

Abstract
Muscle specific receptor tyrosine kinase (MuSK) is an essential postsynaptic transmembrane molecule that mediates clustering of acetylcholine receptors (AChR). MUSK exon 10 is alternatively skipped in human, but not in mouse. Skipping of this exon disrupts a cysteine-rich region (Fz-CRD), which is essential for Wnt-mediated AChR clustering. To investigate the underlying mechanisms of alternative splicing, we exploited block-scanning mutagenesis with human minigene and identified a 20-nucleotide block that contained exonic splicing silencers. Using RNA-affinity purification, mass spectrometry, and Western blotting, we identified that hnRNP C, YB-1 and hnRNP L are bound to MUSK exon 10. siRNA-mediated knockdown and cDNA overexpression confirmed the additive, as well as the independent, splicing suppressing effects of hnRNP C, YB-1 and hnRNP L. Antibody-mediated in vitro protein depletion and scanning mutagenesis additionally revealed that binding of hnRNP C to RNA subsequently promotes binding of YB-1 and hnRNP L to the immediate downstream sites and enhances exon skipping. Simultaneous tethering of two splicing trans-factors to the target confirmed the cooperative effect of YB-1 and hnRNP L on hnRNP C-mediated exon skipping. Search for a similar motif in the human genome revealed nine alternative exons that were individually or coordinately regulated by hnRNP C and YB-1.
Neuromuscular junction (NMJ) is the site of communication between motor neurons and muscle fibers for neuromuscular signal transmission to ensure proper muscle contraction. A series of complex signaling cascades are involved in the formation of the neuromuscular synapse. Prior to the arrival of nerve terminals at muscle fibers, aneural acetylcholine receptor (AChR) clusters form in the central region of muscle fibers1,2,3,4, a phenomenon termed prepatterning. Subsequently, motor axons are guided to prospective prepatterned synaptic regions, and upon its arrival agrin is released from the nerve terminal5,6. Agrin interacts with LRP4 (low-density lipoprotein receptor-related protein 4) and stimulates the association between LRP4 and MuSK (muscle specific receptor tyrosine kinase) to activate MuSK7,8,9. Activation of MuSK induces AChR clustering. AChR clusters are thus highly concentrated in the postsynaptic membrane, where the NMJ forms.
MuSK harbors specific domains to respond to agrin and Wnt ligands. The ectodomain of MuSK has three immunoglobulin (Ig)-like domains (Ig1, Ig2, and Ig3) and a frizzled-like cysteine-rich domain (Fz-CRD)10,11,12 (Fig. 1). Frizzled proteins, receptors for Wnt-ligands, have ten highly conserved cysteine residues forming five disulfide bonds, which are essential for forming a compact folding structure13 (Fig. S1). MuSK Fz-CRD also has ten conserved cysteine residues observed in Frizzled proteins. Crystal structure of MuSK Fz-CRD indeed revealed a five-disulfide-bridged domain structure12. Human Fz-CRD is encoded by exon 10 coding for 6 cysteines and exon 11 coding for 4 cysteines14,15. In human, MUSK generates six splice variants according to ENSEMBL 76, although two are short variants with unknown functional significance. In two of the four remaining splice variants in human, exon 10 encoding 6 out of 10 essential cysteines in Fz-CRD is alternatively skipped (Isoforms C and D, Fig. S2c). In contrast to human, however, mouse Musk exon 10 is constitutively expressed according to the annotations by RefSeq, ENSEMBL 76, and GENCODE M2.
Figure 1. Structures of human and mouse MuSK.
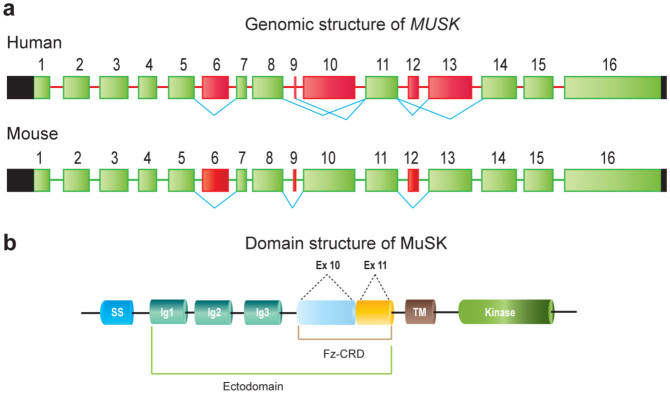
(a) Genomic structures of human MUSK and mouse Musk genes. Constitutive and alternative exons are shown in green and red boxes, respectively. Black boxes indicate untranslated regions (UTRs) and thin lines indicate introns. Alternative skipping of exons annotated in ENSEMBL 76 are shown by blue connecting lines. (b) Domain structure of MuSK. Fz-CRD is a Wnt-responsive domain encoded by exon 10 coding for 6 cysteines (light blue-colored region of Fz-CRD) and exon 11 coding for 4 cysteines (yellow-colored region of Fz-CRD) in human. SS, signal sequence; TM, transmembrane domain.
The first Ig-like domain (Ig1) of MuSK is required for agrin to stimulate MuSK phosphorylation via LRP49. Phosphorylation and activation of MuSK can also be promoted by Wnt proteins by interacting with Fz-CRD. In mouse C2C12 myotubes, Wnt9a and Wnt11 can stimulate MuSK phosphorylation by interacting with Fz-CRD and induce AChR clustering, which requires LRP4, but not agrin16. In another study, Wnt4 has been shown to induce MuSK phosphorylation by interacting with Fz-CRD in COS7 and HEK293T cells and Wnt4 facilitates mouse NMJ formation in vivo17. Muscle prepatterning in zebrafish is also facilitated by interaction of Wnt11r with Fz-CRD of a MuSK homolog, unplugged18, which is mediated by enhancing endocytosis of MuSK19. In zebrafish, unplugged/MuSK has three splice variants: SV1 lacks Ig-like domains 1 to 3 but retains Fz-CRD, and is not responsive to agrin; SV2 lacks Fz-CRD and is not responsive to Wnt; and the full-length isoform that can respond to both agrin and Wnt18. Subsequent studies revealed that agrin-nonresponsive SV1 is expressed in embryos, which is substituted for by the full-length isoform in adults18,20,21. MuSK Fz-CRD additionally plays an important role in motor nerve axon guidance in pathfinding in zebrafish20,21. Partial deletions of Fz-CRD have been analyzed in quail QT-6 fibroblasts22. Artificial deletion of 6 Fz-CRD cysteines or 4 Fz-CRD cysteines caused lack of an activity for MuSK-rapsyn co-clustering22. Amino acids 760 to 820 in the cytoplasmic domain of MuSK, however, have later been shown to be sufficient to confer interaction with rapsyn23. As Fz-CRD is an important and obligatory domain of MuSK for Wnt-mediated AChR clustering in zebrafish18,19,20,21 and mouse C2C12 myotubes16,17, the two discordant reports in zebrafish22,23 may indicate that Fz-CRD has an additional enhancing effect on MuSK-rapsyn interaction or plays another role in Wnt-mediated AChR clustering. Considering the functional significance of 10 cysteines in Fz-CRD, we humans are likely to have acquired an evolutionally novel Wnt-insensitive MuSK isoform lacking 6 essential cysteines, but the underlying mechanisms of alternative skipping of MUSK exon 10 remain unknown.
