

Abstract
Immune homeostasis is a host characteristic that maintains biological balance within a host. Humans have evolved many host defence mechanisms that ensure the survival of individuals upon encountering a pathogenic infection, with recovery or persistence from a viral infection being determined by both viral factors and host immunity. Chronic viral infections, such as hepatitis B virus, hepatitis C virus and HIV, often result in chronic fluctuating viraemia in the face of host cellular and humoral immune responses, which are dysregulated by multi-faceted mechanisms that are incompletely understood. This review attempts to illuminate the mechanisms involved in this process, focusing on immune homeostasis in the setting of persistent viral infection from the aspects of host defence mechanism, including interferon-stimulated genes, apolipoprotein B mRNA editing enzyme catalytic polypeptide 3 (APOBEC3), autophagy and interactions of various immune cells, cytokines and regulatory molecules.
Keywords: hepatitis C virus, HIV, immune homeostasis, viral persistence
Introduction
Immune homeostasis is a host characteristic that maintains biological balance. Humans have evolved many host defence mechanisms that ensure the survival of individuals upon encountering a pathogenic infection. Although healthy individuals coexist in equilibrium with the microbial world, disease can be viewed as a progression away from this immune homeostasis. Microorganisms disturb immune homeostasis because they invade and replicate, causing inflammation and destruction of the tissue, as well as depletion of the host's nutrients, ultimately leading to a chronic infection or death of the host if the infection is not resolved. A well-described example is intestinal homeostasis, which depends on complex interactions between microbes, the intestinal epithelium and the host immune system. Diverse regulatory mechanisms cooperate to maintain intestinal homeostasis, and a breakdown in these pathways may precipitate the chronic inflammatory pathology found in inflammatory bowel disease.1 Hence, a balanced interaction between microbes and the host immune system is required to maintain health.2
In relation to blood-borne viruses that often result in chronic infections, most notably hepatitis C virus (HCV), hepatitis B virus (HBV) and the HIV, their survival in the host may also be a function of immune homeostasis. At the onset of infection, overly rapid viral replication leads to cell lysis, tissue injury and premature host death whereas inadequate viral replication may result in viral latency or clearance (Fig.1). The virus must balance its own growth with the death of host cells and circumvent the immune response to survive. Strategies for viral survival include regulating apoptosis, inhibiting interferon production, modulating the MHC class I/II functions that ultimately affect the cytotoxic lymphocyte and natural killer (NK) responses, and limiting cytokine or chemokine production/function. Long-term viral survival (i.e. viral persistence or latency) requires down-regulation of lytic gene expression, inhibition of apoptosis and minimization of the inflammatory response. In other words, viral persistence requires host immune homeostasis.
Figure 1.
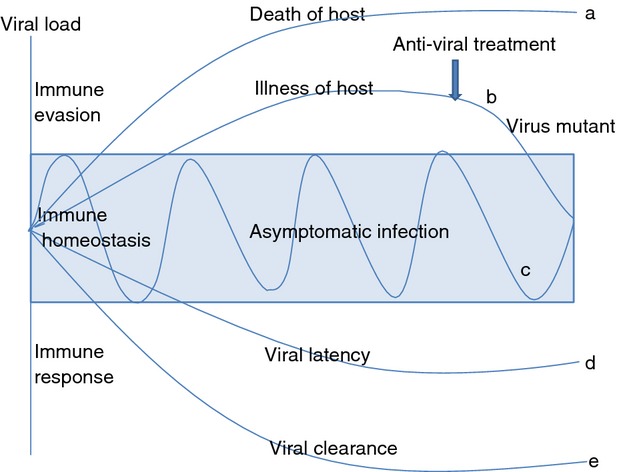
Viral persistence and immune homeostasis. With high viral load infection, virus may evade host immunity, leading to death (a) or illness (b) of the host. With anti-viral treatment, viral load may be reduced, leading to viral clearance or latency. Meanwhile, anti-viral treatment may cause mutation in the virus and drug-resistance (b). When immune homeostasis is maintained, the host exhibits asymptomatic persistent infection, whereby the immune system fight against the virus; and viral load fluctuates during chronic infection (c). Under a strong host immune response, viral infection may become latent (d) or be cleared (e).
Viral factors for persistent infection
Viruses persist in cells because they are able to down-regulate key processes that if left unattended would result in cell death. Specifically, regulation of viral transcription and genomic replication allows for long-term viral persistence. Many viruses (such as HCV, HBV and HIV) that cause persistent infection and chronic disease are successful at in vivo survival because of their cell tropism and ability to auto-regulate their replication efficiently within target cells. Common features of auto-regulation include sensors to the external environment, negative feedback loops, transcriptional enhancers and transcriptional silencers. In some cases, auto-regulation results in steady-state levels of virus replication; in other infections, the virus enters latency only to reactivate intermittently. Viruses have therefore developed numerous strategies for subverting the host defences. The biological characteristics of HCV, HIV and HBV are summarized in Table1, and the viral factors for persistent infection are described below.
Table 1.
Characters of hepatitis C virus (HCV), HIV and hepatitis B virus (HBV)
| Genome | Entry | Translation | Process | Exocytosis | Important molecules in immune evasion | |
|---|---|---|---|---|---|---|
| HCV | Single-stranded, enveloped RNA virus | HCV E1/E2 bind and enter cells via glycosaminoglycans, scavenger receptor class B type 1 (SR-B1), CD81, and low-density lipoprotein (LDL) receptor | HCV RNA is translated by the host cell ribosomes to produce a large viral polyprotein | Polyproteins are cleaved and processed by both host cellular and virus-specific (NS2 and NS3) enzymes. The viral polymerase/replicase (NS5B) copies the viral RNA in the cytoplasm | Mature HCV virions develop and bud through the plasma membrane | Core, E1,E2, NS3/4A, NS5A |
| HIV | Single-stranded RNA virus | HIV gp 120 attaches to the CD4 receptor and the chemokine co-receptor CCR5, fusion of the membranes of virus and cell via the HIV envelope gp 41 | RNA reverse transcription to DNA, called the provirus. The provirus acts as a template to produce the components of new virus particles | The viral proteins are processed and cleaved by HIV protease, in conjunction with cellular proteins | Viral proteins and RNA are assembled and bud from the cell membrane, forming mature HIV particles that can infect other cells | Tat, gp120, Vpu |
| HBV | Partially double-stranded DNA virus | Several regions in the PreS1 domain of the viral large surface protein (LHBs) and the cysteine-rich antigenic loop of the S domain contribute directly to viral attachment | Virus DNA is converted to covalently closed circular cccDNA, which serves as a template to yield two types of RNA: a pre-genomic RNA that undergoes reverse transcription to yield DNA for progeny virus and messenger RNA for structural proteins | The core protein which itself can be phosphorylated by several kinases, forms the basis for the nucleocapsid. Three surface proteins of HBV are secreted as subviral particles | DNA and proteins are assembled into mature virions that are released from the cell | HBsAg, HBx, Pol |
Hepatitis C virus
Binding and entry of HCV is believed to be a multistep process involving HCV envelope glycoproteins E1 and E2 as well as several attachment and entry factors such as CD81, scavenger receptor class B type I, members of the claudin family and occludin.3,4 This process involves several viral and host factors and is targeted by host-neutralizing responses. As the HCV envelope glycoproteins E1 and E2 interact with host cell factors and trigger the conformational changes necessary to initiate infection, they are important targets for virus neutralization. A recent study5 indicated that altered use of the CD81 receptor allowed the virus to escape neutralizing antibodies, and this altered use of CD81 is mediated by residues at positions 447, 458 and 478. Kinetic studies showed that these mutations affect virus–antibody interactions during the post-binding steps of the HCV entry process. Functional studies with a large panel of patient-derived antibodies showed that this mechanism mediates viral escape, leading to persistent infection in general. Another study revealed that mutations in HCV E2 located outside the CD81 binding sites lead to escape from broadly neutralizing antibodies but compromise virus infectivity.6 Recently, it was reported that HCV has evolved mechanisms that antagonize host cell death signals so that viral propagation can continue unabated in infected cells. For example, HCV core protein blocks tumour necrosis factor-α (TNF-α) -mediated apoptosis signalling and inhibits caspase-8 activation.7 A review of how HCV interferes with the different steps of initial antiviral host-response and establishes persistent infections was summarized by Bode et al.8 HCV targets the Toll-like receptor 3 (TLR3), retinoic acid-inducible gene I (RIG-I), and interferon (IFN) signalling pathways as well as the expression and functionality of IFN-stimulated gene (ISG) products and other key sites of host defence mechanisms such as induction of apoptosis within the infected cell. Therefore, HCV might successfully adapt to the host environment and evade the host antiviral response, leading to persistent viral replication and spread.8 The key innate immune evasion strategies used by HCV to establish persistent infection within the liver as well as how host genotype influences the outcome of HCV infection, was reviewed by Horner. 9 The key viral factor involved in the HCV immune evasion programme is the viral NS3/4A protease, which cleaves mitochondrial antiviral signalling protein (MAVS), the RIG-I signalling adaptor protein and Toll-like receptor-domain containing adapter-inducing IFN-β (TRIF), the TLR3 signal transducer. Additionally, there is an increasing role for microRNAs in mediating HCV innate immune evasion strategies, including miR-12210 and miR-21.11
HIV
Chronic HIV infection is linked with a variety of immune evasion strategies beyond the basic integration of the HIV-1 genome into target cells. These include an overall ability to maintain a latent infection and to avoid antibody responses by varying or shielding immunodominant epitopes within the viral envelope glycoprotein. Latent HIV resides in memory T cells or myeloid cells (HIV reservoirs), persisting indefinitely in patients even on potent antiretroviral therapy. The molecular mechanisms of HIV latency are complex and include the absence in resting CD4+ T cells, inhibition of nuclear forms of key host transcriptional factors (e.g. nuclear factor-κB and nuclear factor of activated T cells), the presence of transcriptional repressors, blocking of mRNA splicing, insufficient Tat activity and associated host factors that promote efficient transcriptional elongation, and transcriptional interference, as reviewed previously.12–14
