

Abstract
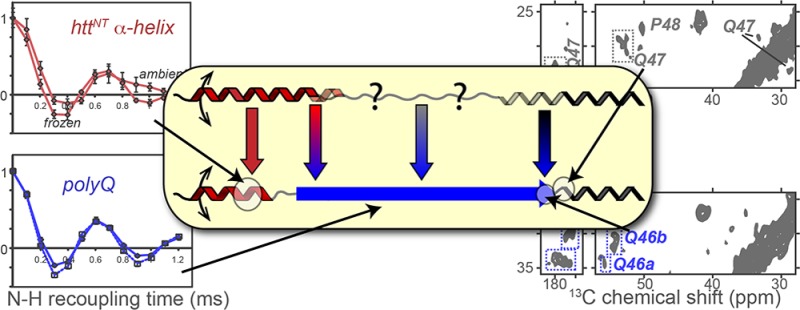
In Huntington’s disease, expansion of a polyglutamine (polyQ) domain in the huntingtin (htt) protein leads to misfolding and aggregation. There is much interest in the molecular features that distinguish monomeric, oligomeric, and fibrillar species that populate the aggregation pathway and likely differ in cytotoxicity. The mechanism and rate of aggregation are greatly affected by the domains flanking the polyQ segment within exon 1 of htt. A “protective” C-terminal proline-rich flanking domain inhibits aggregation by inducing polyproline II structure (PPII) within an extended portion of polyQ. The N-terminal flanking segment (httNT) adopts an α-helical structure as it drives aggregation, helps stabilize oligomers and fibrils, and is seemingly integral to their supramolecular assembly. Via solid-state nuclear magnetic resonance (ssNMR), we probe how, in the mature fibrils, the htt flanking domains impact the polyQ domain and in particular the localization of the β-structured amyloid core. Using residue-specific and uniformly labeled samples, we find that the amyloid core occupies most of the polyQ domain but ends just prior to the prolines. We probe the structural and dynamical features of the remarkably abrupt β-sheet to PPII transition and discuss the potential connections to certain htt-binding proteins. We also examine the httNT α-helix outside the polyQ amyloid core. Despite its presumed structural and demonstrated stabilizing roles in the fibrils, quantitative ssNMR measurements of residue-specific dynamics show that it undergoes distinct solvent-coupled motion. This dynamical feature seems reminiscent of molten-globule-like α-helix-rich features attributed to the nonfibrillar oligomeric species of various amyloidogenic proteins.
Huntington’s disease (HD) is one of several human disorders caused by a genetic mutation that expands CAG repeats within genes encoding several different proteins.1−3 Expansion of the polyglutamine tracts in these proteins beyond a disease-specific threshold correlates to a decrease in the age of pathological onset and an increase in toxicity.2 In HD and other polyQ expansion disorders, disease pathology is associated with protein misfolding and aggregation, including the formation of amyloid-like fibrils. Some experimental evidence supports a toxic role of the polyQ amyloid-like aggregates,4−7 with toxicity being dependent on the nature of the aggregates,7 but others argue for a protective, nontoxic role for the mature aggregates.8 Given the potential impact on disease toxicity and the fact that both polyQ expansion and some level of misfolding, aggregation, or oligomerization are common features among the CAG repeat diseases, much work has been done to characterize the aggregation process. The aggregation propensity of polyQ model peptides increases with Gln repeat length.9 In addition, variations in the flanking sequences can have a dramatic impact on the conformation and aggregation propensity of the polyQ tract and thus may in part explain threshold differences between the CAG expansion disorders.10−14
In HD, Gln expansion occurs in the huntingtin (htt) protein leading to protein misfolding and aggregation. Observed aggregates contain N-terminal fragments of htt15 generated by protease activity16 or aberrant splicing.17 In animal studies, overexpression of the fragment corresponding to mutant htt’s exon 1 leads to neurodegeneration that mimics key features of HD.18 On either side of the polyQ domain, htt exon 1 features flanking segments that act as binding sites for htt-binding proteins and modulate the binding to membranes and subcellular targeting.12,19,20 In cellular studies of mutant htt exon 1, the flanking domains modulate not only the aggregation propensity but also the aggregate morphology, recruitment of other proteins, and toxicity.19,21−23 A Pro-rich domain (PRD) that follows the polyQ domain greatly reduces the aggregation propensity,10−13,24 leading to suggestions that it may in part function to reduce the risk of misfolding and disease.22,25 Preceding the polyQ domain, htt features a 17-residue sequence of mixed hydrophobic and hydrophilic amino acids, termed NT17 or httNT, which in contrast to the PRD initiates and accelerates aggregation.14,26 In doing so, httNT facilitates a distinct multistage aggregation pathway that features α-helix-rich oligomeric species (in contrast to polyQ model peptide aggregation).27−29
Disease toxicity in HD is thought to reflect a gain of toxic function that accompanies conformational changes in the mutant protein. As in other protein misfolding diseases, different conformational states populate the misfolding and aggregation pathway. These different species could all contribute to disease toxicity in their own way, whether by being toxic in their own right, causing toxicity by sequestering vital polyQ-containing proteins, or reducing toxicity by sequestering toxic oligomers or monomers. Therefore, there has been much interest in understanding the conformational changes that occur during aggregation in HD and other deposition-related disorders.
As for other amyloidogenic proteins, nucleation of a β-sheet-rich amyloid-like conformation is a pivotal event in the aggregation of mutant htt N-terminal fragments. This nucleation event can take place in the monomeric or oligomeric state and precedes and allows the formation of antiparallel β-sheet-rich fibrillar aggregates that have all of the features of amyloid-like fibrils. Earlier studies have shown that the C-terminal PRD counteracts the nucleation of β-sheet structure, possibly by inducing polyproline II (PPII) structure in an extended stretch of the preceding polyQ segment.10,11,13,30,31 These observations are primarily based on solution studies of various monomeric species and reflect a backbone-mediated effect of the PPII-structured PRD on the conformational ensemble of the nonaggregated polyQ domain. The degree to which this inferred induction of PPII in the glutamines persists in the oligomeric or fibrillar species has remained unclear. One might expect that also in the fibrils one or more Gln residues at the C-terminal end of the polyQ domain remain outside the amyloid core, perhaps in a PPII conformation.
The N-terminal flanking domain, httNT, is responsible for the formation of oligomeric assemblies.14,26,29,32 The oligomerization precedes the nucleation of β-sheet structure and is instead characterized by extensive α-helical structure.27−29 This α-helicity is largely attributed to the httNT segment, although it may also extend into the polyQ domain,33,34 because the latter has a length-dependent propensity for α-helical structure that is modulated by flanking domains.10,33−40 The oligomer ensemble seems to have an enhanced propensity (compared with the monomers) for β-sheet nucleation, translating into greatly accelerated formation of the β-sheet-rich fibrils.14,26 Thus, the httNT segment effectively counteracts the effects of the PRD and may allow aggregation under conditions where the PRD alone would effectively prevent fibril formation.14 Also in the fibrils, intermolecular interactions among the httNT amphipathic α-helices appear to contribute to the stability and supramolecular structure.41,42 Weakening of such interactions, e.g., by httNT phosphorylation, results in decreases in aggregation propensity, aggregate stability, and disease toxicity.41,43
We previously showed by magic-angle-spinning (MAS) solid-state NMR (ssNMR) and transmission electron microscopy (TEM) that htt peptide fragments aggregate into fibrils with a β-sheet core, but with different secondary structures in the flanking domains.27 Subsequently, electron paramagnetic resonance studies of htt exon 1 outfitted with spin-labels suggested a similar domain assembly of mutant exon 1 fibrils, while also yielding evidence of fibrillar httNT–httNT interactions.42 Our study of fibril formation in the presence of phosphomimetic httNT mutations also indicated a role for intermolecular httNT interactions in the stabilization of fibrillar and oligomeric assemblies,41 with apparent consequences for cellular toxicity.41,43 The connection among aggregate structure, stability, and toxicity7 may in part reflect changes in the access to proteases and chaperones as they interact with oligomeric and aggregated states of htt.26,41
Because of the limited structural data about the fibrils and in particular the oligomeric species, we lack an in-depth understanding of the structural transformations within mutant htt during misfolding and self-assembly. For instance, it is uncertain whether or how the PRD affects the secondary structure of the C-terminal tail end of the polyQ domain in the fibrils. Are the preceding Gln residues in a PPII-like structure, as they seem to be in the monomers, or is this “protective” effect overcome once nucleation and propagation of β-structure have taken place? How accessible and dynamic are the flanking domains in mature fibrils, given their apparent role in stabilizing the various aggregated species? Here, we address these questions through the use of MAS ssNMR, initially on fibrils obtained from a peptide construct (httNTQ30P10K2) that aggregates into fibrils.27 These constructs recapitulate the aggregation behavior of htt exon 144 and have been used in biophysical, mechanistic, and structural studies by us and others.14,20,27,32,41,43−46 This includes several detailed studies of the individual and combined impacts of the flanking domains on aggregation kinetics and aggregate morphology.10,14,27,32 These synthetic constructs facilitate the residue-specific labeling that permits unambiguous identification of specific residues within the various domains. This, in turn, allows for a careful analysis of the structural and dynamic features of these residues. We complement these residue-specific insights with the first ssNMR studies of fibrils prepared from uniformly labeled, full-length mutant htt exon 1. We observe by MAS ssNMR that, in contrast to its effect on the monomeric species, the proximity of the oligoPro flanking domain does not prevent β-sheet structure near the C-terminal end of the polyQ domain. Fibril maturation leads to formation of a relatively well-ordered amyloid core that consists of β-sheet structure and contains most of the polyQ domain. Quantitative site-specific dynamics measurements reveal that, despite their stabilizing role and supramolecular interactions, the httNT α-helices outside this amyloid core undergo molten-globule-like motions.
