < θsat. In domain B, both accumulation and turnover of genetic diversity are observed. Domain B is defined by approximately PI/60 < P < PI, and 4Nμ < θsat. In domain C, genetic diversity cumulates across cycles, and there is almost no variation of genetic diversities within cycles. Domain C is defined by 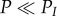 and 4Nμ < θsat. Finally, in domain D, genetic diversity is saturated, 4Nμ > θsat, thus any demographic change will not impact its value.
and 4Nμ < θsat. Finally, in domain D, genetic diversity is saturated, 4Nμ > θsat, thus any demographic change will not impact its value.
3. Species diversification and climatic cycles
An important result of our model is that populations which undergo long-period cycles (domain A) experience a turnover of genetic diversity, whereas populations which undergo short-period cycles (domain C) cumulate genetic diversity. To assess whether these two behaviours could have an impact on species diversification patterns, we investigate the relationship between the known climatic cycles and empirical net diversification rates of animal clades in the Quaternary period (see corresponding reference in the legend of figure 3d); the clades chosen are representative of the main animal orders and include—but are not restricted to—radiations. Recent Quaternary climatic cycles have a periodicity of approximately 100 000 years [49,50]. From this periodicity, we can estimate for each animal the corresponding period P (in generations). Then, we computed the range of diversification rates as a function of the period P (figure 3d).
The range of diversification rates (shaded area figure 3d) decreases with period P (two-sided Spearman correlation test, coefficient −0.76139, p-value < 2.2 × 10−16). The highest diversification rates (the most notorious examples of adaptive radiations: cichlid fishes, Darwin's finches and colubrid snakes) are observed between P = 10 000 and P = 15 000 generations and are associated with the domains B and C where genetic diversity is accumulated across cycles.
These results are expected to be robust to shorter-period cycles (e.g. 20 000 or 41 000 years, as in the early Quaternary [50]) as we use a logarithmic scale for the period, which is weakly affected by a fivefold change. In addition, longer isolation periods than connection periods (as expected in glacial oscillations) will not change the general behaviour of our model. Indeed, the amount of cumulated genetic diversity is mainly determined by the duration of the isolation period and PI (figure 3c) that is defined by model parameters during isolation.
4. Discussion
Although there is increasing evidence of the importance of large-scale environmental changes for the generation and maintenance of genetic diversity, they received few and only recently attention [19,31]. Our study identifies domains where genetic diversities have specific behaviours. In domain A (P > PI and 4Nμ < θsat; figure 3c), genetic diversities undergo large variations, allowing for a high turnover of genetic diversity. The lower limit PI of this domain can be large; for example, we show that for animals, it is of the order of 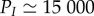 generations for typical mutation rates and a sequence of 10 kb (figure 3d). In this domain, high rates of molecular evolution are expected for animals with a short generation time (as observed for small mammals [51], birds [52] and invertebrates [53]). In domains B and C (P < PI and 4Nμ < θsat; figure 3) genetic diversity cumulates across successive cycles of isolation and connection. Species experiencing such cycles of isolation and connection are predicted to have large amounts of genetic diversity, increasing their adaptive potential [20,21]. Interestingly, species with the highest diversification rates of the Quaternary belong to this domain, for example, the cichlid fishes in Africa [46] and Darwin's finches [44]. We show that successive isolation and connection periods in domain B (figure 2) can also lead to a decrease in genetic diversity when the initial level of genetic diversity is high. The existence of two alternative outcomes could explain why populations which underwent similar isolation and connection events in their history can display different genetic signatures such as found for the meadow grasshopper (0.7–0.9% pairwise sequence difference) and for the hedgehog populations (6–12% pairwise sequence difference) in Europe [1]. In addition, it could partially explain why some clades which experienced short-period cycles of connection and isolation do not display large diversification rates (mammals [42,54]).
