

Abstract
Two subspecies of cynomolgus macaques (Macaca fascicularis) are alleged to co-exist in the Philippines, M. f. philippensis in the north and M. f. fascicularis in the south. However, genetic differences between the cynomolgus macaques in the two regions have never been studied to document the propriety of their subspecies status.
We genotyped samples of cynomolgus macaques from Batangas in southwestern Luzon and Zamboanga in southwestern Mindanao for 15 short tandem repeat (STR) loci and sequenced an 835 bp fragment of the mtDNA of these animals. The STR genotypes were compared with those of cynomolgus macaques from southern Sumatra, Singapore, Mauritius and Cambodia, and the mtDNA sequences of both Philippine populations were compared with those of cynomolgus macaques from southern Sumatra, Indonesia and Sarawak, Malaysia. We conducted STRUCTURE and PCA analyses based on the STRs and constructed a median joining network based on the mtDNA sequences.
The Philippine population from Batangas exhibited much less genetic diversity and greater genetic divergence from all other populations, including the Philippine population from Zamboanga. Sequences from both Batangas and Zamboanga were most closely related to two different mtDNA haplotypes from Sarawak from which they are apparently derived. Those from Zamboanga were more recently derived than those from Batangas, consistent with their later arrival in the Philippines. However, clustering analyses do not support a sufficient genetic distinction of cynomolgus macaques from Batangas from other regional populations assigned to subspecies M. f. fascicularis to warrant the subspecies distinction M. f. philippensis.
Keywords: Macaca fascicularis, subspecies, mitochondrial DNA, short tandem repeat (STR)
Cynomolgus macaques (Macaca fascicularis) probably originated in insular Southeast Asia (Delson, 1980; Fooden, 2006) as early as 3.5 million years ago (mya), based on the most recent and comprehensive genomic evidence (Perelman et al., 2011), after diverging from their common ancestor with the sinica group of macaque species (Li et al., 2009). Their dispersal, which may have ranged as far as the Philippine Islands, reached the mainland, via Sumatra, and was followed by the divergence of rhesus macaques (Smith et al., 2007) as late as 0.9 mya (Osada et al., 2008). A land bridge connected Sumatra, Java, Borneo and the Malay Peninsula for more than 50% of the last 250,000 years, and Sumatra and Java were connected to the Malay Peninsula for two-thirds of this time (Voris, 2000). The current distribution of members of the species is provided in Figure 1.
Figure 1.

Map of the distribution of M. fascicularis illustrating locations of three major and seven minor (on offshore islands) subspecies based on Groves (2001). Names in capital letters with dark arrows indicate the smallest geographical unit (e.g., country, province, island, etc.) within which samples used in this analysis originated. Arrows indicate routes of dispersal of M. f. philippensis, first, across the Palawan Islands and M. f. fascicularis, much later, across the Sulu Archipelago to the Philippines.
Cynomolgus macaques probably followed two separate routes to the Philippines, both of which traversed Sabah, as illustrated by the arrows in Figure 1, but not necessarily at the same time. Macaques may have first reached the Philippines by way of the Palawan Islands, crossing the Mindoro Strait to Mindoro Island of the Philippines. They eventually spread throughout Luzon and evolved a derived morphology featuring their distinctive dark dorsal pelage (Fooden, 1991) that distinguishes them from cynomolgus macaques throughout the remainder of the Philippines and Southeast Asia. Their early arrival in the Philippines is consistent with a longer period of time in isolation to evolve their distinctive morphology. The second route of dispersal probably crossed the Sulu Archipelago at the Sibutu Passage, now 18 miles wide, to the western part of Mindanao. Cynomolgus macaques in western Mindanao closely resemble those in the Sulu Archipelago, Indonesia and Malaysia, exhibiting pale dorsal pelage.
The close morphological similarities between cynomolgus macaques in extreme western Mindanao and the Sulu Archipelago with those in Malaysia and Indonesia assigned to subspecies M. f. fascicularis are consistent with this route being followed much later than that through the Palawan Islands to the northern Islands of the Philippines hypothesized for M. f. philippensis. Fooden’s (1991) survey of pelage color at 128 locations throughout the Philippines revealed exclusively dark pelage in locations throughout the Philippines with light and mixed pelage color largely restricted to the southernmost Island, Mindanao, and the Sulu archipelago. This distribution is consistent with the continued southward dispersal of macaques assigned to subspecies M. f. philippensis to eastern Mindanao followed later by the colonization and limited eastward and northward dispersal of M. f. fascicularis across the Sulu Archipelago. That distribution is more difficult to reconcile with a single dispersal from either the north or the south that should have generated a clear clinal distribution of pelage color.
Dispersal to the Philippines might not have occurred as early as cynomolgus macaques reached the Indochinese mainland, because it is unlikely that a land passage from Sabah to the Philippines, such as that connecting Sundaland to the mainland via Sumatra, ever existed. Certainly, neither the Sibutu Passage nor the Mindoro Strait formed a land bridge to the Philippines during the last glacial maximum (Lambert et al., 2006). This is because the islands of the Philippines do not lie on the Sunda Shelf, having risen de novo from the ocean floor (Esselstyn et al., 2004), and deep waters separate them from Sundaland. Consequently, cynomolgus macaques, being relatively facile swimmers, may have rafted to the southern and northern Philippines across the Sibutu Passage and the Mindoro Strait, respectively (Abegg and Thierry, 2002). These dispersals probably occurred during glacial maxima, the last two of which occurred approximately 170 thousand years ago (kya) and 18 kya, when sea levels fell to within 120 m of their present levels (Heaney, 1986) and the distances across the Sibutu Passage and the Mindoro Strait (Sathiamurthy and Voris, 2006) were shortest. Earlier glacial maxima when rafting would have been most feasible occurred at approximately 200, 240, 325, 400 and 600 kya (Sathiamurthy and Voris, 2006).
Since the distinctive morphology of cynomolgus macaques in the northern and most of the central islands of the Philippines must have required some time to emerge, human intervention is an improbable explanation for the earliest appearance of cynomolgus macaques in the Philippines. Humans could not have transported macaques to the Philippines, as Alfred Wallace speculated (1876), until after approximately 44 kya, the date for Niah Cave in Sarawak (Barker et al., 2002), which represents the earliest known human occupation of the region. Continued dispersal of the late arrivers to Mindanao eastward and northward would have been hindered if the earliest arrivals were already dispersed throughout the Philippines, consistent with the highly restricted distribution of M. f. fascicularis with pale pelage predominantly to the western part of southwest Mindanao.
Whether dispersal occurred by rafting or was facilitated by an unknown land connection, the number of founding members of both dispersals is likely to have been small, probably creating a founder effect in both Philippine subspecies. Such a founder effect is reflected by the genetic structure of the cynomolgus macaques of Mauritius (Lawler et al., 1995; Kanthaswamy et al., 2013; Satkoski Trask et al., 2013) that were transported from Indonesia as pets then released by Portuguese sailors several hundred years ago (Sussman and Tattersall, 1986). We have previously reported evidence of very low genetic diversity of M. f. fascicularis from both Mauritius and unknown locations on Mindanao (Smith et al., 2007; Kanthaswamy et al., 2013). If the ancestors of the modern descendants of the two subspecies arrived in the Philippines at different times, the earliest hypothetical arrivals, now inhabiting Luzon, should exhibit greater genetic differences from Indonesian and Malaysian cynomolgus macaques than those now living in western Mindanao. Moreover, since both migrations to the Philippines must have traversed northeastern Borneo, the descendants of both should more closely resemble modern populations from that region than they resemble each other. Thus, both populations should exhibit evidence of a founder effect, especially the loss of rare alleles, and evidence of derivation from Malaysian cynomolgus macaques of northern Borneo.
