

Abstract
The actin filament-binding and filament-severing activities of the aplyronine, kabiramide and reidispongiolide families of marine macrolides are located within the hydrophobic tail region of the molecule. Two synthetic tail analogs of aplyronine C (SF-01 and GC-04) are shown to bind to G-actin with kd values of 285 +/−33 nM and 132 +/−13 nM, respectively. The crystal structures of actin complexes with GC-04, SF-01 and kabiramide C reveal a conserved mode of tail binding within the cleft that forms between sub-domains (SD) 1 and 3. Our studies support the view that filament severing is brought about by specific binding of the tail region to the SD1/3 cleft on the upper protomer, which displaces loop-D from the lower protomer on the same half-filament. With previous studies showing that GC-04 analog can sever actin filaments, it is argued that the shorter complex lifetime of tail analogs with F-actin would make them more effective at severing filaments compared with plasma gelsolin. Structure-based analyses are used to suggest more reactive or targetable forms of GC-04 and SF-01, which may serve to boost the capacity of the serum actin scavenging system, to generate antibody conjugates against tumor cell antigens, and to reduce sputum viscosity in children with cystic fibrosis.
Keywords: Actin, macrolide analogs, structural biology, chemical synthesis
Introduction
The spatio-temporal regulation of actin filament dynamics is essential for cell motility, cytokinesis and endocytosis, with gelsolin serving as a key regulator of filament length and generator of new barbed ends.[1–5] Marine macrolide natural products, including the aplyronines (e.g. AplC), reidispongiolide A (RedA) and kabiramide C (KabC), are also capable of severing and capping actin filaments.[6–11] Interestingly, a structurally similar dimeric macrolide has been identified within a myxobacterium, which suggests the marine secondary metabolites might actually be produced by symbiotic bacteria.[12] In almost all cases, these natural products consist of a hydrophobic tail, which we have shown to harbor the molecular determinants for actin binding, filament severing and cytotoxicity, and a larger ring that binds to the surface of actin and contributes to the strong binding of the drug to actin.[13] The major focus of the present study is to improve our understanding of the molecular basis of macrolide-mediated binding of actin filaments.[6,7] These studies are important as a number of groups are exploring the use of marine macrolides as anti-cancer drugs. [9,10,11] Unfortunately, their potential as cancer drugs is low, as they are isolated from protected marine organisms, while their chemical synthesis is challenging [14–16]. Nonetheless, the unique specificity and potency of marine macrolides make them useful models for the design of synthetic drugs that target actin.[17–19]
Cytoplasmic actin is released into the serum following stroke, necrosis and trauma, where it polymerizes within fibrin clots at the site of injury to form a viscous mesh.[14] Extracellular actin is normally cleared by the serum actin scavenging system, the most important member of which is plasma gelsolin.[20] However, since plasma gelsolin is capable of carrying out a single severing event in serum,[20] the capacity of the serum actin scavenging system may be significantly reduced following major trauma, thereby compromising patient recovery.[15,16,20,21] Troublingly, a correlation exists between the level of plasma gelsolin and patient survival.[21] Stossel and co-workers have suggested injecting recombinant gelsolin in at-risk patients in order to boost the capacity of their serum actin scavenging systems.[16] An alternative strategy would involve administering a synthetic mimetic of plasma gelsolin; this approach might prove more effective than a protein-based therapeutic, as a small molecule mimetic will have <1% the mass of plasma gelsolin, and could be administered orally. Unfortunately, neither intact KabC nor AplC nor their derivatives are suitable replacements for plasma gelsolin, as they are resource-protected and are potent cytotoxins. [7,8,22]
This study is motivated by our finding that the tail region of RedA accounts for the severing activity of the intact macrolide.[13] This feature could be exploited for the development of a small molecule drug that removes actin filaments associated with diseased states.[14–16,20,21] Tail-region analogs of RedA and AplC (Figure 1) are synthetically much more accessible and contain all the structural determinants required for the binding and severing of actin filaments.[13,17–19] Moreover, these small molecules can accommodate targeting groups to restrict their severing activity to the plasma, boosting the capacity of the actin filament severing system, or they may be linked to antibodies to target tumor cells.
Figure 1. Molecular structures of synthetic tail analogs and macrolides.

Analog 1 and 2, GC-04 and SF-01, aplyronine C (AplC), kabiramide C (KabC) and reidispongiolide A (RedA).
Results and Discussion
Binding studies of Actin with synthetic tail analogs
Previous studies have shown that the tail region of the aplyronine and reidispongiolide families of marine macrolides harbors the actin filament-binding and severing activities of the parent molecule and induces cytotoxicity.[9,13] A quantitative analysis of the interaction between several synthetic tail regions of these macrolides and G-actin is carried out using the Prodan-actin fluorescence assay.[23,24] (Figure 2). Included in these binding studies are two tail analogs that are close to the original length of the AplC tail, called SF-01 and GC-04, two shorter tail-analog versions of AplC, and KabC (Figure 1). A 1 μM solution of Prodan-actin is titrated with 0–2 μM SF-01 (Figure 2A) or GC-04 (Figure 2B), with the binding being measured by a decrease in the intensity of Prodan-actin emission for the SF-01 and GC-04 samples (insets to Figures 2A,B respectively). The shorter tails (analogs 1 and 2) do not change the emission of Prodan-actin even when added at 20 μM (Supplementary figures S1 and S2 respectively). A positive control study shows that KabC (Supplementary figure S3) brings about a significant quenching of Prodan-actin fluorescence that is similar in profile to that recorded for SF-01 and GC-04 (Fig 2A,B). A control study in which small volumes of dimethylformamide (DMF) are added to a solution of Prodan-G-actin shows that the co-solvent has no significant effect on the emission intensity (Supplementary Figure S4). The linear change of the normalized emission intensity of Prodan-actin as a function of SF-01 and GC-04 (insets to Figures 2A,B) indicates stoichiometric binding, which is confirmed by the fact that the plateau value is reached at 1 μM for SF-01 and GC-04. A 1:1 complex is formed in each case as evidenced by the first-order binding curves (Figure 2C).