generations for typical mutation rates and a sequence of 10 kb (figure 3d). In this domain, high rates of molecular evolution are expected for animals with a short generation time (as observed for small mammals [51], birds [52] and invertebrates [53]). In domains B and C (P < PI and 4Nμ < θsat; figure 3) genetic diversity cumulates across successive cycles of isolation and connection. Species experiencing such cycles of isolation and connection are predicted to have large amounts of genetic diversity, increasing their adaptive potential [20,21]. Interestingly, species with the highest diversification rates of the Quaternary belong to this domain, for example, the cichlid fishes in Africa [46] and Darwin's finches [44]. We show that successive isolation and connection periods in domain B (figure 2) can also lead to a decrease in genetic diversity when the initial level of genetic diversity is high. The existence of two alternative outcomes could explain why populations which underwent similar isolation and connection events in their history can display different genetic signatures such as found for the meadow grasshopper (0.7–0.9% pairwise sequence difference) and for the hedgehog populations (6–12% pairwise sequence difference) in Europe [1]. In addition, it could partially explain why some clades which experienced short-period cycles of connection and isolation do not display large diversification rates (mammals [42,54]).
Isolation events might be associated with a reduction of the size of populations [1,55,56]. While strong bottlenecks can cause population extinction, a reduced population size will not affect the relaxation time PI (equation (2.11)). Thus, the limit between the domains (figure 3c) where genetic diversity cumulates or turnover across cycles remains unchanged. However, reduced population size will decrease the relaxation time PW, limiting the size of the domain (C; figure 3c) where genetic diversities remain approximately constant within cycles. Population dynamics within cycle (population growth, generation overlap) are also expected to impact our predictions. Slow recovery from population size changes will decrease PW and strong generation overlap will reduce the transient excess of genetic diversity.
Our results suggest that climatic cycles may drive high genetic diversity accumulation or turnover in populations, leading, respectively, to increased or reduced diversification rates. Although the relationship between genetic diversity and species diversification is supported by some empirical [24,28] and theoretical evidence [57], this hypothesis is still in debate. More generally, there is a lack of comprehensive understanding of the relationship between micro- and macroevolution patterns. Most investigations focused on adaptation and ignored the impact of neutral demographic changes [58,59]. These relationships deserve more investigations, which might answer critical questions in phylogeography, such as the origin of species' pumps [60].
Hughes & Eastwood [61] suggested that extrinsic causes such as geological events, rather than key innovations, could trigger species diversification. Our results propose that such extrinsic causes lead to different dynamics of the genetic diversity depending on species' generation time. We expect that other life-history traits such as dispersal, generation overlap, habitat specialization or mating system might also influence the level of genetic diversity and the rate of molecular evolution [62]. Further investigations in that direction are needed to provide a better understanding of the interaction between extrinsic causes, life-history traits and patterns of diversification.
Our result have implications for the inference of population demographic history from genome polymorphism [63]. We predict that genomic regions having different diversity levels might have different diversity dynamics (e.g. increase and decrease) when experiencing the same cycles of connection and isolation. This effect might be important given the high heterogeneity of polymorphism along genomes [64], or when considering molecular markers with different mutation rates (e.g. allozymes, single nucleotide polymorphisms and microsatellites). In addition, genomic regions might experience different levels of gene flow [65] which could have a direct impact on the dynamics of genetic diversity. These features should be accounted for and could be used to develop robust inference methods. A large range of demographic scenarios, including multiple migration changes, have been implemented in inference methods [63,66], but very few methods account for such heterogeneity [67].
Our results suggest that reconnection events (secondary contacts) after large periods of isolation can provide to populations a large excess of genetic diversity. Thus, they reveal a mechanism that maintains high genetic diversity as unexpectedly observed in some domesticated organisms despite strong selection (e.g. gene flow between domesticated and wild populations, in rice [68] and cats [69]) and also observed—although undesirable—in pathogens [70]. This mechanism—events of isolation and reconnection over a large number of generations—is predominant in pathogens and occurs at different levels and scales. Indeed, pathogens can jump from one species to another after a very long period of isolation and adapt rapidly (e.g. avian flu [15,71] and human immunodeficiency virus [14]). Pathogen admixture can occur between related populations from different isolated geographical regions [13,72]. Pathogens are isolated within hosts in between transmission events and can reconnect through superinfection [73]. Finally, isolation and reconnection events within hosts between genetic compartments (e.g. cerebrospinal fluid, gut mucosa, urine, plasma) are increasingly recognized [74,75]. How pathogen diversity is related to their past demography, their structure or the environment they encountered remain to be explored in more detail. This might be a timely issue, because organisms (invasive species, local adapted population, viruses and other pathogens) have increased opportunities to encounter previously isolated related populations owing to climate change and increased ease of travel worldwide.