Cynomolgus macaques from both Luzon and western Mindanao are employed as animal models in biomedical research in the US. However, the subspecies or region of origin in the Philippines of research subjects is rarely reported. Nor is the extent of genetic differences between the two alleged subspecies known. If the genetic difference between the two Philippine subspecies is comparable to that observed between Indian and Chinese rhesus macaques, two subspecies that are known to exhibit significant differences in their responses to experimental treatment in biomedical research, such as experimental infection of rhesus macaques with SIV (Ling et al., 2002), care should be taken not to include members of both Philippine subspecies in the same experimental protocols. Otherwise, the phenotypic variance in treatment effects may be inflated by genetic variance, requiring larger sample sizes to demonstrate statistically significant differences in response to those treatment effects. Moreover, the two subspecies may not be appropriate as models for studies of the same human diseases as is the case with Indian and Chinese rhesus macaques (Ling et al., 2002). In the present study, we report a genetic comparison between cynomolgus macaques from southern Luzon and western Mindanao where their subspecies affiliations are regarded as unmixed M. f. philippensis and M. f. fascicularis, respectively.
METHODS AND MATERIALS
The research reported in this manuscript complied with protocols approved by the UC Davis Animal Care and Use Committee and adhered to the legal requirements of the United States. Blood samples from 30 cynomolgus macaques from Batangas in southwestern Luzon, 35 from Zamboanga in western Mindanao, 40 from Corregidor Island in Manila Bay, 18 from Cambodia, 79 from Mauritius and 98 from southern Sumatra were obtained directly from incountry breeding farms by Primate Products, Inc. providing the greatest possible authentication of the provenience of all samples. The origin of all animals from Luzon and Mindanao are claimed to be solely from those islands, and the pelage color of the respective animals sampled for the present study is consistent with the absence of admixture in either sample. The animals from Batangas were adults captured wild from multiple troops as breeding stock for a breeding farm on Luzon and are, therefore, representative of the free-ranging populations. Samples were also obtained from 25 cynomolgus macaques wild-caught by the Malaria Research Centre of the University of Malaysia in Sarawak and 29 samples acquired by Maccine Pte. Ltd., Inc. in Singapore with all the required CITES and import permits. The population of cynomolgus macaques from Corregidor were captured in Mindanao for breeding and commercial sale and later released on Corregidor when the venture collapsed (Jerome Nazarino, DVM, Philippine Breeders Association, personal communication). However, because documentation of the antiquity and origin of cynomolgus macaques on Corregidor is lacking, we advise caution in drawing inferences solely based on this population. All samples described above, except those from Sarawak, were genotyped for the 15 unlinked STR loci listed in Table 1 that were carefully selected to ensure against allelic dropout in cynomolgus macaques. The sample from Sarawak was not included in the analyses of STRs due to the insufficient quality of extracted DNA.
Table 1.
Sample size (given in parentheses), Number of alleles (Na) and observed (HO) and expected (HE) heterozygosity for each of 15 STR loci and mean values of each for seven regional populations of cynomolgus macaques.
| Batangas (30) | Zamboanga (35) | Sumatra (98) | Singapore (29) | Cambodia (18) | Corregidor (40) | Mauritius (79) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus | Na | HO | HE | Na | HO | HE | Na | HO | HE | Na | HO | HE | Na | HO | HE | Na | HO | HE | Na | HO | HE |
| D1s548 | 1 | 0 | 0 | 2 | 0.46 | 0.47 | 6 | 0.74 | 0.74 | 5 | 0.46 | 0.63 | 5 | 0.63 | 0.55 | 2 | 0.45 | 0.50 | 6 | 0.56 | 0.74 |
| D3s1768 | 1 | 0 | 0 | 7 | 0.57 | 0.70 | 17 | 0.87 | 0.90 | 13 | 0.88 | 0.85 | 11 | 0.93 | 0.91 | 6 | 0.54 | 0.58 | 5 | 0.16 | 0.66 |
| D4s2365 | 2 | 0.14 | 0.21 | 5 | 0.32 | 0.38 | 14 | 0.79 | 0.84 | 4 | 0.48 | 0.70 | 8 | 0.88 | 0.76 | 7 | 0.67 | 0.72 | 4 | 0.62 | 0.65 |
| D5s1457 | 4 | 0.18 | 0.29 | 6 | 0.51 | 0.57 | 6 | 0.69 | 0.70 | 5 | .068 | 0.66 | 3 | 0.43 | 0.60 | 5 | 0.66 | 0.68 | 3 | 0.53 | 0.50 |
| D6s501 | 4 | 0.68 | 0.69 | 5 | 0.61 | 0.65 | 10 | 0.75 | 0.77 | 7 | 0.76 | 0.82 | 6 | 0.75 | 0.80 | 7 | 0.77 | 0.79 | 5 | 0.67 | 0.61 |
| D7s1826 | 2 | 0 | 0.11 | 9 | 0.69 | 0.77 | 11 | 0.66 | 0.63 | 7 | 0.60 | 0.78 | 8 | 0.94 | 0.86 | 9 | 0.85 | 0.73 | 7 | 0.51 | 0.59 |
| D7s794 | 5 | 0.46 | 0.52 | 8 | 0.53 | 0.69 | 13 | 0.69 | 0.69 | 5 | 0.42 | 0.60 | 5 | 0.44 | 0.60 | 9 | 0.77 | 0.82 | 6 | 0.60 | 0.70 |
| D8s1106 | 5 | 0.80 | 0.59 | 11 | 0.78 | 0.79 | 9 | 0.65 | 0.82 | 8 | 0.72 | 0.68 | 7 | 0.80 | 0.86 | 9 | 0.74 | 0.80 | 6 | 0.67 | 0.77 |
| D8s1466 | 6 | 0.55 | 0.53 | 6 | 0.61 | 0.78 | 7 | 0.64 | 0.69 | 6 | 0.52 | 0.74 | 10 | 0.69 | 0.87 | 7 | 0.61 | 0.83 | 4 | 0.38 | 0.47 |
| D9s921 | 5 | 0.53 | 0.59 | 10 | 0.78 | 0.84 | 9 | 0.73 | 0.78 | 7 | 0.76 | 0.78 | 7 | 0.94 | 0.83 | 11 | 0.84 | 0.85 | 6 | 0.70 | 0.80 |
| D13s765 | 2 | 0.30 | 0.47 | 8 | 0.80 | 0.84 | 20 | 0.75 | 0.83 | 7 | 0.52 | 0.74 | 9 | 0.69 | 0.85 | 8 | 0.82 | 0.80 | 5 | 0.45 | 0.57 |
| D18s861 | 1 | 0 | 0 | 1 | 0 | 0 | 3 | 0.06 | 0.06 | 2 | 0.29 | 0.31 | 3 | 0.50 | 0.60 | 1 | 0 | 0 | 1 | 0 | 0 |
| D19s255 | 1 | 0 | 0 | 3 | 0 | 0.30 | 4 | 0.11 | .038 | 5 | 0.09 | 0.49 | 7 | 0.69 | 0.83 | 1 | 0 | 0 | 2 | 0 | 0.08 |
| 270o7 | 2 | 0.05 | 0.05 | 2 | 0.53 | 0.44 | 6 | 0.62 | 0.65 | 5 | 0.39 | 0.70 | 4 | 0.38 | 0.47 | 3 | 0.15 | 0.23 | 4 | 0.61 | 0.71 |
| 272o12 | 3 | 0.24 | 0.22 | 7 | 0.69 | 0.60 | 8 | 0 .49 | 0.66 | 8 | 0.48 | 0.75 | 8 | 0.69 | 0.87 | 5 | 0.77 | 0.78 | 4 | 0.36 | 0.37 |
| Mean | 2.9 | 0.36 | 0.28 | 6.0 | 0.56 | 0.60 | 9.5 | 0.62 | 0.68 | 6.4 | 0.54 | 0.68 | 6.7 | 0.69 | 0.75 | 6.0 | 0.66 | 0.60 | 4.5 | 0.49 | 0.55 |
For PCR amplification of STR loci, 0.5–1.25 μL of extract was used in each 12.5 μL PCR reaction [67 mM Tris HCl (pH 8.8), 16 mM (NH4)2SO4, 0.01% Tween-20, 0.05 mM each dNTP, 0.2 μM each primer, 1.7 mM MgSO4, 0.025 units/mL (Invitrogen) Platinum Taq]. The annealing temperatures and extension times of the touch-down thermocycler conditions varied for each STR primer pair: 94°C for 3 min, then 60 cycles of 94°C for 20 s denaturing, 54–62°C (decreasing 0.1° C per cycle) annealing, 72°C for 45–90 s extension followed by a final extension at 72°C for 5–60 s, with extension times varying by primer pair. All samples were analyzed on an ABI 3130xl Genetic Analyzer following the recommendations of the manufacturer using the Liz 500 size standard to assign genotypes. Because these markers have been used for assigning parentage to captive cynomolgus macaques without evidence of allelic dropout, we did not routinely confirm genotypes with multiple amplifications. Nevertheless, the data were examined for possible scoring errors due to stutter or allelic dropout, screened for the incidence of null alleles using the program Microchecker version 2.2 (Van Oosterhout et al., 2004) and all suspicious results were confirmed by re-amplification.