Figure 2. Determination of binding stoichiometry and dissociation constants for actin complexes with SF-01 and GC-04.

Fluorescence emission spectra of 1 μM Prodan-G-actin in standard G-buffer as a function of SF-01 (A) and GC-04 (B) at the following concentrations: (a) 0 μM; (b) 0.25 μM; (c) 0.50 μM; (d) 0.74 μM; (e) 0.99 μM; (f) 1.23 μM; (g) 1.48 μM; (h) 1.72 μM; (i) 1.96 μM. Inset figures (Figures. 2a,b) show the normalized decrease in emission intensity of Prodan-G-actin as a function of ligand concentration. The stoichiometry of the complex between Prodan-G-actin and each ligand is indicated as being close to 1:1. (C), Equilibrium binding of SF-01 and GC-04 to Prodan-actin is measured by reducing the concentration of Prodan-G-actin to 200 nM and by varying the concentrations of SF-01 and GC-04 from 20 nM to 2 μM (20, 50, 100, 150, 200, 250, 300, 350, 400, 500, 750, 1000, 1250, 1500, 1750 and 2000 nM) – the spectra and sum of intensities at each data point are shown for SF-01 and GC-04 in the supplementary figures S5 and S6 respectively). The dissociation constants for each complex is calculated by fitting the progress of complex formation (the negative of the difference of the sums of the integrated intensity of the Prodan-G-actin emission spectrum) at function of the free ligand of each ligand, as shown on the y-axis of figure 2C. The best fits for the kd values are 285 +/−33nM nM and 132 +/− 13 nM nM for SF-01 and GC-04 respectively.
Equilibrium binding conditions are achieved for the same interactions by reducing the concentration of Prodan-actin to 200 nM, and by carrying out the titration with SF-01 and GC-04 over a concentration range from 20 nM to 2 μM. The emission spectra for the SF-01 and GC-04 titrations are shown in supplementary figures, S5 and S6 respectively. The dissociation constants (kd) for Prodan-G-actin in complexes with SF-01 and GC-04 are calculated as detailed in the Methods section. In the case of SF-01 the best fit to the binding data is obtained with a kd of 284.7 ± 32.6 nM, while for GC-04 the kd is calculated at 132.1 ± 13.4 nM. The fits to each set of binding data are shown in figure 2C - the associated emission spectra and the integrated intensities of Prodan-G-actin emission for the SF-01 and GC-04 equilibrium studies are shown in supplementary figures S5 and S6, respectively. The calculated kd values of SF-01 and GC-04 are comparable to those measured for G-actin complexes with other small molecule drugs such as cytochalasin D, and for actin-binding proteins including Thymosin β4.[3,8,9,25–27] The kd value calculated for the GC-04 in complex with Prodan-G-Actin in this study is lower than that reported by Perrins [13] (kd values of 132 nM vs 1 μM respectively). We believe the stronger binding found in this study is a result of our adoption of a new G-actin purification procedure, which generates almost 10 times more G-actin per gram compared to the method by Perrins, and yields G-actin with improved binding properties.13 An indication of the improved quality of G-actin is our finding that it can be used for crystallization trials without further purification. The purity of the G-actin preparation is important for kd determinations since the presence of small actin binding proteins will reduce the affinity of GC-04 for G-actin. Another potential contribution to the difference in kd values is the observation that the plot of the Prodan-G-actin/GC-04 interaction shown in Perrins et al (2008) [13] shows evidence of cooperativity ie a Hill coefficient of >1. The fits shown in figure 2C of this study use a Hill coefficient of 1.0, a value mandated by the 1:1 binding stoichiometry.
The kd (sub μM) dissociation constants measured for complexes of G-actin with GC-04 and SF-01 are expected, as their crystal structures show extensive and specific interactions between the polar and apolar groups on the tail analogs with residues lining the cleft between SD1/SD3. Interestingly, the interaction between KabC (and related macrolides) and actin, like plasma gelsolin, is far stronger with kd values in the sub-nM range.[7,13] This latter property accounts for the very long-lived complex (~ hours) between the intact macrolide and the barbed-end of the severed filament, which effectively limits its F-actin severing activity to a single event.[7] Significant to the development of a small molecule mimetic of plasma gelsolin, the weaker binding of SF-01 and GC-04 to actin filaments will shorten the time constant for the actin filament-tail analog complex to 0.35 seconds and 0.76 seconds respectively, assuming an on-rate of 107 M−1s−1. Once dissociated, the free drug may carry out further severing of the filament. We chose to use an on-rate of 107 M−1s−1 because our earlier study indicated that the tail regions of trisoxazole macrolides bind slowly to G-actin.7 In comparison, the time constants calculated using a diffusion-controlled on-rate of 108 M−1s−1 or a slower rate of 106 M−1s−1 for GC-04 binding to G-actin would change to 0.035 seconds and 3.5 seconds, respectively. The range of estimated time constants considered above suggests that drugs derived from the tail region of AplC carry out more severing events per unit time compared to the intact macrolides or gelsolin.