Experimental Procedures
Preparation of Residue-Specifically Labeled Fibrils
Residue-specifically labeled peptides were prepared by solid-phase synthesis at the Small Scale Synthesis facility of the Keck Biotechnology Resource Laboratory of Yale University (New Haven, CT). Site-specific labeling with uniformly 13C- and 15N-labeled residues was performed at various positions as indicated in Table 1. Fmoc- and side-chain-protected 13C- and 15N-labeled amino acids were purchased from Cambridge Isotope Laboratories (Andover, MA) and Isotec (Sigma-Aldrich, St. Louis, MO). The peptides were obtained crude for in-house purification and disaggregation, as previously described.47,48 Aggregation was initiated starting with disaggregated peptide and performed in PBS buffer (pH 7.4) at 37 °C, monitored by a high-performance liquid chromatography-based sedimentation assay, and allowed to proceed to completion.47,48 After being washed with deionized water, mature fibrils were pelleted into 3.2 mm zirconia MAS rotors (Bruker Biospin, Billerica, MA, and CortecNet, Voisins-le-Bretonneux, France) by centrifugation using a home-built ultracentrifugal sample packing tool. Samples were kept hydrated and unfrozen unless otherwise stated.
Table 1. Nomenclature and Site-Specific Labeling of the httNTQ30P10K2 Peptidesa.
| labeling | sequenceb | |
|---|---|---|
| LQP-labeled | U-13C,15N-[L7, Q47, P48] | MATLEKLMKAFESLKSF-QQQ26QQPP9K2 |
| LAQ-labeled | U-13C,15N-[L4, A10, Q46] | MATLEKLMKAFESLKSF-QQQ26QQPP9K2 |
| MA-labeled | U-13C,15N-[M8, A10] | MATLEKLMKAFESLKSF-QQQ26QQPP9K2 |
| LKSQ-labeled | U-13C,15N-[L4, K6, S16, Q19] | MATLEKLMKAFESLKSF-QQQ26QQPP9K2 |
| MF-labeled | U-13C,15N-[M8, F11] | MATLEKLMKAFESLKSF-QQQ26QQPP9K2 |
MAS ssNMR samples contained approximately 8 mg of peptide (with the exception of 1.8 mg in the LKSQ-labeled peptide sample27).
Isotopically labeled residues are underlined.
Preparation of Uniformly Labeled htt Exon 1 Fibrils
To complement the residue-specifically labeled fibrils, we also prepared fibrils from uniformly 13C- and 15N-labeled htt exon 1. To do so, we expressed in Escherichia coli full-length exon 1 fused via its N-terminus to maltose-binding protein (MBP). The employed construct was based on a previously reported construct49 but features a new linker design that avoids the inclusion of extra residues prior to the httNT upon cleavage with factor Xa. The BL21(DE3)pLysS cells (Invitrogen, Grand Island, NY) were incubated in Luria-Bertani medium with ampicillin and chloramphenicol at 37 °C and 250 rpm. The cells were obtained by centrifugation (7200g) and resuspended in M9 medium containing 0.2% (w/v) [U-13C]-d-glucose and 0.1% (w/v) [15N]ammonium chloride (Cambridge Isotope Laboratories, Tewksbury, MA) as carbon and nitrogen sources. The cells were incubated at 37 °C until an optical density (OD600) of 0.6 had been reached and then incubated at 18 °C until an OD600 of 0.7 had been reached. Overexpression was induced by adding 0.5 mM isopropyl β-d-thiogalactopyranoside (RPI Corp., Mt Prospect, IL), supplemented with 0.02% (w/v) [13C]-d-glucose and 0.01% (w/v) [15N]ammonium chloride, and 100 μM ZnSO4. The protein was expressed at 18 °C for 16 h, after which the cells were harvested by centrifugation and the cell pellets resuspended in PBS (pH 7.4) with 1 mM phenylmethanesulfonyl fluoride (ACROS, Fair Lawn, NJ). Cells were opened using a microfluidizer (Microfluidics, Westwood, MA), and cell debris was removed by centrifugation. The fusion protein was purified with a Histrap FF column (GE Healthcare) and assessed for purity by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (12%). Protein identity and isotopic labeling were verified with matrix-assisted laser desorption ionization (MALDI) and electrospray ionization (ESI) time-of-flight (TOF) mass spectrometry at the University of Pittsburgh’s Genomics and Proteomics Core Laboratories. Purified fusion protein was buffer-exchanged to PBS in centrifugal filter units (Millipore, Billerica, MA). To remove the MBP fusion domain, release htt exon 1, and induce fibrillation,49 the fusion protein (at 5.8 mg/mL) was treated with factor Xa protease (0.0028 mg/mL) (Promega, Madison, WI) at 37 °C. The generation and identity of the released htt exon 1 were tested by SDS–PAGE (12%) and ESI-TOF mass spectrometry. Uniformly 13C,15N-labeled htt exon 1 aggregates were pelleted down, resuspended in 1 mL of PBS, and then pelleted into a 3.2 mm MAS rotor using a centrifugal packing tool, as described above.
MAS Solid-State NMR
All experiments were performed with a wide bore Bruker Avance I NMR spectrometer operating at a 1H Larmor frequency of 600 MHz (14.1 T), using either a standard bore 3.2 mm HFCN MAS probe or a 3.2 mm EFree HCN MAS probe from Bruker Biospin. The sample temperature was controlled using a constant flow of cooled gas. Spectra were acquired with the Bruker Topspin software but processed and analyzed with NMRPipe, Sparky, and CCPNMR/Analysis. External referencing to 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) was done indirectly via the 13C signals of adamantane.50 For all spectra shown, additional experimental details can be found in Table S1 of the Supporting Information.
MAS ssNMR Assignments
Residue-specific 13C and 15N assignments were obtained via two-dimensional (2D) 13C–13C and 15N–13C experiments. These experiments involved 1H–13C/15N and 15N–13C cross-polarization (CP) steps combined with 13C–13C transfers via dipolar assisted rotational resonance (DARR)51 mixing (8–15 ms). Typically, 83 kHz two-pulse phase modulation (TPPM52) 1H decoupling was applied during acquisition and evolution. MAS spinning rates (9–10 kHz) and other details can be found in Table S1 of the Supporting Information.
MAS ssNMR Dynamics Measurements
To probe for site-specific dynamics in the httNT and polyQ domains, 15N longitudinal relaxation was measured for backbone 15N sites using a series of 1H–15N CP experiments incorporating a 15N T1 relaxation period, analogous to earlier work.53,54 These measurements were performed at 19–22 kHz MAS, where systematic MAS-dependent measurements had shown 15N–15N spin diffusion to be effectively suppressed. During acquisition, 83 kHz TPPM 1H decoupling was applied. As a complementary indicator of dynamics, we measured the one-bond N–H dipolar couplings of selected labeled sites. This was done at 10 kHz MAS through use of constant-time DIPSHIFT55 experiments involving rotor-synchronous incrementation of an R1817 symmetry sequence (89 kHz RF power)56,57 at the expense of 100 kHz continuous wave 1H decoupling. The experimental data curves were analyzed using numerical simulations with SpinEvolution,58 optimizing both the (apparent) N–H distance and exponential damping during the R sequence mixing time. The apparent N–H distances were converted into dipolar coupling constants (in kilohertz). As a complementary, qualitative measure of dynamics, we performed comparative one-dimensional (1D) 13C experiments comparing 13C direct polarization (DP) to signals obtained via dipolar-based 1H–13C CP, under otherwise identical conditions. These two polarization techniques have differential sensitivities to dynamics, and their comparison has been used to gain insight into local dynamics in various biological systems.
MAS ssNMR Solvent Exposure Measurements
To probe site-specific proximity to the fibril surface and exposure to solvent, we used MAS ssNMR measurements in which protons from highly mobile water molecules are selectively used as a source of polarization. These experiments featured a 1H–13C CP step preceded by a 3 ms T2 relaxation filter and a variable-time 1H–1H spin diffusion period, using a pulse sequence as previously described.59−61 These experiments were performed using a range of spin diffusion periods from 0 to 7 ms, after which the repolarization of the peptide signals was monitored by comparing peak intensities to the unfiltered 1H–13C CP signal. These spectra were obtained at 10 kHz MAS, with 83 kHz TPPM 1H decoupling during acquisition.
Results
Structural Analysis of the Q/P Junction
Previous studies have shown that within monomeric peptides the C-terminal oligoPro domain disrupts the conformational ensemble of the Pro-proximal Gln residues by inducing PPII structure within a stretch of multiple Gln residues.10,11,13,30,31 To probe whether or how this disruption of the polyQ domain is retained in fibrils, we performed MAS ssNMR on aggregates prepared from httNTQ30P10K2 peptides featuring site-specific labeling of residues within and beyond the C-terminal end of the polyQ segment. We previously submitted such aggregates (with differing labeling schemes) to TEM to show their fibrillar morphology.27 Here we examine fibrils prepared from the LQP-labeled peptide (Table 1) that is 13C- and 15N-labeled in Q47 and P48, which constitute the Q/P junction. Using standard 2D ssNMR spectra (e.g., Figure 1a,b), we obtained assignments for the labeled residues. P48, which is the first Pro of the oligoPro domain, is found to have 13C chemical shifts that match typical chemical shifts of Pro residues in a PPII 31 helix.62,63 Figure 2 illustrates the nearly perfect correspondence between the P48 peaks (Figure 2a, black arrows) and the solution NMR chemical shifts (Figure 2c) of a PPII oligoproline segment in the decapeptide, APSYSPPPPP, bound by α-spectrin SH3 (Figure 2d, BMRB entry 1501362). We previously27 found that unlabeled oligoPro in httNTQ30P10K2 aggregates showed the same shifts (Figure 2b), indicating PPII helix structure throughout the oligoPro domain. These ssNMR data on fibrils are in agreement with solution NMR studies of oligoPro in much shorter polyQ-containing peptides in the monomeric state, although no 13C chemical shifts are available for direct comparison.30
Figure 1.