The number of alleles (Na), observed (HO) and expected (HE) levels of heterozygosity and genetic distance between all pairs of populations studied, measured as FST (Weir and Cockerham 1984), were estimated using GENEPOP (version 4.2, Raymond and Rousset, 1995) and PopGene (version 1.32, Yeh and Boyle, 1997). Paired FST values were averaged for each population to reflect that population’s average differentiation from all other populations. GENEPOP 4.2 was also used to test for departure of genotypes from their Hardy-Weinberg equilibrium (HWE) frequencies and for linkage disequilibrium (LD) among pairs of loci in each population at the 0.05 level of probability using Fisher’s exact tests (Raymond and Rousset, 1995). Unbiased P-values were generated for these tests by the Markov Chain Monte Carlo (MCMC) approach (Guo and Thompson, 1992) with 5000 iterations per batch. The statistical significance of the pairwise FST estimates was tested using a probability distribution constructed from permutations of 1000 iterations with a Bonferroni correction of α for multiple comparisons.
Principal component analysis (PCA) was performed with the adegenet 1.2–8 package for R (Jombard, 2008) to identify genetic structure within the data independent of a priori assignment to a particular geographic origin. We also used the program STRUCTURE 2.3.3 (Pritchard et al., 2000; Hubisz et al., 2009) to characterize the structure among the seven regional populations. STRUCTURE uses a Markov Chain Monte Carlo (MCMC) method to compute L(K), the posterior probability that the data fit the hypothesis of K genetically distinct groups. STRUCTURE provides the advantages over PCA that it allows the selection of the most likely number of independent taxonomic units represented by the data and estimates the fractional membership of each animal in each taxon. While estimates of L(K) plateau after reaching the “true” value of K due to increasing variance (Pritchard and Wen, 2003), the height of the modal value of ΔK, a measure of second order rate of change of the STRUCTURE likelihood function [L(K)], is correlated with the strength of the genetic subdivision among the study populations (Evanno et al., 2005). Thus, we used the value of ΔK to select the best value of K. STRUCTURE runs were conducted assuming that between two and seven genetically distinct groups exist among the populations sampled. A total of 5 × 105 MCMC simulations were conducted after a 105 burn-in period. It was assumed that allele frequencies among populations were correlated and that members of each population have ancestors in the others. Each STRUCTURE run was replicated 10 times to assure that group assignments with the highest probabilities were obtained.
An 835 base pair (bp) fragment of mtDNA flanked by nucleotide positions (nps) 15167 and 16050 in the homologous Barbary macaque (M. sylvanus) mtDNA sequence (GenBank accession number AJ309865; Arnason et al., 2000) including the first hypervariable segment (HVS I; nps 15492-16028), approximately one-third of the cytochrome b gene (nps 15193-15357) and the tRNA genes for proline (nps 15424-15491) and threonine (nps 15358-15422) was amplified from each of the samples from Sarawak, Zamboanga, Batangas, Corregidor, southern Sumatra and Cambodia. Primers, cycling parameters, and methods employed were those previously described (Smith & McDonough, 2005; Smith et al., 2007). This primer pair employed was designed to replace a pair of previously-used primers that readily amplified a pseudogene (numtDNA) [Smith and McDonough, 2005]. This pseudogene [Bensasson et al., 2001; Collura & Stewart, 1995] was easily identifiable by its extreme divergence from all rhesus sequences amplified with the redesigned pair of primers, an abundance of transversions and indels, a paucity of polymorphic, but consistently heteroplasmic, sites and the failure of its HVS I to readily align with the reference M. sylvanus sequence. The redesigned primers yielded sequences with specific mutations that provide the haplogroup structure characteristic of true mtDNA sequences, with the majority of the variation in the HVR I region (Smith and McDonough, 2005). This conspicuous difference between the pseudogene and the mtDNA fragments amplified provided confidence that pseudogenes were not included in the sequences analyzed in this study.
Electropherograms were aligned with Sequencher (Gene Codes Corp., Ann Arbor, MI), and mutations identified with reference to the M. sylvanus mtDNA sequence (GenBank accession number AJ309865; Arnason et al., 2000). MtDNA sequences of cynomolgus macaques from Indonesia, Malaysia, Cambodia, Mindanao (unknown locations) and Mauritius that were previously assigned to specific haplogroups (Smith et al., 2007) were included in the analysis as reference samples. The mtDNA sequences were deposited in GenBank (Accession numbers KJ830548-KJ830614 and KJ850509-KJ850587).
The evolutionary history of cynomolgus macaques was also inferred from the mtDNA sequences by using the Maximum Likelihood method in MEGA5 (Tamura et al., 2011) based on the Tamura-Nei model (Tamura and Nei, 1993) to construct a bootstrap consensus tree inferred from 1000 replicates (Felsenstein, 1985). All positions containing gaps and missing data were eliminated. MEGA5 first infers an evolutionary tree by applying the neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach (Tamura et al., 2004), and then it selects the topology with superior log likelihood values. Branch lengths and substitution rate parameters are then optimized for each of six substitution models combined with 4 shape parameters (gamma rate) describing heterogeneity in mutation rates to fit the sequence data, and the model with the highest goodness-of- fit to the data, measured by the Bayesian information criterion (BIC, Schwartz 1978), is selected. Branches corresponding to partitions reproduced in fewer than 50% bootstrap replicates were collapsed. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap (1000 replicates) are shown next to the branches.