Structural analyses of Actin complexes with GC-04 and SF-01
The structure of actin has been described in various complexes with proteins that bind to either G- or F-actin[28] and with small molecule natural products.[6,11,28–31] This study describes the crystal structures of actin complexes with synthetic tail analogs GC-04 and SF-01, and the natural macrolide KabC, which serves as a control in our structural analyses of actin-tail analog complexes. A new approach to generate crystals of actin in complexes with KabC and tail analogs of AplC is introduced that involves treating a concentrated solution of F-actin in F-buffer with a slight molar excess of the drug. Physiological levels of Mg2+ and KCl are maintained at all stages of the crystallization in order to preserve the conformation of an F-actin-like protomer, in part by preserving specific associations between these cations and the actin-drug complex. Subsequent analysis of the three crystal structures of actin in complexes with GC-04, SF-01 and KabC confirms the presence of an F-actin-protomer like conformation rather than the G-actin conformation.[28,32]
A summary of the crystallography statistics for the three complexes is shown in Table 1. The electron densities for GC-04 and SF-01 within the cleft that forms between SD1 and SD3 (Figures. 3a,c) allow us to unambiguously confirm their stereo-chemical configurations for the new synthetic ligands (Figures 3b,d). The structures of actin complexes with GC-04 and SF-01 (Figs. 3a,c) reveal essentially an identical conformation with an RMS deviation between the Cα atoms of actin-GC-04 and actin-SF-01 of only 0.38 Å. Actin in complexes with most proteins or drugs is usually crystallized with a molecule of ATP at the nucleotide site and a disordered loop-D on SD2. Exceptions to this trend include the DNAse-G-actin complex[33] and the TMR-maleimide conjugate of G-actin.[32] Interestingly, the nucleotide-binding site of actin in the complexes with GC-04 and SF-01 is occupied by ADP, even though the ATP was present during all stages of the purification (Figures 3a,c), and ATP is bound to the actin in the KabC complex. A plausible interpretation of this finding is that the macrocyclic ring of KabC helps to trap or prevent hydrolysis of ATP by stabilizing the closed form of the nucleotide-binding site. The similarity in the overall structures of actin in the SF-01, GC-04 and KabC complexes supports a previous study that showed the structures of the ATP and ADP bound actin protomer to be essentially identical.[34]
Table 1.
Statistics for the X-ray data collection and refinement of Actin-SF-01, Actin-GC-04 and Actin-KabC complexes.
| Actin-SF-01 | Actin-GC-04 | Actin-KabC | |
|---|---|---|---|
| A. Data collection | |||
| Wavelength (Å) | 0.97741 | 0.97741 | 0.976 |
| Resolution range (Å) | 50 – 2.90 (2.95 – 2.90) | 50 – 2.95(3.00 – 2.95) | 40 – 1.40 (1.45 – 1.40) |
| Detector Distance (mm) | 310 | 310 | 180 |
| Φ (deg.) collected/ΔΦ (deg.) | 180/1.0 | 360/0.5 | 240/1.0 |
| Exposure time (seconds) | 3 | 3 | 3 |
| Temperature of collect (Kelvin) | 100 | 100 | 100 |
| B. Data statistics | |||
| Space group | P21 | P21 | C2 |
| Unit-Cell parameters (Å) | a=54.65, b=67.34 and c=207.85; β=92.42° | a=54.58, b=70.35 and c=107.67; β=104.7° | a=58.35, b=55.55 and c=104.10; β=92.77° |
| Total reflections | 96340 | 60967 | 269215 |
| Unique reflections | 28609 | 17401 | 59011 |
| Multiplicity | 3.4 (3.3) | 3.5 (2.7) | 4.6 (3.2) |
| Data completeness (%) | 84.2 (83.9) | 98.1 (90.3) | 90.0 (59.7) |
| I/σ(I) | 6.1 (1.64) | 5.96 (1.62) | 8.0 (1.78) |
| Rsyma (%) | 22.8 | 26.7 | 7.6 |
| C. Structure Refinement | |||
| Rfactorb (%) | 22.6 | 22.2 | 16.9 |
| Rfreec (%) | 28.1 | 27.7 | 19.4 |
| RMS from ideal geometry | |||
| Bond lengths (Å) | 0.004 | 0.003 | 0.009 |
| Bond angles (°) | 0.780 | 0.710 | 1.241 |
| Protein residues per AUd | 1480 (4 copies) | 740 (2 copies) | 371 (1 copy) |
| ADP | 4 | 2 | - |
| ATP | - | - | 1 |
| Ca2+ | 4 | 2 | 2 |
| Ramachandran Plot | |||
| Favored region (%) | 97.7 | 97.8 | 98.9 |
| Outliers region (%) | 0 | 0 | 0 |
Rsym = Σhkl Σi|Ii(hkl) − <I(hkl)>|/Σhkl Σi Ii(hkl), where Σhkl denotes the sum over all reflections and Σi is the sum over all equivalent and symmetry-related reflections.
Rfactor = Σ|Fobs − Fcalc|/ΣFobs.
Rfree = Rfactor for 5 % of the data were not included during crystallographic refinement.
AU = Asymmetric unit.
Figure 3. The structures of Actin in complexes with AplC tail analogs.