HnRNP C is a nuclear RNA-binding protein that associates with nascent mRNA transcripts, which plays roles in pre-mRNA splicing24, mRNA stability25, and translational modulation26. HnRNP C has recently been identified as a molecular ruler to classify RNA polymerase II transcripts for export into two categories: a long mRNA and a short uridine-rich small nuclear RNA (U snRNA)27. The Y box-binding protein (YB-1) is a member of the cold shock domain (CSD) protein family, which has binding specificity for both DNA and RNA. YB-1 has multiple roles including transcriptional regulation, translational control, DNA repair, and pre-mRNA splicing28,29. HnRNP L is another nuclear RNA-binding protein and a global splicing regulator30,31,32,33,34,35,36,37,38. It also functions in polyadenylation and mRNA stability39,40.
In the present study, we have dissected the underlying mechanisms of alternative splicing of human MUSK exon 10. We first characterized splicing regulatory cis-elements by scanning mutagenesis. We then identified that the alternative skipping of MUSK exon 10 is coordinately modulated by binding of three splicing suppressors (hnRNP C, YB-1, and hnRNP L) to an exonic splicing silencer (ESS) that is unique to human MUSK exon 10. Remarkably, hnRNP C is the master regulator in this regulatory process, and YB-1 and hnRNP L have additive effects to efficiently achieve splicing suppression.
Results
Alternative splicing of MUSK exon 10 is unique to human
Since exons 9 (7 nucleotides) and 10 (264 nucleotides) of human MUSK are alternatively spliced according to the gene annotation databases, we initially examined the differential selection of these two exons in human skeletal muscle. Using total RNA isolated from human skeletal muscle (Clontech), fragments spanning exons 8 to 11 were amplified by RT-PCR (Fig. S2d). Sequencing of the RT-PCR products revealed three splicing isoforms: (i) exons 9 and 10 included; (ii) exon 10 skipped; and (iii) exons 9 and 10 skipped (Fig. S2f). We could not detect any transcript that skipped only exon 9. We also performed a similar experiment using total RNA isolated from immortalized human myogenic KD3 cells and primary human myoblasts (SkMC), and obtained similar results (Fig. S2d and f). The three observed splicing isoforms are not correctly mapped to the human genome in UCSC Genes, RefSeq, ENCODE/GENCODE Ver. 19 and H-Inv ver. 8.3. As cDNA sequences registered in these annotation databases are correct, a short exon 9 that is comprised of only 7 nucleotides is likely to have precluded correct mapping of cDNAs to the human genome. We also confirmed lack of alternative splicing of mouse Musk exon 10 in 10 different skeletal muscle tissues as well as in C2C12 mouse myoblasts (Figs. S2e and S7c). Lack of alternative splicing of mouse Musk exon 10 is correctly annotated in all of the gene annotation databases shown above. In this study, we investigated the underlying mechanisms of alternative skipping of exon 10 unique to human.
Construction of minigenes for splicing analysis
We first constructed a human MUSK minigene in pcDNA3.1D/V5/His-TOPO expression vector (Invitrogen) spanning exons 8 to 11 (Fig. 2a). Exon 10 in the pcDNA3.1 minigene was alternatively spliced in HeLa cells, as we observed in human skeletal muscle. We next inserted exon 10 and flanking intronic sequences (245 nucleotides in the upstream intron and 200 nucleotides in the downstream intron) into the modified exon-trapping vector pSPL341, which carried two proprietary constitutive exons on each end (Fig. S3a). The pSPL3-human-MUSK minigene (H-iE10i) successfully recapitulated alternative splicing of exon 10 in HeLa cells (Fig. S3a). Serial deletions of intronic nucleotides from both ends of H-iE10i revealed that the shortest minigene (H-iE10i-Δ6) that carried 100 nucleotides in the upstream intron and 60 nucleotides in the downstream intron was still alternatively spliced like H-iE10i in HeLa cells (Fig. S3a). This minigene was termed pSPL3-human-MUSK (pH-wt) (Fig. 2b) and used in the subsequent experiments.
Figure 2. Construction of minigenes and systematic identification of splicing regulatory cis-elements.

(a) Structure of human MUSK minigene in pcDNA3.1D vector (pcDNA-human-MUSK). RT-PCR of this minigene in HeLa and KD3 cells are shown at right. Alternatively and constitutively spliced regions are shown in red and green, respectively. (b) Structure of pSPL3 minigene harboring MUSK/Musk exon 10 and flanking introns originated from human (pSPL3-human-MUSK, termed pH-wt) and mouse (pSPL3-mouse-Musk, termed pm-wt). RT-PCR of these minigenes in HeLa and KD3 cells are shown at right. (c) Schematic of chimeric constructs of pSPL3-human-MUSK minigene partially replaced by corresponding mouse sequences shown in green. RT-PCR of each chimeric minigene in HeLa cells is shown below. (d) Schematic of cis-regulatory block-scanning mutagenesis of MUSK exon 10 in the context of pSPL3-human-MUSK (pH-wt) minigene. A 20-nucleotide heterologous sequence of 5′-TCAGTATGACTCTCAGTATG-3′ is introduced into each block. RT-PCR of pH-wt and 13 block-mutant minigenes in HeLa cells are shown bellow. Arrows point to primer positions in (a), (b), and (c).
We also constructed a similar minigene harboring mouse exon 10 and flanking intronic sequences, pSPL3-mouse-MuSK (pm-wt), and found that exon 10 is constitutively included in this minigene in HeLa cells (Fig. 2b). As the mouse minigene was not alternatively spliced even in human cells, we assumed that nucleotides unique to human enabled alternative splicing of exon 10. We confirmed that our minigene was similarly alternatively spliced in KD3 cells and HeLa cells (Fig. 2a and b). Due to better transfection efficiency, we used HeLa cells and pH-wt/pm-wt in the following studies.
Identification of two exonic splicing silencer (ESS) blocks in human exon 10
We next searched for exonic/intronic segments carrying splicing cis-elements in pH-wt. To this end, we constructed chimeric minigenes made of variable combinations of human and mouse segments (Fig. 2c). We found that the introduction of mouse exon 10 into pH-wt resulted in constitutive splicing (pH-EC in Fig. 2c). We next introduced the first or second half of mouse exon 10 into pH-wt, and found that both constructs resulted in constitutive splicing (pH-FHEC and pH-SHEC in Fig. 2c). To specifically identify exonic splicing cis-elements, we sequentially introduced a 20-nucleotide heterologous sequence block (5′-TCAGTATGACTCTCAGTATG-3′), which was previously reported to have no effect on splicing37,42 (Fig. 2d). We scanned the entire exon 10 by substituting 13 blocks excluding the first and last three nucleotides of the exon. The block scanning mutagenesis detected a potential ESS in two separate blocks (blocks 5 and 12) and a potential exonic splicing enhancer (ESE) in four consecutive blocks (blocks 6, 7, 8, and 9) (Fig. 2d). As skipping of exon 10 has a prospective inhibitory effect on AChR clustering, we dissected the mechanisms associated with ESSs in this communication.
Dissection of ESS blocks on a nucleotide level and detection of trans-acting factors
We next dissected the identified ESS blocks on a nucleotide level. Alignment of human and mouse blocks 5 (ESS5) and 12 (ESS12) revealed three and two discordant nucleotides, respectively. Artificial introduction of discordant nucleotides into ESS5 (pH-mB5) and ESS12 (pH-mB12) resulted in profound and moderate loss of exon skipping, respectively (Fig. 3a and b), which were consistent with the block-scanning mutagenesis experiments (Fig. 2d). Therefore, the splicing suppressive effect of ESS5 was stronger than that of ESS12.
Figure 3. HnRNP C, YB-1 and hnRNP L bind to exon 10 of human MUSK.