The mean numbers of nucleotides in the 835 bp mtDNA fragment studied that differ between all possible pairs of sequences within and between each of five populations (Sumatra, Sarawak, Corregidor, Zamboanga and Batangas) were calculated using the ARLEQUIN v. 2.0 package (Schneider et al., 2000) and used to construct a haplotype median-joining (MJ) network (Bandelt et al., 1999) rooted with a sequence from M. sylvanus, a sister species to all other macaque species (Li et al., 2009). For the haplotype network, all transitions were given equal weight, transversions and gaps associated with single-base indels were given twice the weight of transitions, and all gaps in the poly C regions 15739-15741 and 15719-15721 were treated as missing data. Genetic heterogeneity was expressed as nucleotide diversity (θπ), the average proportion of all nucleotides included in the analysis that differ between all possible pairs of individuals in a given population. This parameter was also estimated for all possible pairs of individuals one each from each of two different populations to assess between-population divergence between pairs of the seven populations whose mtDNA were compared. The number of segregating sites standardized by the harmonic mean of the number of sequences studied ( where S is the total number of segregating sites in the sequence: Waterson, 1975) and the value of ρ, the average number of mutation steps in a MJ haplotype network between each sequence in a population and the basal haplotype from which they are derived (Bandelt et al., 1999), were also calculated. The value of ρ was independently estimated for the Cambodian population as verification of the earlier occupation by cynomolgus macaques in Indonesia than on the mainland. The MJ network from which ρ is estimated predicts the order in which the haplotypes in the network emerged and the geographic region in which the immediate ancestral haplotypes occur. As a measure of within-population diversity, the values of ρ for Batangas and Zamboanga were calculated as the number of mutational steps in the network between each haplotype in the population to the Sarawak haplotype from which it is most closely derived. In equilibrium populations, these three values (θπ, θS and ρ) are expected to be identical and, for mtDNA, provide estimates of θ=Neμ, the population mutation parameter, where θ is proportional to the period of time during which diversity accumulated in each population (Forster et al., 1996). Departures between the values of θS and θπ (θπ − θS) based on mtDNA result from demographic forces and are expressed by Tajima’s D, (standardized by the variance in θπ − θS; Tajima, 1989). In the absence of selection, a statistically significant negative value of D reflects population growth while constant population size is expected to result in a D value of zero. The values of θπ, θS and ρ were compared between the Philippine populations from Zamboanga and Batangas to assess the population history of the two subspecies and whether or not the two regions of the Philippines were likely to have been colonized at approximately the same time.
RESULTS
The genotypes of all of the 15 STR loci were statistically unlinked (p > 0.05) after data from the seven populations were pooled and none of the markers presented any evidence for null alleles at statistically significant levels (p < 0.05). Diversity parameters based on the STR genotypes of the population samples from Batangas, Zamboanga, Corregidor, southern Sumatra, Mauritius, Singapore and Cambodia are given in Table 1. The Batangas samples exhibited approximately half the number of alleles (Na=2.9) and much lower observed heterozygosity (HO=0.36) than the Zamboanga samples (Na=6.0 and HO=0.56, respectively). Genetic diversity of the Batangas sample was also lower than that for the Mauritian sample (Na=4.5 and HO=0.49), whose lack of diversity has been widely reported (Lawler et al., 1995; Kanthaswamy et al., 2013; Satkoski Trask et al., 2013a). Genetic diversity was highest in the Sumatran (Na =9.5 and HO =0.61) and Cambodian (Na =6.7 and HO =0.69) samples while the populations from Singapore and Corregidor exhibited intermediate numbers of alleles (6.0–6.4) but different heterozygosity estimates (0.54 and 0.66, respectively). Inbreeding (FIS) was highest in Singapore (FIS=0.23) and Mauritius (FIS =0.22), lowest in Corregidor (FIS =0.07), Cambodia (FIS =0.09) and Sumatra (FIS =0.10) and intermediate in Batangas (FIS =0.17) and Zamboanga (FIS =0.15).
Genetic distances (FST) between pairs of the seven populations based on STRs, provided in Table 2, were all statistically significant at the 0.05 level of probability. All populations were more distinct from Batangas than from any other population. The Batangas sample (average FST=0.312) was almost twice as genetically distinct from each of the other populations than any other population except Mauritius, the second most distinct population (average FST=0.238). The average genetic distance between pairs of the remaining five populations ranged from 0.144 (Singapore) to 0.171 (Cambodia). The Mauritian population was least distant from Sumatra (FST=0.128), the alleged recent homeland of Mauritian cynomolgus macaques (Tosi and Coke, 2007), and the population from Batangas was least distant from Corregidor (FST=0.188), the population most geographically proximate to it. The populations from Corregidor and Zamboanga were more similar to each other (FST=0.076) than either population was to any other population, as were the populations from Singapore and Cambodia (FST=0.072), the two mainland populations. Sumatra, the alleged homeland of cynomolgus macaques (Tosi and Coke, 2007), was least distant from the two mainland populations (Cambodia FST=0.093; Singapore FST=0.096). The remaining pair of populations, with an FST lower than 0.10, was Singapore/Zamboanga (FST=0.096). The values of FIS, FST and FIT among all seven populations were 0.125, 0.181 and 0.283, respectively, signifying relatively high levels of both inbreeding and population structure.
Table 2.
Pairwise and average genetic distance (FST) and average FIS based on 15 STR loci in seven regional populations of cynomolgus macaques
| Average* | Paired FST Estimates | |||||||
|---|---|---|---|---|---|---|---|---|
| FIS | FST | Zamboanga | Corregidor | Sumatra | Singapore Cambodia | Mauritius | ||
| Batangas | 0.17 | 0.31 | 0.30 | 0.19 | 0.33 | 0.30 | 0.36 | 0.39 |
| Zamboanga | 0.15 | 0.16 | 0.08 | 0.14 | 0.10 | 0.12 | 0.23 | |
| Corregidor | 0.07 | 0.16 | 0.16 | 0.11 | 0.15 | 0.25 | ||
| Sumatra | 0.10 | 0.16 | 0.10 | 0.09 | 0.13 | |||
| Singapore | 0.23 | 0.14 | 0.07 | 0.20 | ||||
| Cambodia | 0.09 | 0.17 | 0.23 | |||||
| Mauritius | 0.22 | 0.24 | ||||||
| All Populations Combined | ||||||||
| FIS = 0.125 | ||||||||
| FST = 0.181 | ||||||||
| FIT = 0.283 | ||||||||
The first two columns of data provide population-specific FIS and FST values averaged over all six other populations with which a population is paired.
The results of our PCA, provided in Figure 2, in which the first and second dimensions account for 41.5% and 23.8% of the variance, respectively, and spheres represent 95% confidence limits, are consistent with the values of paired FST. All populations are approximately equally distributed in PC1 with Mauritius and Batangas exhibiting the greatest separation from each other and each separated equally far from the other populations in PC2.
Figure 2.

Principal Coordinates Analysis (PCA) based on genotypes at 15 STR loci in seven regional populations of cynomolgus macaques. Ellipses indicate the 95% confidence limits for each regional population and grid intervals are in units 0f 0.5.
The results of the STRUCTURE analyses are illustrated in Figures 3 and are consistent with the PCA and FST values. When only the three Philippine populations were included in a STRUCTURE analysis, provided in Figure 3A, ΔK values support a K value of 2. In the STRUCTURE plot, Batangas and Zamboanga are well differentiated from each other while the Corregidor results could suggest admixture of the two populations in approximate proportions of 25% and 75%, respectively, although such evidence could actually reflect shared ancestry of all three populations in Sabah with discretely different patterns of drift. A value of K=3 was strongly supported by ΔK values when all seven populations were included in a STRUCTURE analysis. As shown in Figure 3B, when K=3, the Mauritian samples are discretely differentiated from those in all of the other populations, the three Philippine samples resemble one another, but exhibit minor resemblance to both mainland populations, Cambodia and Singapore, which themselves more closely resemble the Sumatran population.
Figure 3.