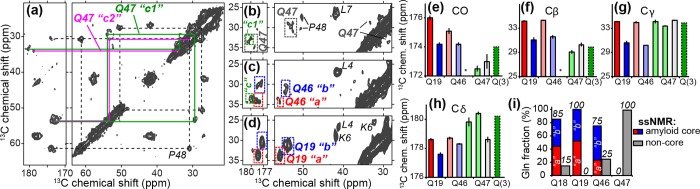
13C chemical shift assignments via 2D MAS ssNMR spectra. (a) Aliphatic-carbonyl (left) and intra-aliphatic (right) spectral regions from an 8 ms DARR 13C–13C 2D spectrum on LQP-labeled httNTQ30P10K2 fibrils at 275 K. (b–d) Comparison of individual Gln residues in different parts of the polyQ domain: (b) Q47 (15 ms DARR, 9.8 kHz MAS), (c) Q46 (8 ms DARR, 10 kHz MAS), and (d) Q19 (25 ms DARR, 13 kHz MAS, data from ref (27)). In these spectra, color-coded lines, boxes, and quoted letters (“a”, “b”, etc.) mark the Gln conformers discussed in the text. In panels b–d, peaks from labeled residues L4, K6, L7, and P48 are indicated. (e) Backbone and (f–h) side-chain 13C chemical shifts for Gln in httNTQ30P10K2 fibrils. Red/blue color coding indicates conformers “a” and “b” (see refs (27) and (48)). Newly observed resonances for Q46 and Q47 are shown with paler color to indicate the conformers, where applicable. Green conformers for Q46 and Q47 resemble a “Q(3)” conformer reported in polyQ fibrils,64 with a Cδ shift near 180 ppm. Missing resonances are indicated with asterisks. (i) Fractions of residues Q18, Q19, Q46, and Q47 that feature the amyloid core signature, estimated from ssNMR 2D peak volumes.
Figure 2.
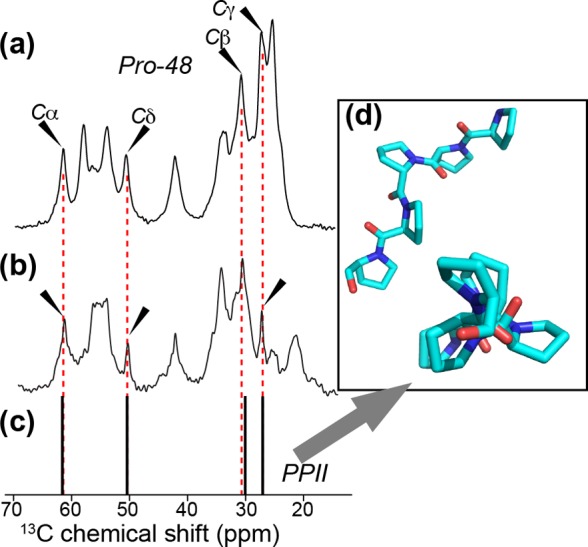
13C chemical shifts for Pro in httNTQ30P10K2 fibrils. (a) 1H–13C 1D CP MAS spectrum of LQP-labeled fibrils at 9.8 kHz MAS, showing the peaks due to the first Pro (P48, black arrows). (b) Natural abundance Pro signals (black arrows) in unlabeled httNTQ30P10K2 fibrils at 10 kHz MAS.27 Unmarked peaks are due to other residues, either isotopically enriched (in panel a) or at natural abundance. Panel (b) adapted with permission from ref (27). Copyright 2011 American Chemical Society. (c) Chemical shifts of Pro in an example of PPII-structured oligoPro (shown in panel d), from previously published solution NMR on a chimera of the peptide APSYSPPPPP and α-spectrin SH3.62
Next, we examined the extent to which Gln residues preceding oligoPro are affected by the proximity to the oligoPro segment and the end of the polyQ domain. Previously, we have identified a signature chemical shift pattern that characterizes polyQ amyloid core residues.48 This pattern features two coexisting equal populations of Gln residues that have different NMR signals. As such chemical shift differences imply distinct conformations, we have referred to these Gln as conformers “a” and “b”. They feature unique Cβ and Cγ chemical shifts that are highly uncommon in other protein structures27,48 but thus far have been seen in all ssNMR studies of polyQ amyloid.27,41,48,64 Figure 1d shows a representative 2D MAS ssNMR spectrum of httNTQ30P10K2 aggregates27 in which Q19 (the second Gln in the polyQ segment) is uniformly isotopically labeled. The sets of peaks corresponding to the two amyloid core conformers “a” and “b” are highlighted with red and blue boxes, as well as matching colored labels Q19 “a” and Q19 “b”, respectively. We adopt the same conformer-specific color coding in subsequent figures. The corresponding sets of 13C chemical shifts are also shown as red and blue bar graphs in Figure 1e.
The peaks for the 13C- and 15N-labeled Q47 (green and magenta lines in Figure 1a; dashed boxes in Figure 1b), directly preceding P48, clearly lack these chemical shifts that identify the β-sheet polyQ core. Q47 displays some structural heterogeneity, with two major conformers that are marked as “c1” and “c2” in Figure 1. The Q47 Cβ and Cγ chemical shifts near ∼30 and ∼34 ppm, respectively, are very different from the polyQ core signature and more similar to typical Gln shifts found in globular proteins. Thus, Q47 is structurally heterogeneous and not participating in the polyQ amyloid core. Given the multiresidue effect of the PPII-structured oligoPro flanking sequence in the monomeric state, one may expect a similar conformation for residues further into the polyQ segment. To investigate this, we prepared a fibril sample with isotopic labels in Q46 [alongside labeling of L4 and A10 in the httNT segment (Table 1)]. Figure 1c shows results from a 13C–13C 2D experiment on this sample, again highlighting in color-coded, dashed boxes the peaks assigned to Q46. Unlike Q47, the majority population of this residue does feature the 13C amyloid core shifts (compare to Q19 in Figure 1d) and is thus found in the amyloid core.27,41,48,64 A small fraction (∼25%) of Q46 adopts chemical shifts that resemble those of Q47, indicating that in some of the peptide chains this residue is outside the core. This contrasts with Q19, which lacks detectable signals with non-core shift values, but resembles Q18, the first residue of the polyQ domain, which also exhibits a small fraction of Gln outside the amyloid core (Figure 1i). Thus, in these peptide fibrils, we see a highly localized conformational disruption of the amyloid core at both ends of the polyQ domain, which is most pronounced in the single residue just prior to the oligoPro flanking segment.
Solvent Accessibility and Mobility in the Q/P Junction
Having established that the Q/P junction constitutes a remarkably tight transition between two secondary structure elements, we performed MAS ssNMR experiments to assess the solvent accessibility of this structural motif that acts as a recognition site for certain htt-binding proteins.19,21,65,66 First, we directly probed water accessibility via T2-filtered 13C MAS ssNMR,59 previously used on other amyloid fibrils and membrane proteins.60,61,67 After signals from (rigid) protein protons had been eliminated (Figure 3b, red), time-dependent 1H polarization transfer from mobile water reflects the proximity of specific residues to the fibril surface.67 We observe a differential repolarization of the residues in the different peptide domains (Figure 3c). The fastest transfer occurs to L7, which is outside the amyloid core and in the N-terminal α-helix (see below). At the Q/P junction, Q47 and P48 both show increased water accessibility compared to that of the dehydrated amyloid core.27
Figure 3.

Solvent exposure and dynamics at the Q/P junction of httNTQ30P10K2 fibrils. (a) 1H–13C CP spectrum of the LQP-labeled sample. (b) A 3 ms T2 filter eliminates virtually all signals (red). 1H–1H mixing (7 ms, black) allows 1H polarization transfer from mobile waters to the fibrils. The most prominent peaks are due to L7 in the httNT helix, followed by P48 and Q47 (each Cα marked with an arrow). (c) Polarization buildup curves of Cα sites (marked in (panel b) as a function of 1H–1H mixing, normalized relative to the unfiltered 1H–13C CP signal in panel a. (d) Comparison of 1H–13C CP (black) and 13C DP (cyan) spectra on LQP-labeled fibrils. Unlike L7 peaks, various Q47 peaks are notably narrower in the DP data, reflecting locally increased dynamics. The DP spectrum is rescaled up to allow better comparison of the line widths.
To evaluate local dynamics, we compared the peak intensities of CP-based and DP 13C ssNMR spectra. The dipolar mediated 1H–13C transfer in CP experiments is less effective for mobile sites, while the slow relaxation recovery of rigid 13C signals suppresses the most rigid sites in the DP spectra. Thus, mobile and rigid residues can be distinguished and even spectroscopically “filtered out”. At the htt Q/P junction, these experiments reveal populations of Q47 with different levels of dynamics. Upon examination of the side-chain Cγ (Figure 3d), the c2 conformer of Q47 is suppressed in the 13C DP experiment (i.e., is rigid), while conformer c1 is present and even reveals a quite narrow line width (i.e., slower T2 relaxation due to faster dynamics). The absolute peak intensities (and their differences between the CP and DP experiments) for both these Q47 conformers are indicative of dynamics significantly greater than that of the amyloid core (Figure S1 of the Supporting Information). Thus, Q47 features increased mobility and solvent exposure, consistent with its location outside, or at the surface of, the polyQ amyloid core.
Uniformly 13C- and 15N-Labeled Exon 1 Aggregates
To complement the residue-specific insights into the Gln and Pro domains allowed by the use of our synthetic peptide aggregates, we also prepared uniformly 13C- and 15N-labeled htt exon 1 by overexpression in E. coli. Removal of the MBP fusion domain by factor Xa treatment released htt exon 1 with a 44-residue polyQ domain and a C-terminal His tag. Because of a new linker design, factor Xa cleavage did not leave additional residues N-terminal to the httNT segment, as validated by ESI TOF mass spectrometry data showing the released exon 1 to have the gene-encoded start Met-Ala-Thr. Under conditions analogous to those used in our study of the residue-specifically labeled samples, 1.1 mg of the fibrillized uniformly 13C- and 15N-labeled protein was analyzed by 2D 13C–13C ssNMR spectroscopy. The obtained spectrum (Figure 4) is dominated by the signals from the most rigid parts of the sample: the 44-residue polyQ domain and the many prolines in the C-terminal PRD segment. These signals match those obtained from the residue-specific labels discussed in the synthetic htt exon 1 model peptide samples. This can be readily appreciated from Figure 4b, in which spectra from the fully labeled and residue-specifically labeled fibrils are overlaid (see also Figure S2 of the Supporting Information). Thus, it appears that the NMR signals observed for specific residues labeled in the polyQ core of the httNTQ30P10K2 fibrils (i.e., Q18, Q19, and Q46) similarly apply to the bulk of the polyQ domain of htt exon 1. Analogously, the signals of P48 (Figure 1) match those of the uniformly 13C- and 15N-labeled PRD in Figure 4. These data support our earlier analysis of the bulk natural abundance 13C signals of both Gln and Pro in httNTQ30P10K2 fibrils.27 The htt exon 1 spectra are dominated by the large numbers of Gln and Pro residues, as may be expected. Consistent with its dynamic nature (see below), in the current size-limited sample it is difficult to detect the signals from httNT.