(a, b) Mouse nucleotides are introduced into ESS5 (a) and ESS12 (b) of pH-wt to generate pH-mB5 and pH-mB12, respectively. RT-PCR of each mutated minigene in HeLa cells is compared with that of pH-wt. Primer positions are shown by arrows. (c) Sequences of ESS5 RNA probes carrying human (H-B5), mouse (m-B5), and partially deleted (H-B5Δ5) sequences. (d)32P-labeled H-B5 or m-B5 RNA probe was incubated in the presence or absence of HeLa nuclear extract (NuEX) and resolved on a native polyacrylamide gel to observe free and protein-bound RNA species (arrows). (e) Coomassie blue staining of RNA affinity-purified products from HeLa nuclear extract using the indicated biotinylated RNA probes. Three proteins of ~70, ~50, and ~40 kDa (arrows) are differentially associated with H-B5 compare to m-B5 and H-B5Δ5. Mass spectrometry analysis revealed that the three proteins are HnRNP L (L), YB-1, and hnRNP C (C). (f) Immunoblotting (IB) of RNA affinity purified proteins in panel (e) with the indicated antibodies.
The vast majority of splicing enhancer and silencer sequences have been reported to function through the binding of cognate regulatory proteins43. To determine if potential trans-acting factors stably interact with the ESS sequences, binding reactions were performed with a HeLa nuclear extract and an RNA probe harboring human or mouse ESS5 sequence (Fig. 3c). An RNA mobility shift assay with a native gel showed three slow migrating complexes with the 32P-labeled human ESS5 RNA probe (H-B5) (Fig. 3d). In contrast, the two slow migrating complexes observed with H-B5 were not visible with the mouse ESS5 RNA probe (m-B5). We also performed a similar experiment with human/mouse ESS12 RNA probes, but could not detect any differentially associated complex (Fig. S3b and c). We thus focused on identification of the proteins bound to ESS5.
HnRNP C, YB-1, and hnRNP L are bound to human ESS5
We next performed an RNA affinity purification assay using a HeLa nuclear extract and a biotinylated ESS5 RNA probe (Fig. 3c). Three distinct bands of ~70, ~50, and ~40 kD were associated with the wild-type human ESS5 RNA probe (H-B5) but not with the mouse probe (m-B5) or a partially deleted human probe (H-B5Δ5) (Fig. 3e). Mass spectrometry analysis of the excised bands disclosed that the identified bands were hnRNP L, YB-1, and hnRNP C, respectively, which were confirmed by immunoblotting using respective antibodies [anti-hnRNP L 4D11 (sc-32317, Santa Cruz Biotechnology), anti-YB1 (A303-230A, Bethyl Laboratories), and anti-hnRNP C1/C2 4F4 (SC-32308, Santa Cruz Biotechnology)] (Fig. 3f). Similar analysis with ESS12 detected no differentially associated molecule (Fig. S3d).
HnRNP C, YB-1, and hnRNP L coordinately enhance skipping of human MUSK exon 10
We next examined the effects of the identified trans-factors on skipping of exon 10 by siRNA-mediated downregulation of the individual factors in HeLa cells. We first confirmed efficient downregulation of each factor (Fig. 4a). Downregulation of hnRNP C resulted in a significant loss of exon skipping (Fig. 4b). Similarly, downregulation of YB-1 and hnRNP L caused a loss of exon skipping, but to a lesser extent compared to hnRNP C. Downregulation of all three trans-factors exerted a more prominent effect than hnRNP C alone. Thus, YB-1 and hnRNP L are likely to have additive effects on exon skipping. We observed similar alterations in alternative splicing patterns with a second set of siRNAs targeting different sites of each mRNA (Fig. S3e).
Figure 4. HnRNP C, YB-1, and hnRNP L coordinately promote skipping of MUSK exon 10.

(a) Immunoblotting (IB) using the indicated antibodies after gene knockdown with siRNA against control (siCont), hnRNP C (siC), YB-1 (siYB-1), and hnRNP L (siL) in HeLa cells. (b) RT-PCR of pSPL3-human-MUSK (pH-wt) minigene in HeLa cells treated with the indicated siRNAs. The mean and SD (n = 3) of the ratio of exon skipping in each treatment is shown below the gel image. (c) Immunoblotting (IB) with the indicated antibodies after cDNA overexpression of hnRNP C, YB-1, and hnRNP L in HeLa cells. Endogenous (endo) and exogenous (exon) YB-1 proteins are pointed by arrows. (d) RT-PCR of pH-wt minigene in HeLa cells co-transfected with the indicated cDNAs. The mean and SD (n = 3) of the ratio of exon skipping in each treatment is shown below the gel image.
We next overexpressed cDNA of each trans-factor in HeLa cells. We first confirmed the expression of each cDNA by immunoblotting (Fig. 4c). As expected, overexpression of hnRNP C induced skipping of exon 10 (Fig. 4d). A similar increase in exon skipping was also observed with overexpression of YB-1 and hnRNP L, but to a lesser extent compared to hnRNP C. The most prominent skipping was observed when all three factors were overexpressed together, which was consistent with the knockdown results.
HnRNP C is a critical regulator in inducing exon skipping, whereas YB-1 and hnRNP L have augmenting effects
Having identified the critical cis-element and their cognate-binding partners, we next analyzed the molecular basis of specific binding of each trans-factor to ESS5. HnRNP C prefers to bind to poly-T stretch motifs24,44 and both hnRNP L and YB-1 prefer to bind to C/A-rich motifs29,32,45. ESS5 carries a stretch of five T's in the first half of the block and a C/A-rich sequence in the second half of the block (H-B5 in Fig. 5a and b). In vitro SELEX studies of hnRNP L demonstrated that CACA and ACAC sequences confer high-affinity binding motifs and that CAAC and CACC confer low-affinity binding motifs for hnRNP L32, where motifs present in ESS5 are underlined. On the contrary, in vitro SELEX studies of YB-1 revealed that CATC and CACC sequences confer high-affinity binding motifs for YB-129, where a motif present in ESS5 is underlined. Therefore, the second half of ESS5 harbors overlapping binding motifs of both YB-1 and hnRNP L. To characterize the precise binding sites of the associated factors, we introduced a series of artificial point mutations into the human ESS5 RNA probe and checked the binding of each factor by RNA-affinity purification followed by Western blotting (Fig. 5a and b). We observed that poly-T stretch-disrupting mutations in the first half indeed abolished the binding of hnRNP C (Fig. 5a, lanes 3, 4, and 5), and four or more consecutive T-nucleotides are necessary for hnRNP C binding. To our surprise, we noticed that binding of YB-1 and hnRNP L was also compromised along with disruption of the hnRNP C binding (Fig. 5a, lanes 3, 4 and 5). This suggested that binding of YB-1 and hnRNP L was dependent on poly T-stretch. On the other hand, introduction of mutations in the second half (C/A- rich sequences) compromised binding of YB-1 and hnRNP L, but not of hnRNP C (Fig. 5b). In addition, characterization of essential nucleotides for binding of hnRNP L (CAACA) and YB-1 (ACACCT) revealed that binding motifs of hnRNP L and YB-1 indeed overlap (CAACACCT) in the second half of ESS5, where the overlapping nucleotides are underlined. Considering the overall findings (Fig. 5c), we predicted that binding of hnRNP C to the poly-T stretch facilitates the binding of YB-1 and hnRNP L to the adjacent downstream site. To test this hypothesis, we depleted hnRNP C from a HeLa nuclear extract using a specific antibody (Fig. S4a) and performed RNA affinity purification assays. As we had expected, depletion of hnRNP C nullified the binding of YB-1 and hnRNP L (Fig. S4b).
Figure 5. Binding of hnRNP C, YB-1, and hnRNP L to specific motifs enhances coordinated skipping of MUSK exon 10.

