

Abstract
Cytotoxic T lymphocytes (CTL) are a major factor in the control of HIV replication. CTL arise in acute infection, causing escape mutations to spread rapidly through the population of infected cells. As a result, the virus develops partial resistance to the immune response. The factors controlling the order of mutating epitope sites are currently unknown and would provide a valuable tool for predicting conserved epitopes. In this work, we adapt a well-established mathematical model of HIV evolution under dynamical selection pressure from multiple CTL clones to include partial impairment of CTL recognition, 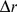 , as well as cost to viral replication,
, as well as cost to viral replication, 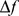 . The process of escape is described in terms of the cost-benefit tradeoff of escape mutations and predicts a trajectory in the cost-benefit plane connecting sequentially escaped sites, which moves from high recognition loss/low fitness cost to low recognition loss/high fitness cost and has a larger slope for early escapes than for late escapes. The slope of the trajectory offers an interpretation of positive correlation between fitness costs and HLA binding impairment to HLA-A molecules and a protective subset of HLA-B molecules that was observed for clinically relevant escape mutations in the Pol gene. We estimate the value of
. The process of escape is described in terms of the cost-benefit tradeoff of escape mutations and predicts a trajectory in the cost-benefit plane connecting sequentially escaped sites, which moves from high recognition loss/low fitness cost to low recognition loss/high fitness cost and has a larger slope for early escapes than for late escapes. The slope of the trajectory offers an interpretation of positive correlation between fitness costs and HLA binding impairment to HLA-A molecules and a protective subset of HLA-B molecules that was observed for clinically relevant escape mutations in the Pol gene. We estimate the value of 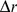 from published experimental studies to be in the range (0.01–0.86) and show that the assumption of complete recognition loss (
from published experimental studies to be in the range (0.01–0.86) and show that the assumption of complete recognition loss (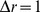 ) leads to an overestimate of mutation cost. Our analysis offers a consistent interpretation of the commonly observed pattern of escape, in which several escape mutations are observed transiently in an epitope. This non-nested pattern is a combined effect of temporal changes in selection pressure and partial recognition loss. We conclude that partial recognition loss is as important as fitness loss for predicting the order of escapes and, ultimately, for predicting conserved epitopes that can be targeted by vaccines.
) leads to an overestimate of mutation cost. Our analysis offers a consistent interpretation of the commonly observed pattern of escape, in which several escape mutations are observed transiently in an epitope. This non-nested pattern is a combined effect of temporal changes in selection pressure and partial recognition loss. We conclude that partial recognition loss is as important as fitness loss for predicting the order of escapes and, ultimately, for predicting conserved epitopes that can be targeted by vaccines.
Author Summary
Like many viruses, HIV has evolved mechanisms to evade the host immune response. As early as a few weeks after infection is initiated, mutations appear in the viral genome that reduce the ability of cytotoxic T lymphocytes (CTL) to control virus replication. However, of the many mutations in the viral genome that could potentially mediate viral escape from the CTL response, a specific subset are typically observed. This suggests that some mutations either entail too high a fitness cost for the virus, or are relatively inefficient escape mutations. A successful vaccine would target the CTL response to these regions in such a way that escape would not be possible. We use a computational model of HIV infection in order to study the factors that determine whether a given escape mutation will occur, how long it will be maintained in the population, and how these changes in the viral genome will affect the CTL response. Our analysis highlights the important role of partial recognition loss conferred by a mutation in producing the complex dynamics of escape that are observed during the course of infection.
Introduction
HIV replication continues for years despite a highly active immune response. Depletion of cytotoxic CD8+ T cells (CTL) in SIV infected animals causes rapid increase in viremia [1], [2] showing that CTL control HIV/SIV replication; that this response is antigen-specific is evident from rapid genetic evolution of HIV in antigenically important regions. Antigenic escape is one of the major mechanisms of HIV resilience in face of an active immune response, impeding effective vaccine design [3] and implicated in the progression to AIDS [4]. Shortly after infection is initiated, many CTL clones arise to target the transmitted virus strain [5]–[7], each clone recognizing a distinct 8–10 amino acid viral peptide (epitope) presented on the surface of an infected cell by MHC molecules. Escape mutations in CTL epitopes begin to be selected within a month of infection and continue to be selected throughout chronic infection, sometimes causing a decrease in the intrinsic replication rate of the virus (fitness cost) [8]–[10]. However, despite a sustained CTL response, not all targeted epitopes escape. Moreover, among epitopes that do escape, the rate of escape slows dramatically over the first 100 days post infection. It remains unclear which parameters decide the timing and rate of escape in a given epitope as well as which epitopes escape and which are preserved throughout chronic infection [11]–[13].
Mathematical models of HIV evolution in the presence of multiple CTL clones have been applied to study the emergence of late escape mutations [14], [15] and the effect of distributed CTL pressure on the rate of escape [12], [16]. Previous work has emphasized two parameters, the mutation cost (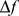 ) and the number of active epitopes (n). It can be inferred that escape mutations come at a cost from the observation of occasional reversion of escape mutations upon transmission between MHC mismatched individuals [8], [9], [17], [18], as well as the frequent acquisition of compensatory mutations outside escaping epitopes. [19], [20]. Common escape mutations have been shown experimentally to have wide ranging fitness costs [10], [21], [22]. However, fitness costs and the number of CTL clones acting on the virus are not the sole determinants of the dynamics of escape. The degree of escape conferred by a mutation is equally important. It has been observed in HIV infected individuals [23] and SIV infected animals [24] that CTL are capable of recognizing different variants of an epitope with different efficiencies. Thus, in general, an escape mutant does not fully abrogate recognition of the corresponding CTL clone.