(a,c,e) The structures of Actin-SF-01, Actin-GC-04 and Actin-GS-1[47] in a cartoon representation respectively. (b,d) Stereoviews of the electron densities for the SF-01 and GC-04 tail analogs of AplC and RedA. The 2mFo-DFc maps are calculated with σA-weighted coefficients and contoured at 1 σ. (f) Superposition of the structures of Actin-SF-01, Actin-GC-04 and Actin-GS-1. The SF-01 and GC-04 ligands are shown bound to the cleft between SD1 and SD3. The same binding site between SD1/3 is identified for gelsolin domain 1 and loop-D of SD2 of adjacent longitudinal actin protomer in the filament.
The structure of GC-04 bound to actin (Figure 3c and Figure 4a) is closely matched to those reported for the tail regions of RedA[10] and KabC.[6] A total of 14 residues located within the SD1/SD3 cleft of actin are found to make hydrophobic contacts with the GC-04 tail region. The N-vinyl formamide group of GC-04, which is buried deep within the cleft, interacts with residues Tyr133, Val139, Tyr143, Gly168 and Tyr169 from SD1/SD3 of actin. The aliphatic region of the GC-04 tail interacts with residues Tyr143, Arg147, Thr148, Leu346, Leu349, Thr351 and Met355. The cyclic PMP acetal group of GC-04 interacts with the main chain of the residues Tyr143, Ala144, Gly146 and Ile345 (Figure 4a). Of considerable interest is the finding that the binding site of the PMP group of GC-04 (Figure 4c) is similar to the locus occupied by the ester linkages between the tail regions and the macrolide rings of KabC and RedA. These structural studies support our previous study that suggest the PMP group may mimic, in small part, the binding of the macrolide ring of RedA to the entrance to the SD1/SD3 cleft.[13]
Figure 4. Tail-analogs interactions with actin.
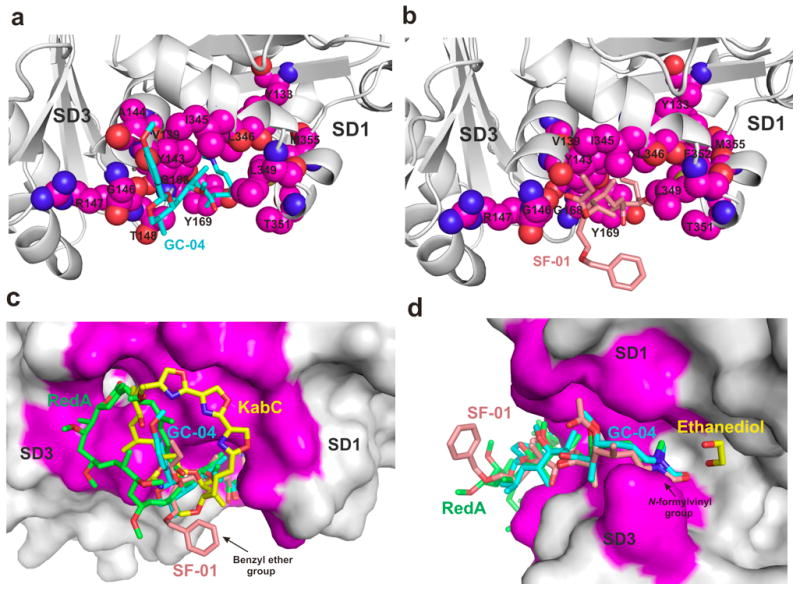
(a,b) Binding site between SD1/SD3 for AplC tail analogs GC-04 and SF-01, respectively. Similar hydrophobic contacts are observed for the aliphatic regions of GC-04 and SF-01. The tail analogs are shown as a stick representation and the residues contacting the ligands from SD1/SD3 cleft are shown as spheres. (c) The PMP group of GC-04 is located at a similar position to the locus occupied by the ester linkage between the tail region and the macrolide ring. The structure of actin-GC-04 supports the view that the cyclic PMP acetal group can mimic in small part the binding of the macrocyclic ring of RedA, AplC and KabC to the entrance to the SD1/SD3 cleft. Also, the position of the macrocyclic ring can be used to improve the binding affinity of these drugs, as observed for the fully elaborated macrolide rings in RedA, AplC and KabC. The benzyl ether group from SF-01 projects out of the cleft and is exposed to the solvent. The solvent exposed phenyl ring of SF-01 is conveniently positioned to place a targeting groups or reactive groups to the tail without impacting the binding of the derivative to actin. (d) The structure of GC-04 and SF-01 bound to actin closely matches that found for the tail-regions of AplA[35], RedA[10] and KabC. An ethanediol molecule was found between reidispongiolide A and actin. The position of ethanediol could be used to create a new set of ligands around the N-vinyl formamide group.
The structure of the actin-SF-01 complex (Figure 3a and Figure 4b) shows that the tail region engages in similar interactions to those found for GC-04, with a total of 13 residues within the SD1/SD3 cleft forming hydrophobic contacts with SF-01. The N-vinyl formamide group of SF-01 interacts with residues Tyr133, Val139, Tyr143, Gly168 and Tyr169, while the aliphatic portion interacts with Tyr143, Gly146, Arg147, Tyr169, Leu346, Leu349, Thr351, Phe352 and Met355. The propionate group at C-11 interacts with Tyr143, Gly146 and Ile345 (Figure 4b). Unexpectedly, the actin-SF-01 complex shows that the entrance to the cleft between SD1 and SD3 accommodates the propionate group at C-11, which follows the same path as the PMP group. In contrast, the other oxygen, which is part of the benzyl ether group, projects out of the cleft and is exposed to the aqueous solvent (Figure 3a and Figure 4b,d). While this configuration is unfavorable, it only has a modest effect on the kd value of the actin complex with SF-01 compared to GC-04 (Fig. 2C). Interestingly, the location of the benzyl group of SF-01 (Figures 4c,d) could serve as a convenient and non-perturbing solvent-exposed site to introduce targeting or reactive groups onto the tail analog. For example, the introduction of a maleimide group at this site could be used to link SF-01 to a cancer-targeting antibody, or a PEG group to restrict the drug to the plasma where it may sever extracellular actin filaments.