(a, b) Scanning mutagenesis to map the binding motifs of hnRNP C, YB-1, and hnRNP L in ESS5 (H-B5). RNA probe sequences are shown in the upper panels, where discordant nucleotides between human and mouse are underlined in H-B5 (ESS5). Artificial mutations are shown in red. RNA affinity-purified products are detected by immunoblotting in the lower panels. The results are indicated on the right side by “+” and “−” for positive and negative binding to each probe, respectively. (c) The resulting binding site of each factor from panels (a) and (b) is schematically shown. Essential binding nucleotides are indicated by large green letters and are underlined. (d) Schematic of a reporter minigene (pSPL3-human-MUSK-MS2-PP7). MS2 coat protein-binding hairpin RNA (blue) is substituted for the first half of ESS5 (binding site of hnRNP C). Similarly, PP7 coat protein-binding hairpin RNA (orange) is substituted for the second half of ESS5 (binding sites of hnRNP L and YB-1). (e) RT-PCR of pSPL3-human-MUSK-MS2-PP7 minigene in HeLa cells that are co-transfected with the indicated effectors. Blue and orange letters match to those in (d). The mean and SD (n = 3) of the ratio of exon skipping in each treatment is shown below the gel image.
Binding of hnRNP C, YB-1, and hnRNP L to ESS5, but not to the other site, induces skipping of MUSK exon 10
Having characterized a coordinated regulation of hnRNP C, YB-1, and hnRNP L on skipping of exon 10, we next examined the additive effect of YB-1 or hnRNP L on hnRNP C-mediated exon skipping. To this end, we made a reporter minigene (pSPL3-human-MUSK-MS2-PP7), in which the bacteriophage MS2 coat protein-binding site was substituted for the native ‘TTTTTCT’ sequence in the first half of ESS5 (the binding site of hnRNP C), and the bacteriophage PP7 coat protein-binding site was substituted for the native ‘CAACACCTC’ sequence in the second half of ESS5 (the binding site of YB-1 and hnRNP L) (Fig. 5d). We also made cDNA fusion constructs, hnRNP C-MS2, YB-1-PP7, and hnRNP L-PP7, to artificially tether splicing trans-factors to the respective sites. As expected, tethering of MS2-tagged hnRNP C alone efficiently induced exon skipping of 79% (Fig. 5e, lane 3). Tethering of PP7-tagged YB-1 and hnRNP L without tethering MS2-tagged hnRNP C induced exon skipping of 52% (Fig. 5e, lane 5) and 36% (Fig. 5e, lane 8), respectively. In contrast, hnRNP C, YB-1, or hnRNP L without a tethering tag did not induce exon skipping (Fig. 5e, lanes 2, 4 and 7), suggesting that ESS5 was the only site where these factors were able to bind and function. We also confirmed that MS2- or PP7-tagged factor has no effect on a minigene lacking MS2- or PP7-binding site (Fig. S4d). Simultaneous recruitment of hnRNP C with either YB-1 or hnRNP L further induced exon skipping (Fig. 5e, lanes 6 and 9), which reconfirmed the additive effects of YB-1 and hnRNP L on hnRNP C-mediated splicing suppression.
RNA-dependent interaction of hnRNP C, YB-1, and hnRNP L and search for similar targets in other human genes
We next examined the molecular interaction between the three trans-factors. Co-immunoprecipitation revealed that HnRNP C and hnRNP L were bound in an RNA-dependent manner (Fig. S5a), whereas hnRNP C and YB-1 were not (Fig. S5b). Similarly, hnRNP L and YB-1 were bound in an RNA-dependent manner (Fig. S5c), which was consistent with a previous report45.
We further asked if coordinated splicing regulation by hnRNP C, YB-1, hnRNP L is unique to MUSK exon 10. As hnRNP L has highly degenerative SELEX motifs, which are overlapping with YB-1 motifs, we analyzed coordinated splicing only by hnRNP C and YB-1. Search for adjacent hnRNP C- and YB-1-binding motifs in human alternative cassette exons and flanking introns detected 378 candidate sites. We randomly selected 37 exons and analyzed alternative splicing in HeLa cells in the presence of siCont, siC, siYB-1, and siC/siYB-1. We found that alternative splicing events of 9 exons were affected by hnRNP C and/or YB-1 in HeLa cells, whereas 13 exons were not expressed and 15 exons were not alternatively spliced or affected by knockdown (Fig. S6). Among the 9 exons, 3 exons were coordinately skipped by hnRNP C and YB-1 (Fig. S6a).
Expressions of splicing repressing hnRNP C and YB-1 are reduced with muscle differentiation
As MuSK is a muscle-specific receptor protein having an important role in muscle development and function, we next examined the splicing profile of human MUSK exon 10 in different stages of myogenic differentiation. We cultured immortalized human myogenic KD3 cells in differentiation medium for four days to make myotubes (Fig. 6a). RT-PCR spanning endogenous MUSK exon 10 at different time points revealed that skipping of exon 10 was suppressed on and after differentiation day 3 (Fig. 6b, and Supplementary Table 1). Similarly, myotube differentiation suppressed skipping of MUSK exon 10 in primary human myoblasts (SkMC) (Figs. S7a, b, and 6c), and skipping of exon 10 constitutes only 12% in human skeletal muscle (Fig. 6c and Supplementary Table 1). Thus, skipping of exon 10 is a minor event in any differentiation stages of myogenic and muscle cells.
Figure 6. Suppression of hnRNP C and YB-1 expressions in myogenic differentiation enhances inclusion of MUSK exon 10.

(a) Phase-contrast images showing a temporal profile of differentiation of KD3 cells. (b) RT-PCR showing alternative splicing of endogenous MUSK exon 10 at different differentiation days of KD3 cells. (c) Real-time RT-PCR to quantify endogenous human MUSK transcripts. The absolute copy numbers of each isoform (Isoforms A-D, as shown in Fig. S2f) were estimated using total RNA isolated from KD3 cells, primary human myoblasts (SkMC) and human skeletal muscle (HSKM). Then the ratio of exon 10-skipping (including both isoforms C and D as shown in Fig. S2f) was determined (% exon skipping). UD, undifferentiated cells; DM, differentiated myotubes (at day 4 for KD3 and at day 8 for SkMC). Transcripts levels were normalized against GAPDH values. Bars represent mean and SD of three independent experiments. (d, e) Endogenous expression of splicing suppressors by RT-PCR (d) and immunoblotting (e) at different differentiation days of KD3 cells. Expressions of myogenin and GAPDH are shown as internal controls in panels (b), (d), and (e). The same gel image of GAPDH is used in panels (b) and (d).
In KD3 cells, we found that the expressions of hnRNP C and YB-1 were indeed decreased on and after day 3, whereas the expression level of hnRNP L was not significantly changed at the mRNA (Fig. 6d) and protein (Fig. 6e) levels. Therefore, reduction of hnRNP C and YB-1 in the course of muscle differentiation causes reduction of skipping of human MUSK exon 10 to produce a Wnt-sensitive MuSK isoform.
Discussion
In this study, we have identified splicing regulatory cis-elements and cognate trans-factors that drive alternative splicing of human MUSK exon 10. HnRNP C enhances exon skipping in coordination with YB-1 and hnRNP L by binding to a regulatory exonic splicing silencer, ESS5. Two splicing suppressive cis-elements, ESS5 and ESS12, are recognized by block-scanning mutagenesis, although the ESS activity of ESS12 is not as conspicuous as that of ESS5. Most splicing regulatory ESSs and ESEs function through the binding of cognate regulatory proteins43. Indeed, ESS5 is recognized by the three splicing trans-factors, but no associated molecules are detected for ESS12. In contrast, the splicing suppressive activity of ESS12 is likely to be regulated by a local secondary structure of pre-mRNA. Alternatively, a binding affinity of a splicing trans-factor to ESS12 is too low to be detected by the RNA mobility shift assay and the RNA affinity purification assay.