Supplementary Material
Acknowledgements
The authors thank the editor Hans Heesterbeek, the associate editor Wieslaw Babik, Thomas Lenormand and four anonymous reviewers for their valuable comments and suggestions.
Data accessibility
This study involves no original data.
Funding statement
This project was supported by the Swiss National Science Foundation (grants no. PZ00P3_139421/1 and 31003A-130065), by an Interdisciplinary grant from the Faculty of Biology and Medicine (FBM) of the University of Lausanne and by the Department of Ecology and Evolution of the University of Lausanne.
References
- 1.Hewitt G. 2000. The genetic legacy of the Quaternary ice ages. Nature 405, 907–913. ( 10.1038/35016000) [DOI] [PubMed] [Google Scholar]
- 2.Szabo B, Haynes C, Jr, Maxwell TA. 1995. Ages of Quaternary pluvial episodes determined by uranium-series and radiocarbon dating of lacustrine deposits of eastern Sahara. Palaeogeogr. Palaeoclimatol. Palaeoecol. 113, 227–242. ( 10.1016/0031-0182(95)00052-N) [DOI] [Google Scholar]
- 3.Heller R, Brüniche-Olsen A, Siegismund HR. 2012. Cape buffalo mitogenomics reveals a Holocene shift in the African human-megafauna dynamics. Mol. Ecol. 21, 3947–3959. ( 10.1111/j.1365-294X.2012.05671.x) [DOI] [PubMed] [Google Scholar]
- 4.Brown DM, et al. 2007. Extensive population genetic structure in the giraffe. BMC Biol. 5, 57 ( 10.1186/1741-7007-5-57) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnett R, Yamaguchi N, Barnes I, Cooper A. 2006. The origin, current diversity and future conservation of the modern lion (Panthera leo). Proc. R. Soc. B 273, 2119–2125. ( 10.1098/rspb.2006.3555) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seehausen O. 2002. Patterns in fish radiation are compatible with pleistocene desiccation of lake Victoria and 14,600 year history for its cichlid species flock. Proc R. Soc. Lond. B 269, 491–497. ( 10.1098/rspb.2001.1906) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nevado B, Mautner S, Sturmbauer C, Verheyen E. 2013. Water-level fluctuations and metapopulation dynamics as drivers of genetic diversity in populations of three tanganyikan cichlid fish species. Mol. Ecol. 22, 3933–3948. ( 10.1111/mec.12374) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zink RM. 1996. Comparative phylogeography in North American birds. Evolution 50, 308–317. ( 10.2307/2410802) [DOI] [PubMed] [Google Scholar]
- 9.Hewitt GM. 1996. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 58, 247–276. ( 10.1111/j.1095-8312.1996.tb01434.x) [DOI] [Google Scholar]
- 10.Avise JC. 1992. Molecular population structure and the biogeographic history of a regional fauna: a case history with lessons for conservation biology. Oikos 63, 62–76. ( 10.2307/3545516) [DOI] [Google Scholar]
- 11.Patarnello T, Volckaert FAMJ, Castilho R. 2007. Pillars of hercules: is the Atlantic Mediterranean transition a phylogeographical break? Mol. Ecol. 16, 4426–4444. ( 10.1111/j.1365-294X.2007.03477.x) [DOI] [PubMed] [Google Scholar]
- 12.Gillespie RG, Roderick GK. 2014. Evolution: geology and climate drive diversification. Nature 509, 297–298. ( 10.1038/509297a) [DOI] [PubMed] [Google Scholar]
- 13.Burke DS. 1997. Recombination in HIV: an important viral evolutionary strategy. Emerg. Infect. Dis. 3, 253 ( 10.3201/eid0303.970301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Korber B, Gaschen B, Yusim K, Thakallapally R, Kesmir C, Detours V. 2001. Evolutionary and immunological implications of contemporary HIV-1 variation. Br. Med. Bull. 58, 19–42. ( 10.1093/bmb/58.1.19) [DOI] [PubMed] [Google Scholar]
- 15.Flahault A, Zylberman P. 2010. Influenza pandemics: past, present and future challenges. Public Health Rev. 32, 319–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagylaki T. 1979. The island model with stochastic migration. Genetics 91, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitlock MC. 1992. Temporal fluctuations in demographic parameters and the genetic variance among populations. Evolution 46, 608–615. ( 10.2307/2409631) [DOI] [PubMed] [Google Scholar]