Structure-guided design of macrolide-tail analogs
The results from our structural studies on actin complexes with GC-04 and SF-01 support our view that the tail region of AplC and related macrolides can function as a small mimetic of plasma gelsolin. The severing activity of these tail analogs may be improved however by increasing their affinity for actin by introducing additional interacting groups onto the tail. Using the high-resolution structure of the actin-KabC complex as our guide, and the position of the PMP group of GC-04 as a starting point, we identified a region at the entrance of the SD1 and SD3 cleft that could accommodate a hydrophobic extension to the cyclic PMP acetal group without impacting the binding of the tail region to the SD1/SD3 cleft (Figures 4c,d). The additional groups on this extended tail region could engage in van der Waals contacts with residues Gly23, Asp24, Asp25 and Ile341 on SD1, and residues Pro-332, Glu-334 found on the surface of SD3. These residues at the entrance of the SD1 and SD3 cleft take part in hydrophobic interactions with the macrocyclic rings of RedA and KabC (Figures 4c,d). Another possibility to increase the affinity of the tail for actin is to introduce polar groups close to the N-vinyl formamide group of GC-04 and SF-01. Since the N-vinyl formamide group does not make any direct hydrogen bonds with polar residues on actin, additional hydrogen bonds between ligand-protein could potentially improve the binding affinity. We also note the structure of the Actin/AplC complex[35] identifies an ethanediol solvent molecule that should engage in hydrogen bonds with the nitrogen atom from the main chain of Ile136 and with the hydroxyl group from Tyr133, both of which are located in a cavity that exists between the N-vinyl formamide group of AplC and actin (Figure 4d). Thus, the affinity of the tail analog for actin could be improved by introducing polar groups close to the N-vinyl formamide group to mimic the interaction between ethanediol and actin. A recent study by Kita and co-workers showed that modifications to the N-formyl vinyl group of AplA do not overly perturb the interaction of the adduct with actin.[36]
The structure of the actin-SF-01 complex reveals the terminal benzyl group projects out of the cleft and is exposed to the solvent. While energetically unfavorable, this geometry could prove useful in the design of a new class of cell-targetable tail region drugs. In particular, an activated or reactive group such as an acrylamide, maleimide, NHS-ester, or an azide or alkyne introduced via a carboxy-benzyl version of SF-01 or a further optimized form of that tail, would make it possible to prepare monoclonal antibody conjugates of the tail analog that recognize a tumour marker. A related SF-01 derivative capable of being aerosolized could form the basis of a drug treatment to reduce actin filaments that form in the sputum of children with cystic fibrosis.
Conclusion
Actin-targeting marine macrolides are widely used to inhibit actin filament dynamics in cells, resulting in apoptosis.[6,8] This property has led several research groups to suggest that AplA, RedA, KabC and related macrolides could be used as anti-tumour drugs.[9] Unfortunately, actin-targeted macrolides are invariably cell permeable and cytotoxic undermining their potential as anti-cancer drugs. Moreover, these macrolides are isolated from protected marine organisms, while their chemical synthesis is challenging, and so it is highly unlikely they will ever be considered for clinical trials. We have shown through biochemical, cell biological[13] and structural studies herein that synthetic versions of the AplC tail region harbor the determinants for the actin-binding and severing activities of the parent molecule. The weaker binding of the tail analog with actin compared to the intact macrolide reduces the lifetime of their complexes with G-actin to sub-second time scales. This property should allow SF-01 and GC-04 to carry out multiple severing events on a filament per unit time compared to the tighter binding parent macrolide or plasma gelsolin. The actin binding and filament severing properties of tail analogs could be further improved by adding targeting groups to restrict the distribution of the tail to the serum or to cavities in the lung, or to link the tail to antibodies that recognize specific tumor markers. These improvements could be used to boost the greatly reduced levels of plasma gelsolin in patients recovering from stroke, trauma or transplantation or to target actin filaments in the sputum of children with cystic fibrosis.