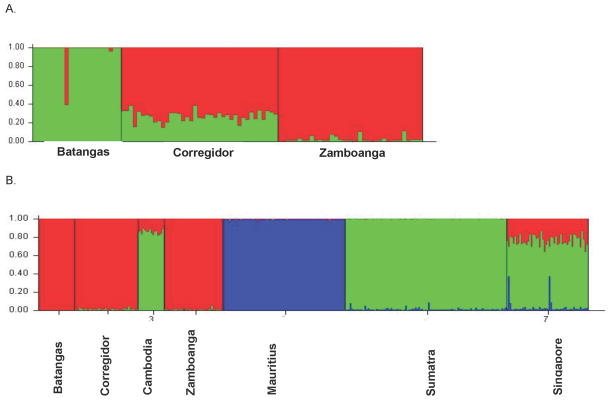
A. STRUCTURE plot of three Philippine populations (Batangas, Corregidor and Zamboanga) with K=2 based on genotypes at 15 STR loci. Batangas and Zamboanga are well differentiated in the plot with Corregidor showing admixture of approximately 25% and 75% with each of the two populations. B. STRUCTURE plot of all seven populations of cynomolgus macaques based on STRs when K=3. Note that the three Philippine populations for a single group that is well differentiated from populations from Sumatra and Mauritius.
Because the mtDNA sequences from Batangas were foreshortened at both ends due to poor quality sequence, only 697 base pairs of sequence were available for all five populations employed in the phylogenetic analysis. The lack of ambiguity (e.g., double peaks) in the electropherograms of all sequences, including reference samples, the complete absence of indels, premature stop codons or frame shift mutations in the cytochrome b, tRNAPro and tRNAThr coding sequences and the predominance of mutations at the third positions of codons (48 of 70) in these coding regions are consistent with our assumptions that none of the sequences amplified represent pseudogenes. The numbers of complete, trimmed 697 bp fragments include 14, 24, 37, 25 and 15 sequences from Batangas, Zamboanga, Corregidor, Sarawak and Sumatra, respectively. A total of 65 segregating sites were identified in the three Philippine populations, or 1, 37 and 49 sites respectively, in Batangas, Zamboanga and Corregidor. These include one insertion, two transversions and 62 transitions that define 2, 8 and 13 different haplotypes, respectively, in the three populations. Corregidor was the only population to share more than a single haplotype with another population, one each with Zamboanga and Batangas, and the latter two were the only other populations to share any haplotypes (one each with Corregidor) with another population. The Batangas and Zamboanga populations were fixed for different alleles at 11 positions, all of which were polymorphic in the Corregidor population, clearly indicating the affiliation of each of Corregidor’s 37 haplotypes with either Zamboanga (M. f. fascicularis) or Batangas (M. f. philippensis).
The ML tree based on the 697 bp sequence of mtDNA is provided in Figure 4. This represents an abridged version of a much larger tree from which redundant haplotypes were removed to improve legibility of the tree. The tree first divides into two major clades, Fas1 and Fas2, as previously reported (Smith et al., 2007), with the latter clade containing all Philippine samples and the former containing all sequences from the mainland. All samples from the Batangas, Zamboanga and Sarawak populations fell in the Fas2a haplogroup of the Fas2 clade of the tree. This haplogroup includes subhaplogroups Fas2a1, Fas2a2, Fas2a3 and Fas2a4 that were previously defined (Smith et al., 2007), reference samples of which are included in the tree.
Figure 4.

Maximum Likelihood tree for a 697 bp fragment of mtDNA of five regional populations of cynomolgus macaques based on the Tamura-Nei model with 500 replicates. Branches corresponding to partitions reproduced in fewer than 50% bootstrap replicates were collapsed. Bootstrap values are provided for each branch. Sequences with “Fas” prefixes are reference sequences representing previously identified mtDNA haplogroups in cynomolgus macaques (Smith et al., 2007). Each such reference is followed by the identification number of the sample representative of that haplogoup whose prefix identifies the country of origin of that sample. Sequences with prefixes “Cor”, “Luz”, “Zam”, “Sar”, “Sum”, “Phi”, “Viet”, “Mcy”, “Maur”, and “Imcy” are sequences of samples from Corregidor, Luzon (Batangas), Zamboanga, Sarawak, Sumatra, Philippines (Mindanao), Vietnam, Malaysia, Mauritius and Indonesia, respectively. The tree is rooted with a sequence from M. sylvanus.
All fourteen mtDNA sequences from Batangas represented only two haplotypes that differed by a single mutation and clustered together with 16 haplotypes from Corregidor and an Indonesian sample (Genbank accession number DQ373508) from subhaplogroup Fas2a4 with 99% bootstrap support. The samples from Zamboanga were much more genetically diverse than those from Batangas, as shown in Figure 5, and represent the three of the four subhaplogroups of Fas2a (Fas2a1, Fas2a2 and Fas2a3) that are not represented in Batangas. Thus, Zamboanga and Batangas share no haplogroups in common. Subhaplogroups Fas2a1 and Fas2a2 represent the majority (12 of 14) of the Zamboanga haplotypes. These include two samples from Zamboanga and 4 Philippine samples from unknown locations in Mindanao previously assigned to haplogroup Fas2a1 that form a clade with 99% support and one sample from Zamboanga that cluster with three samples from Corregidor, and three samples (two from Malaysia and one from an unknown location in Mindanao) previously assigned to haplogroup Fas2a2 with 90% bootstrap support. Fas2a1 and Fas2a2 are closely related and together join with a haplotype from Sarawak with 93% bootstrap support. The remaining three sequences from Zamboanga were shared with three Malaysian and one Indonesian cynomolgus macaque sequence assigned to subhaplogroup Fas2a3 with 100% support. The remaining sequences from Sarawak were very heterogeneous (see Figure 5) and most were included in two Fas2 clades not closely related to any Philippine samples or any known subhaplotype of the Fas2 clade, but with relatively low (77%) bootstrap support. All 30 Cambodian sequences, represented by three haplotypes labeled VietC (because they were originally believed to have originated in Vietnam) in Figure 4, belonged to the Fas1 major clade, represented among the five populations only in Sumatra, and comprise 26 haplotypes, three of which are shown in Figure 4. However, all 30 Cambodian sequences belonged to the Fas1a subclade while all Fas1 sequences from Sumatra belong to the Fas1c subclade.
Figure 5.

Median-Joining haplotype network based on mtDNA sequences employed in Figure 5, above, and rooted with a sequence from M. sylvanus. The reference sequences are identified by both their sample numbers and haplogroup acronym of each reference sample. Note: The sequence of sample number 10 (**) from Zamboanga is identical to the haplogroup reference sequence Fas2a2 and is shared with Corregidor and the sequence of sample number 50 from Batangas is shared with Corregidor.
Levels of nucleotide diversity within each population and between all pairs of populations are shown in Table 3. The Sumatran population exhibited at least twice the level of nucleotide diversity (θπ=0.049) than any other population. Sarawak and Corregidor had intermediate levels of nucleotide diversity (θπ=0.022 and θπ=0.023, respectively), Zamboanga had relatively low levels of diversity (θπ=0.013), but the level of mtDNA diversity in the Batangas population was much lower (θπ=0.0005) than that in any other population. The value of θS was identical to that of θπ in Batangas, slightly higher than θπ in Zamboanga and lower than θπ in Corregidor. The values of Tajima’s D for Batangas, Zamboanga and Corregidor were 0.00, − 0.52 and 0.62, respectively, (p> 0.70 in all three populations), indicating D is not statistical significance from a value of 0 and suggesting a lack of population expansion in all three populations.
Table 3.