- 18.Alcala N, Streit D, Goudet J, Vuilleumier S. 2013. Peak and persistent excess of genetic diversity following an abrupt migration increase. Genetics 193, 953–971. ( 10.1534/genetics.112.147785) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jesus F, Wilkins J, Solferini V, Wakeley J. 2006. Expected coalescence times and segregating sites in a model of glacial cycles. Genet. Mol. Res. 5, 466–474. [PubMed] [Google Scholar]
- 20.Hermisson J, Pennings PS. 2005. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics 169, 2335–2352. ( 10.1534/genetics.104.036947) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrett RDH, Schluter D. 2008. Adaptation from standing genetic variation. Trends Ecol. Evol. 23, 38–44. ( 10.1016/j.tree.2007.09.008) [DOI] [PubMed] [Google Scholar]
- 22.Hernandez RD, Kelley JL, Elyashiv E, Melton SC, Auton A, McVean G, Project G, Sella G, Przeworski M. 2011. Classic selective sweeps were rare in recent human evolution. Science 331, 920–924. ( 10.1126/science.1198878) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones FC, et al. 2012. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484, 55–61. ( 10.1038/nature10944) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abbott R, et al. 2013. Hybridization and speciation. J. Evol. Biol. 26, 229–246. ( 10.1111/j.1420-9101.2012.02599.x) [DOI] [PubMed] [Google Scholar]
- 25.Frankham R. 1996. Relationship of genetic variation to population size in wildlife. Conserv. Biol. 10, 1500–1508. ( 10.1046/j.1523-1739.1996.10061500.x) [DOI] [Google Scholar]
- 26.Edmands S. 2007. Between a rock and a hard place: evaluating the relative risks of inbreeding and outbreeding for conservation and management. Mol. Ecol. 16, 463–475. ( 10.1111/j.1365-294X.2006.03148.x) [DOI] [PubMed] [Google Scholar]
- 27.Barrier M, Baldwin BG, Robichaux RH, Purugganan MD. 1999. Interspecific hybrid ancestry of a plant adaptive radiation: allopolyploidy of the Hawaiian silversword alliance (asteraceae) inferred from floral homeotic gene duplications. Mol. Biol. Evol. 16, 1105–1113. ( 10.1093/oxfordjournals.molbev.a026200) [DOI] [PubMed] [Google Scholar]
- 28.Seehausen O. 2004. Hybridization and adaptive radiation. Trends Ecol. Evol. 19, 198–207. ( 10.1016/j.tree.2004.01.003) [DOI] [PubMed] [Google Scholar]
- 29.Gavrilets S, Losos JB. 2009. Adaptive radiation: contrasting theory with data. Science 323, 732–737. ( 10.1126/science.1157966) [DOI] [PubMed] [Google Scholar]
- 30.Bezault E, Mwaiko S, Seehausen O. 2011. Population genomic tests of models of adaptive radiation in lake Victoria region cichlid fish. Evolution 65, 3381–3397. ( 10.1111/j.1558-5646.2011.01417.x) [DOI] [PubMed] [Google Scholar]
- 31.Aguilée R, Claessen D, Lambert A. 2013. Adaptive radiation driven by the interplay of eco-evolutionary and landscape dynamics. Evolution 67, 1291–1306. ( 10.1111/j.1558-5646.2012.01816.x) [DOI] [PubMed] [Google Scholar]
- 32.Latter BD. 1973. The island model of population differentiation: a general solution. Genetics 73, 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crow JF. 1986. Basic concepts in population, quantitative, and evolutionary genetics. New York, NY: WH Freeman and Company. [Google Scholar]
- 34.Fisher RA. 1930. The genetical theory of natural selection. Oxford, UK: Clarendon Press. [Google Scholar]
- 35.Wright S. 1931. Evolution in Mendelian populations. Genetics 16, 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimura M, Crow JF. 1964. The number of alleles that can be maintained in a finite population. Genetics 49, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nei M. 1973. Analysis of gene diversity in subdivided populations. Proc. Natl Acad. Sci. USA 70, 3321–3323. ( 10.1073/pnas.70.12.3321) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maruyama T. 1970. Effective number of alleles in a subdivided population. Theor. Popul. Biol. 1, 273–306. ( 10.1016/0040-5809(70)90047-X) [DOI] [PubMed] [Google Scholar]