Figure 4.
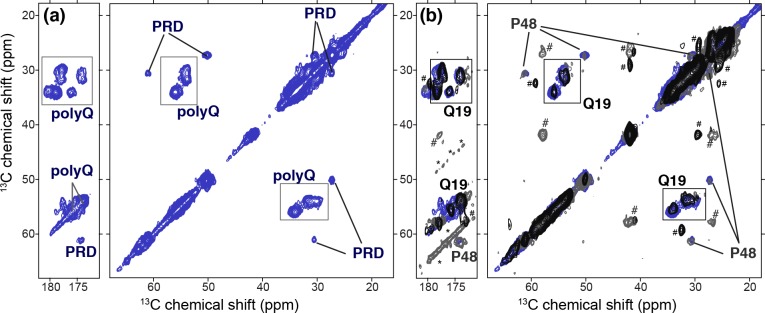
PolyQ and PRD bulk signals. (a) 2D 13C–13C spectrum of 1.1 mg of uniformly 13C- and 15N-labeled fibrillar htt exon 1 featuring a 44-residue polyQ domain. The predominant peaks are due to the Gln in the polyQ amyloid core and the Pro residues of the PRD. (b) These peaks match those of the residue-specifically labeled httNTQ30P10K2 fibrils from panels a and d of Figure 1: Q19 signals (black spectrum; Figure 1d) match the signal of the uniformly 13C- and 15N-labeled polyQ; P48 signals (dark gray; Figure 1a) match those of the fully labeled PRD prolines. Asterisks indicate spinning sidebands. Hash marks indicate httNT peaks that are visible in the httNTQ30P10K2 sample, but not detectable in the current htt exon 1 samples (see the text). All data are obtained at 600 MHz, using 15 or 25 ms DARR (see Table S1 of the Supporting Information).
Dynamics of the httNT α-Helix Backbone
We have previously identified the presence of a defined α-helical segment within httNT in the httNTQ30P10K2 fibrillar aggregates.27 Those initial measurements, which for the first time demonstrated the presence of an α-helix in the fibrils and delineated its exact sequence location, were limited to qualitative measurements of dynamics because of small sample sizes. Here, we performed more detailed and quantitative ssNMR experiments on several new httNTQ30P10K2 samples containing more peptide and new labeling patterns (Table 1). Via 2D MAS ssNMR experiments on the mature aggregates, we obtained assignments for five httNT residues: L4, K6, L7, M8, and A10 (e.g., Figure S3 and Table S2 of the Supporting Information; BMRB entry 25146). The obtained assignments validated and reproduced several previous assignments,27,41 and in all cases, the chemical shifts of the labeled httNT sites are indicative of α-helical secondary structure. Moreover, the new assignments allow for application of more quantitative torsion angle analysis (with TALOS+68), yielding the following α-helical dihedral angles for L7: ϕ = −63 ± 3°, and ψ = −42 ± 5° (Figure S3 of the Supporting Information). These new samples also allow for quantitative analysis of the dynamics of the httNT α-helix, through the use of several complementary MAS ssNMR measurements. First, we probed domain dynamics by measuring the 15N longitudinal relaxation rates for the backbone nitrogens of residues in the α-helix. On the basis of our published27 and current (Figure S3 of the Supporting Information) ssNMR data, we know that both M8 and A10 form part of the httNT α-helix retained in the fibrils. Consistent with them undergoing similar (domain) motion, both residues have similar 15N backbone R1 values, i.e., 0.11 ± 0.07 s–1 (Figure 5a). This value exceeds the 15N R1 of structured regions of, e.g., crystalline GB1 and are closer to the R1 of solvent-exposed loop regions.54 To test how this compares to the rigidity of the polyQ core, we also measured the backbone 15N R1 rate for site-specifically uniformly 13C- and 15N-labeled Q10 in K2Q11PGQ11D2 fibrils (see also ref (48)). The two distinct NMR signals seen for this single amyloid core residue have backbone 15N R1 values of 0.02–0.05 s–1 (Figure 5b), which resemble the structured regions in crystalline proteins.
Figure 5.

Dynamics measurements. (a and b) 15N longitudinal relaxation for (a) residues in the httNT α-helix of httNTQ30P10K2 and (b) residue Q10 of the polyQ amyloid core of K2Q11PGQ11D2. (a) Relaxation curves are shown for the unfrozen sample (red) and frozen sample (blue). (b) 15N longitudinal relaxation curves at 275 K for conformers “a” and “b” of Q10 in unfrozen polyQ peptide K2Q11PGQ11D2 (described in ref (48)). Data were obtained as 1D 15N spectra at 19–22 kHz MAS. (c and d) N–H dipolar recoupling curves for M8 and A10 in the httNT of httNTQ30P10K2 fibrils, obtained on unfrozen (287 K, red) or frozen (250 K, blue) samples, respectively. (e) N–H coupling measurements of the polyQ amyloid core, showing separate curves for both conformers of the uniformly 13C- and 15N-labeled residue Q10 in unfrozen (275 K) fibrillar K2Q11PGQ11D2. Panels c–e used an R1817 symmetry sequence at 10 kHz MAS. All data were acquired at 600 MHz (1H).
Order Parameters Validate httNT α-Helix Dynamics
One concern with a reliance on T1 NMR relaxation in MAS NMR as a measure of dynamics is that the T1 relaxation may contain significant nonmotional contributions, for instance because of the proximity of methyl groups that act as relaxation sinks.69−71 This is one of the reasons why the comparison of 13C CP and DP spectra that we previously applied27 is primarily a qualitative measurement. To address this concern, we took two alternative and complementary approaches: order parameter measurements and a determination of the temperature dependence of the MAS ssNMR dynamics measurements.
NMR order parameters provide a measure of dynamics that is based on the fact that, e.g., N–H dipolar couplings are reduced (“averaged”) by local dynamics. We probed the backbone N–H dipolar couplings in the N-terminal α-helix using R sequence-based DIPSHIFT experiments (see Experimental Procedures). As shown in Figure 5c–e, the time-dependent oscillations are different for residues in the httNT α-helix (Figure 5c,d) compared to those in a β-sheet polyQ core residue (Figure 5e). Data analysis via numerical simulations indicates significant differences in the corresponding N–H dipolar order parameters. Simulations of rigid sites (the polyQ core residue and data below) reproduced the experimental data very well and indicated dipolar coupling constants of ∼11.2 ± 0.4 kHz. These are consistent with an essentially rigid state, as also seen in other amyloid-like fibrils.72 Residues M8 and A10 in the httNT α-helix feature a decreased dipolar coupling strength (9.5 ± 1.5 kHz) and increased relaxation damping, indicative of pronounced molecular motion. For these dynamic sites, the fit quality was poorer, presumably because of an inability to fully capture the effects of anisotropic and heterogeneous dynamics. We note that the fits above did not explicitly consider the 1H chemical shift anisotropy (CSA).73 In test simulations using standard CSA values,73,74 the trend in coupling constants seen for the httNT and amyloid core residues was unchanged, even though the exact fit optima were dependent on the precise CSA values employed.
httNT α-Helix Motion Is Coupled to the Solvent
To further test the connection between these NMR parameters and true dynamics, we also examined the effect of sample temperature on both the relaxation and order parameter measurements. In particular, we reduced the sample temperature until the solvent froze, as validated by observation of the broadening of the water 1H NMR signals. As a result, the 15N longitudinal relaxation of both labeled httNT α-helix residues slowed to R1 values of 0.02 ± 0.03 s–1 (for M8) and 0.03 ± 0.04 s–1 (A10), which match those of residues in a structured polyQ core (Figure 5a, blue). In addition, a dramatic change in the N–H dipolar recoupling experiments is observed (Figure 5c,d, blue). The low-temperature N–H dipolar coupling data feature larger dipolar coupling constants (∼10.5 ± 0.3 kHz) and reduced relaxation damping, resulting in curves that more closely resemble those of the rigid polyQ core (compare to Figure 5e).
The effects of freezing the sample are also manifest in 13C-based ssNMR experiments, where we observe an increased 1H–13C CP intensity and a larger CP–DP difference in backbone and aromatic sites (Figure 6). This indicates rigidification of the backbones and side chains and also shows that these difference experiments are indeed reflective of increased dynamics. In other words, previously noted differences between the polyQ amyloid core and httNT segment27 are not simply due to the absence and presence of methyl groups in either domain.
Figure 6.
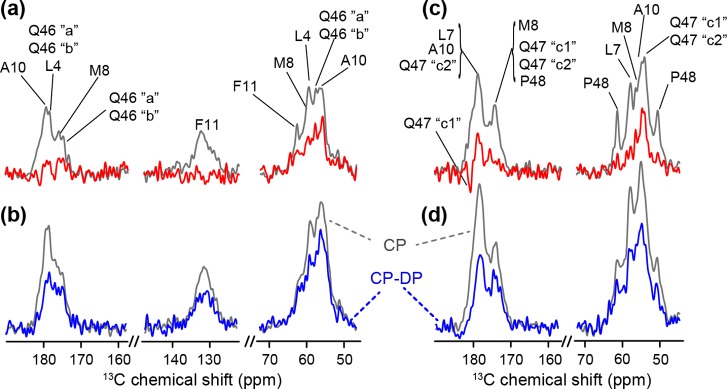
1D 13C ssNMR of fibrils of mixed (1:1) MF-labeled and LAQ-labeled peptides at (a) 287 K and (b) 265 K and mixed MA-labeled and LQP-labeled peptides at (c) 287 K and (d) 270 K. A CP–DP difference spectrum is colored red and blue, with the corresponding 1H–13C CP spectrum colored gray. The relative increase in intensity in the difference spectra, when the solvent freezes, indicates a rigidification that applies to the backbone as well as (hydrophobic) side chains (e.g., F11 in panel b). This implies solvent-coupled dynamics that extend to the hydrophobic face of the amphipathic α-helix (see Figure S3 of the Supporting Information).