) and the number of active epitopes (n). It can be inferred that escape mutations come at a cost from the observation of occasional reversion of escape mutations upon transmission between MHC mismatched individuals [8], [9], [17], [18], as well as the frequent acquisition of compensatory mutations outside escaping epitopes. [19], [20]. Common escape mutations have been shown experimentally to have wide ranging fitness costs [10], [21], [22]. However, fitness costs and the number of CTL clones acting on the virus are not the sole determinants of the dynamics of escape. The degree of escape conferred by a mutation is equally important. It has been observed in HIV infected individuals [23] and SIV infected animals [24] that CTL are capable of recognizing different variants of an epitope with different efficiencies. Thus, in general, an escape mutant does not fully abrogate recognition of the corresponding CTL clone.
In the present work, we address the process of antigenic escape in terms of a cost-benefit diagram. The benefit of an escape mutation is a partial CTL recognition loss and the mutation cost is a partial reduction in viral replication rate. We extend the basic model introduced by Althaus and De Boer [14] to include partially effective escape mutations and investigate how the two opposing evolutionary forces together determine the observed rate of escape from the CTL response [12], [25]. The model predicts that a positive correlation between recognition and fitness losses emerges during sequential escape mutations, the strength of which changes over time as pressure from the immune system wanes. We compare our model predictions with existing data showing a correlation between fitness and recognition losses in clinically relevant escape mutations from the Pol gene [26] and estimate the range of recognition losses that occurs in commonly observed escape mutations from three published studies [27]–[29].
Furthermore, the inclusion of partially effective escape mutations in the model can reproduce the diverse patterns of intra-epitope escape that are routinely observed in HIV infected patients. During the majority of escape mutations that have been studied with time-resolved viral sequencing, 2–10 distinct epitope sequences grow in number to replace the transmitted sequence and eventually one mutant epitope spreads to the entire population. Furthermore, the dominant mutated epitope sequence changes over time [11], [30], [31]. Although, in some epitopes, mutations are added at new sites in a nested fashion, in a larger number of epitopes, mutations at new sites replace mutations at previous sites leading to a non-nested pattern. This pattern is atypical for models assuming constant selection pressure. The analysis below illustrates how time-dependent selection due to the changing CTL pressure and partial CTL recognition loss can produce the non-nested pattern of escape (see [32] for review).
Results
Phases of HIV infection
In order to study escape from the immune response we consider a model that includes target cells, infected cells and multiple CTL clones which recognize regions in the viral genome (epitopes) with equal avidities (Figure 1A and Materials and Methods ). The model predicts three distinct phases of HIV infection (Figure 1B), as follows. Phase 1: The transmitted HIV strain expands in the population of target cells. Phase 2: All CTL clones that recognize cells infected by the transmitted strain are activated, expand, and reduce the number of infected cells. A steady state is obtained with constant levels of infected cells and CTL, which represents chronic HIV infection (see Equations S1–S3). Phase 3: Escape mutations in viral epitopes emerge, changing the genetic composition of the population of infected cells and the clonal composition of the CTL population but only weakly affecting their overall sizes.
Figure 1. A computational model of the interaction between HIV and multiple CTL clones.

(A) The model given by Equations 6 to 8 comprises three interacting cell compartments: target cells (T), infected cells (I) and multiple CTL clones (E). Viral genomes contain multiple epitopes, which can mutate to partially abrogate CTL recognition. An escape mutation is denoted by an X. Each CTL clone recognizes a single viral epitope and is stimulated to divide at a rate proportional to the number of infected cells with recognizable epitopes. The model is designed to study the rate of escape in epitopes when CTL pressure is distributed across multiple epitopes, as well as study intra-epitope escape patterns when CTL respond dynamically to the infected cells that they recognize. Black arrows: flux of cells from one compartment to another. Blue arrows: dependence of the rate of flux from one compartment on another. Dotted lines represent attenuation of the interaction strength. (B) Simulation example showing three phases of HIV evolution. A single virus strain initiates the infection (transmitted strain, black). In response to the growing number of infected cells, multiple CTL clones are activated (colored lines), and the system reaches a steady state. Finally, virus strains with escape mutations (dashed, colored lines) replace the transmitted strain. In response to lowered activation signals, some CTL clones decline. The escape strains are colored to match the CTL against which an escape was most recently acquired. Model parameters: number of epitopes, 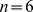 ; number of sites per epitope,
; number of sites per epitope, 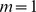 . Epitopes 1–3 have parameters that allow escape
. Epitopes 1–3 have parameters that allow escape 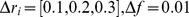 , epitopes 4–6 have parameters that prohibit escape,
, epitopes 4–6 have parameters that prohibit escape, 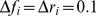 . Other parameters are listed in Table 1.
. Other parameters are listed in Table 1.