- 39.Maynard Smith J. 1970. Population size, polymorphism, and the rate of non-Darwinian evolution. Am. Nat. 104, 231–237. ( 10.1086/282657) [DOI] [Google Scholar]
- 40.Nei M, Takahata N. 1993. Effective population size, genetic diversity, and coalescence time in subdivided populations. J. Mol. Evol. 37, 240–244. [DOI] [PubMed] [Google Scholar]
- 41.Durrett R. 2002. Probability models for DNA sequence evolution. New York, NY: Springer. [Google Scholar]
- 42.Stanley SM. 1979. Macroevolution. San Francisco, CA: W. H. Freeman. [Google Scholar]
- 43.Ricklefs RE. 2006. Global variation in the diversification rate of passerine birds. Ecology 87, 2468–2478. ( 10.1890/0012-9658(2006)87[2468:GVITDR]2.0.CO;2) [DOI] [PubMed] [Google Scholar]
- 44.Grant PR, Grant BR. 1992. Hybridization of bird species. Science 256, 193–197. ( 10.1126/science.256.5054.193) [DOI] [PubMed] [Google Scholar]
- 45.Irschick DJ, Vitt LJ, Zani PA, Losos JB. 1997. A comparison of evolutionary radiations in mainland and Caribbean anolis lizards. Ecology 78, 2191–2203. ( 10.1890/0012-9658(1997)078[2191:ACOERI]2.0.CO;2) [DOI] [Google Scholar]
- 46.Kocher TD. 2004. Adaptive evolution and explosive speciation: the cichlid fish model. Nat. Rev. Genet. 5, 288–298. ( 10.1038/nrg1316) [DOI] [PubMed] [Google Scholar]
- 47.Guryev V, Smits BMG, van de Belt J, Verheul M, Hubner N, Cuppen E. 2006. Haplotype block structure is conserved across mammals. PLoS Genet. 2, e121 ( 10.1371/journal.pgen.0020121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lynch M. 2010. Evolution of the mutation rate. Trends Genet. 26, 345–352. ( 10.1016/j.tig.2010.05.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hays J, Imbrie J, Shackleton N. 1976. Variations in the Earth's orbit: pacemaker of the ice ages. Science 194, 1121–1132. ( 10.1126/science.194.4270.1121) [DOI] [PubMed] [Google Scholar]
- 50.Elderfield H, Ferretti P, Greaves M, Crowhurst S, McCave I, Hodell D, Piotrowski A. 2012. Evolution of ocean temperature and ice volume through the mid-Pleistocene climate transition. Science 337, 704–709. ( 10.1126/science.1221294) [DOI] [PubMed] [Google Scholar]
- 51.Ohta T. 1993. An examination of the generation-time effect on molecular evolution. Proc. Natl Acad. Sci. USA 90, 10 676–10 680. ( 10.1073/pnas.90.22.10676) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mooers AO, Harvey PH. 1994. Metabolic rate, generation time, and the rate of molecular evolution in birds. Mol. Phylogenet. Evol. 3, 344–350. ( 10.1006/mpev.1994.1040) [DOI] [PubMed] [Google Scholar]
- 53.Thomas JA, Welch JJ, Lanfear R, Bromham L. 2010. A generation time effect on the rate of molecular evolution in invertebrates. Mol. Biol. Evol. 27, 1173–1180. ( 10.1093/molbev/msq009) [DOI] [PubMed] [Google Scholar]
- 54.Bininda-Emonds ORP, et al. 2007. The delayed rise of present-day mammals. Nature 446, 507–512. ( 10.1038/nature05634) [DOI] [PubMed] [Google Scholar]
- 55.Young A, Boyle T, Brown T. 1996. The population genetic consequences of habitat fragmentation for plants. Trends Ecol. Evol. 11, 413–418. ( 10.1016/0169-5347(96)10045-8) [DOI] [PubMed] [Google Scholar]
- 56.Lorenzen ED, Heller R, Siegismund HR. 2012. Comparative phylogeography of African savannah ungulates. Mol. Ecol. 21, 3656–3670. ( 10.1111/j.1365-294X.2012.05650.x) [DOI] [PubMed] [Google Scholar]
- 57.Gavrilets S. 2003. Perspective: models of speciation: what have we learned in 40 years? Evolution 57, 2197–2215. ( 10.1111/j.0014-3820.2003.tb00233.x) [DOI] [PubMed] [Google Scholar]