Average proportion of nucleotide differences (θπ) in mtDNA within each population (on diagonal in bold print), between (off diagonal) each pair of populations, corresponding values of θS and ρ and average of θπ (Σ θπ/4) over the other four populations of cynomolgus macaques.
| Sumatra | Sarawak | Corregidor | Zamboanga | Batangas | |
|---|---|---|---|---|---|
| Sumatra | 0.0490 | 0.0553*** | 0.0690 | 0.0637 | 0.0737 |
| Sarawak | 0.0217 | 0.0339 | 0.0303 | 0.0331 | |
| Corregidor | 0.0226 | 0.0297 | 0.0175 | ||
| Zamboanga | 0.0125 | 0.0390 | |||
| Batangas | 0.0005 | ||||
| Σ θπ /4 | 0.0654 | 0.0382 | 0.0375 | 0.0407 | 0.0408 |
| θS | - | - | 0.0168 | 0.0142 | 0.0005 |
| D | - | - | 0.62 | -0.52 | 0.00 |
| p | - | - | 0.77 | 0.70 | 1.00 |
| ρ | 0.0303**** 0.0125**** | - | 0.0059* | 0.0112** | |
value of ρ for distance between 1) Corregidor sequences H10 (the Fas2a2 haplotype that Corregidor and Zamboanga share), H66 and H71, and Zamboanga sequences H54-H60 and 2) the closest ancestral Sarawak sequence, H22.
value of ρ for distance between 1) Corregidor sequences H61-H65, H67-H70, the Batangas sequence H51 and sequence H52 (that Batangas and Corregidor share in common) and 2) the closest ancestral Sarawak sequence, H28.
average value of θπ between each Sumatran and each Sarawak sequence
average value of ρ between haplotypes in each population and the basal haplotype in the MJ network
The Sumatran population least resembled all other populations, with an average between-population θπ value of 0.065, while Batangas and Zamboanga were equally distant from all other populations (θπ =0.041). The population from Sarawak most closely (θπ =0.0550), and that from Batangas (θπ =0.0737) least closely, resembled the Sumatran population. The population from Sarawak most closely resembled that from Zamboanga (θπ =0.030) but only slightly less than it resembled that from Batangas (θπ =0.033) and Corregidor (θπ =0.034). The closest resemblance was that between Batangas and Corregidor (θπ =0.018), the two most geographically proximate of the populations.
The MJ network with M. sylvannus as its root (sample number 1) is provided in Figure 5. Three Sumatran sequences (samples 40, 46 and 47) that cluster with haplogroup Fas2d (reference sample 19) are closest to the root (M. sylvannus), hence older than all other cynomolgus macaque sequences, followed by five sequences from Sarawak (samples 20, 22, 25, 26 and 31) that are collectively ancestral to all Philippine sequences. A haplotype of haplogroup Fas2c (reference sample 18) and a Sumatran sequence (sample 37) are immediately ancestral to all Fas1 sequences (reference samples 2–8) suggesting that the latter major mtDNA clade of sequences is derived from Fas2c, a relatively rare subclade that we have not found outside Indonesia (Smith et al., 2007). Both haplotypes from Batangas clustered closely with 10 of the 13 haplotypes from Corregidor, one of which (sample 50*) is shared with Batangas, are members of haplogroup Fas2a4 (reference sample 12) and may have been introduced to Luzon via Palawan. No reticulations occurred in this cluster of 11 haplotypes. All haplotypes in this star-like cluster clearly derive from the basal haplotype, i.e., that un-sampled haplotype closest to Corregidor haplotype 65 in the MJ network. Eight of the 10 haplotypes from Zamboanga clustered with three of the 13 haplotypes from Corregidor, one of which (sample 10) is two steps removed from both the basal (unsampled) haplotype of this cluster belonging to haplogroups Fas2a1 (reference sample 9) and Fas2a2 (reference sample 10, shared with Zamboanga) and a haplotype (sample 22) from Sarawak. One reticulation appears in this cluster of 12 haplotypes. Those 2 Corregidor haplotypes (samples 66 and 71) closely related to the 8 Zamboanga haplotypes and to sample 10, the basal haplotype of the Fas2a2 cluster that is shared in common between Zamboanga and Corregidor, might have been introduced to Mindanao across the Sulu Archipelago. The remaining two Zamboanga sequences (samples 52 and 53) were members of haplogroup Fas2a3 (reference sample 11) and fell 9–10 mutational steps from haplotype 10.
The value of ρ was calculated as the average number of mutational steps from each of the 11 haplotypes in the cluster containing the two Batangas haplotypes and each of the 11 haplotypes containing all but two of the Zamboanga haplotypes to their nearest ancestral sequence from Sarawak, sequence 28 for the haplotypes in the Batangas cluster and sequences 20 (for samples 52 and 53) and 22 (for the remaining samples) for the haplotypes in the two Zamboanga clusters. The resulting values of ρ for Batangas and Zamboanga were ρ=0.011 and ρ=0.006, respectively, suggesting that haplotypes from Batangas have been diverging longer than those from Zamboanga. The values of ρ for the sequences from Sumatra, Cambodia and Sarawak were ρ=0.0303, ρ=0.0233 and ρ=0.0125, all estimated as the average number of mutational steps from each haplotype to the basal haplotype in their individual MJ networks, suggesting that cynomolgus macaques reached mainland SE Asia before they traversed what is now Borneo.
DISCUSSION
Haplotypes from Sumatra were closest to the root of the MJ network in Figure 5, and all Fasl haplotypes are derivative of Fas2 haplotypes, suggesting that mainland haplotypes, all of which belong to the Fas1 clade, are younger than, and derived from, haplotypes that originated in insular Southeast Asia, the apparent homeland of cynomolgus macaques. In Figure 5 the Fas1a subclades of Indochina are most directly derived from subclade Fas2c, a relatively rare haplotype reported only from Indonesia. The highest level of within-population diversity in both mtDNA (θπ) and STRs (Na) in Sumatra, its greatest mtDNA differentiation from all other populations (Σ θπ /4 in Table 3) and highest value of ρ, which is linear in time, are also consistent with an Indonesian homeland for cynomolgus macaques.
The predominant restriction of pale and intermediate pelage color of cynomolgus macaques to Mindanao suggests the dispersal of, followed by admixture between, the ancestors of two separate founder populations of cynomolgus macaques, one in the north, who arrive earlier and dispersed southward, and another in the south, who arrived later and dispersed northward. Based on Fooden’s (1991) subspecies distinction between cynomolgus macaques in the north and south of the Philippines, the distribution of pelage color throughout the Philippines and the hypothesis that the colonization of Luzon by cynomolgus macaques predates that of western Mindanao, we predicted that cynomolgus macaques from Batangas, on Luzon, would exhibit greater genetic differentiation from Malaysian and Indonesian cynomolgus macaques than those from Zamboanga, in western Mindanao, which was in fact the case. We also predicted that the haplotypes of both Batangas and Zamboanga would more closely resemble haplotypes of Sarawak than those of each other (between population θπ in Table 3), which they did. Because rafting and/or storm dispersal would have been required of cynomolgus macaques to reach the Philippines, which are not a part of the Sunda Shelf and were never connected to it by land, via either route, we predicted that levels of genetic diversity would be low in both Philippine populations, which they were, as we previously reported for cynomolgus macaques from unknown localities in Mindanao (Smith et al., 2007; Kanthaswamy et al., 2013). Because the cynomolgus macaques from Batangas were wild caught from multiple troops, circumstances of captivity cannot be responsible for their very low level of genetic diversity. The somewhat greater influence of founder effect on mtDNA than on the number of STR alleles in Batangas might have resulted from the greater sieving effect of mtDNA due to its lower (by three-quarters) effective population size than that of nuclear loci. We also predicted that haplotypes of cynomolgus macaques from Batangas would be both older (furthest from the root of the haplotype network) and more diverse (higher value of θπ) than those of cynomolgus macaques from Zamboanga, which they were.