The HIV envelope glycoprotein that drives HIV entry is highly variable. Its plasticity allows HIV to escape the immune system, and its variability is associated with HIV tropism, fitness and replicative capacity. Entry inhibitors represent a new generation of antiviral for the treatment of HIV infection. Several compounds which block the attachment of HIV gp120 to either the CD4+ T-cell receptor or the CCR5/CXCR4 co-receptors are currently in clinical development. Changes within a 10-amino-acid segment encompassing residues 36–45 within the HR1 region of gp41, however, may cause resistance to the entry inhibitor, enfuvirtide, while multiple changes in different gp120 domains (V1, V2, V3, C2 and C4) are associated with loss of susceptibility to other entry inhibitors.15 Resistance to antibodies by point mutation on the V2/V3 loop and N-linked carbohydrate glycans on gp120 has been indicated as hindering antibody neutralization.16 Recently, one study found that CD4+ T memory stem cells harbour high per-cell levels of HIV-1 DNA and make increasing contributions to the total viral CD4+ T-cell reservoir over time. Hence, HIV-1 may exploit the stem cell characteristics of cellular immune memory to promote long-term viral persistence.17
Hepatitis B virus
During active replication, HBV produces enormous viral loads in the blood and a massive surplus of subviral surface antigen particles in the serum of infected patients without killing hepatocytes. Together with the use of a reverse transcriptase during replication, it provides an enormous genetic flexibility for the selection of viral mutants upon selective pressure.18 Escape mutations in the major hydrophilic region of hepatitis B surface antigen (HBsAg) are reported widely, and these mutations lead to diagnostic problems, emergence of vaccine-escape mutants, and hepatitis B immunoglobulin prophylaxis failure.19 For example, mutations in the preS gene can result in undetectable HBsAg even when HBV is replicating. Surface gene mutations lead to decreased binding affinity to anti-HBs, which are associated with a vaccine escape mutant that is occasionally found in clinical cases. Mutations in the basal core promoter are associated with increased HBV replication and high incidence of progressive liver diseases. HBx protein can disrupt RIG-I-mediated IFN-β induction by down-regulating MAVS20 and acts as a de-ubiquitinating enzyme to de-ubiquitinate RIG-I and other molecules.21 One research study suggests that HBV polymerase (Pol) blocks IFN regulatory factor (IRF) signalling, indicating that HBV Pol may be the viral molecule that effectively counteracts host innate immune response in the early phase of the infection.22 It appears that HBV has evolved numerous strategies to counteract defence mechanisms and maintain persistence by actively manipulating the host innate immune response.23,24
Host defence mechanisms for persistent infection
For many viruses, it has been shown that innate immune responses, most notably induction of type I and III IFNs, are the first line of defence limiting viral replication and spread, so contributing to the outcome of an infection. Given the importance of this defence, viruses have evolved numerous counteracting strategies including the blockade of IFN induction, interference with signalling triggered by IFNs, inhibition of the action of ISGs, or disturbance of the action of IFN-induced antiviral proteins.25,26
Hepatitis C virus and interferon
Interferon-α is an essential component of innate antiviral immunity and of treatment regimens for chronic HCV infection. Resistance to IFN might be important for HCV persistence and failure of IFN-based therapies. Several in vitro escape experiments led to the identification of HCV envelope mutations resulting in increased viral fitness and conferring IFN-α resistance.27 Hepatitis C virus can shut down the host IFN responses by using the viral NS3/4A protease to cleave MAVS/virus-induced signaling adapter28 and TRIF,29 two key adaptor molecules essential for interferon signalling activation. Recently, a study showed that NS4B can block the interaction of stimulator of interferon genes and Tank-binding kinase 1 to evade host innate immunity.30 A strong association with viral clearance was found for a single-nucleotide polymorphism (SNP) in the promoter/enhancer region of the IL28B gene (encoding IFN-λ3). The strongest association was found for SNP rs12979860, which is located about 3 kb upstream of the IL28B coding region.
Patients with a CC genotype at this SNP were more than twice as likely to achieve a sustained virological response as patients with a CT or TT genotype.31 Both the success rate of treatment and the rate of spontaneous virus clearance were shown to increase profoundly in patients with the C/C genotype.31–34 Recently, researchers discovered a new transiently induced region upstream of IFNL3 (IL28B) on chromosome 19q13.13 that harbours a dinucleotide variant ss469415590 (TT or ΔG) which is in high linkage disequilibrium with rs12979860, a genetic marker strongly associated with HCV clearance. Compared with rs12979860, ss469415590 is more strongly associated with HCV clearance in individuals of African ancestry, although it provides comparable information in Europeans and Asians.35 Moreover, the IFNL4 ss469415590 variant was identified as a better predictor than IFNL3 (IL28B) rs12979860 of pegylated IFN-α/ribavirin therapy failure in HCV/HIV-1 co-infected patients.36
HIV/hepatitis B virus and interferon
HIV persistence is linked with the ability of the virus to dysregulate and evade the innate immune response. The IRFs are transcription factors that play major roles in innate immunity. Rustagi and Gale reviewed the interplay between HIV-1 and IRF3.37 It was reported that Vpu plays a role in antagonism of IRF3 function.38 However, Vpu was later reported to inhibit the activation of the IFN-β promoter by blocking nuclear factor-κB signalling instead of IRF3.39 A frequent functional TLR7 polymorphism, TLR7 Gln11Leu, resulting in significantly less IFN-α production, has been associated with higher viral loads and accelerated disease progression to advanced immune suppression in HIV patients and may also be associated with increased HIV susceptibility.40 The expression of HBV polymerase (Pol) in human hepatic cell lines was also found to inhibit the induction of IFN-stimulated genes and to result in the weakened antiviral activity of IFN-α. This provided a possible molecular mechanism by which HBV resists IFN therapy and maintains its persistence.41 Kimkong et al.42 showed that the A allele of 1823G/A SNP within the IFNA1 gene was significantly associated with an increased risk of chronic HBV infection compared with healthy individuals and the self-limited HBV group. Furthermore, HBV induces the expression of interleukin-8 (IL-8), which in turn reduces HBV sensitivity to IFN-α.43 All HBV protein components can reportedly impair type I IFN transcription in different ways. The molecular mechanism underlying the HBV-induced suppression of type I IFN-mediated antiviral immunity was reviewed by Guo. 44
Receptors and ISGs
Numerous reviews have detailed the host immune system control of viral infection.45–47 The innate immune system constitutes the first line of defence against viruses, initiating an antiviral response. Viruses are recognized by this system primarily through detection of their nucleic acids, either their packaged genome or viral replication intermediates within the infected cell.48 Three classes of receptors, designated RIG-I-like receptors, TLRs and nucleotide oligomerization domain-like receptors, sense viral components such as double-stranded RNA, single-stranded RNA and DNA.49 These types of recognition induce the transcription of pro-inflammatory cytokines and type I IFNs, inducing the expression of hundreds of ISGs, which engage in counteracting virus replication and spread. However, viruses have evolved a fine-tuned mechanism to evade detection by the immune system or to interfere with the resulting signalling pathways as described previously.50–53
APOBEC3
The apolipoprotein B mRNA-editing enzyme catalytic polypeptide 3 (APOBEC3) subgroup plays an important role in the innate immune system, acting in host defence against exogenous viruses and endogenous retro-elements. The role of APOBEC3 proteins in the inhibition of viral infection was first described for HIV-1. However, in the past few years many studies have also shown evidence of APOBEC3 action on other viruses associated with human diseases, including HCV, HBV, human T-lymphotropic virus, human papillomavirus, herpes simplex virus-1, and Epstein–Barr virus. APOBEC3 inhibits these viruses through a series of editing-dependent and -independent mechanisms. However, many viruses have evolved mechanisms to counteract APOBEC effects.54
Autophagy
Autophagy is an evolutionarily conserved intracellular process by which bulk cytoplasm is enveloped inside a double-membraned vesicle and shuttled to lysosomes for degradation. Autophagy is essential for tissue homeostasis and development, and defective autophagy is associated with a number of diseases. With respect to their role in antiviral responses, the autophagy proteins function in targeting viral components or virions for lysosomal degradation in a process termed xenophagy, and they also play a role in the initiation of innate and adaptive immune system responses to viral infections. Consistent with this antiviral role of host autophagy, some viruses encode virulence factors that interact with the host autophagy machinery and block the execution of the process. In contrast, other viruses appear to use components of the autophagic machinery to foster their own intracellular growth or non-lytic cellular egress.55
Autophagy has virus-specific roles relating to viral replication, host innate and adaptive immune responses, virus-induced cell death programmes, and viral pathogenesis.56 Autophagy has been proposed as a protective mechanism against viral infection by degrading the pathogens within autolysosomes. This is strengthened by the fact that several proteins involved in IFN signalling pathways are linked to autophagy regulation. Again, several viruses have evolved strategies to divert IFN-mediated pathways and autophagy to their own benefit.57 A recent study showed that HCV perturbs mitochondrial dynamics by promoting mitochondrial fission followed by mitophagy, which attenuates HCV-induced apoptosis and contributes to persistent HCV infection.58
Interactions of immune cells
Dendritic cells
Chronic HBV or HCV infection is the result of an inadequate immune response towards the virus. Dendritic cells (DC) of patients with chronic HBV or HCV are impaired in their maturation and function, resulting in more tolerogenic rather than immunogenic responses, which may contribute to viral persistence. However, the experimental findings are controversial, some researchers finding no quantitative, phenotypic, or functional impairment of myeloid DC (mDC) or plasmacytoid DC (pDC) in chronic HBV or HCV.