Experimental Section
Actin Purification
Actin is purified from rabbit muscle by using an improvement to the method by Spudich and Watt.[27] In particular, actin is extracted from the same acetone powder in six sequential cycles involving stirring with standard G-buffer (2 mM Tris, 0.2 mM CaCl2, 0.2 mM ATP and 1 mM DTT and adjusted to pH 8.0 using 1M KOH) for 5 minutes followed by centrifugation at 10,000 g for 10 minutes. The bulk of the actin is obtained from the 2nd through the 5th extractions. The pooled extracts are passed through multiple 0.22 um filters (Millipore) to remove any debris or protein aggregates. The final G-actin solution is polymerized by adding MgCl2 and KCl to 2 mM and 100 mM, respectively, and is incubated for 2 hours at 20°C. KCl to a final concentration of 0.6 M is added to the F-actin solution and the sample is centrifuged at 100,000 g for 1 hour. The pellet is resuspended in G-buffer and dialyzed overnight at 4°C. The protein solution is centrifuged at 100,000 g for 1 hour - the supernatant is subjected to two further cycles of polymerization and depolymerization. The final G-actin solution is adjusted to 5 mg/ml in G-buffer and analysed by SDS-PAGE. The G-actin obtained from this purification procedure is sufficiently pure to carry out a crystallization screen. We note that the Prodan-G-actin prepared using this new procedure exhibits a higher affinity for GC-04 compared to the G-actin preparation used by Perrins 13. A 10 ml solution of G-actin is polymerized for 3 hours by adding MgCl2 and KCl to 2 mM and 0.1 M respectively. The extremely viscous F-actin preparation is treated with a 1.2 molar equivalent of SF-01, GC-04 or KabC, which results in a decrease in viscosity that is complete within 60 minutes. These samples are centrifuged at 100,000 g for 30 minutes and the supernatants are used for crystallization screens.
Synthesis of tail region analogs of AplC and RedA
The AplC and RedA tail analogs (Analog 1, Analog 2, SF-01 and GC-04; Figure 1) are prepared according to Paterson and co-workers.[18,19] The compounds were dissolved in DMSO to a concentration of 2 mM and stored at −20 °C.
Binding of tail region analogs to Prodan-G-actin
Purified G-actin is labelled with the thiol-reactive probe Acrylodan (Invitrogen) in a G-buffer lacking DTT.[24,37,38] Protein concentrations are measured by using the Bradford assay[39] and labelling efficiencies are measured by using an UV-1601PC UV-Visible Spectrophotometer (Shimadzu), using an extinction coefficient of actin at 290 nm (A290nm of 0.63 = 1mg/ml) [37] and extinction coefficients of Prodan at 375 nm of 1.85 × 104 M−1 cm−1 and at 290 nm of 1.85 × 104 M−1 cm−1.[24] Labelling efficiencies (Prodan:actin) are typically measured at ~30%. The fluorescence emission spectrum of Prodan-G-actin in standard G-buffer is measured on an Aminco-Bowman Series 2 (AB2) Fluorimeter (ThermoSpectronic). Fluorescence based titrations of Prodan-G-actin binding to drugs are carried out on a solution of 1 μM Prodan-G-actin in standard G-buffer in the presence of different concentrations of each tail-analog or KabC, as indicated in the legend to figure 2A–C. The progress of complex formation is recorded by using the integrated intensity or a normalized change in intensity of the Prodan-G-actin emission spectrum at each concentration of ligand.
Determination of dissociation constants
The dissociation constants (kd) for actin in complex with SF-01 and GC-04 are calculated by using an iterative unweighted least-squares routine to the Hill equation
The binding data is fit using Igor Pro (Wavemetrics) where y is the amount of G-actin bound to the tail analog and is recorded using the negative of the difference in the integrated intensity of Prodan-G-actin spectrum as a function of the free concentration of SF-01 and GC-04. Further, n is fixed to 1.0 to infer first-order ligand binding, a is the base value at y=0, b is the maximum value at full saturation, and kd is the dissociation constant in nM.
Calculation of complex lifetimes
The time constants or average lifetimes of G-actin complexes with SF-01 and GC-04 are estimated as follows. The off-rate (k−1) for each complex is calculated using the equation where k1 is the on-rate and kd is the dissociation constant. Given a kd value of 132 nM for the GC-04-Prodan-G-actin complex, and assuming an on-rate (k1) for the binding of GC-04 to Prodan-G-actin of 107 M−1s−1, which is based on our previous study that showed the tail region binds slowly to actin,7 we calculate an off-rate (k−1) of 1.32 s−1. The time constant or average lifetime for the complex is the reciprocal of k−1 ie 0.76 seconds. In the case of SF-01 the kd is calculated at 285 nM, the off-rate is 2.85 s−1 and its complex with G-actin has a time constant of 0.35 seconds.
Crystallization of actin complexes with GC-04 and SF-01
The actin-SF-01, Actin-GC-04 and actin-KabC complexes are concentrated and further dialyzed against F-buffer (2 mM Tris buffer pH 8.0 containing 0.2 mM ATP, 0.2 mM CaCl2, 2 mM MgCl2, 0.1M KCl, 1mM DTT and 1mM NaN3). Actin-SF-01, actin-GC-04 and actin-KabC are concentrated to 15 mg ml−1 and screened using the sparse matrix method[40] with a Phoenix Robot (Art Robbins Instruments, Sunnyvale, CA) using the following crystallization screens: Berkeley Screen (set of solution created at Lawrence Berkeley National Laboratory, Berkeley, CA) Crystal Screen I and II, Index, PEG/Ion, SaltRx and PEGRx (Hampton Research, Aliso Viejo, CA). The optimum condition for crystallization of actin-SF-01 and actin-GC-04 is found in 0.1 M BIS-TRIS pH 5.5, 25% of Polyethylene glycol 3,350. The actin-KabC complex is crystallized in 0.1 M MES pH 5.5, 0.1 M CaCl2, 12% 1,6-Hexanediol and 17 % of Polyethylene glycol 1,500. Crystals are obtained after 2 days by the sitting-drop vapor-diffusion method with the drops consisting of a mixture of 0.7 μl of protein solution and 0.5 μl of reservoir solution.