We have characterized the mutual coordination between the three trans-factors. HnRNP C binds to a poly-T stretch in the first half of ESS5, whereas binding motifs of hnRNP L and YB-1 are overlapped in the second half of ESS5. In addition, binding of YB-1 and hnRNP L is dependent on hnRNP C. One possible mechanism for this is that overlapping binding motifs between hnRNP L and YB-1 provoke competitive binding of the two factors. A third molecule, hnRNP C, may rearrange RNA conformation to stabilize the binding of either hnRNP L or YB-1. Similar RNA-mediated stabilization of binding of another RNA-binding protein by a specific RNA-binding protein has been reported in other genes. In CD45, hnRNP L stabilizes the binding of hnRNP A1 in exon 4, in which binding of hnRNP A1 and the subsequent splicing suppression is dependent on hnRNP L34. The authors demonstrate that hnRNP L interacts with hnRNP A1, which is lost by RNase. In Tpm1, splicing of exon 3 is coordinately repressed by PTB and Raver 146. Interaction between PTB and Raver 1, which requires the target RNA, results in a conformational change of the tertiary complex to bring the repressor domain of both molecules in close apposition to synergistically promote exon skipping. Therefore RNA-dependent molecular interaction and coordinated splicing suppression is unlikely to be unique to ESS5 and is likely to be functional in many other alterative splicing events.
Binding of YB-1 and hnRNP L to the same target and the subsequent coordinated splicing regulation have been previously reported45. The authors report that overexpression and depletion of either YB-1 or hnRNP L is sufficient to repress and derepress splicing, respectively. In their report, YB-1 exerts a stronger effect than hnRNP L, as in our MUSK exon 10. To the best of our knowledge, MUSK exon 10 is the first target where coordinated splicing regulation by YB-1 and hnRNP L is dependent on hnRNP C. RT-PCR analysis of 37 targets, where an hnRNP C-binding ‘TTTT' motif and a YB-1-binding ‘CATC/CACC' motif are adjacent to each other in human alternative cassette exons and flanking introns, revealed three alternative cassette exons that are coordinately skipped by hnRNP C and YB-1. Although detailed functional coordination of hnRNP C and YB-1 on these targets, and involvement of hnRNP L, remain unknown, MUSK exon 10 is unlikely to be the only target where hnRNP C, YB-1, and hnRNP L coordinately induce exon skipping.
Recent studies of alternative splicing in different cellular and physiological states have broadened our understanding of the molecular basis for functional selection of a certain isoform in a tissue-specific and developmental stage-specific manner. These studies demonstrated multiple intriguing features of splicing regulatory factor(s) including cell-type specific expression, intracellular localization, post-translational modification on different cellular stimuli, etc. Precise regulation of cellular differentiation is indispensable for the proper development of vertebrate embryo and deregulated differentiation results in diverse human congenital abnormalities including cancers. In human myogenic cells (KD3), skipping of MUSK exon 10 is significantly reduced from ~41% to ~22% upon myogenic differentiation, which is in parallel with the reduced expression of hnRNP C and YB-1 (Fig. 6). We also observed a similar expression profile of MUSK exon 10 in primary human myoblasts (SkMC) upon myogenic differentiation (Figs. S7a, b and 6c). Reduced expression of hnRNP C, YB-1 and even hnRNP L upon myogenic differentiation is also reported in mouse myoblast cells (C2C12)47,48. Therefore, human has evolutionally acquired ESS5 to skip MUSK exon 10, whereas downregulation of its trans-factors, hnRNP C and YB-1, in muscle differentiation remains unchanged in the course of evolution. It is interesting to note that YB-1 is a negative regulator of C2C12 myoblast differentiation48. YB-1 cooperatively interacts with MSX1 that inhibits the expression of MyoD by binding to the core enhancer region (CER) of the MyoD promoter48. Therefore, reduction of YB-1 expression in the course of differentiation is physiologically relevant to facilitating myogenic differentiation.
Physiological and evolutional significance of acquisition of an exon 10-skipped Wnt-insensitive MuSK isoform in human remains elusive, although an exon 10-skipped MuSK isoform constitutes a minor fraction in human skeletal muscle. An exon 10-skipped isoform is deficient of Fz-CRD function16,17. In CHRNA1 encoding AChR α subunit, only humans and great apes have acquired alternative inclusion of a non-functional exon P3A, which we have shown to be regulated by hnRNP H41, PTB49, and hnRNP L38. Although the evolutional significance of having acquired a non-functional exon P3A remains unsolved, skipping of MUSK exon 10 is the second defective splicing event that is unique to human. In mammals, CHRNE encoding the AChR ε subunit is expressed only at NMJ to promote endplate-specific expression of AChR50,51,52. Similarly, Colq encoding collagen Q has two promoters that are activated in slow- and fast-twitch muscles, respectively. In fast-twitch muscles, the Colq-1a promoter is activated only at NMJ53. Differentiation-induced inclusion of MUSK exon 10, as well as NMJ-specific expression of CHRNE and COLQ, suggest that human might have acquired NMJ-specific expression of a Wnt-responsive exon 10-included MUSK isoform to suppress extra-synaptic formation of AChR clusters. To achieve such beneficial expression, exonic splicing cis-elements have evolved to utilize spatiotemporally regulated expressions of splicing trans-factors that were already functional in lower mammals.
Methods
Sources of human skeletal muscle RNA
To scrutinize MUSK isoforms in human skeletal muscle, we purchased human skeletal muscle total RNA (Clontech). We also purchased primary human skeletal muscle cells (SkMC, Lonza). Immortalized human myogenic cells (KD3) were kindly provided by Dr. Naohiro Hashimoto (National Center for Geriatrics and Gerontology, Japan)54,55,56.
Cell culture and transfection
HeLa and C2C12 cells were cultured in DMEM (Sigma-Aldrich) with 10% fetal bovine serum (FBS, Sigma-Aldrich). SkMC cells were maintained in SkGM medium (Lonza). KD3 cells were grown in high-glucose (4.5 g/ml) DMEM (hDMEM) media containing 20% FCS and 2% Ultroser G serum substitute (PALL). To induce myogenic differentiation in C2C12 or SkMC, the culture medium of confluent cells was switched to DMEM supplemented with 2% horse serum. For myogenic differentiation of KD3 cells, we cultured confluent cells in hDMEM containing 5 μg/ml holo-transferrin (bovine), 10 μg/ml insulin, and 10 nM selenite (TIS, Gibco), as well as 2% FCS. HeLa and KD3 cells were transfected by FuGENE 6 (Roche) and Avalanche (EZT-HSKM-1, EZ Biosystems), respectively.
Construction of MUSK minigene for splicing analysis
We constructed human MUSK minigene spanning exons 8 to 11 in pcDNA3.1D/V5-HiS-TOPO vector (Invitrogen) using a proofreading DNA polymerase (PrimeSTAR, Takara). Four PCR products were first amplified: exon 8 to intron 8 (IVS8+389); intron 8 (IVS8-148) to intron 9 (IVS9+121); intron 9 (IVS9-243) to intron 10 (IVS10+202); and intron 10 (IVS10-77) to exon 11. The PCR primers carried additional 5′ sequences that matched to the neighboring amplicons. Amplicons 1/2 and 3/4 were first ligated each other, respectively, and the generated fragments 1/2 and 3/4 were ligated to generate a single fragment to be cloned into pcDNA3.1D/V5-HiS-TOPO vector. The final product, pcDNA-human-MUSK, thus lacked 13865, 5248, and 7426 nucleotides in the middle of introns 8, 9, and 10, respectively.