The PCA analysis that revealed an apparent cline from Sumatra to mainland southeast Asia and Mindanao to a terminus in Luzon is not inconsistent with the hypothesis that all Philippine cynomolgus macaques derive from a single expansion from Sabah via the Sulu Archipelago and dispersed northward to Luzon where they became isolated and phenotypically distinct. However, the Batangas haplotypes derive from a Sarawak, not Zamboangan, haplotype that is very different from that Sarawak haplotype from which the Zamboangan haplotypes derive, and the haplotypes from Batangas are older than, not younger than, those from Zamboanga. A single entry and southward dispersal of cynomolgus macaques from Luzon should have reveal a Zamboangan, not Batangan, terminus of the cline.
The exceptionally low genetic diversity in Batangas was unexpected but might result from its geographic location at the very populous southwestern-most extreme of the Island of Luzon not far from Manila. The extirpation of cynomolgus macaques associated with rapid human population growth, habitat destruction and industrialization may have caused a genetic bottleneck from which the population on Corregidor was spared. Accordingly, haplotypes 61–65 and 67–70 from Corregidor, that closely clustered with the two haplotypes from Batangas but far removed from all Zamboangan haplotypes, might be, or alternatively might once have been, widespread throughout most of Luzon. The most common haplotype on Batangas, that most likely to survive a genetic bottleneck, was also the most common lineage on Corregidor and only one mutational step from the basal haplotype of one of two well-defined but genetically distant clusters of haplotypes on Corregidor in the MJ haplotype network. While the origin of cynomolgus macaques on Corregidor is problematical and not germane to comparisons between haplotypes of Luzon and Mindanao, the presence there of two very different clusters of haplotypes derived from Sarawak, haplogroups Fas2a4 (Batangas) and Fas2a1/Fas2a2 (Zamboanga) that are otherwise exclusively found in, but not shared between, Batangas and Zamboanga, provides support for the hypothesis that cynomolgus macaques on Corregidor include animals derived from both the earlier and the later expansion of cynomolgus macaques to the Philippines. All haplotypes identified in the Corregidor population clustered closely with those of either Batangas or Zamboanga, which themselves were very divergent, and only two Philippine haplotypes (two from Zamboanga) were not closely related to either of these two clusters. Thus, the inclusion of the Corregidor haplotypes in the present analysis provides a more comprehensive estimate of genetic diversity derived from the two different founder populations of cynomolgus macaques in the Philippines, especially the earlier of the two to arrive. We regard haplotypes 61–65 and 67–70 to be indigenous to Corregidor and haplotypes 10, 66 and 71 to represent cynomolgus macaques bred in Mindanao then recently abandoned on Corregidor. Alternatively, the founding population of cynomolgus macaques in western Mindanao might have been much larger and/or their rate of gene flow with Malaysia much higher than for the founders of Luzon. Studies of other cynomolgus macaque populations more distant from dense population centers on Luzon and in the islands intervening between Luzon and Mindanao are needed to select between these two alternative hypotheses and further evaluate the hypothesis of dual origins of Philippine cynomolgus macaques at different times.
Based on the entire 835 bp mtDNA fragment, 697 bp of which were included in the present analysis, Smith et al. (2007) estimated the nucleotide divergence between Indian and Chinese rhesus macaques, the two most divergent regional populations of rhesus macaques which many regard as different subspecies, to be θπ=0.076, a value very close to that (θπ=0.074) differentiating cynomolgus macaques from Sumatra and Batangas. Smith et al. (2006) estimated FST between Indian and Chinese rhesus macaques based on 24 STR loci that extensively overlap those employed in the present analysis to be FST=0.083. In contrast, most cynomolgus populations were far more distant from each other than the two subspecies of rhesus macaques, with FST=0.33 for Batangas and Sumatra. Batangas was approximately twice as genetically distant from all populations assigned to subspecies M. f. fascicularis, including Zamboanga, than any of the latter populations were to each other based on STR loci, and Zamboanga was equally distant from any population other than Batangas. When Mauritius and Corregidor are regarded as anomalous cases due to loss of diversity through founders effect and gain of diversity due to inter-subspecies admixture, respectively, and not considered, the FST estimates for divergence of Batangas from other M. f. fascicularis populations and divergence between pairs of M. f. fascicularis populations are FST=0.32 and FST=0.11, respectively. However, Batangas may appear anomalous, as does Mauritius from Sumatra, due to its drastic loss of alleles through a founder effect and/or subsequent genetic bottleneck, which in the comparison based on mtDNA was mitigated by the inclusion of haplotypes from Corregidor absent in, but clearly derive from, those in Batangas. Moreover, our PCA exhibited a clear clinal distribution from the homeland of cynomolgus macaques in Indonesia to Luzon and our STRUCTURE analyses and pairwise FST statistics revealed Sumatra, the homeland of the species, as the most distant from all other populations, results similar to those for population structure of rhesus macaques (Hasan et al., 2014). While, as with Chinese and Indian rhesus macaques, both regional variants of Philippine cynomolgus macaques are employed as subjects in biomedical research and care should be taken to determine and report which variant is used in any given study and to avoid admixture of the two variants in captive breeding facilities, a distinctive subspecific assignment of the Batangas cynomolgus macaques seems unjustified.
Acknowledgments
Grant Sponsor: NIH grants RR05090 and RR025871 to D.G.S.
This study was supported by the California National Primate Research Center base grant (No. RR000169-48), and by grants number RR005090 and RR025781 to DGS from the National Center for Research Resources (NCRR) of the National Institutes of Health (NIH). This research adhered to the American Society of Primatologists principles for the ethical treatment of nonhuman primates and the animals employed have been managed in compliance with Institutional Animal Care and Use Committee (IACUC) regulations or in accordance with the National Institutes of Health guidelines or the US Department of Agriculture regulations prescribing the humane care and use of laboratory animals. The authors wish to thank the staff of Primate Products, Inc. of Immokalee, Florida for acquiring blood samples from the animals and documenting their provenience and the staff of Simian Conservation and Breeding Center (SICONBREC) and Delmundo Trading in the Philippines for their help in sample collection and document processing and shipping.