The dynamics of infected cells and CTL during the escape phase (Phase 3) depends on the number of epitopes that are targeted, n, and on the degree of recognition loss per mutation, 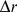 (Table 1). In the simplest case, if a single CTL clone is present in steady state and an escape mutation arises which completely abrogates CTL recognition (
(Table 1). In the simplest case, if a single CTL clone is present in steady state and an escape mutation arises which completely abrogates CTL recognition (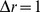 ), the CTL clone contracts and the population of infected cells containing the escape mutation grows uncontrolled until target cells are depleted (whether and for how long the escape mutation is maintained after disappearance of the CTL clone depends on the fitness cost of the mutation). In contrast, if the recognition loss conferred by the escape mutation is partial (
), the CTL clone contracts and the population of infected cells containing the escape mutation grows uncontrolled until target cells are depleted (whether and for how long the escape mutation is maintained after disappearance of the CTL clone depends on the fitness cost of the mutation). In contrast, if the recognition loss conferred by the escape mutation is partial (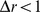 ), the population of infected cells grows only transiently, and the CTL clone expands until a new steady state is reached. When a group of CTL clones with similar avidity target the infected cell population (as shown in Figure 1B), the spread of either partly or fully effective escape mutation causes the corresponding CTL clone to contract, since it recognizes infected cells less efficiently than other CTL clones (see Equation S5). Because infected cells continue to be recognized by other CTL clones, the population of infected cells does not grow out of control following the escape mutation. Thus, the model reproduces the three-phase dynamics that can be inferred from kinetic data in HIV infected individuals including the waning of CTL responses to escaped epitopes [11], [13], [30], [31].
), the population of infected cells grows only transiently, and the CTL clone expands until a new steady state is reached. When a group of CTL clones with similar avidity target the infected cell population (as shown in Figure 1B), the spread of either partly or fully effective escape mutation causes the corresponding CTL clone to contract, since it recognizes infected cells less efficiently than other CTL clones (see Equation S5). Because infected cells continue to be recognized by other CTL clones, the population of infected cells does not grow out of control following the escape mutation. Thus, the model reproduces the three-phase dynamics that can be inferred from kinetic data in HIV infected individuals including the waning of CTL responses to escaped epitopes [11], [13], [30], [31].
Table 1. Model parameters.
| Parameter | Realistic Value | Description | Reference |
| n | 1–8 | Number of epitopes recognized during first 100 days | [11], [13], [30], [31] |
| m | 2–10 | Number of sites per epitope important for recognition | [11], [13], [30], [31] |
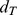
|
1 
|
Rate at which activated target cells transition out of the highly infectable phase | [46], [68] |
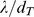
|
 cells cells |
Activated target cell level | [47], [69], [70] |
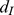
|
1 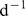
|
Virus-induced infected cell death rate | [55] |
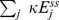
|
4 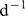
|
CTL-induced infected cell death rate | [47], [69], [70] |
| β |
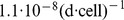
|
Basic efficiency of target cell infection | [47], [69], [70] |
| s |
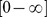
|
Intrinsic mutation cost | [10], [21], [22], [27]–[29] |
| α |
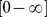
|
Reduction of CTL recognition | [27]–[29] |
|
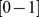
|
Fractional reduction in intrinsic replication rate | |
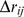
|
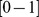
|
Fractional reduction of CTL recognition | |

|
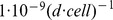
|
CTL killing efficiency. Gives 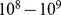 CTLs in chronic infection CTLs in chronic infection |
[45] |
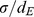
|
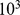 cells cells |
Initial population of CTLs | [69] |
| c | 1 
|
Maximum growth rate of effector cells | [69] |
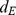
|
0.1 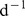
|
Death rate of effector cells | [47], [69], [70] |
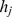
|
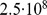 cells cells |
Number of recognized infected cells for half maximal proliferation of CTL (inverse avidity). Gives an infected cell level of 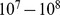 in chronic infection in chronic infection |
[44] |
Model parameters for the model of escape from multiple CTL shown in Figure 1.
The escape rate of a mutant strain is determined by the loss of CTL recognition and the loss of viral fitness
Escape mutations begin to be selected once CTL reach sufficiently high levels (third phase in Figure 1B). Mutations in all targeted epitopes begin to grow simultaneously, albeit with different rates due to variation in the amounts of fitness and recognition losses caused by a mutation. The exponential growth rates (escape rates) of mutant strains determine which mutant strain will grow to dominate the population. Here we describe the growth of a mutated strain (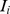 ) with a single mutation in epitope 1. An escape mutation results in both a fractional fitness cost,
) with a single mutation in epitope 1. An escape mutation results in both a fractional fitness cost, 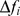 , and a fractional loss of CTL recognition,
, and a fractional loss of CTL recognition, 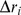 . The mutant strain begins to grow as
. The mutant strain begins to grow as 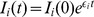 , with initial growth rate
, with initial growth rate 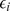 :
:
| (1) |
where 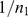 is the fraction of CTL population recognizing epitope 1 (see Equation 7 in Materials and Methods and Text S1). Thus, the escape rate reflects the balance between the partial recognition loss and the partial fitness loss, which determines whether a mutant strain has a selective advantage (
is the fraction of CTL population recognizing epitope 1 (see Equation 7 in Materials and Methods and Text S1). Thus, the escape rate reflects the balance between the partial recognition loss and the partial fitness loss, which determines whether a mutant strain has a selective advantage (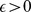 ). In order to infer, for example, the fitness cost of an escape mutation, it is necessary to measure not only the escape rate, but also the CTL recognition loss.