It has been demonstrated that both HBV particles and purified HBsAg have an immune modulatory capacity and may directly contribute to the dysfunction of mDC in patients with chronic HBV.59 HBsAg inhibited the production of IFN-α by pDC through the induction of monocytes that secrete TNF-α and IL-10, and through the down-regulation of TLR9 expression on pDC.60 The direct immune regulatory effect of HBV and circulating HBsAg particles on the function of DC can be considered as part of the mechanism by which HBV escapes immunity.
Likewise, HCV inhibits cell surface expression of HLA-DR, prevents DC maturation, and induces IL-10 production. HCV can have direct and/or indirect inhibitory effects on antigen-presenting cells, resulting in reduction of antigen-specific T-cell activation.61 Studies have revealed that different HCV proteins used distinct mechanisms to down-regulate DC functions. Individual HCV proteins (Core, NS3, NS4, NS5) as well as fused polyprotein (Core-NS3-NS4) were found to impair functions of both immature DC and mature DC by regulating the expression of co-stimulatory and antigen presentation molecules, strikingly reducing IL-12 secretion, inducing the expression of Fas ligand to mediate apoptosis, interfering with allostimulatory capacity, inhibiting TLR signalling, and inhibiting nuclear translocation of nuclear factor-κB in DC.62 In addition, DC inhibition is connected to exhaustion of CD8+ T-cell functionality, including production of IFN-γ, IL-2, TNF-α and CD107a mobilization during chronic HCV infection.63 Myeloid DC from patients with chronic HCV infection expressed up-regulated levels of two inhibitory ligands, Fas ligand and the ligand 2 of programmed death-1 (PD-L2), compared with mDC from healthy people. The mDC of HCV patients had cytotoxic effects on autologous patient T cells and allogeneic healthy T cells. These results indicate that the cytotoxic activity of mDC is up-regulated to kill T cells during chronic HCV infection, which represents a novel mechanism of HCV immune evasion.64 All of these effects may account for or contribute to the low overall level of immunogenicity of HCV observed in chronically HCV-infected patients.
Dendritic cells are key regulators of the host response to HIV-1 infection. They are among the first cells to encounter HIV-1 at mucosal sites where they are co-opted by HIV-1 to facilitate transmission.65 The DC not only encounter the virus in the sexual mucosa, but, more importantly, they are also able to transfer the virus to its primary target cells, CD4+ T cells, either within the mucosa or after migration to lymph nodes. This results in explosive viral replication that is much greater than that resulting from direct infection of T cells.66 HIV-1 directly and indirectly modulates DC function to hinder the formation of effective antiviral immunity and fuel immune activation. HIV-1 evades innate immune sensing by mDC, resulting in suboptimal maturation and poor generation of antiviral adaptive responses and contributing to regulatory T (Treg) cell development.
Plasmacytoid DC interactions with HIV-1 are pleotropic, modulating immune responses on an axis between immunostimulatory and immunosuppressive. The pDC promote immune activation through an altered phenotype of persistent type I IFN secretion and weak antigen presentation capacity. Conversely, HIV-1 stimulates secretion of indoleamine 2,3 dioxygenase by pDC, resulting in Treg cell induction.67 This not only serves to blunt anti-HIV immune responses but also may dampen chronic immune activation. Dysfunction of DC in chronic persistent infectious disease is multifactorial, implicating a combination of viral and non-viral factors. We presume that viral persistence is the result of the host's insufficient antiviral immune responses and viral evasion.
Natural killer cells
Natural killer cells are important antiviral effectors of innate immunity because of their contribution to virus elimination via direct killing of infected cells and cytokine (IFN-γ and TNF-α) production; their regulation depends on a finely tuned balance between inhibitory and activating receptors. Recent research highlights the fact that NK cells are also regulatory cells engaged in reciprocal interactions with DC, macrophages, T cells and endothelial cells. Hence, NK cells can limit or exacerbate immune responses.68,69
In the last few years major progress has been made in better understanding the role of NK cells in HCV infection. This includes multiple pathways by which HCV impairs or limits NK cell activation. Many studies showed that chronic HCV infection is associated with dysfunctional or biased NK cell phenotypes. Compelling studies suggested that peripheral NK cells are activated during chronic HCV infection, most likely by IFN-α/β signalling. These activated peripheral NK cells display an increase in cytotoxicity with elevated expression of NKG2D, NKp46, and TNF-related apoptosis inducing ligand (TRAIL) and elevated activation of signal transducer and activator of transcription 1 (STAT1) along with a decrease in IFN-γ production.70,71 The elevated cytotoxicity level of NK cells may contribute to liver injury, whereas the decreased production of IFN-γ may facilitate the inability to clear HCV.72
Previous studies showed that the cross-linking of the E2 protein of HCV with CD81 on NK cells inhibits their activation, cytokine production, cytotoxic granule release and proliferation. These results implicate HCV-E2-mediated inhibition of NK cells as an efficient HCV evasion strategy targeting the early antiviral activities of NK cells and allowing the virus to establish itself as a chronic infection. Although the data are still controversial,73 studies suggested that genes encoding the inhibitory NK cell receptor KIR2DL3 and its human leucocyte antigen C group 1 (HLA-C1) ligand directly influence resolution of HCV infection.74 The specific combination of KIR2DL3 and HLA-C1 was protective against chronic HCV infection. Natural killer cells contribute to anti-HCV defence in vivo in the earliest stages of infection, providing innate protection from HCV acquisition.75 Genetic and clinical studies have suggested that NK cells not only play an important role in both spontaneous and IFN-α therapy-based HCV clearance, but may also contribute to hepatocellular damage in viral hepatitis.76,77
Like NK cells in HCV patients, NK cells in HBV patients enhance cytolytic activity and dysfunctional cytokine production, which may contribute to virus persistence78,79 and liver inflammation by TRAIL-mediated death of hepatocytes.80,81 Recent data have highlighted that NK cells are capable of exerting antiviral and immunoregulatory functions while also contributing to the pathogenesis of liver injury via death receptor pathways like a double-edged sword.82,83
Studies have suggested that NK cells can contribute to the control of HIV-1 infection through the recognition of virus-infected cells by both activating and inhibitory killer immunoglobulin-like receptors (KIRs) like KIR3DS1 and KIR3DL1.84,85 The NK cells expressing KIR3DS1 showed strong, significant dose- and cell contact-dependent inhibition of HIV-1 replication in target cells expressing HLA-B Bw4-80I compared with NK cells that did not express KIR3DS1.86 However, studies demonstrated that KIR+ NK cells can place immunological pressure on HIV-1 and that the virus can evade such NK cell-mediated immune pressure by selecting for sequence polymorphisms. Therefore, NK cells may play a role in contributing to viral evolution.87 Several key NK cell receptors have been associated with HIV-1 disease progression and/or transmission, implying that NK cells might contribute markedly to the control of HIV-1 infection88 while HIV-1-encoded proteins can facilitate evasion from NK cell recognition.89
Regulatory T cells
Surviving an infection requires the generation of an immune response that controls the invading pathogen while limiting collateral damage to self tissues that may result from an exuberant immune response. Various populations of regulatory cells, including CD4+ CD25+ Foxp3+ Treg cells, have been shown to play a central role in the establishment of these controlled immune responses. Treg cells are involved in the control of immune-tolerance by regulating immune homeostasis and limiting immune activation. Defects in Treg cell numbers or function have been related to the development of human autoimmune diseases, while increases in Treg cell numbers or activity could limit anti-tumour immune responses.90
In contrast to these two scenarios, a much more complex picture emerges for the role of Treg cells in infectious diseases. Treg-cell-mediated inhibition of antimicrobial immune responses could lead to ineffective clearance of the pathogen contributing to the chronicity of the infection. Conversely, Treg cells participate in terminating immune responses, so preventing exacerbated and potentially harmful immune activation and immune-mediated injury. The balance between regulatory T cells and effector immune functions influences the outcome of host–virus coexistence and appears tuned to maintain immune homeostasis.91
HIV infection is associated with a progressive CD4+ T-cell lymphopenia, and defective HIV-specific CD8+ T-cell responses are known to play a key role in the control of viral replication. Persistent immune activation is a hallmark of HIV infection and is involved in disease progression independent of viral load. The consequences of Treg cell expansion, observed in HIV infection, could be either beneficial by suppressing generalized T-cell activation or detrimental by weakening HIV-specific responses and so contributing to viral persistence. The resulting balance between these contrasting outcomes might have critical implications in pathogenesis.92,93 Treg cell heterogeneity analysis in the context of HIV infection was reviewed by Simonetta and Bourgeois.94 They discussed how the identification of naive and effector Treg cell subsets modulates understanding of Treg cell biology during HIV infection and the potential impact of HIV infection on mechanisms governing peripheral differentiation of adaptive Treg cells.