We also inserted MUSK/Musk exon 10 and flanking intronic sequences (245 nucleotides of intron 9 and 200 nucleotides of intron 10) of both human and mouse in the modified exon-trapping vector, pSPL341. The PCR product carried NotI and PacI sites on each for cloning into pSPL3. Artificial mutations and block replacement were engineered into the pSPL3 minigenes using the QuikChange site-directed mutagenesis kit (Stratagene) (Supplementary Table 2).
We made chimeric constructs of pSPL3-human-MUSK carrying human and mouse segments using the megaprimer method57. At first, we PCR-amplified the desired mouse segment with primers that carried 20 to 25 complementary nucleotides to pSPL3-human-MUSK at the 5′ ends. The PCR amplicon was used as a megaprimer for the QuikChange site-directed mutagenesis kit to make chimeric pSPL3-human-MUSK minigenes. The entire inserts were sequenced for all the generated clones to ensure absence of PCR artifacts.
RT-PCR and real-time RT-PCR
Total RNA was extracted 40 h after transfection using Trizol (Invitrogen), followed by DNase I treatment (Qiagen). cDNA was synthesized with an oligo-dT primer (Invitrogen) using ReverTra Ace reverse transcriptase (Toyobo). RT-PCR was performed using GoTaq (Promega) (Supplementary Table 3).
Real-time RT-PCR was performed using LightCycler 480 II (Roche) and the SYBR Premix Ex Taq II (Takara) to quantify endogenous human MUSK transcripts. The absolute copy numbers of each of MUSK isoforms were estimated using specific primers (Supplementary Table 3) and cDNA fragments cloned into pGEM-T as references.
RNA-electrophoretic mobility shift assays (RNA-EMSA)
32P-labeled RNA probes comprised of 20 nucleotides were transcribed in vitro with the T7 RiboMAX large-scale RNA production system (Promega) using 32P-UTP as previously reported58. The template DNA for transcription was generated by overlap extension PCR using two overlapping primers (Supplementary Table 4), where T7 promoter sequence was introduced at the 5' end of the forward primer. Binding reactions were carried out using HeLa nuclear extract (CilBiotech) and radiolabeled RNA probes at 30°C for 15 min in a 15-μl reaction mixture, with a final concentration of 3.0 mM MgCl2, 50 mM KCl, 20 mM Tris-HCl (pH 7.5), 0.1 mM EDTA, 0.1% (v/v) Triton X-100, 10% (v/v) glycerol, 3 μg of BSA, 1 μg of tRNA, 4 U RNasin (Promega). RNA-protein complexes were analyzed by 5% polyacrylamide gel electrophoresis (PAGE) at 4°C using 0.5× Tris-borate-EDTA (TBE) buffer. Dried gels were subjected to autoradiography.
RNA affinity purification assay
We synthesized 20-nucleotide biotinylated RNA probes with the T7 RiboMAX large-scale RNA production system (Promega) using 3.0 mM Biotin-14-CTP (Invitrogen) as described previously38. The template DNA was generated as described for RNA-EMSA.
The RNA affinity purification method was modified from the previously adopted protocol38. Biotinylated RNAs (0.75 nmol) and HeLa nuclear extract (30 μl) (CilBiotech) were mixed in a 500-μl binding buffer [20 mM HEPES, pH 7.8, 150 mM KCl, 0.1 mM EDTA, 1 mM DTT, 1 mM PMSF, 0.05% Triton X, 1× Protease Inhibitor Cocktail (Active Motif)], and were incubated at 30°C for 3 h with gentle agitation. In parallel, 50 μl streptavidin-conjugated beads (Streptavidin-sepharose, GE Healthcare) were blocked with a 1:1 mixture of 1 ml binding buffer containing yeast tRNA (0.1 mg/100 μl of beads) and 1 ml PBS containing 4% BSA at 4°C with rotation for 1 h. The beads were mixed with the binding solution for 2 h at 4°C with gentle rotation. After washing the beads four times with 1 ml binding buffer, RNA-bound proteins were eluted in SDS loading buffer by boiling at 95°C for 5 min. The isolated proteins were fractionated on a 10% SDS-polyacrylamide gel and stained with Coomassie blue or by immunoblotting.
Mass spectrometry
Mass spectrometry was performed as previously described38.
Depletion of hnRNP C from nuclear extract
Antibody-mediated depletion of hnRNP C from HeLa cell nuclear extract was performed using Protein G HP spin trap (GE Healthcare) according to the manufacturer's instructions.
siRNA knockdown and minigene splicing
We synthesized the following human siRNAs (Sigma Genosys):
5'-CAACGGGACUAUUAUGAUATT-3' for hnRNP C;
5'-CCACGCAAUUACCAGCAAATT-3' for YB-1; and
5'-GAAUGGAGUUCAGGCGAUGTT-3' for hnRNP L.
We also synthesized a second set of human siRNAs:
5'-GUAGAGAUGAAGAAUGAUATT-3' for hnRNP C,
5'-AGAAGGUCAUCGCAACGAATT -3' for YB-1, and
5'-CUACGAUGACCCGCACAAATT-3' for hnRNP L.
The control siRNA was AllStar Negative Control siRNA (1027281) by Qiagen. Cells were plated 24 h before transfection in six-well culture plates (1.5 × 105 cells/well). The transfection reagent included each siRNA duplex at a final concentration of 30 nM, 1 μl Lipofectamine 2000 (Invitrogen), and 500 ng minigene in 100 μl Opti-MEM medium. The cells were harvested three days after transfection for RT-PCR and immunoblotting analyses.
cDNA overexpression and minigene splicing
Human hnRNP C cDNA was amplified with total RNA of human skeletal muscle (Clontech), and cloned into pCDNA3.1D/V5-His TOPO (Invitrogen) to make pcDNA-hnRNP C. We previously constructed pcDNA-hnRNP L38. The human YB-1 expression vector (pCMV-YB-1-myc-nuc) was kindly provided by Dr. Akira Yokomizo (Kyushu University, Japan)59. Cells were plated 24 h prior to transfection in a six-well culture plate (1.5 × 105 cells/well) and transfected with 1 μg of expression construct(s), 500 ng of the minigene, and 6.0 μl of FuGENE 6 (Roche) in 100 μl Opti-MEM medium. The cells were harvested three days after transfection for RT-PCR and immunoblotting analyses.
Harvesting cells for immunoblotting
Cells were washed twice in PBS and harvested in PBS with 1× Protease Inhibitor Cocktail. After centrifugation at 2,000 × g for 5 min, the pellets were resuspended in buffer A [10 mM HEPES-NaOH (pH 7.8), 10 mM KCl, 0.1 mM EDTA, 1 mM DTT, 0.5 mM PMSF, 0.1% Nonidet P-40, 1× Protease Inhibitor Cocktail] and kept for 30 min on ice. After sonication, samples were centrifuged at 20,000 × g for 5 min to remove cell debris. The total cell lysate was subjected to immunoblotting.
Tethered function assay
Tethered function assay was performed by co-transfection of a reporter minigene carrying MS2- and PP7-binding sites and effector construct(s) fused to either MS2 or PP7 coat protein. To make pcDNA-hnRNP C-MS2, an insert encoding MS2 was isolated from pcDNA-hnRNP L-MS238 using XhoI and XbaI restriction enzymes, purified, and cloned into the respective sites of pcDNA-hnRNP C. We purchased a vector harboring PP7 cDNA (pET22HT-PP7delFG) from Addgene. Using this vector, a PCR product spanning PP7 cDNA was amplified with primers having XhoI and XbaI sites at the 5′ ends. This was subsequently cloned into XhoI/XbaI sites of pcDNA-hnRNP L38 to obtain pcDNA-hnRNP L-PP7. Using the In-Fusion cloning kit (Clontech), we introduced PP7 cDNA into pCMV-YB-1-myc-nuc to produce pCMV-YB-1-PP7-myc-nuc. The absence of artifacts was confirmed by sequencing the entire inserts. We previously made pcDNA-MS2 harboring only MS2 cDNA38. We also cloned only PP7 cDNA into pcDNA3.1D/V5-HiS-TOPO to make pcDNA-PP7.