). In order to infer, for example, the fitness cost of an escape mutation, it is necessary to measure not only the escape rate, but also the CTL recognition loss.
When multiple epitopes are targeted and recognition and fitness losses vary across epitopes, the growth rates of escape mutants vary as well (Figure 1B). In data from HIV infected individuals, it is observed that the rate of escape slows dramatically over the first 100 days post infection [11]–[13]. The rise of escape mutants predicted by the model is consistent with these findings (Figure S1). Furthermore, assuming only a minor fitness cost, the entire observed variation of escape rates over time and across sites can be simulated from the variation of the recognition loss, 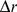 .
.
The trajectory of escape mutations in the cost-benefit plane (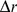 ,
, 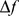 )
)
The process of gradual viral escape from the immune response of an infected host continues for years. During this time, the virus shows a limited number of detectable CTL responses against different sites ( [33]) where a total of 5–30 escape mutations are selected [11], [30]. We investigate the trajectory connecting these escape mutations in the cost-benefit plane and predict how the average fitness costs and recognition losses incurred by an escape mutation will change as escape progresses. For this aim, we use a simplified version of the main model (see
Materials and Methods
), focused only on the order of escape mutations (i.e., model dynamics are not considered explicitly). Parameters
[33]) where a total of 5–30 escape mutations are selected [11], [30]. We investigate the trajectory connecting these escape mutations in the cost-benefit plane and predict how the average fitness costs and recognition losses incurred by an escape mutation will change as escape progresses. For this aim, we use a simplified version of the main model (see
Materials and Methods
), focused only on the order of escape mutations (i.e., model dynamics are not considered explicitly). Parameters 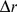 and
and 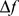 are randomly generated for a genome with multiple epitopes and multiple sites per epitope (10 epitopes, with 10 sites per epitope in Figure 2) and epitope sites are ranked in the descending order of escape rate (Equation 1). When many random runs are compared, a correlation between
are randomly generated for a genome with multiple epitopes and multiple sites per epitope (10 epitopes, with 10 sites per epitope in Figure 2) and epitope sites are ranked in the descending order of escape rate (Equation 1). When many random runs are compared, a correlation between 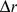 and
and 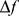 for escape mutations of a given rank is observed (Figure 2A). The average trajectory of escape in the cost-benefit plane moves from high recognition loss, low fitness cost to low recognition loss, high fitness cost. The maximum escape rate per epitope decreases over many rounds of escape (Figure 2B) and each epitope escapes at more than one site. This prediction is consistent with experimental observation, where the majority of escaping epitopes undergo more than one mutation [13], [30].
for escape mutations of a given rank is observed (Figure 2A). The average trajectory of escape in the cost-benefit plane moves from high recognition loss, low fitness cost to low recognition loss, high fitness cost. The maximum escape rate per epitope decreases over many rounds of escape (Figure 2B) and each epitope escapes at more than one site. This prediction is consistent with experimental observation, where the majority of escaping epitopes undergo more than one mutation [13], [30].
Figure 2. The escape trajectory in the cost-benefit plane bends over time due to CTL decay.

Fitness costs and recognition losses are randomly generated for 100 sites (10 epitopes with 10 sites per epitope) in order to study the sequence of escaped sites (black line) in the whole genome without CTL decay (A,B) or with CTL decay (C,D) for 1000 simulation runs. (A) For each site that escapes, the fractional fitness cost, 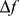 , multiplied by the number of epitopes,
, multiplied by the number of epitopes, 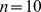 , and fractional recognition loss,
, and fractional recognition loss, 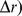 (Equation 1 and Table 1) is shown. Colors show the predicted rank of escape mutations, from early escape mutations (blue) to late escape mutations (red). The average trajectory over all runs (black) moves from high recognition loss, low fitness cost to low recognition loss, high fitness cost. Inset: The best-fit slope for each escape rank. A positive correlation is observed between the fitness and recognition losses for all epitopes that escape at a given rank. (B) The maximum escape rate of any epitope site for all 10 epitopes for a representative simulation run. (C–D) As in (A–B), except including CTL decay. CTL decay is simulated by reducing recognition losses for all epitope sites in epitopes that have partially escaped according to
(Equation 1 and Table 1) is shown. Colors show the predicted rank of escape mutations, from early escape mutations (blue) to late escape mutations (red). The average trajectory over all runs (black) moves from high recognition loss, low fitness cost to low recognition loss, high fitness cost. Inset: The best-fit slope for each escape rank. A positive correlation is observed between the fitness and recognition losses for all epitopes that escape at a given rank. (B) The maximum escape rate of any epitope site for all 10 epitopes for a representative simulation run. (C–D) As in (A–B), except including CTL decay. CTL decay is simulated by reducing recognition losses for all epitope sites in epitopes that have partially escaped according to 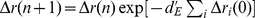 , summing over all i sites in the epitope that have escaped with
, summing over all i sites in the epitope that have escaped with 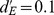 per escape. When CTLs decay in response to an escape in an epitope, the immune pressure on all other sites in that epitope is decreased. The result is that the average trajectory in the cost-benefit plane bends towards the horizontal axis.
per escape. When CTLs decay in response to an escape in an epitope, the immune pressure on all other sites in that epitope is decreased. The result is that the average trajectory in the cost-benefit plane bends towards the horizontal axis.