Treg cells also appear to play a role in the control of chronic viral hepatitis.95 Patients with either chronic HBV or HCV infections have accumulated circulating Treg cells that express CD45RO, high intracellular cytotoxic T-lymphocyte antigen 4 (CTLA-4) and PD-1 and which have the capacity to inhibit proliferation of both CD4+ and CD8+ T cells as well as T helper type 1 (Th1) cytokine production. An increased number of circulating Treg lymphocytes has been shown in patients with chronic HBV infection when compared with those who have cleared HBV.96 Similar findings have been found in chronic HCV infection when compared with healthy controls.97 Of particular interest is that viral load and the number of Treg cells are closely related. Therefore, chronic HBV or HCV patients harbour an increased percentage of Treg cells in peripheral blood compared with controls. Treg cells have an immunosuppressive effect on HBV/HCV-specific T helper cells. The presence of Treg cells could contribute to an inadequate immune response against the virus, leading to chronic infection. Treg cells generated in chronic viral infection may be part of a normal host immune response to infection with a natural evolution from pro-inflammatory responses to immune regulation, where neither are related directly to virus infection and where the benefit would be in preventing uncontrolled tissue destruction. Treg cells could, in that situation, be regarded as protective, with viral persistence and less liver damage, a by-product of that protection. However, viral induction of the Treg cell response favours persistent infection and survival of the organism. One could speculate that the hepatitis viruses skew the natural immune response towards regulation rather than inflammation as part of evolutionary development.95 One consequence of the modulation of excessive immune responses and control of immunopathology by Treg cells is enhanced pathogen survival and possibly long-term viral persistence.
Hence, pathogen persistence may represent a compromise reached between the pathogen and the host immune response. Activation of Treg cells may contribute to the maintenance of immunity to chronic infections in which pathogen persistence is required for the development of memory. A balance between effector and regulatory mechanisms may determine the outcome of an infection, and this may, in some cases, be beneficial for both the host and the pathogen.98,99 In conclusion, Treg cells contribute to the viral persistence and maintain immune homeostasis.
Regulatory B cells
Emerging significant evidence indicates that B cells can actively modulate immune responses through mechanisms that do not directly involve the production of antibodies. B cells appear to have the capacity to both induce and suppress immune effector mechanisms, and they exert these functions both by contact-dependent interactions and through the secretion of cytokines. These B cells are functionally defined ‘regulatory B cells’, or Breg cells.100 Breg cells may act earlier than Treg cells and may play an important role in autoimmune and allergic diseases. The Breg subtypes may be homologous to the Treg subtypes (Br1 cells expressing IL-10, Br3 cells expressing transforming growth factor-β, and B-Foxp3 cells), although the Br1 subtype seems to predominate. Nevertheless, differences with Treg cells may exist: Breg cell activation may chiefly involve the TLRs rather than the antigen receptor, and Breg cells act earlier, facilitating the recruitment of Treg cells and then disappearing once the Treg cells become operational.101,102 In autoimmunity, tumour immunology, and some viral infections, Breg cells modulate T-cell function via IL-10 production. Studies indicate that Breg cells contribute to HIV infection-associated immune dysfunction by T-cell impairment via IL-10 and possibly PD-L1 expression.103
Interactions of cytokines and regulatory molecules
Programmed death-1
Viral infection may modulate many molecules to impair specific cytotoxic T lymphocyte reactivity through induction of either a virus-associated tolerogenic-like state or apoptosis. Virus-specific CD8+ T-cell exhaustion may represent a mechanism of viral persistence. One key pathway is the inhibitory receptor programmed death-1 (PD-1) binding to one of its ligands, PD-L1 or PD-L2, transmitting a negative signal to the T cells expressing PD-1, reducing cytokine production and proliferation, and regulating the balance between T-cell activation, tolerance and immunopathology.104 PD-1 has been reported to be up-regulated in exhausted CD8+ T cells during chronic viral infections, such as HBV, HCV and HIV. Expression of PD-1, which is driven by viral replication and associated with T-cell dysfunction, was also up-regulated on virus-specific CD4+ T cells.105 Expression of PD-1/PD-L1 was also up-regulated in monocytes/macrophages in HCV-infected subjects compared with HCV-resolved or healthy subjects. Up-regulation of PD-1 was inversely associated with the degree of IL-12 inhibition in HCV infection.106 Our research data suggested that HCV core/gC1qR engagement on monocytes/macrophages triggers the expression of PD-1 and suppressor of cytokine signalling 1 (SOCS-1), which can associate to deliver negative signalling to TLR-mediated pathways controlling expression of IL-12, a key cytokine linking innate and adaptive immunity.107 The inhibitory immunoregulatory receptor, CTLA-4, was also up-regulated in HIV-specific CD4+ T cells but not CD8+ T cells in all categories of HIV-infected subjects.108
Blocking PD-1/PD-L1 interaction with an anti-PD-L1 antibody improved the capacity of expansion of virus-specific CD4+ CD8+ T cells. However, highly PD-1-positive intrahepatic CD8+ T cells were more phenotypically exhausted with increased CTLA-4 and reduced CD28 and CD127 expression, suggesting that active antigen-specific stimulation in the liver induces a profound functional exhaustion not reversible by PD-1/PD-L blockade alone.109 A PD-1+ CD127− phenotype associated with anergic features and apoptosis in chronic HCV patients has been identified.110 In the case of CD4+ T cells, blockade of PD-L1/2, IL-10 and transforming growth factor-β1 increased expansion of CD4+ T cells in patients with chronic HCV, restoring HCV-specific production of IL-2, IFN-γ and TNF-α.111
Likewise, intrahepatic HBV-specific CD8+ T cells expressed higher levels of PD-1 and lower levels of CD127 than their peripheral counterparts. Blockade of PD-1/PD-L1 interaction increased CD8+ T-cell proliferation, IFN-β and IL-2 production by circulating intrahepatic lymphocytes, even though anti-PD-L1 had a stronger effect on intrahepatic compared with peripheral T cells.112 Anti-PD-L1 might be a good therapeutic candidate for chronic viral infection.
Tim-3
Notably, not all dysfunctional T cells express PD-1, nor are they all rescued by blockade of the PD-1/PD-1 ligand pathway. The expression of T-cell immunoglobulin and mucin domain-containing protein 3 (Tim-3) is increased on CD4+ and CD8+ T cells in chronic virus infections such as HBV, HCV and HIV. Tim-3 expression correlates with a dysfunctional and senescent phenotype (CD127low CD57high), a central rather than effector memory profile (CD45RAnegative CCR7high), and reduced Th1/Tc1 cytokine production.113 Up-regulation of Tim-3 and inhibition of IL-12 are also observed in monocytes/macrophages incubated with HCV-expressing hepatocytes, as well as in primary monocytes/macrophages or monocytic THP-1 cells incubated with HCV core protein. Tim-3 blockade reduces HCV core-mediated expression of the negative immunoregulators PD-1 and SOCS-1 and increases STAT-1 phosphorylation. Conversely, blocking PD-1 or silencing SOCS-1 gene expression also decreases Tim-3 expression and enhances IL-12 secretion and STAT-1 phosphorylation.114
The role of PD-1 and Tim-3 expression in T-cell exhaustion during HCV/HIV co-infection has been investigated. HCV-specific T cells in HCV/HIV co-infection show elevated frequencies of dual Tim-3/PD-1 expression that correlate with liver disease progression.115 Several markers of immune exhaustion such as PD-1, Lymphocyte Activation Gene-3, Tim-3 and CTLA-4, which are also negative regulators of immune activation, are preferentially up-regulated on T cells during HIV infection,116 and in vitro blockade of PD-1 and Tim-3 restores specific T-cell responses.