To construct a reporter minigene, we substituted the bacteriophage MS2 coat protein-binding hairpin RNA sequence (5'-ACATGAGGATCACCCATGT-3')38 for the first half of the ESS5 sequence (5'-TTTTTCT-3') and PP7 coat protein-binding hairpin RNA sequence (5'-GGCACAGAAGATATGGCTTCGTGCC-3')60 for the second half of ESS5 sequence (5'-CAACACCTC-3') in MUSK exon 10 in pSPL3 minigene using the QuikChange site-directed mutagenesis kit.
Antibodies
Antibodies used in this study were anti-hnRNP C1/C2 4F4 (SC-32308, Santa Cruz Biotechnology), anti-YB1 (A303-230A, Bethyl Laboratories), anti-hnRNP L 4D11 (sc-32317, Santa Cruz Biotechnology), anti-His-tag (D291-3, Medical & Biological Laboratories), anti-GAPDH (G9545, Sigma-Aldrich), anti-β-actin C4 (sc-47778, Santa Cruz Biotechnology), anti-myogenin (M-225) (sc-576, Santa Cruz Biotechnology), and anti-U2AF65 MC3 (sc-53942, Santa Cruz Biotechnology).
Co-immunoprecipitation
Protein-protein interactions were studied by the co-immunoprecipitation (Co-IP) experiment using the Nuclear Complex Co-IP kit (Active Motif) according to the manufacturer's instructions, in the presence or absence of RNase (RNase cocktail enzyme mix, Ambion). We incubated 100 μg of HeLa nuclear extract with 2 μg of anti-hnRNP C or anti-YB-1 antibody. We included isotype-matched normal IgG as a control. The binding buffer contained 150 mM NaCl. The protein G beads with bound molecules were boiled and run on a polyacrylamide gel followed by immunoblotting.
In silico search for binding sites of hnRNP C and YB-1 at alternative cassette exons in the human genome
We first searched for the hnRNP C motif (‘TTTT’) in human alternative cassette exons and flanking introns according to ENSEMBL 76. We then searched for the YB-1 SELEX motifs (CATC and CACC) within 5 to 16 nucleotides upstream and downstream of the hnRNP C site. We similarly identified hnRNP C-binding sites in iCLIP data24,44. After filtering the iCLIP sites to alternative cassette exons and flanking introns, we searched for the YB-1 SELEX motifs (CATC and CACC) within 20 nucleotides upstream and downstream of the iCLIP sites.
Author Contributions
Ki.O. conceived the project. F.N., M.A.R. and A.M. designed experiments; F.N. performed most of the experiments; M.A.R. contributed to RNA affinity purification assay and mass spectrometry analysis; Ke.O. and J.T. contributed to RNA-EMSA and in silico analysis, respectively. F.N., M.A.R. and Ki.O. wrote the paper.
Supplementary Material
Supplementary Information
Acknowledgments
We are grateful to Dr. Akira Yokomizo (Kyushu University, Japan) for providing pCMV-YB-1-myc-nuc expression vector, to Dr. Naohiro Hashimoto (National Center for Geriatrics and Gerontology, Japan) for providing KD3 cells, and to Dr. Kentaro Taki (Nagoya University) for technical assistance on the mass spectrometry analysis. This work was supported by Grants-in-Aid from the MEXT and MHLW of Japan.
References
- Yang X., Li W., Prescott E. D., Burden S. J. & Wang J. C. DNA topoisomerase II beta and neural development. Science 287, 131–134 (2000). [DOI] [PubMed] [Google Scholar]
- Yang X. et al. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron 30, 399–410 (2001). [DOI] [PubMed] [Google Scholar]
- Lin W. et al. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 410, 1057–1064 (2001). [DOI] [PubMed] [Google Scholar]
- Arber S., Burden S. J. & Harris A. J. Patterning of skeletal muscle. Curr Opin Neurobiol 12, 100–103 (2002). [DOI] [PubMed] [Google Scholar]
- Panzer J. A., Song Y. & Balice-Gordon R. J. In vivo imaging of preferential motor axon outgrowth to and synaptogenesis at prepatterned acetylcholine receptor clusters in embryonic zebrafish skeletal muscle. J Neurosci 26, 934–947 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N. & Burden S. J. MuSK controls where motor axons grow and form synapses. Nat Neurosci 11, 19–27 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N. et al. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 135, 334–342 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B. et al. LRP4 serves as a coreceptor of agrin. Neuron 60, 285–297 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Coldefy A. S., Hubbard S. R. & Burden S. J. Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). J Biol Chem 286, 40624–40630 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masiakowski P. & Yancopoulos G. D. The Wnt receptor CRD domain is also found in MuSK and related orphan receptor tyrosine kinases. Curr Biol 8, R407–R407 (1998). [DOI] [PubMed] [Google Scholar]
- Xu Y. K. & Nusse R. The Frizzled CRD domain is conserved in diverse proteins including several receptor tyrosine kinases. Curr Biol 8, R405–R406 (1998). [DOI] [PubMed] [Google Scholar]
- Stiegler A. L., Burden S. J. & Hubbard S. R. Crystal structure of the frizzled-like cysteine-rich domain of the receptor tyrosine kinase MuSK. J Mol Biol 393, 1–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszmusz E., Patthy A., Trexler M. & Patthy L. Localization of disulfide bonds in the frizzled module of Ror1 receptor tyrosine kinase. J Biol Chem 276, 18485–18490 (2001). [DOI] [PubMed] [Google Scholar]
- Jennings C. G., Dyer S. M. & Burden S. J. Muscle-specific trk-related receptor with a kringle domain defines a distinct class of receptor tyrosine kinases. Proc Natl Acad Sci U S A 90, 2895–2899 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela D. M. et al. Receptor Tyrosine Kinase Specific for the Skeletal-Muscle Lineage - Expression in Embryonic Muscle, at the Neuromuscular-Junction, and after Injury. Neuron 15, 573–584 (1995). [DOI] [PubMed] [Google Scholar]
- Zhang B. et al. Wnt proteins regulate acetylcholine receptor clustering in muscle cells. Mol Brain 5, 7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strochlic L. et al. Wnt4 participates in the formation of vertebrate neuromuscular junction. PLoS One 7, e29976 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L., Lefebvre J. L., Gordon L. R. & Granato M. Wnt signals organize synaptic prepattern and axon guidance through the zebrafish unplugged/MuSK receptor. Neuron 61, 721–733 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon L. R., Gribble K. D., Syrett C. M. & Granato M. Initiation of synapse formation by Wnt-induced MuSK endocytosis. Development 139, 1023–1033 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L., Gordon L. R., Shtibin E. & Granato M. Temporal and spatial requirements of unplugged/MuSK function during zebrafish neuromuscular development. PLoS One 5, e8843 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Lefebvre J. L., Zhao S. & Granato M. Zebrafish unplugged reveals a role for muscle-specific kinase homologs in axonal pathway choice. Nat Neurosci 7, 1303–1309 (2004). [DOI] [PubMed] [Google Scholar]
- Zhou H., Glass D. J., Yancopoulos G. D. & Sanes J. R. Distinct domains of MuSK mediate its abilities to induce and to associate with postsynaptic specializations. J Cell Biol 146, 1133–1146 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antolik C., Catino D. H., Resneck W. G. & Bloch R. J. The tetratricopeptide repeat domains of rapsyn bind directly to cytoplasmic sequences of the muscle-specific kinase. Neuroscience 141, 87–100 (2006). [DOI] [PubMed] [Google Scholar]