The dynamic interplay between CTL and partially effective escape mutations shape the overall course of HIV escape. As mentioned above, an escape mutation results in the decay of the cognate CTL population. In turn, the CTL decay causes the potential benefit of other escape mutations in the escaping epitope to decrease, because the overall CTL pressure on the epitope is lessened. The presence or absence of CTL decay has observable consequences for the trajectory of escape mutations in the cost-benefit plane. Without the CTL decay, the average slope of the trajectory stays constant over time until no more escapes are possible (Figure 2A). In contrast, when CTL clones decay in response to the recognition loss, the trajectory bends towards X-axis (Figure 2C) as early escape in an epitope causes escape mutations on the other sites in that epitope to become progressively less advantageous. Therefore, the total number of escape mutations decreases when CTL decay is included (from 55 in Figure 2A to 20 in Figure 2), which is the result of fewer escape mutations per epitope (fewer steps in Figure 2D compared to Figure 2B).
Weak correlation between  and
and 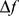 has been observed in the Pol gene
has been observed in the Pol gene
The simulation example in Figure 2 offers an interpretation of the work by Mostowy et al [26], where a weak but statistically significant correlation was observed between fitness costs and HLA binding losses in clinically derived Pol sequences. The existence of the correlation follows from Equation 1, which states that more costly mutations will not appear unless they confer a large benefit to the virus. The model relates the weak strength of the correlation (slope = −0.12 between the fitness decrease and the impairment of HLA binding) to the large number of active epitopes. The existence of the CTL decay caused by escape can further reduce the slope: mutations sampled during the acute phase of HIV infection are predicted to have a much larger slope than those sampled during the chronic phase (Figure 3B). Since the majority of database sequences used in the cited work fall into the latter category, the small slope of the correlation observed can partially be caused by CTL decay.
Figure 3. Fitness loss and recognition losses of escape mutations are predicted to correlate positively, with a slope that deceases in time.

Sites are randomly sampled from the simulation described in the caption to Figure 2 in order to simulate the effect of acute samples (high ranking sites), late chronic samples (low ranking sites) or patient samples from random times (random ranks). (A) No CTL decay. The slope of the correlation from acute (blue) or late chronic (red) sampled escape mutations is the same, however, it is lower for escape mutations sampled at random times (green). (B) CTL decay causes the slope of the correlation to decrease in time, due to decrease of CTL selection pressure. CTL decay is introduced as described in the caption to Figure 2 and Model.
There is an important caveat which must be considered when comparing the model with the cited work [26]. Loss of CTL recognition in the model is given by  , which is a composite parameter comprising changes in antigen processing, presentation and recognition, whereas Mostowy et al use the computationally predicted loss of HLA-epitope binding,
, which is a composite parameter comprising changes in antigen processing, presentation and recognition, whereas Mostowy et al use the computationally predicted loss of HLA-epitope binding,  . To convert the units, we analyzed data from several publications in which both overall loss of CTL recognition as well as HLA binding impairment was measured [27]–[29]. We found a strong correlation between
. To convert the units, we analyzed data from several publications in which both overall loss of CTL recognition as well as HLA binding impairment was measured [27]–[29]. We found a strong correlation between 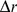 and
and 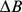 expressed as a linear relationship
expressed as a linear relationship 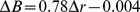 (Figure S2, Equations S10–S14), which justifies our comparison between the model predictions and this data.
(Figure S2, Equations S10–S14), which justifies our comparison between the model predictions and this data.
Three patterns of antigenic escape in an epitope with two sites
Epitopes with many sites can produce different combinations of escape mutations (haplotypes) in response to CTL pressure. In [30] it was observed that the mutated sequence changed over time in the majority of epitopes that were studied longitudinally and, interestingly, the order in which escape haplotypes appeared varied from epitope to epitope. We divide the epitopes into three characteristic patterns, based on the order of dominant haplotypes: “simple”, “nested” and “leapfrog”. We can illustrate these patterns in an epitope with two sites, in which four haplotypes are possible. The presence or absence of an escape mutation at each site is denoted by a 1 or a 0, respectively. The infrequently observed “simple” pattern is characterized by a single escape haplotype. For example, in an epitope with two sites, the sequence of haplotypes observed is 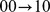 . The “nested” escape adds a new mutation sequentially to a previously mutated sequence:
. The “nested” escape adds a new mutation sequentially to a previously mutated sequence: 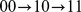 . This pattern is predicted if mutations at both sites are under a constant, positive selection pressure (
. This pattern is predicted if mutations at both sites are under a constant, positive selection pressure (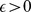 throughout the course of infection). The “leapfrog” pattern is characterized by a switch in the dominant single-site mutation:
throughout the course of infection). The “leapfrog” pattern is characterized by a switch in the dominant single-site mutation:  . In the present model, the leapfrog pattern of escape arises due to time-dependent CTL selection pressure. Below we use the model to determine the distribution of fitness and recognition losses within an epitope that produces the leapfrog pattern of escape.
. In the present model, the leapfrog pattern of escape arises due to time-dependent CTL selection pressure. Below we use the model to determine the distribution of fitness and recognition losses within an epitope that produces the leapfrog pattern of escape.