In addition, the Tim-3 pathway appears to control Treg cell and effector T (Teff) cell balance by altering cell proliferation and apoptosis during HCV infection.117 Blockade of Tim-3 on CD4+ CD25+ T cells promoted expansion of Teff cells more substantially than Treg cells through improved STAT-5 signalling, so correcting the imbalance of Foxp3+ Treg Foxp3− Teff cells that was induced by HCV infection. Beyond T cells, there was a significant increase of Tim-3 expression in peripheral blood mononuclear cells, circulating NK cells, and liver infiltrating lymphocytes from chronic HBV patients compared with healthy controls, which may in turn suppress NK cell functions in chronic HBV patients.118
NKG2A
With regard to NK cells, expression levels of NKG2A were up-regulated on NK cells from individuals with chronic HCV. Treatment with pegylated IFN and ribavirin resulted in a down-regulation of NKG2A expression on CD56dim NK cells whereas individuals with a sustained virological response had greater numbers of NKG2A-positive, KIR-negative NK cells than those without sustained virological response.119 In addition, infection with HBV increases levels of the inhibitory receptor NKG2A on NK cells in humans and reduces their ability to clear HBV. Reagents designed to block the interaction between NKG2A and HLA-E in peripheral NK cells from patients with active chronic HBV increased their cytotoxicity in vitro.120 Higher NKG2A expression levels in the cytotoxic NK subset were found in the later stages of HIV infection compared with patients at an early stage of infection. A reverse association between the percentage of NKG2A-positive cells in the cytotoxic NK subset and the CD4 cell count was observed in all HIV-1-infected groups.121 NKG2A seems to be a target of viral persistence and a modulator of NK immune activation.
KLRG1
We demonstrated that killer cell lectin-like receptor subfamily G member 1 (KLRG1), a transmembrane protein preferentially expressed on T cells, is highly expressed on CD56+ NK cells, which are significantly reduced in their numbers and functions in the peripheral blood of patients with chronic HCV infection compared with subjects without infection. Importantly, blockade of KLRG1 signalling significantly recovered the impaired IFN-γ production by NK cells from HCV-infected subjects.122 We also demonstrated that KLRG1 was over-expressed on CD4+ T cells from HCV-infected, HBV vaccine non-responders compared with HBV vaccine responders. These results may partly explain why HBV vaccine responses in HCV-infected individuals are often blunted when compared with uninfected populations.123
Interleukin-17
Interleukin-17A (also called IL-17) is the prototypic cytokine of the IL-17 family, which includes six members: IL-17A, B, C, D, E and F. IL-17A and IL-17F have similar functions: they induce the production of pro-inflammatory cytokines, chemokines and metalloproteinases from various tissues and cell types. Researchers found that peripheral blood Th17 cell levels in patients with chronic hepatitis B were significantly higher than those in the healthy controls associated with elevated serum levels of IL-17A.124,125 Increased IL-17-secreting cells in the liver and peripheral Th17 cells among patients with chronic hepatitis B are associated with greater fibrosis and higher degrees of inflammation compared with healthy controls.126 In vitro, IL-17 together with IL-17-activated monocytes were able to promote the activation of stellate cells, which in turn exacerbates liver fibrosis and the inflammatory response.127 Interleukin-17-mediating liver neutrophil recruitment via induction of IL-8 may be one potential mechanism of liver injury in patients with chronic HBV.128
Both Th17 frequency and plasma IL-17A levels inversely correlated with viral load in chronic HBV infected patients.129 Wang et al. demonstrated that IL-17A effectively suppressed HBV replication in a non-cytopathic manner, and the over-expression of myxovirus resistance A and oligoadenylatesynthetase mRNA was involved in the suppression of HBV replication by IL-17A.130 All these findings suggest that IL-17 participates in the liver injury process and viral clearance after HBV infection, which seems paradoxical. Other researchers found that the frequency of Th17 cells display no correlation with serum HBV DNA loads or alanine aminotransferase levels, although the frequency of Th17 cells in peripheral blood was significantly higher in paediatric patients with CHB compared with healthy controls.131
Serum IL-17 levels are higher in chronic HCV-infected patients, and HCV-specific Th17 cells correlated with liver damage but not HCV viral replication.132 However, novel intriguing data indicate that a Th17 boost could be associated with spontaneous HCV clearance.133 It is possible that Th17 could play a dual role (both beneficial and harmful) and that an imbalance of regulating factors (chemokines, transcription factors, receptor expression) rather than the lymphocyte itself could tip the Th17 immune response one way or the other. The role of Th17 cells in host anti-HCV defence is beginning to emerge, and one has to focus upon its potential beneficial aspects and not only on its destructive potential.134
During the course of HIV infection, Th17 cells are lost very early and their loss has been shown to correlate with bacterial translocation.135 Interleukin-17 production was significantly lower in patients with detectable plasma viraemia when compared with successfully treated HIV-infected patients and healthy controls.136 A significant negative correlation between virus-specific Th17 cells and HIV-1 plasma viral load (pVL) indicates a gradual loss of Th17 cells with HIV-1 disease progression.137 The IL-17 production in HIV-infected patients could be recovered through a sustained suppression of viral replication in the peripheral blood through combined anti-retroviral therapy. When Th17 differentiation cytokines were given along with lamivudine to the cultures with established HIV infection, a Th17 response was fully restored and virus replication was suppressed.136,138,139 All these findings indicate the potential use of immunotherapeutic modalities to supplement anti-retroviral drugs for restoring Th17 response in chronically infected patients.
In summary, diverse regulatory mechanisms cooperate to maintain immune homeostasis, and a breakdown in these pathways may facilitate viral persistence. Use of immunotherapeutic modalities to supplement anti-retroviral drugs may contribute to immune homeostasis and prevent viral damage to the host in chronically infected patients.
Acknowledgments
This study was supported by grants from the Natural Science Foundation of China (NO. 31170877) and the Shaanxi Provincial Science and Technology Management Program (NO. 2012KTCL03-19). Yun Zhou, a joint PhD student, was supported partly by the China Scholarship Council (CSC 201306590012).
Disclosures
No conflicting financial interests exist.
References
- 1.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 2.Harrison OJ, Maloy KJ. Innate immune activation in intestinal homeostasis. J Innate Immun. 2011;3:585–93. doi: 10.1159/000330913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Helle F, Dubuisson J. Hepatitis C virus entry into host cells. Cell Mol Life Sci. 2008;65:100–12. doi: 10.1007/s00018-007-7291-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.von Hahn T, Rice CM. Hepatitis C virus entry. J Biol Chem. 2008;283:3689–93. doi: 10.1074/jbc.R700024200. [DOI] [PubMed] [Google Scholar]
- 5.Fofana I, Fafi-Kremer S, Carolla P, et al. Mutations that alter use of hepatitis C virus cell entry factors mediate escape from neutralizing antibodies. Gastroenterology. 2012;143:223–33. doi: 10.1053/j.gastro.2012.04.006. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keck ZY, Li SH, Xia J, et al. Mutations in hepatitis C virus E2 located outside the CD81 binding sites lead to escape from broadly neutralizing antibodies but compromise virus infectivity. J Virol. 2009;83:6149–60. doi: 10.1128/JVI.00248-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim H, Ray R. Evasion of TNF-α-mediated apoptosis by hepatitis C virus. Methods Mol Biol. 2014;1155:125–32. doi: 10.1007/978-1-4939-0669-7_11. [DOI] [PubMed] [Google Scholar]