Discussion
Secondary Structure of Fibrillar PolyQ and Its Flanking Domains
In monomeric polypeptides, the oligoPro flanking segment adopts a PPII conformation that modulates the structure of multiple preceding Gln residues,10,11,13,30,31 presumably allowed in part by Gln’s innate propensity for PPII structure31,75 (Figure 7). Our MAS ssNMR data (Figures 1 and 2) show that the oligoPro segment in our httNTQ30P10K2 fibrils also adopts a PPII helix, which starts from the very first Pro residue. The same resonances are also seen in Figure 4 for Pro residues in uniformly 13C- and 15N-labeled htt exon 1 fibrils. We also found that the Gln residue preceding the PRD fails to adopt the canonical β-sheet polyQ amyloid conformation. This is indicated by the complete absence of the characteristic polyQ ssNMR signature (Figure 1) as well as the increased motion and solvent accessibility of this residue (Figure 3). Remarkably, unlike the case in earlier solution-state studies, we find that in the fibrils this oligoPro-induced non-β-structure is mostly limited to a single Gln. In the fibrils, the penultimate Gln, Q46, does display the chemical shift signature of the polyQ amyloid core. A minor population (∼25%) of Q46 is found to show some of the characteristic side-chain chemical shifts that are seen for Q47 and attributed to mobile residues outside the amyloid core64 (see also below). In the uniformly 13C- and 15N-labeled htt exon 1 fibrils, we find that the bulk of the polyQ domain features the same two sets of ssNMR signals. Thus, the amyloid core is formed by Gln residues throughout most of the polyQ segment. These data are in line with our previous analysis27 of the bulk Gln signals in unlabeled httNTQ30P10K2 fibrils. They are also reminiscent of the results of the ssNMR study of aggregated uniformly labeled polyQ without htt flanking domains.64 Thus, once formation of amyloid structure is initiated (either by spontaneous nucleation or via seeded elongation by preformed polyQ amyloid), β-sheet structure propagates throughout essentially the entire polyQ domain. As may be expected, there is no indication of β-sheet structure in the oligoPro segment, in contrast to the formation of some β-structure in the final residues of httNT (see ref (27)). Thus, the “protective” ability of the oligoPro segment to induce PPII structure within multiple preceding Gln residues is specific to the monomeric (and possibly oligomeric) species and is not retained in the mature amyloid aggregates.
Figure 7.
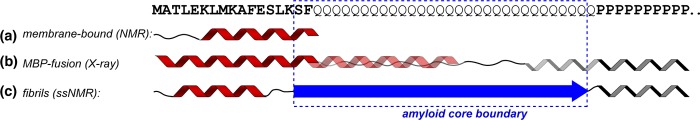
Secondary structure of htt N-terminal fragments under differing conditions. (a) NMR studies have shown extensive α-helicity in isolated httNT upon interacting with a lipid bilayer.77 (b) In crystals formed from a MBP fusion construct, httNT is fully α-helical, followed by partial α-helicity in the N-terminal part of the polyQ domain.33 The oligoPro segment forms a PPII helix, with extended or PPII structure for several preceding Gln residues. (c) Mature fibrils feature a relatively well-defined β-sheet amyloid core (dashed blue box; see also Figure 8), formed at the expense of any α-helical or PPII structure within the polyQ. A short α-helix in httNT helps stabilize the fibrils via molten-globule-like assemblies that may be inherited from non-β oligomers (see the text).
By ssNMR, we find in fibrils an α-helix in httNT that spans only part of this N-terminal segment (Figure 7, Figure S3 of the Supporting Information, and refs (27) and (41)) and undergoes extensive dynamics (e.g., Figure 5). Although various studies have proposed or discussed α-helical structure for httNT,12,33,34,39,76 these studies did not take place in the context of fibrils. An intriguing feature of the fibrillar httNT α-helix is that it is shorter than α-helices seen in experimental studies in the context of non-oligomers, membranes, and the crystal lattice of a chimeric construct (Figure 7).28,29,33,77 The detailed conformation of the oligomers remains uncertain, but it is clear that interactions between httNT α-helices are critical for their formation.14,27−29,32,44 At the same time, intermolecular interactions seem to be required to have httNT form stable α-helical structure.14,29,32
Previous studies have reported that the polyQ domain may have a propensity for α-helical content,33,34,39 especially in the N-terminal polyQ portion when attached to an α-helical flanking domain.10,33,35,78 As one example (Figure 7b), extensive α-helical structure is seen within the polyQ domain of N-terminal htt fragments fused to MBP.33 Once fibril formation has reached completion, our use of MAS ssNMR on fibrils shows that no α-helical structure remains in the polyQ domain.27,41 Indeed, this β-structure actually extends into the C-terminal end of the httNT segment, at its C-terminal junction with the polyQ (Figure 7).
Boundaries and Alignment of the PolyQ Amyloid Core
We previously described48 how the β-sheet polyQ amyloid core can be identified on the basis of its unique chemical shift signature featuring the two conformers “a” and “b” (as illustrated in Figure 1 in red and blue, respectively). Through our use of synthetic peptides and residue-specific labeling, we have shown that these two conformers are observed even for a single labeled residue independent of its position in the polyQ domain. In httNTQ30P10K2 aggregates, this signature was seen when we labeled the first two Gln residues directly following the httNT flanking domain.27 Here, we show it is also present near the C-terminal end of the polyQ domain, providing further support to our hypothesis that this doubled signature is a general and intrinsic feature of Gln throughout the β-strands of polyQ amyloid. Moreover, we also showed that the same chemical shifts are seen for the Gln residues that make up the bulk of the polyQ amyloid core in the uniformly 13C- and 15N-labeled htt exon 1 fibrils (Figure 4). This complements and confirms our previous analysis of unlabeled httNTQ30P10K2 fibrils, based on spectroscopic filtering of the rigid amyloid core.27 Work by the Baldus group on polyQ fibrils lacking htt-like flanking domains reproduced our assignments of the amyloid core signals, even though, because of their labeling approach, it was not apparent that the doubled signature is an attribute of each individual amyloid core residue.64 We are pursuing further ssNMR studies into the conformational underpinnings of this polyQ-specific amyloid48 signature, which we here employ to establish the presence or absence of specific residues within the amyloid core.
In many amyloids, one finds well-defined boundaries for the fibrils’ β-sheet amyloid core. In these amyloids, β-strand-based “unit cells” within the β-sheets stack into a single well-defined register that balances the alignment of hydrophobic residues, the formation of Asn or Gln “ladders”, and the interactions of charged residues. This is perhaps most obvious in the commonly found parallel in-register amyloid structures, where the β-sheets are defined by translation of a unit cell featuring a single β-strand.79−81 Also more complex cases, such as the fibrils formed by the fungal prion HET-s82 or the antiparallel fibrils made by Iowa mutant Aβ,83 feature a strictly aligned repeating unit within the fibrillar β-sheets (see Figure 8a,b). Misalignment of new β-strands that are added to the elongating fibril has a significant energetic penalty, resulting in the templated elongation into structurally homogeneous fibrils. However, during elongation of polyQ amyloid, the alignment constraints on incoming polypeptide chains are much less rigorous because of the repetitive and degenerate nature of the primary sequence. This translates into an ability to incorporate polyQ domains of other lengths and from other proteins, a phenomenon previously described as “promiscuous” polyQ amyloid growth.42,84−87 Even when a homogeneous pool of polyQ peptides is forming amyloid, the penalty of having incoming peptide chains not lining up perfectly with the existing amyloid core is likely to be relatively small, especially for longer polyQ lengths. Thus, this would allow for a “misalignment” (from the point of view of residue number, if not of residue type) of the incoming peptide chains with the existing polyQ amyloid core. This translates into having peptide chains in various residue number “registers” inside each individual fibril. Among other effects, this kind of chain misalignment could lead to the generation of disordered loops and tails decorating the amyloid core, with lengths and locations that vary from peptide to peptide, even within one fibril. A previous in silico study has noted this kind of imperfect alignment and how it resulted in disordered tails at the ends of the polyQ peptide.37 In ssNMR experiments, such disordered non-β-segments would manifest themselves as Gln signals lacking the amyloid core signature and featuring enhanced dynamics. Such signals were observed in a previous ssNMR study64 of polyQ fibrils lacking htt-based flanking domains (see also below). Although these were discussed in the context of flexible loops, they would also be expected for “tail ends” of disordered Gln beyond the polyQ core. Here, we find in htt context that the uniformly 13C- and 15N-labeled fibrils also contain this type of signal, with it accounting for 11% of the Gln based on an integration of the Cδ−Cγ/Cβ cross-peaks. In the site-specifically labeled httNTQ30P10K2 fibrils, we observe that the final Gln (Q47) has chemical shifts that closely match those of the disordered glutamines. On the other hand, for the very first Gln residue of the polyQ segment (Q18)27 and also the penultimate Gln (Q46), both of which form part of the amyloid core, we find only minor populations of the disordered Gln residues (∼15 and 25%, respectively). The second Gln residue, Q19, lacks such a disordered population (Figure 1i). These observations seem consistent with a very limited degree of residue number misalignment within the polyQ amyloid core. It remains unclear whether this is the full explanation of these observations. Generally, we note that the proportion of disordered glutamines in these fibril samples seems to be lower than that reported for polyQ fibrils lacking htt flanking domains.64 This might imply an ordering effect of the non-β httNT and oligoPro flanking sequences, which could effectively help “set the register” of the polyQ amyloid core.32 Alternatively, this may also in part reflect a dependence on the polyQ length, because in the study mentioned above the disordered Gln pool seemed to be most pronounced in the longest polyQ.64 Moreover, the degree of structural and dynamic disorder within the polyQ domains of the fibrils can be affected by the aggregation conditions, as previously discussed for htt exon 1 aggregates.7
Figure 8.
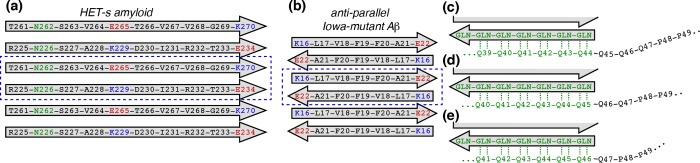
Peptide-chain (mis)alignment during amyloid elongation. The amyloid cores of (a) HET-s and (b) Iowa mutant Aβ are assembled from strictly aligned repeating “unit cells” (dashed rectangles).82,83 Incoming peptide chains align their charged (red/blue) and hydrogen-bonding side chains (Asn/Gln; green). (c–e) The antiparallel β-sheet polyQ amyloid is more forgiving of misalignment of incoming peptide chains, because a different alignment with the amyloid core (green) still allows for similar backbone- and side-chain hydrogen bonding to the core (dashed lines). This would leave varying numbers of non-amyloid Gln residues at the N- and C-terminal polyQ ends (as shown in panel c–e) and in flexible loops. This may explain the localized disorder in Gln at or near the polyQ end (Figure 1i). These non-amyloid signals are largely (for 75% of the fibrillized httNTQ30P10K2 peptides) limited to the final Gln, consistent with the schematic shown in panel e.