LITERATURE CITED
- Abegg C, Thierry B. Macaca evolution and dispersal in insular south-east Asia. Biol J Linn Soc. 2002;75:555–576. [Google Scholar]
- Arnason U, Gullberg A, Burquete AS, Janke A. Molecular estimates of primate divergences and new hypotheses for primate dispersal and the origin of modern humans. Hereditas. 2000;133:217–228. doi: 10.1111/j.1601-5223.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- Bandelt H-J, Forster P, Roehl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Barker G, Barton H, Beavitt P, Bird M, Daly P, Doherty C, Gilbertson D, Hunt C, Krigbaum J, Lewis H, Manser J, McLaren S, Paz V, Piper P, Pyatt B, Rabett R, Reynolds T, Rose J, Rushworth G, Stephens M. Prehistoric foragers and farmers in southeast Asia: renewed investigations at Niah Cave, Sarawak. Proc Prehist Soc. 2002;68:147–164. [Google Scholar]
- Delson E. Fossil macaques, phyletic relationships and a scenario of deployment. In: Lindburg DG, editor. The macaques. Studies in ecology, behavior and evolution. New York: van Nostrand Rheinhold; 1980. pp. 10–30. [Google Scholar]
- Esselstyn JA, Widmann P, Heaney LR. The mammals of Palawan Island, Philippines. Proc Biol Soc Wash. 2004;117:271–302. [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 2005;14:2611–20. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP (Phylogeny Inference Package) version 3.6b. Department of Genome Sciences, University of Washington; Seattle: 2004. Distributed by the author. [Google Scholar]
- Fooden J. Systematic Review of Philippine Macaques (Primates, Cercopithecidae: Macaco, fascicularis subspp.) Fieldiana:Zoology, N.S. 1991;(64):1–44. Field Museum of Natural History. [Google Scholar]
- Fooden J. Systematic Review of the Rhesus Macaque, Macaca mulatta (Zimmermann, 1780) Fieldiana Zoology. 2000;96:1–180. [Google Scholar]
- Fooden J. Comparative review of fascicularis-group species of macaques (primates: Macaca) Fieldiana Zoology. 2006;107:1–43. [Google Scholar]
- Forster P, Harding R, Torroni A, Bandelt H-J. Origin and evolution of Native American mtDNA variation: a reappraisal. Amer J Hum Genet. 1996;59:935–945. [PMC free article] [PubMed] [Google Scholar]
- Heaney LR. Biogeography of mammals in SE Asia: estimates of rates of colonization, extinction and speciation. Biol J Linn Soc. 1986;28:127–165. [Google Scholar]
- Hubisz MJ, Falush D, Stephens M, Pritchard JK. Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour. 2009;9:1322–32. doi: 10.1111/j.1755-0998.2009.02591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart T. Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- Kanthaswamy S, Trask JS, George D, Kou A, Erickson B, Smith D. Hybridization and stratification of nuclear genetic variation in Macaca mulatta and M. fascicularis. Int J Primatol. 2008;29:1295–311. doi: 10.1007/s10764-008-9295-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanthaswamy S, Trask JS, George DA, Kou AK, Hoffman LN, Doherty TB, Erickson BJ-A, Houghton P, Smith DG. The genetic composition of cynomolgus macaque populations (Macaca cynomolgus) used in biomedical research. J Med Primatol. 2013;42:120–131. doi: 10.1111/jmp.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AB, Meñez C, Villanoy L, David LT. Movement of Water Across Passages Connecting Philippine Inland Sea Basins. Marine Science Institute, University of the Philippines; 2006. [Google Scholar]
- Lawler SH, Sussman RW, Taylor LL. Mitochondrial DNA of the Mauritian macaques (Macaca fascicularis): an example of the founder effect. Am J Phys Anthropol. 1995;96:133–41. doi: 10.1002/ajpa.1330960203. [DOI] [PubMed] [Google Scholar]
- Li J, Han K, Xing J, Kim H-S, Rogers J, Ryder AOA, Disotell T, Bisong Yue B, Batzer MA. Phylogeny of the macaques (Cercopithecidae: Macaca) based on Alu elements. Gene. 2009;448:242–249. doi: 10.1016/j.gene.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling B, Veazey RS, Luckay A, Penedo C, Xu K, Lifson JD, Marx P. SIVmac pathogenesis in rhesus macaques of Chinese and Indian origin compared with primary HIV infections in humans. AIDS. 2002;16:1489–96. doi: 10.1097/00002030-200207260-00005. [DOI] [PubMed] [Google Scholar]
- Malhi RS, Sickler B, Lin D, et al. MamuSNP: a resource for rhesus macaque (Macaca mulatta) genomics. PLoS ONE. 2007;2:e438. doi: 10.1371/journal.pone.0000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada N, Hashimoto K, Kameoka Y, Hirata M, Tanuma R, Uno Y, Inoue I, Hida M, Suzuki Y, Sugano S, Terao K, Kusuda J, Takahashi I. Large-scale analysis of Macaca fascicularis transcripts and inference of genetic divergence between M. fascicularis and M. mulatta. BMC Genomics. 2008;9:90. doi: 10.1186/1471-2164-9-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelman P, Johnson WE, Roos C, Seuanez HN, Horvath JE, et al. A Molecular Phylogeny of Living Primates. PLoS Genet. 2011;7(3):e1001342. doi: 10.1371/journal.pgen.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard J, Wen W. Documentation for STRUCTURE Software: Version 2. Chicago: Department of Human Genetics, University of Chicago; 2003. [Google Scholar]
- Raymond M, Rousset F. GENEPOP: Population genetics software for exact tests and ecumenicism. J Hered. 1995;86:248–249. [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Sathiamurthy E, Voris HK. Pleistocene Sea Level Maps for the Sunda Shelf. The Field Museum; Chicago, IL: 2006. [Google Scholar]
- Satkoski J, George D, Smith DG, Kanthaswamy S. Genetic characterization of wild and captive rhesus macaques in China. J Med Primatol. 2008a;37:67–80. doi: 10.1111/j.1600-0684.2007.00228.x. [DOI] [PubMed] [Google Scholar]
- Satkoski J, Malhi RS, Kanthaswamy S, Tito RY, Malladi V, Smith DG. Pyrosequencing as a method for SNP identification in the rhesus macaque (Macaca mulatta) BMC Genomics. 2008b;9:256. doi: 10.1186/1471-2164-9-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satkoski Trask JA, Garnica WT, Kanthaswamy S, Malhi RS, Smith DG. 4040 SNPs for genomic analysis in the rhesus macaque (Macaca mulatta) Genomics. 2011;98:352–358. doi: 10.1016/j.ygeno.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satkoski Trask JA, Houghton P, George DA, Kanthaswamy S, Smith DG. Population and Landscape Genetics of an Introduced Species (M. fascicularis) on the Island of Mauritius. PLoS ONE. 2013a;8(1):e53001. doi: 10.1371/journal.pone.0053001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satkoski Trask JA, Smith DG, Houghton P, Lerche N, Kanthaswamy S. Single Nucleotide Polymorphisms Reveal Patterns of Allele Sharing across a Species Boundary in Macaca mulatta and M. fascicularis. Amer J Primatol. 2013b;75:135–144. doi: 10.1002/ajp.22091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S, Roessli D, Excofflier L. Laboratory of Genetics and Biometry. University of Geneva; Switzerland: 2000. ARLEQUIN 2.001: a software for population genetic data analysis. [Google Scholar]
- Smith DG, George DA, Kanthaswamy S, McDonough J. Identification of country of origin and admixture between Indian and Chinese rhesus macaques. Intern J Primatol. 2006;27:881–898. [Google Scholar]
- Smith DG, McDonough JW, George DA. Mitochondrial DNA variation within and among regional populations of longtail macaques (Macaca fascicularis) in relation to other species of the fascicularis group of macaques. Amer J Primatol. 2007;69:182–198. doi: 10.1002/ajp.20337. [DOI] [PubMed] [Google Scholar]
- Sussman RW, Tattersall I. Distribution, abundance, and putative ecological strategy of Macaca fascicularis on the island of Mauritius, southwestern Indian Ocean. Folia Primatolog. 1986;46:28–43. [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–596. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molec Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Nat Acad Sciences (USA) 2004;101:11030–11035. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosi AJ, Morales JC, Melnick DJ. Paternal, maternal, and biparental molecular markers provide unique windows onto the evolutionary history of macaque monkeys. Evolution. 2003;57:1419–1435. doi: 10.1111/j.0014-3820.2003.tb00349.x. [DOI] [PubMed] [Google Scholar]
- Tosi AJ, Coke CS. Comparative phylogenetics offer new insights into the biogeographic history of Macaca fascicularis and the origin of the Mauritian macaques. Mol Phylogenet Evol. 2007;42:498–504. doi: 10.1016/j.ympev.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P. Micro-Checker: Software for Identifying and Correcting Genotyping Errors in Microsatellite Data. Mol Ecol Notes. 2004;4:535–538. [Google Scholar]
- Voris HK. Maps of Pleistocene Sea Levels in Southeast Asia: Shorelines, River Systems and Time Durations. J Biogeography. 2000;27:1153–1167. [Google Scholar]
- Wallace AR. The geographical distribution of animals. London: Macmillan; 1876. [Google Scholar]
- Watterson GA. On the number of segregating sites in genetical models without recombination. Theoret Pop Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Yeh FC, Boyle TJB. Population genetic analysis of co-dominant and dominant markers and quantitative traits. Belgian J Botany. 1997;129:157. [Google Scholar]