- 8.Bode JG, Brenndorfer ED, Haussinger D. Subversion of innate host antiviral strategies by the hepatitis C virus. Arch Biochem Biophys. 2007;462:254–65. doi: 10.1016/j.abb.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 9.Horner SM. Activation and evasion of antiviral innate immunity by hepatitis C virus. J Mol Biol. 2014;426:1198–209. doi: 10.1016/j.jmb.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimakami T, Yamane D, Jangra RK, Kempf BJ, Spaniel C, Barton DJ, Lemon SM. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc Natl Acad Sci USA. 2012;109:941–6. doi: 10.1073/pnas.1112263109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Chen J, Wang H, Shi J, Wu K, Liu S, Liu Y, Wu J. HCV-induced miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1. PLoS Pathog. 2013;9:e1003248. doi: 10.1371/journal.ppat.1003248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siliciano RF, Greene WC. HIV latency. Cold Spring Harb Perspect Med. 2011;1:a007096. doi: 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donahue DA, Wainberg MA. Cellular and molecular mechanisms involved in the establishment of HIV-1 latency. Retrovirology. 2013;10:11. doi: 10.1186/1742-4690-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Battistini A, Sgarbanti M. HIV-1 latency: an update of molecular mechanisms and therapeutic strategies. Viruses. 2014;6:1715–58. doi: 10.3390/v6041715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Briz V, Poveda E, Soriano V. HIV entry inhibitors: mechanisms of action and resistance pathways. J Antimicrob Chemother. 2006;57:619–27. doi: 10.1093/jac/dkl027. [DOI] [PubMed] [Google Scholar]
- 16.Kamp W, Berk MB, Visser CJ, Nottet HS. Mechanisms of HIV-1 to escape from the host immune surveillance. Eur J Clin Invest. 2000;30:740–6. doi: 10.1046/j.1365-2362.2000.00697.x. [DOI] [PubMed] [Google Scholar]
- 17.Buzon MJ, Sun H, Li C, et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat Med. 2014;20:139–42. doi: 10.1038/nm.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glebe D, Bremer CM. The molecular virology of hepatitis B virus. Semin Liver Dis. 2013;33:103–12. doi: 10.1055/s-0033-1345717. [DOI] [PubMed] [Google Scholar]
- 19.Purdy MA. Hepatitis B virus S gene escape mutants. Asian J Transfus Sci. 2007;1:62–70. doi: 10.4103/0973-6247.33445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Li Y, Mao A, Li C, Tien P. Hepatitis B virus X protein suppresses virus-triggered IRF3 activation and IFN-β induction by disrupting the VISA-associated complex. Cell Mol Immunol. 2010;7:341–8. doi: 10.1038/cmi.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang J, Tang H. Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell. 2010;1:1106–17. doi: 10.1007/s13238-010-0141-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Ryu WS. Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: implications for immune evasion. PLoS Pathog. 2010;6:e1000986. doi: 10.1371/journal.ppat.1000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Revill P, Yuan Z. New insights into how HBV manipulates the innate immune response to establish acute and persistent infection. Antivir Ther. 2013;18:1–15. doi: 10.3851/IMP2542. [DOI] [PubMed] [Google Scholar]
- 24.Han Q, Zhang C, Zhang J, Tian Z. The role of innate immunity in HBV infection. Semin Immunopathol. 2013;35:23–38. doi: 10.1007/s00281-012-0331-y. [DOI] [PubMed] [Google Scholar]
- 25.Haller O, Weber F. Pathogenic viruses: smart manipulators of the interferon system. Curr Top Microbiol Immunol. 2007;316:315–34. doi: 10.1007/978-3-540-71329-6_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008;89(Pt 1):1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 27.Serre SB, Krarup HB, Bukh J, Gottwein JM. Identification of IFN-α induced envelope mutations of hepatitis C virus in vitro associated with increased viral fitness and interferon resistance. J Virol. 2013;87:12776–93. doi: 10.1128/JVI.00901-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA. 2005;102:17717–22. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li K, Foy E, Ferreon JC, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA. 2005;102:2992–7. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding Q, Cao X, Lu J, Huang B, Liu YJ, Kato N, Shu HB, Zhong J. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J Hepatol. 2013;59:52–8. doi: 10.1016/j.jhep.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 31.Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 32.Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-α and ribavirin therapy. Nat Genet. 2009;41:1100–4. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-α and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–9. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 34.Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013;45:164–71. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franco S, Aparicio E, Parera M, Clotet B, Tural C, Martinez MA. IFNL4 ss469415590 variant is a better predictor than ILF3 (IL28B) rs12979860 of pegylated interferon-α/ribavirin therapy failure in hepatitis C virus/HIV-1 coinfected patients. AIDS. 2014;28:133–6. doi: 10.1097/QAD.0000000000000052. [DOI] [PubMed] [Google Scholar]
- 37.Rustagi A, Gale M., Jr Innate antiviral immune signaling, viral evasion and modulation by HIV-1. J Mol Biol. 2014;426:1161–77. doi: 10.1016/j.jmb.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doehle BP, Chang K, Rustagi A, McNevin J, McElrath MJ, Gale M., Jr Vpu mediates depletion of interferon regulatory factor 3 during HIV infection by a lysosome-dependent mechanism. J Virol. 2012;86:8367–74. doi: 10.1128/JVI.00423-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hotter D, Kirchhoff F, Sauter D. HIV-1 Vpu does not degrade interferon regulatory factor 3. J Virol. 2013;87:7160–5. doi: 10.1128/JVI.00526-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oh DY, Baumann K, Hamouda O, et al. A frequent functional toll-like receptor 7 polymorphism is associated with accelerated HIV-1 disease progression. AIDS. 2009;23:297–307. doi: 10.1097/QAD.0b013e32831fb540. [DOI] [PubMed] [Google Scholar]
- 41.Chen J, Wu M, Zhang X, et al. Hepatitis B virus polymerase impairs interferon-α-induced STA T activation through inhibition of importin-alpha5 and protein kinase C-δ. Hepatology. 2013;57:470–82. doi: 10.1002/hep.26064. [DOI] [PubMed] [Google Scholar]
- 42.Kimkong I, Tangkijvanich P, Hirankarn N. Association of interferon-α gene polymorphisms with chronic hepatitis B virus infection. Int J Immunogenet. 2013;40:476–81. doi: 10.1111/iji.12055. [DOI] [PubMed] [Google Scholar]
- 43.Pollicino T, Bellinghieri L, Restuccia A, Raffa G, Musolino C, Alibrandi A, Teti D, Raimondo G. Hepatitis B virus (HBV) induces the expression of interleukin-8 that in turn reduces HBV sensitivity to interferon-α. Virology. 2013;444:317–28. doi: 10.1016/j.virol.2013.06.028. [DOI] [PubMed] [Google Scholar]
- 44.Guo P. Suppression of interferon-mediated antiviral immunity by hepatitis B virus: an overview of research progress. Scand J Immunol. 2013;78:230–7. doi: 10.1111/sji.12086. [DOI] [PubMed] [Google Scholar]
- 45.Thimme R, Binder M, Bartenschlager R. Failure of innate and adaptive immune responses in controlling hepatitis C virus infection. FEMS Microbiol Rev. 2012;36:663–83. doi: 10.1111/j.1574-6976.2011.00319.x. [DOI] [PubMed] [Google Scholar]
- 46.Quaranta MG, Mattioli B, Vella S. Glances in immunology of HIV and HCV infection. Adv Virol. 2012;2012:434036. doi: 10.1155/2012/434036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mogensen TH, Melchjorsen J, Larsen CS, Paludan SR. Innate immune recognition and activation during HIV infection. Retrovirology. 2010;7:54. doi: 10.1186/1742-4690-7-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iwasaki A. A virological view of innate immune recognition. Annu Rev Microbiol. 2012;66:177–96. doi: 10.1146/annurev-micro-092611-150203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–81. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 51.Bonjardim CA, Ferreira PC, Kroon EG. Interferons: signaling, antiviral and viral evasion. Immunol Lett. 2009;122:1–11. doi: 10.1016/j.imlet.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pichlmair A, Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27:370–83. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 53.Schroder M, Bowie AG. An arms race: innate antiviral responses and counteracting viral strategies. Biochem Soc Trans. 2007;35(Pt 6):1512–4. doi: 10.1042/BST0351512. [DOI] [PubMed] [Google Scholar]
- 54.Vieira VC, Soares MA. The role of cytidine deaminases on innate immune responses against human viral infections. Biomed Res Int. 2013;2013:683095. doi: 10.1155/2013/683095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kudchodkar SB, Levine B. Viruses and autophagy. Rev Med Virol. 2009;19:359–78. doi: 10.1002/rmv.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin LT, Dawson PW, Richardson CD. Viral interactions with macroautophagy: a double-edged sword. Virology. 2010;402:1–10. doi: 10.1016/j.virol.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Espert L, Codogno P, Biard-Piechaczyk M. Involvement of autophagy in viral infections: antiviral function and subversion by viruses. J Mol Med (Berl) 2007;85:811–23. doi: 10.1007/s00109-007-0173-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim SJ, Syed GH, Khan M, Chiu WW, Sohail MA, Gish RG, Siddiqui A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci USA. 2014;111:6413–8. doi: 10.1073/pnas.1321114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Op den Brouw ML, Binda RS, van Roosmalen MH, Protzer U, Janssen HL, van der Molen RG, Woltman AM. Hepatitis B virus surface antigen impairs myeloid dendritic cell function: a possible immune escape mechanism of hepatitis B virus. Immunology. 2009;126:280–9. doi: 10.1111/j.1365-2567.2008.02896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shi B, Ren G, Hu Y, Wang S, Zhang Z, Yuan Z. HBsAg inhibits IFN-α production in plasmacytoid dendritic cells through TNF-α and IL-10 induction in monocytes. PLoS ONE. 2012;7:e44900. doi: 10.1371/journal.pone.0044900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saito K, Ait-Goughoulte M, Truscott SM, et al. Hepatitis C virus inhibits cell surface expression of HLA-DR, prevents dendritic cell maturation, and induces interleukin-10 production. J Virol. 2008;82:3320–8. doi: 10.1128/JVI.02547-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krishnadas DK, Ahn JS, Han J, Kumar R, Agrawal B. Immunomodulation by hepatitis C virus-derived proteins: targeting human dendritic cells by multiple mechanisms. Int Immunol. 2010;22:491–502. doi: 10.1093/intimm/dxq033. [DOI] [PubMed] [Google Scholar]