The mobile, non-amyloid core Gln residues that were identified as the “Q(3)” population by the Baldus group64 feature a distinct chemical shift pattern. It can be distinguished from the polyQ core residues, by its Cδ chemical shift near 180 ppm (Figure 1e, green bars at the far right) or the presence of an off-diagonal Cβ–Cγ peak in 13C–13C 2D spectra (Figure 1b). A close inspection of several other Gln-labeled fibrils that we have studied (refs (27), (41), and (48) and unpublished results) reveals weak signals with similar chemical shifts. This includes both the uniformly 13C- and 15N-labeled exon 1 fibrils’ bulk Gln signals and specifically the first and last Gln residues in our httNTQ30P10K2 fibrils. Perhaps most strikingly, this applies to residue Q47, which has peaks (in particular conformer c1) that closely resemble these chemical shift values (Figure 1e). The most notable deviation is in the 13CO chemical shift, but this is likely due to the well-established effect of the adjacent Pro.88 The Q47 chemical shifts also bear a strong resemblance to those of Gln preceding Pro in intrinsically disordered proteins (IDPs), which have a characteristic set of backbone chemical shifts of ∼174 ppm (CO) and ∼53.5 ppm (Cα). These differ significantly from both random coil shifts and non-pre-proline IDP Gln residues. Therefore, the final Gln is indeed disordered and affected by the PPII oligoPro segment. We speculate that Q47 might feature a PPII-like structure that resembles the conformation of the oligoPro-flanked polyQ tail prior to β-nucleation.
htt-Binding Proteins and Their Effect on Aggregation
Thus, we localized at the Q/P junction a transition from β-sheet to PPII structure, occurring remarkably abruptly at the final Gln residue, which lacks the amyloid core signal and instead features increased dynamics and water exposure. The PPII-structured oligoPro elements are known to provide the binding sites for certain htt-binding proteins, some of which can bind to fibrils.21 One of these, the PPII-binding WW domain protein FE65 is thought to specifically recognize this particular Q/P junction, which would require the observed accessibility (i.e., solvent exposure) of said junction.19 The anti-oligoproline antibody MW7 can bind aggregates from mice,65,89−91 and it can also destabilize preexisting fibrils and even interfere with fibril formation.91 Our experiments indicate that one of its oligoPro-binding sites occurs at the very edge of the amyloid core. Optimal tight binding of MW7 to this site may thus interfere with the formation of the polyQ amyloid core or at least reduce the stability of the fibrillar assembly.
Molten-Globule-like Dynamics of the α-Helical httNT Segment
Intermolecular httNT interactions play a crucial role in accelerating the aggregation of htt fibrils, by allowing the formation of α-helix-rich oligomers.14,26,29,32 httNT–httNT interactions also contribute to the stability of the mature fibrils.41,42 Our MAS ssNMR data show that in mature fibrils the α-helical segments nonetheless remain at least partly solvent-exposed. Moreover, despite the structural role in the fibrils,41,42 the httNT α-helix experiences solvent-coupled dynamics, in both its backbone and its hydrophobic side chains. This is reflected in greater 15N and 13C longitudinal relaxation rates and reduction of N–H dipolar coupling. The pronounced effect of freezing the solvent shows that this mobility is coupled closely to the solvent dynamics. Solvent freezing impacts even the hydrophobic face of the amphipathic helix (e.g., Phe ring in Figure 6), which would form the hydrophobic core of clustering of the amphipathic α-helices within the fibrils. Thus, our ssNMR data indicate that the interacting httNT α-helices in the fibrils are in a molten-globule-like state, in which amphipathic α-helices are formed and interact but remain flexible and loosely packed. Although we still lack a good understanding of the structure and dynamics of htt-related oligomers, such molten-globule-like httNT assemblies remind us of the proposed molten-globule-like intermediate states of other amyloid-forming proteins.92−96 If the α-helix-rich oligomers formed by htt N-terminal fragments turn out to be dynamic, then the dynamic httNT α-helices in the fibrils might even trace their origins to the initial interactions in the self-assembly of the oligomeric precursors.
Conclusions
Extensive structural changes are common during the aggregation of amyloidogenic proteins. This includes the loss of native secondary and tertiary structure elements as well as the formation of the highly rigid, intermolecularly hydrogen-bonded β-sheets that characterize amyloid-like fibrils. Through the use of MAS ssNMR spectroscopy, we have gained new insights into the structural features of the polyQ domain and its flanking segments after the aggregation of htt N-terminal fragments. Following nucleation, a β-sheet-based amyloid core is formed in the polyQ domain, which includes Gln residues very close to the C-terminal oligoPro segment. The latter forms a PPII helix that extends to, but fails to significantly disrupt, the polyQ amyloid core in mature fibrils. This contrasts with its effect prior to fibril formation, when it modulates the conformational ensemble of multiple preceding glutamines sufficiently to suppress nucleation. The β-sheet amyloid core extends slightly beyond the N-terminal end of the polyQ segment, stopping short of the amphipathic α-helix close to the very N-terminus.27 Here, we found that this amphipathic helix displays pronounced solvent-coupled dynamics, seemingly reflecting a molten-globule-like assembly that is reminiscent of some oligomeric amyloid precursors. While α-helix-rich and dynamic oligomeric intermediates are implicated in other amyloid-forming proteins and peptides,95−98 htt-related fibrils may be relatively unusual for such proteins in that they retain a substantial part of these structural and dynamic features in the mature fibrils. The obtained new insights into the structure of htt-related fibrils may also help explain reported inhibitory effects and aggregate binding characteristics of antibodies and other htt-binding proteins.
Acknowledgments
We thank Mike Delk, Guy Uechi, Ravindra Kodali, and Bankanidhi Sahoo for technical assistance and helpful discussions.
Glossary
Abbreviations
- CP
cross-polarization
- CSA
chemical shift anisotropy
- DARR
dipolar-assisted rotational resonance
- DP
direct polarization
- DSS
4,4-dimethyl-4-silapentane-1-sulfonic acid
- HD
Huntington’s disease
- htt
huntingtin
- httNT
htt N-terminal segment (MATLEKLMKAFESLKSF)
- IDP
intrinsically disordered protein
- MAS
magic angle spinning
- MBP
maltose-binding protein
- oligoPro
oligoproline
- polyQ
polyglutamine
- PPII
polyproline type II
- PRD
proline-rich domain
- ssNMR
solid-state NMR
- TPPM
two-pulse phase modulation.
Supporting Information Available
Tables S1 and S2 and Figures S1–S3. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ K.K.: Indian Institute of Technology Jodhpur, Jodhpur, Rajasthan, India.
Author Present Address
|| Z.H.: Russell H. Morgan Department of Radiology & Radiological Science, Johns Hopkins University School of Medicine, Baltimore, MD 21287-0006.
This work was supported by funds from the University of Pittsburgh and the National Institutes of Health (Grants R01 AG019322 to R.W. and P.C.A.v.d.W., R01 GM099718 to R.W., and T32 GM088119 to C.L.H.) and Grant UL1 RR024153 from the National Center for Research Resources (NCRR).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ross C. A. (2002) Polyglutamine pathogenesis: Emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron 35, 819–822. [DOI] [PubMed] [Google Scholar]
- Bates G. P., and Benn C. (2002) The polyglutamine diseases. In Huntington’s Disease, 3rd ed., pp 429–472, Oxford University Press, Oxford, U.K. [Google Scholar]
- Zuccato C.; Valenza M.; Cattaneo E. (2010) Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 90, 905–981. [DOI] [PubMed] [Google Scholar]
- Kar K.; Arduini I.; Drombosky K. W.; van der Wel P. C. A.; Wetzel R. (2014) d-Polyglutamine amyloid recruits l-polyglutamine monomers and kills cells. J. Mol. Biol. 426, 816–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieri L.; Madiona K.; Bousset L.; Melki R. (2012) Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 102, 2894–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; Dunlap J. R.; Andrews R. B.; Wetzel R. (2002) Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum. Mol. Genet. 11, 2905–2917. [DOI] [PubMed] [Google Scholar]
- Nekooki-Machida Y.; Kurosawa M.; Nukina N.; Ito K.; Oda T.; Tanaka M. (2009) Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 9679–9684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M.; Finkbeiner S. (2012) Protein aggregates in Huntington’s disease. Exp. Neurol. 238, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Ferrone F. A.; Wetzel R. (2002) Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl. Acad. Sci. U.S.A. 99, 11884–11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A. M.; Thakur A. K.; Chellgren V. M.; Thiagarajan G.; Williams A. D.; Chellgren B. W.; Creamer T. P.; Wetzel R. (2006) Oligoproline effects on polyglutamine conformation and aggregation. J. Mol. Biol. 355, 524–535. [DOI] [PubMed] [Google Scholar]
- Darnell G.; Orgel J. P. R. O.; Pahl R.; Meredith S. C. (2007) Flanking polyproline sequences inhibit β-sheet structure in polyglutamine segments by inducing PPII-like helix structure. J. Mol. Biol. 374, 688–704. [DOI] [PubMed] [Google Scholar]
- Rockabrand E.; Slepko N.; Pantalone A.; Nukala V. N.; Kazantsev A.; Marsh J. L.; Sullivan P. G.; Steffan J. S.; Sensi S. L.; Thompson L. M. (2007) The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum. Mol. Genet. 16, 61–77. [DOI] [PubMed] [Google Scholar]