- 58.Hansen TF, Martins EP. 1996. Translating between microevolutionary process and macroevolutionary patterns: the correlation structure of interspecific data. Evolution 50, 1404–1417. ( 10.2307/2410878) [DOI] [PubMed] [Google Scholar]
- 59.Uyeda JC, Hansen TF, Arnold SJ, Pienaar J. 2011. The million-year wait for macroevolutionary bursts. Proc. Natl Acad. Sci. USA 108, 15 908–15 913. ( 10.1073/pnas.1014503108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rull V. 2011. Neotropical biodiversity: timing and potential drivers. Trends Ecol. Evol. 26, 508–513. ( 10.1016/j.tree.2011.05.011) [DOI] [PubMed] [Google Scholar]
- 61.Hughes C, Eastwood R. 2006. Island radiation on a continental scale: exceptional rates of plant diversification after uplift of the Andes. Proc. Natl Acad. Sci. USA 103, 10 334–10 339. ( 10.1073/pnas.0601928103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bazin E, Glémin S, Galtier N. 2006. Population size does not influence mitochondrial genetic diversity in animals. Science 312, 570–572. ( 10.1126/science.1122033) [DOI] [PubMed] [Google Scholar]
- 63.Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M. 2013. Robust demographic inference from genomic and SNP data. PLoS Genet. 9, e1003905 ( 10.1371/journal.pgen.1003905) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abecasis G, et al. 2010. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073. ( 10.1038/nature09534) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bierne N, Gagnaire P-A, David P. 2013. The geography of introgression in a patchy environment and the thorn in the side of ecological speciation. Curr. Zool. 59, 72–86. [Google Scholar]
- 66.Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 5, e1000695 ( 10.1371/journal.pgen.1000695) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roux C, Fraïsse C, Castric V, Vekemans X, Pogson GH, Bierne N. 2014. Can we continue to neglect genomic variation in introgression rates when inferring the history of speciation? A case study in a Mytilus hybrid zone. J. Evol. Biol. 27, 1662–1675. ( 10.1111/jeb.12425) [DOI] [PubMed] [Google Scholar]
- 68.Caicedo AL, et al. 2007. Genome-wide patterns of nucleotide polymorphism in domesticated rice. PLoS Genet. 3, e163 ( 10.1371/journal.pgen.0030163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lecis R, Pierpaoli M, Biro Z, Szemethy L, Ragni B, Vercillo F, Randi E. 2006. Bayesian analyses of admixture in wild and domestic cats (Felis silvestris) using linked microsatellite loci. Mol. Ecol. 15, 119–131. ( 10.1111/j.1365-294X.2005.02812.x) [DOI] [PubMed] [Google Scholar]
- 70.Lawrence JG. 2005. Common themes in the genome strategies of pathogens. Curr. Opin. Genet. Dev. 15, 584–588. ( 10.1016/j.gde.2005.09.007) [DOI] [PubMed] [Google Scholar]
- 71.Garten RJ, et al. 2009. Antigenic and genetic characteristics of swine origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325, 197–201. ( 10.1126/science.1176225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leroy T, Le Cam B, Lemaire C. In press. When virulence originates from non-agricultural hosts: new insights into plant breeding. Infect. Genet. Evol. [DOI] [PubMed] [Google Scholar]
- 73.Conway DJ, Roper C, Oduola AM, Arnot DE, Kremsner PG, Grobusch MP, Curtis CF, Greenwood BM. 1999. High recombination rate in natural populations of Plasmodium falciparum. Proc. Natl Acad. Sci. USA 96, 4506–4511. ( 10.1073/pnas.96.8.4506) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trono D, Van Lint C, Rouzioux C, Verdin E, Barré-Sinoussi F, Chun T-W, Chomont N. 2010. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science 329, 174–180. ( 10.1126/science.1191047) [DOI] [PubMed] [Google Scholar]
- 75.Renzette N, et al. 2013. Evolution of the influenza A virus genome during development of Oseltamivir resistance in vitro. J. Virol. 88, 272–281. ( 10.1128/JVI.01067-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study involves no original data.