- 63.Rodrigue-Gervais IG, Rigsby H, Jouan L, Sauve D, Sekaly RP, Willems B, Lamarre D. Dendritic cell inhibition is connected to exhaustion of CD8+ T cell polyfunctionality during chronic hepatitis C virus infection. J Immunol. 2010;184:3134–44. doi: 10.4049/jimmunol.0902522. [DOI] [PubMed] [Google Scholar]
- 64.Zhao L, Tyrrell DL. Myeloid dendritic cells can kill T cells during chronic hepatitis C virus infection. Viral Immunol. 2013;26:25–39. doi: 10.1089/vim.2012.0058. [DOI] [PubMed] [Google Scholar]
- 65.Borrow P. Innate immunity in acute HIV-1 infection. Curr Opin HIV AIDS. 2011;6:353–63. doi: 10.1097/COH.0b013e3283495996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harman AN, Kim M, Nasr N, Sandgren KJ, Cameron PU. Tissue dendritic cells as portals for HIV entry. Rev Med Virol. 2013;23:319–33. doi: 10.1002/rmv.1753. [DOI] [PubMed] [Google Scholar]
- 67.Miller E, Bhardwaj N. Dendritic cell dysregulation during HIV-1 infection. Immunol Rev. 2013;254:170–89. doi: 10.1111/imr.12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–10. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 69.Caligiuri MA. Human natural killer cells. Blood. 2008;112:461–9. doi: 10.1182/blood-2007-09-077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Edlich B, Ahlenstiel G, Zabaleta Azpiroz A, et al. Early changes in interferon signaling define natural killer cell response and refractoriness to interferon-based therapy of hepatitis C patients. Hepatology. 2012;55:39–48. doi: 10.1002/hep.24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Golden-Mason L, Stone AE, Bambha KM, Cheng L, Rosen HR. Race- and gender-related variation in natural killer p46 expression associated with differential anti-hepatitis C virus immunity. Hepatology. 2012;56:1214–22. doi: 10.1002/hep.25771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ahlenstiel G. The natural killer cell response to HCV infection. Immune Netw. 2013;13:168–76. doi: 10.4110/in.2013.13.5.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Szabo G, Chang S, Dolganiuc A. Altered innate immunity in chronic hepatitis C infection: cause or effect? Hepatology. 2007;46:1279–90. doi: 10.1002/hep.21938. [DOI] [PubMed] [Google Scholar]
- 74.Khakoo SI, Thio CL, Martin MP, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science. 2004;305:872–4. doi: 10.1126/science.1097670. [DOI] [PubMed] [Google Scholar]
- 75.Golden-Mason L, Cox AL, Randall JA, Cheng L, Rosen HR. Increased natural killer cell cytotoxicity and NKp30 expression protects against hepatitis C virus infection in high-risk individuals and inhibits replication in vitro. Hepatology. 2010;52:1581–9. doi: 10.1002/hep.23896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cheent K, Khakoo SI. Natural killer cells and hepatitis C: action and reaction. Gut. 2011;60:268–78. doi: 10.1136/gut.2010.212555. [DOI] [PubMed] [Google Scholar]
- 77.Bozzano F, Marras F, Biassoni R, De Maria A. Natural killer cells in hepatitis C virus infection. Expert Rev Clin Immunol. 2012;8:775–88. doi: 10.1586/eci.12.71. [DOI] [PubMed] [Google Scholar]
- 78.Oliviero B, Varchetta S, Paudice E, et al. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology. 2009;137:1151–60. doi: 10.1053/j.gastro.2009.05.047. e1–7. [DOI] [PubMed] [Google Scholar]
- 79.Mondelli MU, Varchetta S, Oliviero B. Natural killer cells in viral hepatitis: facts and controversies. Eur J Clin Invest. 2010;40:851–63. doi: 10.1111/j.1365-2362.2010.02332.x. [DOI] [PubMed] [Google Scholar]
- 80.Dunn C, Brunetto M, Reynolds G, et al. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J Exp Med. 2007;204:667–80. doi: 10.1084/jem.20061287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Z, Zhang S, Zou Z, et al. Hypercytolytic activity of hepatic natural killer cells correlates with liver injury in chronic hepatitis B patients. Hepatology. 2011;53:73–85. doi: 10.1002/hep.23977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maini MK, Peppa D. NK cells: a double-edged sword in chronic hepatitis B virus infection. Front Immunol. 2013;4:57. doi: 10.3389/fimmu.2013.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Busca A, Kumar A. Innate immune responses in hepatitis B virus (HBV) infection. Virol J. 2014;11:22. doi: 10.1186/1743-422X-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martin MP, Qi Y, Gao X, et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet. 2007;39:733–40. doi: 10.1038/ng2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martin MP, Gao X, Lee JH, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet. 2002;31:429–34. doi: 10.1038/ng934. [DOI] [PubMed] [Google Scholar]
- 86.Alter G, Martin MP, Teigen N, et al. Differential natural killer cell-mediated inhibition of HIV-1 replication based on distinct KIR/HLA subtypes. J Exp Med. 2007;204:3027–36. doi: 10.1084/jem.20070695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alter G, Heckerman D, Schneidewind A, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature. 2011;476:96–100. doi: 10.1038/nature10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jost S, Altfeld M. Control of human viral infections by natural killer cells. Annu Rev Immunol. 2013;31:163–94. doi: 10.1146/annurev-immunol-032712-100001. [DOI] [PubMed] [Google Scholar]
- 89.Jost S, Altfeld M. Evasion from NK cell-mediated immune responses by HIV-1. Microbes Infect. 2012;14:904–15. doi: 10.1016/j.micinf.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Belkaid Y. Role of Foxp3-positive regulatory T cells during infection. Eur J Immunol. 2008;38:918–21. doi: 10.1002/eji.200738120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Belkaid Y, Tarbell K. Regulatory T cells in the control of host-microorganism interactions (*) Annu Rev Immunol. 2009;27:551–89. doi: 10.1146/annurev.immunol.021908.132723. [DOI] [PubMed] [Google Scholar]
- 92.Chevalier MF, Weiss L. The split personality of regulatory T cells in HIV infection. Blood. 2013;121:29–37. doi: 10.1182/blood-2012-07-409755. [DOI] [PubMed] [Google Scholar]
- 93.Imamichi H, Lane HC. Regulatory T cells in HIV-1 infection: the good, the bad, and the ugly. J Infect Dis. 2012;205:1479–82. doi: 10.1093/infdis/jis238. [DOI] [PubMed] [Google Scholar]
- 94.Simonetta F, Bourgeois C. CD4+ FOXP3+ Regulatory T-Cell Subsets in Human Immunodeficiency Virus Infection. Front Immunol. 2013;4:215. doi: 10.3389/fimmu.2013.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rushbrook SM, Hoare M, Alexander GJ. T-regulatory lymphocytes and chronic viral hepatitis. Expert Opin Biol Ther. 2007;7:1689–703. doi: 10.1517/14712598.7.11.1689. [DOI] [PubMed] [Google Scholar]
- 96.Stoop JN, van der Molen RG, Baan CC, van der Laan LJ, Kuipers EJ, Kusters JG, Janssen HL. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology. 2005;41:771–8. doi: 10.1002/hep.20649. [DOI] [PubMed] [Google Scholar]
- 97.Cabrera R, Tu Z, Xu Y, Firpi RJ, Rosen HR, Liu C, Nelson DR. An immunomodulatory role for CD4+ CD25+ regulatory T lymphocytes in hepatitis C virus infection. Hepatology. 2004;40:1062–71. doi: 10.1002/hep.20454. [DOI] [PubMed] [Google Scholar]
- 98.Belkaid Y, Rouse BT. Natural regulatory T cells in infectious disease. Nat Immunol. 2005;6:353–60. doi: 10.1038/ni1181. [DOI] [PubMed] [Google Scholar]
- 99.Vahlenkamp TW, Tompkins MB, Tompkins WA. The role of CD4+CD25+ regulatory T cells in viral infections. Vet Immunol Immunopathol. 2005;108:219–25. doi: 10.1016/j.vetimm.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 100.Vitale G, Mion F, Pucillo C. Regulatory B cells: evidence, developmental origin and population diversity. Mol Immunol. 2010;48:1–8. doi: 10.1016/j.molimm.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 101.Berthelot JM, Jamin C, Amrouche K, Le Goff B, Maugars Y, Youinou P. Regulatory B cells play a key role in immune system balance. Joint Bone Spine. 2013;80:18–22. doi: 10.1016/j.jbspin.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 102.Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221–41. doi: 10.1146/annurev-immunol-020711-074934. [DOI] [PubMed] [Google Scholar]