In order to study intra-epitope dynamics, we use the main model (Equations 6 to 8) and consider a genome composed of a large number of two-site epitopes. This simple case can be studied in detail, and the results can illustrate the general case, where epitopes are comprised of many sites. We use Equation 1 to determine the escape rates for each of the three mutant haplotypes has for a given level of CTL clone 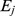 recognizing epitope
recognizing epitope 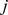 :
:
| (2) |
| (3) |
| (4) |
where 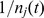 is the time dependent fraction of the total CTL population comprised by CTL clone
is the time dependent fraction of the total CTL population comprised by CTL clone 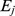 and we have neglected the small parameter
and we have neglected the small parameter 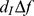 (Table 1). We consider the case where the initial escape rate of haplotype 10 in epitope
(Table 1). We consider the case where the initial escape rate of haplotype 10 in epitope 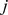 (
( ) is higher than the initial escape rate of haplotype 01 (
) is higher than the initial escape rate of haplotype 01 (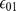 ).
).
Once an escape haplotype has reached a sufficiently high frequency, CTL clone 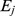 will decay monotonically in time, but the virus load will stay stable, since the population of infected cells will be controlled by the other CTL clones. As
will decay monotonically in time, but the virus load will stay stable, since the population of infected cells will be controlled by the other CTL clones. As 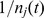 decreases and the CTL-induced pressure on the epitope declines, the fitness losses at the two sites,
decreases and the CTL-induced pressure on the epitope declines, the fitness losses at the two sites,  , begin to dominate the escape rates (Equations 2–4), and the favored haplotype will change. Eventually, the decay of the CTL clone will cause the transmitted haplotype (00) to regain fitness advantage and become a dominant strain again (the existence of compensatory mutations may cause stabilization of the last escaped clone, see
Discussion
). The sequence of dominant haplotypes can follow one of the patterns, as follows:
, begin to dominate the escape rates (Equations 2–4), and the favored haplotype will change. Eventually, the decay of the CTL clone will cause the transmitted haplotype (00) to regain fitness advantage and become a dominant strain again (the existence of compensatory mutations may cause stabilization of the last escaped clone, see
Discussion
). The sequence of dominant haplotypes can follow one of the patterns, as follows:
Simple pattern
The simple pattern of escape (Figure 4A) can occur if the second site cannot escape ( , Equation 3). It can also occur if the second site is weakly advantageous, yet the double mutant (11) does not have time to grow to appreciable frequency before the transmitted haplotype (00) regains the advantage in growth rate. Observation of this pattern implies a large difference in the fitness costs and/or recognition loss magnitudes between the two sites in an epitope.
, Equation 3). It can also occur if the second site is weakly advantageous, yet the double mutant (11) does not have time to grow to appreciable frequency before the transmitted haplotype (00) regains the advantage in growth rate. Observation of this pattern implies a large difference in the fitness costs and/or recognition loss magnitudes between the two sites in an epitope.
Figure 4. Dynamical selection pressure from CTLs causes three possible patterns of intra-epitope escape: Example for an epitope with two sites.

Intra-epitope escape in one epitope with two sites is studied for the model shown in Figure 1 and Model. The sequence in which haplotypes are selected depends on the distribution of fitness and recognition losses within an epitope. The fraction of the infected cell population containing each of the four haplotypes in the escaping epitope is shown in “simple” (A), “leapfrog” (C) and “nested” pattern (E). For each pattern, the dependence of the escape rate (Equations 2–4) for each haplotype on the fraction of CTLs responding to the epitope (B, D, F). The inset shows CTL dynamics: the size of the CTL clone to escaping epitope (red) and the total CTL number (black). Parameters: (A,B) 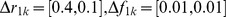 , (C,D)
, (C,D) 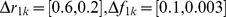 , (E,F)
, (E,F) 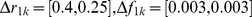 with
with 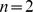 ,
,  for all panels; other parameters are given in Table 1.
for all panels; other parameters are given in Table 1.
Leapfrog pattern
The leapfrog pattern can occur if haplotype 01 gains the advantage over 10 before the 10 loses the advantage over 00 (Figure 4C and D). In order for this to occur, site 1 must have both a greater fitness and a greater recognition loss than site 2:
| (5) |
The CTL clone to the escaping epitope decays at a faster rate during the time period when variant 10 dominates the population than than during the period when 01 dominates the population. The difference is due to the relatively higher recognition loss of the mutation at the first site (inset Figure 4D). The exact conditions required for this pattern are given in Text S3.
Nested pattern
When both single-mutant haplotypes continue to have a positive growth rate throughout the course of infection, as in the case of constant selection pressure, the double-mutant haplotype has the highest escape rate (Figure 4E, F). The nested pattern may occur if the escape rates of both single-mutant haplotypes are similar (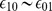 ).
).