- Zarnack K. et al. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell 152, 453–466 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty S. Regulation of urokinase receptor mRNA stability by hnRNP C in lung epithelial cells. Mol Cell Biochem 272, 107–118 (2005). [DOI] [PubMed] [Google Scholar]
- Lee E. K. et al. hnRNP C promotes APP translation by competing with FMRP for APP mRNA recruitment to P bodies. Nat Struct Mol Biol 17, 732–739 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCloskey A., Taniguchi I., Shinmyozu K. & Ohno M. hnRNP C tetramer measures RNA length to classify RNA polymerase II transcripts for export. Science 335, 1643–1646 (2012). [DOI] [PubMed] [Google Scholar]
- Kohno K., Izumi H., Uchiumi T., Ashizuka M. & Kuwano M. The pleiotropic functions of the Y-box-binding protein, YB-1. Bioessays 25, 691–698 (2003). [DOI] [PubMed] [Google Scholar]
- Wei W. J. et al. YB-1 binds to CAUC motifs and stimulates exon inclusion by enhancing the recruitment of U2AF to weak polypyrimidine tracts. Nucleic Acids Res 40, 8622–8636 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung L. H. et al. Diverse roles of hnRNP L in mammalian mRNA processing: a combined microarray and RNAi analysis. RNA 14, 284–296 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossbach O. et al. Crosslinking-immunoprecipitation (iCLIP) analysis reveals global regulatory roles of hnRNP L. RNA Biol 11, 146–155 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui J. et al. Intronic CA-repeat and CA-rich elements: a new class of regulators of mammalian alternative splicing. EMBO J 24, 1988–1998 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui J., Stangl K., Lane W. S. & Bindereif A. HnRNP L stimulates splicing of the eNOS gene by binding to variable-length CA repeats. Nat Struct Biol 10, 33–37 (2003). [DOI] [PubMed] [Google Scholar]
- Chiou N. T., Shankarling G. & Lynch K. W. hnRNP L and hnRNP A1 induce extended U1 snRNA interactions with an exon to repress spliceosome assembly. Mol Cell 49, 972–982 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- House A. E. & Lynch K. W. An exonic splicing silencer represses spliceosome assembly after ATP-dependent exon recognition. Nat Struct Mol Biol 13, 937–944 (2006). [DOI] [PubMed] [Google Scholar]
- Motta-Mena L. B., Heyd F. & Lynch K. W. Context-dependent regulatory mechanism of the splicing factor hnRNP L. Mol Cell 37, 223–234 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong A., Nguyen J. & Lynch K. W. Differential expression of CD45 isoforms is controlled by the combined activity of basal and inducible splicing-regulatory elements in each of the variable exons. J Biol Chem 280, 38297–38304 (2005). [DOI] [PubMed] [Google Scholar]
- Rahman M. A. et al. HnRNP L and hnRNP LL antagonistically modulate PTB-mediated splicing suppression of CHRNA1 pre-mRNA. Sci Rep 3, 2931 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang S., Felthauser A. M. & Mertz J. E. Binding of hnRNP L to the pre-mRNA processing enhancer of the herpes simplex virus thymidine kinase gene enhances both polyadenylation and nucleocytoplasmic export of intronless mRNAs. Mol Cell Biol 25, 6303–6313 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui J., Reither G. & Bindereif A. Novel functional role of CA repeats and hnRNP L in RNA stability. RNA 9, 931–936 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda A. et al. hnRNP H enhances skipping of a nonfunctional exon P3A in CHRNA1 and a mutation disrupting its binding causes congenital myasthenic syndrome. Hum Mol Genet 17, 4022–4035 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaal T. D. & Maniatis T. Multiple distinct splicing enhancers in the protein-coding sequences of a constitutively spliced pre-mRNA. Mol Cell Biol 19, 261–273 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black D. L. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72, 291–336 (2003). [DOI] [PubMed] [Google Scholar]
- Konig J. et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17, 909–915 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. et al. A complex network of factors with overlapping affinities represses splicing through intronic elements. Nat Struct Mol Biol 20, 36–45 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideau A. P. et al. A peptide motif in Raver1 mediates splicing repression by interaction with the PTB RRM2 domain. Nat Struct Mol Biol 13, 839–848 (2006). [DOI] [PubMed] [Google Scholar]
- Bland C. S. et al. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res 38, 7651–7664 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y. J. & Lee H. YB1/p32, a nuclear Y-box binding protein 1, is a novel regulator of myoblast differentiation that interacts with Msx1 homeoprotein. Exp Cell Res 316, 517–529 (2010). [DOI] [PubMed] [Google Scholar]
- Bian Y. et al. Tannic acid facilitates expression of the polypyrimidine tract binding protein and alleviates deleterious inclusion of CHRNA1 exon P3A due to an hnRNP H-disrupting mutation in congenital myasthenic syndrome. Hum Mol Genet 18, 1229–1237 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike S., Schaeffer L. & Changeux J. P. Identification of a DNA element determining synaptic expression of the mouse acetylcholine receptor delta-subunit gene. Proc Natl Acad Sci U S A 92, 10624–10628 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duclert A., Savatier N., Schaeffer L. & Changeux J. P. Identification of an element crucial for the sub-synaptic expression of the acetylcholine receptor epsilon-subunit gene. J Biol Chem 271, 17433–17438 (1996). [DOI] [PubMed] [Google Scholar]
- Ohno K., Anlar B. & Engel A. G. Congenital myasthenic syndrome caused by a mutation in the Ets-binding site of the promoter region of the acetylcholine receptor epsilon subunit gene. Neuromuscul Disord 9, 131–135 (1999). [DOI] [PubMed] [Google Scholar]
- Lee H. H. et al. Transcriptional regulation of acetylcholinesterase-associated collagen ColQ: differential expression in fast and slow twitch muscle fibers is driven by distinct promoters. J Biol Chem 279, 27098–27107 (2004). [DOI] [PubMed] [Google Scholar]
- Shiomi K. et al. CDK4 and cyclin D1 allow human myogenic cells to recapture growth property without compromising differentiation potential. Gene Ther 18, 857–866 (2011). [DOI] [PubMed] [Google Scholar]
- Wada M. R., Inagawa-Ogashiwa M., Shimizu S., Yasumoto S. & Hashimoto N. Generation of different fates from multipotent muscle stem cells. Development 129, 2987–2995 (2002). [DOI] [PubMed] [Google Scholar]
- Hashimoto N. et al. Immortalization of human myogenic progenitor cell clone retaining multipotentiality. Biochem Biophys Res Commun 348, 1383–1388 (2006). [DOI] [PubMed] [Google Scholar]
- Ohno K. et al. Myasthenic syndromes in Turkish kinships due to mutations in the acetylcholine receptor. Ann Neurol 44, 234–241 (1998). [DOI] [PubMed] [Google Scholar]
- Mayeda A. & Krainer A. R. Mammalian in vitro splicing assays. Methods Mol Biol 118, 315–321 (1999). [DOI] [PubMed] [Google Scholar]
- Shiota M. et al. Y-box binding protein-1 promotes castration-resistant prostate cancer growth via androgen receptor expression. Endocr Relat Cancer 18, 505–517 (2011). [DOI] [PubMed] [Google Scholar]
- Chao J. A., Patskovsky Y., Almo S. C. & Singer R. H. Structural basis for the coevolution of a viral RNA-protein complex. Nat Struct Mol Biol 15, 103–105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information