- 103.Siewe B, Stapleton JT, Martinson J, Keshavarzian A, Kazmi N, Demarais PM, French AL, Landay A. Regulatory B cell frequency correlates with markers of HIV disease progression and attenuates anti-HIV CD8+ T cell function in vitro. J Leukoc Biol. 2013;93:811–8. doi: 10.1189/jlb.0912436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.D'Souza M, Fontenot AP, Mack DG, et al. Programmed death 1 expression on HIV-specific CD4+ T cells is driven by viral replication and associated with T cell dysfunction. J Immunol. 2007;179:1979–87. doi: 10.4049/jimmunol.179.3.1979. [DOI] [PubMed] [Google Scholar]
- 106.Ma CJ, Ni L, Zhang Y, et al. PD-1 negatively regulates interleukin-12 expression by limiting STAT-1 phosphorylation in monocytes/macrophages during chronic hepatitis C virus infection. Immunology. 2011;132:421–31. doi: 10.1111/j.1365-2567.2010.03382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang Y, Ma CJ, Ni L, et al. Cross-talk between programmed death-1 and suppressor of cytokine signaling-1 in inhibition of IL-12 production by monocytes/macrophages in hepatitis C virus infection. J Immunol. 2011;186:3093–103. doi: 10.4049/jimmunol.1002006. [DOI] [PubMed] [Google Scholar]
- 108.Kaufmann DE, Kavanagh DG, Pereyra F, et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol. 2007;8:1246–54. doi: 10.1038/ni1515. [DOI] [PubMed] [Google Scholar]
- 109.Nakamoto N, Kaplan DE, Coleclough J, et al. Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology. 2008;134:1927–37. doi: 10.1053/j.gastro.2008.02.033. e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Radziewicz H, Ibegbu CC, Fernandez ML, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. 2007;81:2545–53. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Raziorrouh B, Ulsenheimer A, Schraut W, et al. Inhibitory molecules that regulate expansion and restoration of HCV-specific CD4+ T cells in patients with chronic infection. Gastroenterology. 2011;141:1422–31. doi: 10.1053/j.gastro.2011.07.004. e1–6. [DOI] [PubMed] [Google Scholar]
- 112.Fisicaro P, Valdatta C, Massari M, et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology. 2010;138:682–93. doi: 10.1053/j.gastro.2009.09.052. e1–4. [DOI] [PubMed] [Google Scholar]
- 113.Golden-Mason L, Palmer BE, Kassam N, et al. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol. 2009;83:9122–30. doi: 10.1128/JVI.00639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang Y, Ma CJ, Wang JM, Ji XJ, Wu XY, Jia ZS, Moorman JP, Yao ZQ. Tim-3 negatively regulates IL-12 expression by monocytes in HCV infection. PLoS ONE. 2011;6:e19664. doi: 10.1371/journal.pone.0019664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vali B, Jones RB, Sakhdari A, et al. HCV-specific T cells in HCV/HIV co-infection show elevated frequencies of dual Tim-3/PD-1 expression that correlate with liver disease progression. Eur J Immunol. 2010;40:2493–505. doi: 10.1002/eji.201040340. [DOI] [PubMed] [Google Scholar]
- 116.Khaitan A, Unutmaz D. Revisiting immune exhaustion during HIV infection. Curr HIV/AIDS Rep. 2011;8:4–11. doi: 10.1007/s11904-010-0066-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Moorman JP, Wang JM, Zhang Y, et al. Tim-3 pathway controls regulatory and effector T cell balance during hepatitis C virus infection. J Immunol. 2012;189:755–66. doi: 10.4049/jimmunol.1200162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ju Y, Hou N, Meng J, et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) mediates natural killer cell suppression in chronic hepatitis B. J Hepatol. 2010;52:322–9. doi: 10.1016/j.jhep.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 119.Harrison RJ, Ettorre A, Little AM, Khakoo SI. Association of NKG2A with treatment for chronic hepatitis C virus infection. Clin Exp Immunol. 2010;161:306–14. doi: 10.1111/j.1365-2249.2010.04169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li F, Wei H, Gao Y, Xu L, Yin W, Sun R, Tian Z. Blocking the natural killer cell inhibitory receptor NKG2A increases activity of human natural killer cells and clears hepatitis B virus infection in mice. Gastroenterology. 2013;144:392–401. doi: 10.1053/j.gastro.2012.10.039. [DOI] [PubMed] [Google Scholar]
- 121.Zhang R, Xu J, Hong K, et al. Increased NKG2A found in cytotoxic natural killer subset in HIV-1 patients with advanced clinical status. AIDS. 2007;21(Suppl. 8):S9–17. doi: 10.1097/01.aids.0000304691.32014.19. [DOI] [PubMed] [Google Scholar]
- 122.Wang JM, Cheng YQ, Shi L, Ying RS, Wu XY, Li GY, Moorman JP, Yao ZQ. KLRG1 negatively regulates natural killer cell functions through the Akt pathway in individuals with chronic hepatitis C virus infection. J Virol. 2013;87:11626–36. doi: 10.1128/JVI.01515-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shi L, Wang JM, Ren JP, et al. KLRG1 impairs CD4+ T cell responses via p16ink4a and p27kip1 pathways: role in hepatitis B vaccine failure in individuals with hepatitis C virus infection. J Immunol. 2014;192:649–57. doi: 10.4049/jimmunol.1302069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ge J, Wang K, Meng QH, Qi ZX, Meng FL, Fan YC. Implication of Th17 and Th1 cells in patients with chronic active hepatitis B. J Clin Immunol. 2010;30:60–7. doi: 10.1007/s10875-009-9328-2. [DOI] [PubMed] [Google Scholar]
- 125.Yang B, Wang Y, Zhao C, Yan W, Che H, Shen C, Zhao M. Increased Th17 cells and interleukin-17 contribute to immune activation and disease aggravation in patients with chronic hepatitis B virus infection. Immunol Lett. 2013;149:41–9. doi: 10.1016/j.imlet.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 126.Zhang JY, Zhang Z, Lin F, et al. Interleukin-17-producing CD4+ T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology. 2010;51:81–91. doi: 10.1002/hep.23273. [DOI] [PubMed] [Google Scholar]
- 127.Sun HQ, Zhang JY, Zhang H, Zou ZS, Wang FS, Jia JH. Increased Th17 cells contribute to disease progression in patients with HBV-associated liver cirrhosis. J Viral Hepat. 2012;19:396–403. doi: 10.1111/j.1365-2893.2011.01561.x. [DOI] [PubMed] [Google Scholar]
- 128.Ye Y, Xie X, Yu J, et al. Involvement of Th17 and Th1 effector responses in patients with hepatitis B. J Clin Immunol. 2010;30:546–55. doi: 10.1007/s10875-010-9416-3. [DOI] [PubMed] [Google Scholar]
- 129.Xue-Song L, Cheng-Zhong L, Ying Z, Mo-Bin W. Changes of Treg and Th17 cells balance in the development of acute and chronic hepatitis B virus infection. BMC Gastroenterol. 2012;12:43. doi: 10.1186/1471-230X-12-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang B, Zhao XP, Fan YC, Zhang JJ, Zhao J, Wang K. IL-17A but not IL-22 suppresses the replication of hepatitis B virus mediated by over-expression of MxA and OAS mRNA in the HepG2.2.15 cell line. Antiviral Res. 2013;97:285–92. doi: 10.1016/j.antiviral.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 131.Zhu S, Zhang H, Dong Y, et al. The correlation between T helper type 17 cells and clinical characters in Chinese paediatric patients with chronic hepatitis B. Clin Exp Immunol. 2013;171:307–12. doi: 10.1111/cei.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chang Q, Wang YK, Zhao Q, Wang CZ, Hu YZ, Wu BY. Th17 cells are increased with severity of liver inflammation in patients with chronic hepatitis C. J Gastroenterol Hepatol. 2012;27:273–8. doi: 10.1111/j.1440-1746.2011.06782.x. [DOI] [PubMed] [Google Scholar]
- 133.Seetharam AB, Borg BB, Subramanian V, Chapman WC, Crippin JS, Mohanakumar T. Temporal association between increased virus-specific Th17 response and spontaneous recovery from recurrent hepatitis C in a liver transplant recipient. Transplantation. 2011;92:1364–70. doi: 10.1097/TP.0b013e31823817f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Balanescu P, Ladaru A, Voiosu T, Nicolau A, Ene M, Balanescu E. Th17 and IL-17 immunity in chronic hepatitis C infection. Rom J Intern Med. 2012;50:13–8. [PubMed] [Google Scholar]
- 135.Bixler SL, Mattapallil JJ. Loss and dysregulation of Th17 cells during HIV infection. Clin Dev Immunol. 2013;2013:852418. doi: 10.1155/2013/852418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zaffiri L, d'Ettorre G, Ceccarelli G, et al. Recovery of interleukin-17 production from interleukin-15-stimulated CD4+ mononuclear cells in HIV-1-infected patients with sustained viral suppression. J Interferon Cytokine Res. 2014;34:35–40. doi: 10.1089/jir.2012.0011. [DOI] [PubMed] [Google Scholar]
- 137.Singh A, Vajpayee M, Ali SA, Mojumdar K, Chauhan NK, Singh R. HIV-1 diseases progression associated with loss of Th17 cells in subtype ‘C’ infection. Cytokine. 2012;60:55–63. doi: 10.1016/j.cyto.2012.06.288. [DOI] [PubMed] [Google Scholar]
- 138.Alvarez Y, Tuen M, Nadas A, Hioe CE. In vitro restoration of Th17 response during HIV infection with an antiretroviral drug and Th17 differentiation cytokines. AIDS Res Hum Retroviruses. 2012;28:823–34. doi: 10.1089/aid.2011.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kim CJ, McKinnon LR, Kovacs C, et al. Mucosal Th17 cell function is altered during HIV infection and is an independent predictor of systemic immune activation. J Immunol. 2013;191:2164–73. doi: 10.4049/jimmunol.1300829. [DOI] [PubMed] [Google Scholar]