X-ray data collection and structure determination
Crystals are placed in a reservoir solution containing 20% glycerol and then flash-frozen in liquid nitrogen. Native data sets for the actin-SF-01, actin-GC-04 and Actin-KabC complexes were collected at the Berkeley Center for Structural Biology beamline 5.0.1 of the Advanced Light Source at Lawrence Berkeley National Laboratory (LBNL). The diffraction data are recorded using an ADSC-Q315r detector. The data sets are collected using 180° of oscillation, with Δφ = 1° and a wavelength of 0.97741 Å. The datasets are processed using the program HKL-2000.[41]
The crystal structures of actin-SF-01, actin-GC-04 and actin-KabC are determined by the molecular-replacement method with the program PHASER[42] within the Phenix suite[43], using as a search model the previous structure of actin-KabC (PDB code 1QZ5).[6] The atomic positions obtained from molecular replacement and the resulting electron density maps are used to build the Actin-SF-01 and Actin-GC-04 structures and initiate crystallographic refinement and model rebuilding. Structure refinement is performed using the phenix.refine program.[44] Translation-libration-screw (TLS) refinement with each protein chain as a single TLS group is used in the process. Manual rebuilding using COOT[45] and addition of water molecules allowed for construction of the final models. Five percent of the data are randomly selected for cross validation. Root-mean-square deviation differences from ideal geometries for bond lengths, angles and dihedrals are calculated with PHENIX.[43] The overall stereochemical quality of the final models are assessed using the program MOLPROBITY.[46]
Supplementary Material
Acknowledgments
This work is supported by grants awarded to GM from the National Institutes of Health (5R01EB005217-02), and to IP from the EPSRC (GR/S19929/01). We are grateful to the staff of the Berkeley Center for Structural Biology at the Advanced Light Source of Lawrence Berkeley National Laboratory. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health and National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Author Contributions
J.H.P and G.M. wrote the manuscript with significant contributions from all other authors. G.M. and C.P. carried out the purification of actin complexes and the actin binding studies on the AplC tail-region analogs. ACH carried out analyses of binding data. S.J.F., G.C. and I.P carried out the synthesis of AplC and RedA tail regions. J.H.P., N.W.M. and P.D.A carried out the X-ray crystallography experiments. G.M. led the project. All the authors made contributions to the final manuscript.
Conflict of interest
The authors declare they have no conflict of interest.
Data deposition: The atomic coordinates and structure factors of actin-GC-04, actin-SF-01 and actin-KabC are deposited in the Protein Data Bank with accession codes 4K43, 4K42 and 4K41, respectively.
References
- 1.Pollard TD, Borisy GG. Cell. 2003;112(4):453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 2.Choidas A, Jungbluth A, Sechi A, Murphy J, Ullrich A, Marriott G. Eur J Cell Biol. 1998;77:81–90. doi: 10.1016/S0171-9335(98)80075-7. [DOI] [PubMed] [Google Scholar]
- 3.Roy P, Raijfur Z, Jones D, Marriott G, Loew L, Jacobson K. J Cell Biol. 2001;153:1035–1048. doi: 10.1083/jcb.153.5.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldschmidt-Clermont PJ, Furman MI, Wachsstock D, Safer D, Nachmias VT, Pollard TD. Mol Biol Cell. 1992;3:1015–1024. doi: 10.1091/mbc.3.9.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartwig JH, Bokoch GM, Carpenter CL, Janmey PA, Taylor LA, Toker A, Stossel TP. Cell. 1995;82(4):643–653. doi: 10.1016/0092-8674(95)90036-5. [DOI] [PubMed] [Google Scholar]
- 6.Klenchin VA, Allingham JS, Kin R, Tanaka J, Marriott G, Rayment I. Nat Struct Biol. 2003;10:1058–1063. doi: 10.1038/nsb1006. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka J, Yan Y, Choi J, Bai J, Klenchin VA, Rayment I, Marriott G. Proc Natl Acad Sci USA. 2003;100:13851–13856. doi: 10.1073/pnas.2233339100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fenteany G, Zhu S. Curr Topics Med Chem. 2003;3:593–616. doi: 10.2174/1568026033452348. [DOI] [PubMed] [Google Scholar]
- 9.Kigoshi H, Suenaga K, Takagi M, Akao A, Kanematsu K, Kamei N, Okugawa Y, Yamada K. Tetrahedron. 2002;58:1075–1102. [Google Scholar]
- 10.Allingham JS, Zampella A, D’Auria MV, Rayment I. Proc Natl Acad Sci USA. 2005;102:14527–14532. doi: 10.1073/pnas.0502089102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Auria MV, Paloma LG, Minale L, Zampella A, Verbist JF, Roussakis C, Debitus C, Patissou J. Tetrahedron. 1994;50:4829–4834. [Google Scholar]
- 12.Jansen R, Steinmetz H, Sasse F, Schubert WD, Hagelüken G, Albrecht SC, Müller R. Tetrahedron Letters. 2008;49:5796–5799. [Google Scholar]