- Darnell G. D.; Derryberry J.; Kurutz J. W.; Meredith S. C. (2009) Mechanism of cis-inhibition of polyQ fibrillation by polyP: PPII oligomers and the hydrophobic effect. Biophys. J. 97, 2295–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur A. K.; Jayaraman M.; Mishra R.; Thakur M.; Chellgren V. M.; Byeon I.-J. L.; Anjum D. H.; Kodali R. B.; Creamer T. P.; Conway J. F.; Gronenborn A. M.; Wetzel R. (2009) Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 16, 380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M.; Sapp E.; Chase K. O.; Davies S. W.; Bates G. P.; Vonsattel J. P.; Aronin N. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. [DOI] [PubMed] [Google Scholar]
- Lunkes A.; Lindenberg K. S.; Ben-Haïem L.; Weber C.; Devys D.; Landwehrmeyer G. B.; Mandel J.-L.; Trottier Y. (2002) Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell 10, 259–269. [DOI] [PubMed] [Google Scholar]
- Sathasivam K.; Neueder A.; Gipson T. A.; Landles C.; Benjamin A. C.; Bondulich M. K.; Smith D. L.; Faull R. L. M.; Roos R. A. C.; Howland D.; Detloff P. J.; Housman D. E.; Bates G. P. (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 110, 2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L.; Sathasivam K.; Seller M.; Cozens B.; Harper A.; Hetherington C.; Lawton M.; Trottier Y.; Lehrach H.; Davies S. W.; Bates G. P. (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506. [DOI] [PubMed] [Google Scholar]
- Chow W. N. V.; Luk H. W.; Chan H. Y. E.; Lau K.-F. (2012) Degradation of mutant huntingtin via the ubiquitin/proteasome system is modulated by FE65. Biochem. J. 443, 681–689. [DOI] [PubMed] [Google Scholar]
- Burke K. A.; Kauffman K. J.; Umbaugh C. S.; Frey S. L.; Legleiter J. (2013) The interaction of polyglutamine peptides with lipid membranes is regulated by flanking sequences associated with huntingtin. J. Biol. Chem. 288, 14993–15005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z.-H.; Wang Y.; Sapp E.; Cuiffo B.; Wanker E.; Hayden M. R.; Kegel K. B.; Aronin N.; DiFiglia M. (2004) Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J. Neurosci. 24, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duennwald M. L.; Jagadish S.; Muchowski P. J.; Lindquist S. L. (2006) Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc. Natl. Acad. Sci. U.S.A. 103, 11045–11050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duennwald M. L.; Jagadish S.; Giorgini F.; Muchowski P. J.; Lindquist S. L. (2006) A network of protein interactions determines polyglutamine toxicity. Proc. Natl. Acad. Sci. U.S.A. 103, 11051–11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbach B.; Scherzinger E.; Schweiger K.; Lurz R.; Lehrach H.; Wanker E. E. (1999) Aggregation of truncated GST-HD exon 1 fusion proteins containing normal range and expanded glutamine repeats. Philos. Trans. R. Soc., B 354, 991–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel R., and Mishra R. (2014) Structural biology: order, disorder and conformational flux. In Huntington’s Disease (Bates G. P., Tabrizi S., and Jones L., Eds.) 4th ed., Oxford University Press, Oxford, U.K. [Google Scholar]
- Tam S.; Spiess C.; Auyeung W.; Joachimiak L.; Chen B.; Poirier M. A.; Frydman J. (2009) The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 16, 1279–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanandam V. N.; Jayaraman M.; Hoop C. L.; Kodali R.; Wetzel R.; van der Wel P. C. A. (2011) The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 133, 4558–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra R.; Jayaraman M.; Roland B. P.; Landrum E.; Fullam T.; Kodali R.; Thakur A. K.; Arduini I.; Wetzel R. (2012) Inhibiting the nucleation of amyloid structure in a huntingtin fragment by targeting α-helix-rich oligomeric intermediates. J. Mol. Biol. 415, 900–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman M.; Kodali R.; Sahoo B.; Thakur A. K.; Mayasundari A.; Mishra R.; Peterson C. B.; Wetzel R. (2012) Slow amyloid nucleation via α-helix-rich oligomeric intermediates in short polyglutamine-containing huntingtin fragments. J. Mol. Biol. 415, 881–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellgren B. W.; Miller A.-F.; Creamer T. P. (2006) Evidence for polyproline II helical structure in short polyglutamine tracts. J. Mol. Biol. 361, 362–371. [DOI] [PubMed] [Google Scholar]
- Chellgren B. W.; Creamer T. P. (2004) Short sequences of non-proline residues can adopt the polyproline II helical conformation.. Biochemistry 43, 5864–5869. [DOI] [PubMed] [Google Scholar]
- Jayaraman M.; Mishra R.; Kodali R.; Thakur A. K.; Koharudin L. M. I.; Gronenborn A. M.; Wetzel R. (2012) Kinetically competing huntingtin aggregation pathways control amyloid polymorphism and properties. Biochemistry 51, 2706–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. W.; Chelliah Y.; Kim S. W.; Otwinowski Z.; Bezprozvanny I. (2009) Secondary structure of Huntingtin amino-terminal region. Structure 17, 1205–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Długosz M.; Trylska J. (2011) Secondary structures of native and pathogenic huntingtin N-terminal fragments. J. Phys. Chem. B 115, 11597–11608. [DOI] [PubMed] [Google Scholar]
- Nagai Y.; Inui T.; Popiel H. A.; Fujikake N.; Hasegawa K.; Urade Y.; Goto Y.; Naiki H.; Toda T. (2007) A toxic monomeric conformer of the polyglutamine protein. Nat. Struct. Mol. Biol. 14, 332–340. [DOI] [PubMed] [Google Scholar]
- Davies P.; Watt K.; Kelly S. M.; Clark C.; Price N. C.; McEwan I. J. (2008) Consequences of poly-glutamine repeat length for the conformation and folding of the androgen receptor amino-terminal domain. J. Mol. Endocr. 41, 301–314. [DOI] [PubMed] [Google Scholar]
- Laghaei R.; Mousseau N. (2010) Spontaneous formation of polyglutamine nanotubes with molecular dynamics simulations. J. Chem. Phys. 132, 165102. [DOI] [PubMed] [Google Scholar]
- Nakano M.; Watanabe H.; Rothstein S. M.; Tanaka S. (2010) Comparative characterization of short monomeric polyglutamine peptides by replica exchange molecular dynamics simulation. J. Phys. Chem. B 114, 7056–7061. [DOI] [PubMed] [Google Scholar]
- Williamson T. E.; Vitalis A.; Crick S. L.; Pappu R. V. (2010) Modulation of polyglutamine conformations and dimer formation by the N-terminus of huntingtin. J. Mol. Biol. 396, 1295–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobelmann M. D.; Murphy R. M. (2011) Location trumps length: polyglutamine-mediated changes in folding and aggregation of a host protein.. Biophys. J. 100, 2773–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra R.; Hoop C. L.; Kodali R.; Sahoo B.; van der Wel P. C. A.; Wetzel R. (2012) Serine phosphorylation suppresses huntingtin amyloid accumulation by altering protein aggregation properties.. J. Mol. Biol. 424, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugg C. W.; Isas J. M.; Fischer T.; Patterson P. H.; Langen R. (2012) Structural features and domain organization of huntingtin fibrils.. J. Biol. Chem. 287, 31739–31746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X.; Greiner E. R.; Mishra R.; Kodali R. B.; Osmand A. P.; Finkbeiner S.; Steffan J. S.; Thompson L. M.; Wetzel R.; Yang X. W. (2009) Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 64, 828–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo B.; Singer D.; Kodali R.; Zuchner T.; Wetzel R. (2014) Aggregation behavior of chemically synthesized, full-length huntingtin exon1. Biochemistry 53, 3897–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley C. B.; Perevozchikova T.; Berthelier V. (2011) Structural formation of huntingtin exon 1 aggregates probed by small-angle neutron scattering.. Biophys. J. 100, 2504–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perevozchikova T.; Stanley C. B.; McWilliams-Koeppen H. P.; Rowe E. L.; Berthelier V. (2014) Investigating the structural impact of the glutamine repeat in huntingtin assembly. Biophys. J. 107, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Nuallain B.; Thakur A. K.; Williams A. D.; Bhattacharyya A. M.; Chen S.; Thiagarajan G.; Wetzel R. (2006) Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay. Methods Enzymol. 413, 34–74. [DOI] [PubMed] [Google Scholar]
- Kar K.; Hoop C. L.; Drombosky K. W.; Baker M. A.; Kodali R.; Arduini I.; van der Wel P. C. A.; Horne W. S.; Wetzel R. (2013) β-hairpin-mediated nucleation of polyglutamine amyloid formation. J. Mol. Biol. 425, 1183–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier M. A.; Li H.; Macosko J.; Cai S.; Amzel M.; Ross C. A. (2002) Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 277, 41032–41037. [DOI] [PubMed] [Google Scholar]
- Harris R. K.; Becker E. D.; De Menezes S. M.; Granger P.; Hoffman R. E.; Zilm K. W. (2008) Further conventions for NMR shielding and chemical shifts (IUPAC Recommendations 2008). Magn. Reson. Chem. 46, 582–598. [DOI] [PubMed] [Google Scholar]
- Takegoshi K.; Nakamura S.; Terao T. (2001) 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 344, 631–637. [Google Scholar]
- Bennett A. E.; Rienstra C. M.; Auger M.; Lakshmi K. V.; Griffin R. G. (1995) Heteronuclear decoupling in rotating solids. J. Chem. Phys. 103, 6951–6958. [Google Scholar]
- Torchia D. A.; Szabo A. (1982) Spin-lattice relaxation in solids.. J. Magn. Reson. 49, 107–121. [Google Scholar]
- Lewandowski J. R.; van der Wel P. C. A.; Rigney M.; Grigorieff N.; Griffin R. G. (2011) Structural complexity of a composite amyloid fibril. J. Am. Chem. Soc. 133, 14686–14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munowitz M.; Griffin R. G.; Bodenhausen G.; Huang T. H. (1981) Two-dimensional rotational spin-echo nuclear magnetic resonance in solids: Correlation of chemical shift and dipolar interactions. J. Am. Chem. Soc. 103, 2529–2533. [Google Scholar]