We use numeric computation to determine which pattern of escape is observed over a range of recognition and fitness losses at each of the two epitope sites. The number of epitopes is fixed, n, the escape rate for the first haplotype, 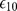 , and the ratio of the fitness costs in the two epitope sites,
, and the ratio of the fitness costs in the two epitope sites, 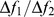 , at values that are representative of acute infection (Figure 5) or chronic infection (Figure S3) (see Figure S1 for the range of escape rates observed in HIV infected patients). For large escape rates, the leapfrog pattern can be observed for large values of
, at values that are representative of acute infection (Figure 5) or chronic infection (Figure S3) (see Figure S1 for the range of escape rates observed in HIV infected patients). For large escape rates, the leapfrog pattern can be observed for large values of 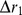 and a broad range of
and a broad range of 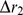 (Figure 5). Smaller values of
(Figure 5). Smaller values of 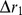 produce smaller escape rates, and the leapfrog is observed in a narrow range of small
produce smaller escape rates, and the leapfrog is observed in a narrow range of small 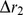 (Figure S3). In some cases, haplotype 11 is observed as a short intermediate between haplotypes 10 and 01 (labeled “nested leapfrog” in Figure 5). The time interval during which a given haplotype dominates the population depends on parameters of loss and recognition at epitope site,
(Figure S3). In some cases, haplotype 11 is observed as a short intermediate between haplotypes 10 and 01 (labeled “nested leapfrog” in Figure 5). The time interval during which a given haplotype dominates the population depends on parameters of loss and recognition at epitope site, 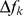 and
and 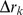 . The less costly haplotype can dominate the population for months to years (shown as
. The less costly haplotype can dominate the population for months to years (shown as 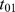 in the inset of Figures AS3 and Figure 5). CTL to the escaping epitope will decay at the fastest rate when variant 11 dominates the population (inset Figure 4F).
in the inset of Figures AS3 and Figure 5). CTL to the escaping epitope will decay at the fastest rate when variant 11 dominates the population (inset Figure 4F).
Figure 5. The pattern of emergence of escape variants in a single epitope contains information about the fraction of recognition and fitness lost by single-site mutations in the epitope.

Using simulation of the model (Figure 1A, Equations 6 to 8) with two sites per epitope,  , the pattern of escape is calculated for a range of recognition and fitness losses. The pattern that is obtained is plotted as a function of the parameters of recognition loss at the first and second site (
, the pattern of escape is calculated for a range of recognition and fitness losses. The pattern that is obtained is plotted as a function of the parameters of recognition loss at the first and second site (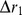 and
and 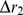 , respectively). In each panel, certain parameters are fixed in order to focus on the effect of recognition loss. Fixed parameters are: the escape rate of the first haplotype (
, respectively). In each panel, certain parameters are fixed in order to focus on the effect of recognition loss. Fixed parameters are: the escape rate of the first haplotype (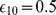 ) and the number of targeted epitopes (
) and the number of targeted epitopes ( ), values which correspond to escape mutations that occur in acute infection (see Figure S3 for parameters that correspond to later in infection). Fitness costs are chosen such that the second site is less costly than the first:
), values which correspond to escape mutations that occur in acute infection (see Figure S3 for parameters that correspond to later in infection). Fitness costs are chosen such that the second site is less costly than the first: 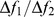 equal to 3 (A) or much less costly than the first,
equal to 3 (A) or much less costly than the first, 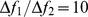 (B). Other parameters given in Table 1. Mostowy: 2012iv Equations S6 (red line) and S9 (blue line) determine the region where the leapfrog pattern can be observed. Regions that require
(B). Other parameters given in Table 1. Mostowy: 2012iv Equations S6 (red line) and S9 (blue line) determine the region where the leapfrog pattern can be observed. Regions that require  are not allowed by definition (magenta line). The shaded regions between these three lines correspond to regions of parameter space where both sites escape. The corresponding patterns are: “leapfrog” (
are not allowed by definition (magenta line). The shaded regions between these three lines correspond to regions of parameter space where both sites escape. The corresponding patterns are: “leapfrog” ( , Figure 4C), “nested” (
, Figure 4C), “nested” (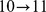 , Figure 4E), “nested leapfrog” (
, Figure 4E), “nested leapfrog” (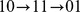 ). Observation of the leapfrog pattern in an epitope tightly constrains the fraction of CTL recognition loss conferred by sites in an epitope. The inset shows the length of time during which haplotype 01 is dominant in the escaping epitope.
). Observation of the leapfrog pattern in an epitope tightly constrains the fraction of CTL recognition loss conferred by sites in an epitope. The inset shows the length of time during which haplotype 01 is dominant in the escaping epitope.
Discussion
The recent availability of time-resolved, deep-sequencing data from HIV infected patients have illuminated the complexity of interaction between CTL clones and the genetically diverse HIV population. Using a model including multiple CTL clones, we demonstrate that the rate of viral escape depends on both the partial recognition losses and fitness costs associated with an escape mutation as well as the number of active CTL clones. Furthermore, the model predicts that CTL populations change their relative sizes in response to viral escape, which has observable consequences for the sequence and number of escape mutations that are possible over time. Changing selection pressure on an epitope due to declining CTL levels can also cause the dominant escape variant in an epitope to change in a non-nested fashion. The model contains several assumptions and simplifications, as follows.
Pre-existing mutations
We assume that the population of infected cells in acute infection and steady state is large, so that all single escape mutants exist in the population before the rise of CTL (as in [14], [15]). The assumption is supported by estimates of the large effective HIV population size [4], [34], [35]. We can also infer the fact of preexistence of CTL escape mutations from the observed preexistence of drug-resistant mutations evident in the early emergence of resistance to mono-therapy (e.g. [36]). Furthermore, since the number of escape mutations per epitope is typically larger than the number of drug resistance sites per drug, the mutation cost of the least costly escape mutation can be assumed to be lower – and the frequency of preexisting mutations higher – for escape mutations. The preexistence of single mutants casts doubts on the interpretation of very late escape mutations as the result of the late appearance of an escape mutation not initially present in the population [37].