- 13.Perrins RD, Cecere G, Paterson I, Marriott G. Chem & Biol. 2008;15:287–294. doi: 10.1016/j.chembiol.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 14.Dahl B, Schiødt FV, Ott P, Gvozdenovic R, Yin HL, Lee WM. Shock. 1999;12(2):102–104. doi: 10.1097/00024382-199908000-00002. [DOI] [PubMed] [Google Scholar]
- 15.DiNubile MJ, Stossel TP, Ljunghusen OC, Ferrara JL, Antin JH. Blood. 2002;100:4367–4371. doi: 10.1182/blood-2002-06-1672. [DOI] [PubMed] [Google Scholar]
- 16.Christofidou-Solomidou M, Scherpereel A, Solomides CC, Christie JD, Stossel TP, Goelz S, DiNubile MJ. Journal of investigative medicine. 2002;50:54–60. doi: 10.2310/6650.2002.33518. [DOI] [PubMed] [Google Scholar]
- 17.Yeung KS, Paterson I. Angew Chem Int Ed. 2002;41:4632–4653. doi: 10.1002/anie.200290057. [DOI] [PubMed] [Google Scholar]
- 18.Paterson I, Ashton K, Britton R, Cecere G, Chouraqui G, Florence GJ, Knust H, Stafford J. Chem Asian J. 2008;3:367–387. doi: 10.1002/asia.200700357. [DOI] [PubMed] [Google Scholar]
- 19.Paterson I, Fink SJ, Lee LYW, Atkinson SJ, Blakey SB. Org Lett. 2013;15:3118–3121. doi: 10.1021/ol401327r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith DB, Janmey PA, Sherwood JA, Howard RJ, Lind SE. Blood. 1988;72:214–218. [PubMed] [Google Scholar]
- 21.Mounzer KC, Moncure M, Smith YR, Dinubile MJ. Am J Respir Crit Care Med. 1999;160:1673–1681. doi: 10.1164/ajrccm.160.5.9807137. [DOI] [PubMed] [Google Scholar]
- 22.Matsunaga S, Fusetani N, Hashimoto K, Koseki K, Noma M. J Am Chem Soc. 1986;108:847–849. [Google Scholar]
- 23.Petchprayoon C, Suwanborirux K, Tanaka J, Yan YL, Sakata T, Marriott G. Bioconjugate Chem. 2005;16:1382–1389. doi: 10.1021/bc050006j. [DOI] [PubMed] [Google Scholar]
- 24.Marriott G, Zechel K, Jovin TM. Biochemistry. 1988;27:6214–6220. doi: 10.1021/bi00417a004. [DOI] [PubMed] [Google Scholar]
- 25.Carlier MF, Pantaloni D. Journal of Biological Chemistry. 2007;282(32):23005–23009. doi: 10.1074/jbc.R700020200. [DOI] [PubMed] [Google Scholar]
- 26.Lal AA, Korn ED. Journal of Biological Chemistry. 1985;260(18):10132–10138. [PubMed] [Google Scholar]
- 27.Goldschmidt-Clermont PJ, Machesky LM, Baldassare JJ, Pollard TD. Science. 1990;247:1575–1578. doi: 10.1126/science.2157283. [DOI] [PubMed] [Google Scholar]
- 28.Dominguez R. Trends Biochem Sci. 2004;29:572–578. doi: 10.1016/j.tibs.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Allingham JS, Tanaka J, Marriott G, Rayment I. Org Lett. 2004;6:597–599. doi: 10.1021/ol036458y. [DOI] [PubMed] [Google Scholar]
- 30.Klenchin VA, King R, Tanaka J, Marriott G, Rayment I. Chemistry & Biology. 2005;12:287–291. doi: 10.1016/j.chembiol.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 31.Hagelueken G, Albrecht SC, Steinmetz H, Jansen R, Heinz DW, Kalesse M, Schubert WD. Angewandte Chemie International Edition. 2009;48:595–598. doi: 10.1002/anie.200802915. [DOI] [PubMed] [Google Scholar]
- 32.Otterbein LR, Graceffa P, Dominguez R. Science. 2001;293:708–711. doi: 10.1126/science.1059700. [DOI] [PubMed] [Google Scholar]
- 33.Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC. Nature. 1990;347:37– 44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- 34.Rould MA, Wan Q, Joel PB, Lowey S, Trybus KM. Journal of Biological Chemistry. 2006;281(42):31909–31919. doi: 10.1074/jbc.M601973200. [DOI] [PubMed] [Google Scholar]
- 35.Hirata K, Muraoka S, Suenaga K, Kuroda T, Kato K, Tanaka H, Yamamoto M, Takata M, Yamada K, Kigoshi H. J Mol Biol. 2006;356:945–954. doi: 10.1016/j.jmb.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 36.Kita M, Yoneda K, Hirayama Y, Yamagishi K, Saito Y, Sugiyama Y, Kigoshi H. ChemBioChem. 2012;13(12):1754–1758. doi: 10.1002/cbic.201200385. [DOI] [PubMed] [Google Scholar]
- 37.Miyata H, Kinosita K, Marriott G. Journal of Biochemistry. 1997;121(3):527–533. doi: 10.1093/oxfordjournals.jbchem.a021618. [DOI] [PubMed] [Google Scholar]
- 38.Marriott G, Miyata H, Kinosita K., Jr Biochemistry Int. 1992;126:943–951. [PubMed] [Google Scholar]
- 39.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 40.Jancarik J, Kim SH. J Appl Cryst. 1991;24:409–411. [Google Scholar]
- 41.Otwinowski Z, Minor W. Methods in Enzymology: Macromolecular Crystallography. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 42.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Acta Crystallogr D. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P, Cowtan K. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 46.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendal WB, III, Soeyink J, Richardson JC, Richardson DC. Nucleic Acids Research. 2007;35:375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McLaughlin PJ, Gooch JT, Mannherz HG, Weeds AG. Nature. 1993;364:685–692. doi: 10.1038/364685a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.