- Carravetta M.; Edén M.; Zhao X.; Brinkmann A.; Levitt M. H. (2000) Symmetry principles for the design of radiofrequency pulse sequences in the nuclear magnetic resonance of rotating solids. Chem. Phys. Lett. 321, 205–215. [Google Scholar]
- Zhao X.; Edén M.; Levitt M. H. (2001) Recoupling of heteronuclear dipolar interactions in solid-state NMR using symmetry-based pulse sequences. Chem. Phys. Lett. 342, 353–361. [Google Scholar]
- Veshtort M.; Griffin R. G. (2006) SPINEVOLUTION: A powerful tool for the simulation of solid and liquid state NMR experiments. J. Magn. Reson. 178, 248–282. [DOI] [PubMed] [Google Scholar]
- Kumashiro K. K.; Schmidt-Rohr K.; Murphy O. J.; Ouellette K. L.; Cramer W. A.; Thompson L. K. (1998) A novel tool for probing membrane protein structure: solid-state NMR with proton spin diffusion and X-nucleus detection. J. Am. Chem. Soc. 120, 5043–5051. [Google Scholar]
- Andronesi O. C.; Bergen M. v.; Biernat J.; Seidel K.; Griesinger C.; Mandelkow E.; Baldus M. (2008) Characterization of Alzheimer’s-like paired helical filaments from the core domain of tau protein using solid-state NMR spectroscopy. J. Am. Chem. Soc. 130, 5922–5928. [DOI] [PubMed] [Google Scholar]
- Huster D.; Yao X.; Hong M. (2002) Membrane protein topology probed by 1H spin diffusion from lipids using solid-state NMR spectroscopy. J. Am. Chem. Soc. 124, 874–883. [DOI] [PubMed] [Google Scholar]
- Candel A. M.; Conejero-Lara F.; Martinez J. C.; van Nuland N. A. J.; Bruix M. (2007) The high-resolution NMR structure of a single-chain chimeric protein mimicking a SH3–peptide complex. FEBS Lett. 581, 687–692. [DOI] [PubMed] [Google Scholar]
- Kricheldorf H. R.; Müller D. (1984) Secondary structure of peptides: 15. 13C NMR CP/MAS study of solid elastin and proline-containing copolyesters. Int. J. Biol. Macromol. 6, 145–151. [Google Scholar]
- Schneider R.; Schumacher M. C.; Mueller H.; Nand D.; Klaukien V.; Heise H.; Riedel D.; Wolf G.; Behrmann E.; Raunser S.; Seidel R.; Engelhard M.; Baldus M. (2011) Structural characterization of polyglutamine fibrils by solid-state NMR spectroscopy.. J. Mol. Biol. 412, 121–136. [DOI] [PubMed] [Google Scholar]
- Southwell A. L.; Khoshnan A.; Dunn D. E.; Bugg C. W.; Lo D. C.; Patterson P. H. (2008) Intrabodies binding the proline-rich domains of mutant huntingtin increase its turnover and reduce neurotoxicity.. J. Neurosci. 28, 9013–9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.-G.; Yang H.; Zhao J.; Jiang Y.-J.; Hu H.-Y. (2014) Autoinhibitory structure of the WW domain of HYPB/SETD2 regulates its interaction with the proline-rich region of huntingtin.. Structure 22, 378–386. [DOI] [PubMed] [Google Scholar]
- Ader C.; Schneider R.; Seidel K.; Etzkorn M.; Becker S.; Baldus M. (2009) Structural rearrangements of membrane proteins probed by water-edited solid-state NMR spectroscopy. J. Am. Chem. Soc. 131, 170–176. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Delaglio F.; Cornilescu G.; Bax A. (2009) TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalk A.; Berendsen H. J. C. (1976) Proton magnetic relaxation and spin diffusion in proteins. J. Magn. Reson. 24, 343–366. [Google Scholar]
- Chevelkov V.; Zhuravleva A. V.; Xue Y.; Reif B.; Skrynnikov N. R. (2007) Combined analysis of 15N relaxation data from solid- and solution-state NMR spectroscopy. J. Am. Chem. Soc. 129, 12594–12595. [DOI] [PubMed] [Google Scholar]
- Fry E. A.; Sengupta S.; Phan V. C.; Kuang S.; Zilm K. W. (2011) CSA-enabled spin diffusion leads to mas rate-dependent T1’s at high field. J. Am. Chem. Soc. 133, 1156–1158. [DOI] [PubMed] [Google Scholar]
- Helmus J. J.; Surewicz K.; Surewicz W. K.; Jaroniec C. P. (2010) Conformational flexibility of Y145Stop human prion protein amyloid fibrils probed by solid-state nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 132, 2393–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou G.; Paramasivam S.; Yan S.; Polenova T.; Vega A. J. (2013) Multidimensional magic angle spinning NMR spectroscopy for site-resolved measurement of proton chemical shift anisotropy in biological solids. J. Am. Chem. Soc. 135, 1358–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L.; Grishaev A.; Cornilescu G.; Bax A. (2010) The impact of hydrogen bonding on amide 1H chemical shift anisotropy studied by cross-correlated relaxation and liquid crystal NMR spectroscopy. J. Am. Chem. Soc. 132, 10866–10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly M. A.; Chellgren B. W.; Rucker A. L.; Troutman J. M.; Fried M. G.; Miller A.-F.; Creamer T. P. (2001) Host–guest study of left-handed polyproline II helix formation. Biochemistry 40, 14376–14383. [DOI] [PubMed] [Google Scholar]
- Atwal R. S.; Xia J.; Pinchev D.; Taylor J.; Epand R. M.; Truant R. (2007) Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Genet. 16, 2600–2615. [DOI] [PubMed] [Google Scholar]
- Michalek M.; Salnikov E. S.; Bechinger B. (2013) Structure and topology of the huntingtin 1−17 membrane anchor by a combined solution and solid-state NMR approach. Biophys. J. 105, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popiel H. A.; Nagai Y.; Onodera O.; Inui T.; Fujikake N.; Urade Y.; Strittmatter W. J.; Burke J. R.; Ichikawa A.; Toda T. (2004) Disruption of the toxic conformation of the expanded polyglutamine stretch leads to suppression of aggregate formation and cytotoxicity. Biochem. Biophys. Res. Commun. 317, 1200–1206. [DOI] [PubMed] [Google Scholar]
- Margittai M.; Langen R. (2008) Fibrils with parallel in-register structure constitute a major class of amyloid fibrils: Molecular insights from electron paramagnetic resonance spectroscopy. Q. Rev. Biophys. 41, 265–297. [DOI] [PubMed] [Google Scholar]
- van der Wel P. C. A. (2012) Domain swapping and amyloid fibril conformation. Prion 6, 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R.; Wickner R. B. (2013) Molecular structures of amyloid and prion fibrils: consensus versus controversy.. Acc. Chem. Res. 46, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmer C.; Lange A.; Van Melckebeke H.; Siemer A. B.; Riek R.; Meier B. H. (2008) Amyloid fibrils of the HET-s(218–289) prion form a β solenoid with a triangular hydrophobic core. Science 319, 1523–1526. [DOI] [PubMed] [Google Scholar]
- Qiang W.; Yau W.-M.; Luo Y.; Mattson M. P.; Tycko R. (2012) Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 109, 4443–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito R. R.; Ron D. (2000) Conformational disease. Nat. Cell Biol. 2, E207–E209. [DOI] [PubMed] [Google Scholar]
- Ferrigno P.; Silver P. A. (2000) Polyglutamine expansions: Proteolysis, chaperones, and the dangers of promiscuity. Neuron 26, 9–12. [DOI] [PubMed] [Google Scholar]
- Thakur A. K.; Wetzel R. (2002) Mutational analysis of the structural organization of polyglutamine aggregates. Proc. Natl. Acad. Sci. U.S.A. 99, 17014–17019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slepko N.; Bhattacharyya A. M.; Jackson G. R.; Steffan J. S.; Marsh J. L.; Thompson L. M.; Wetzel R. (2006) Normal-repeat-length polyglutamine peptides accelerate aggregation nucleation and cytotoxicity of expanded polyglutamine proteins. Proc. Natl. Acad. Sci. U.S.A. 103, 14367–14372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Bigam C. G.; Holm A.; Hodges R. S.; Sykes B. D. (1995) 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 5, 67–81. [DOI] [PubMed] [Google Scholar]
- Ko J.; Ou S.; Patterson P. H. (2001) New anti-huntingtin monoclonal antibodies: Implications for huntingtin conformation and its binding proteins. Brain Res. Bull. 56, 319–329. [DOI] [PubMed] [Google Scholar]
- Khoshnan A.; Ko J.; Patterson P. H. (2002) Effects of intracellular expression of anti-huntingtin antibodies of various specificities on mutant huntingtin aggregation and toxicity. Proc. Natl. Acad. Sci. U.S.A. 99, 1002–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legleiter J.; Lotz G. P.; Miller J.; Ko J.; Ng C.; Williams G. L.; Finkbeiner S.; Patterson P. H.; Muchowski P. J. (2009) Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J. Biol. Chem. 284, 21647–21658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safar J.; Roller P. P.; Gajdusek D. C.; Gibbs C. J. (1994) Scrapie amyloid (prion) protein has the conformational characteristics of an aggregated molten globule folding intermediate.. Biochemistry 33, 8375–8383. [DOI] [PubMed] [Google Scholar]
- Staniforth R. A.; Giannini S.; Higgins L. D.; Conroy M. J.; Hounslow A. M.; Jerala R.; Craven C. J.; Waltho J. P. (2001) Three-dimensional domain swapping in the folded and molten-globule states of cystatins, an amyloid-forming structural superfamily. EMBO J. 20, 4774–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skora L.; Becker S.; Zweckstetter M. (2010) Molten globule precursor states are conformationally correlated to amyloid fibrils of human β-2-microglobulin.. J. Am. Chem. Soc. 132, 9223–9225. [DOI] [PubMed] [Google Scholar]
- Bemporad F.; Chiti F. (2012) Protein misfolded oligomers: experimental approaches, mechanism of formation, and structure-toxicity relationships.. Chem. Biol. 19, 315–327. [DOI] [PubMed] [Google Scholar]
- Gerber R.; Tahiri-Alaoui A.; Hore P. J.; James W. (2006) Oligomerization of the human prion protein proceeds via a molten globule intermediate. J. Biol. Chem. 282, 6300–6307. [DOI] [PubMed] [Google Scholar]
- Narayanan S.; Reif B. (2005) Characterization of chemical exchange between soluble and aggregated states of β-amyloid by solution-state NMR upon variation of salt conditions. Biochemistry 44, 1444–1452. [DOI] [PubMed] [Google Scholar]
- Vendrely C.; Valadié H.; Bednarova L.; Cardin L.; Pasdeloup M.; Cappadoro J.; Bednar J.; Rinaudo M.; Jamin M. (2005) Assembly of the full-length recombinant mouse prion protein I. Formation of soluble oligomers. Biochim. Biophys. Acta 1724, 355–366. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.