Eventual reversion to the wild type is prevented by compensatory mutations
Mutations compensating for fitness losses were documented for drug-resistance mutations (e.g. [38]–[40]) and for immune-escape mutations [10], [19], [20] (For an overview of compensatory mutations in HIV see [41], [42]). However, the dynamics of compensation remains poorly understood. Therefore, our model does not explicitly include compensatory mutations and, as a result, predicts the eventual reversion of any escape mutation with a fitness cost. Instead of including compensation, we calculate the period of time that a given mutation would be maintained in the population before reversion occurs, which is the same time interval where compensation would be necessary in order to prevent reversion (inset of Figures 5 and S3). When the non-nested pattern of escape is observed in infected patients, the first escape variant is typically short-lived compared to the second escape variant, and the transmitted variant is not observed after the initial escape [11], [30], [31]. Our interpretation is that the fitness cost of the first variant is large relative to the second variant and does not have time to be compensated before the second variant gains the advantage. The second variant either has a very small fitness cost or is compensated gradually during its lifetime (compensation is not explicitly simulated). Our calculation of the lifetimes of different escape variants in an epitope is a first step towards understanding the timescales associated with compensation.
Multiple CTL clones of equal avidity
The present analysis is focused on the first year post-infection, the time interval in which most escape mutations occur. Therefore, we consider a group of CTL clones with similar avidities that are present initially in similar numbers. This is a reasonable assumption after escape mutations have occured in the first few immunodominant epitopes during the resolution of acute viremia. At this time, a large number of CTL clones are activated and are maintained for many months at simular levels [6], [33]. Once all CTL clones are activated, our model predicts that the steady state viral load is proportional to the inverse avidity of the most avid CTL clone (Equation S2). Kadolsky and Asquith [43] estimated that the average viral load increases by only 0.051 log copies/ml per CTL escape. This small increase, which we interpret to be the average difference avidity spacing between CTL clones, justifies our assumption that CTL avidities are, indeed, very closely spaced. Our analysis shows that, given closely spaced avidities, it is recognition and fitness losses that govern the order and timing of escape mutations, rather than variation in CTL avidity.
Other assumptions
The model includes additional simplifying assumptions, as follows. i) The proliferation rate of CTL clones saturates with the infected cell number, but not with the total CTL level. In the original model [14], the authors postulate that the total number of CTL limits the growth of individual CTL clones, which causes clones to interact and enables the co-existence of multiple CTL clones with different avidities. We believe that further study is needed to verify the existence and the possible origin of the interclonal interaction. (Note that the target availability may not be the cause of interclonal interaction, because most CTL are not bound to their targets even at the peak of infection, and the overall effector to target ratio is less than two [44].) ii) CTL are short-lived in the absence of antigen, with an average lifetime of 10 days, as is consistent with early studies of CTL dynamics in SIV system [45], [46] and mathematical modeling of these data [47]. iii) We ignore recombination, which may increase the rate of emergence of escape mutations among different epitopes [15] as well as the long term rate of evolution [48], [49]. For the intra-epitope dynamics of escape, recombination between neighboring sites will be a small correction. (iv) CTL clones against escape mutants are not included in the model. By including partial rather than full recognition loss, we allow CTL to escaped epitopes to continue to exert selection pressure without incorporating additional CTL clones. Though clones to escape mutants may exist, introducing extra CTL clones (5 clones for each two-site epitope, instead of 1) would complicate analysis of the model without changing the essential results. (v) Small fitness costs are assumed. Equation 1 states that costly escapes 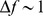 cannot arise when recognition losses are partial and many CTL clones target the viral genome. We choose to focus on the time interval corresponding to the first year after the resolution of acute infection when these assumptions are fulfilled. Escape mutations entailing a high fitness cost can only escape when only one or two CTL clones dominate strongly, which we estimate from previous data to be during the 3–4 weeks post-infection. (vi) Fitness costs are positive. A fraction of mutations that are transmitted to an individual that fall outside the individuals HLA-restricted epitopes revert (i.e. may have a negative fitness cost). However, the rate of reversion is very small [17] and hardly interferes with the faster dynamics of within-epitope mutations. (vii) We consider escape processes in different epitopes separately, because typically they do not overlap much in time. In general, however, linkage effects (clonal interference, background selection) between epitope mutations and compensatory mutations of same or different epitopes may complicate the picture (see [32] for review of recent research in this area).
cannot arise when recognition losses are partial and many CTL clones target the viral genome. We choose to focus on the time interval corresponding to the first year after the resolution of acute infection when these assumptions are fulfilled. Escape mutations entailing a high fitness cost can only escape when only one or two CTL clones dominate strongly, which we estimate from previous data to be during the 3–4 weeks post-infection. (vi) Fitness costs are positive. A fraction of mutations that are transmitted to an individual that fall outside the individuals HLA-restricted epitopes revert (i.e. may have a negative fitness cost). However, the rate of reversion is very small [17] and hardly interferes with the faster dynamics of within-epitope mutations. (vii) We consider escape processes in different epitopes separately, because typically they do not overlap much in time. In general, however, linkage effects (clonal interference, background selection) between epitope mutations and compensatory mutations of same or different epitopes may complicate the picture (see [32] for review of recent research in this area).
Thus, our model has demonstrated that partial recognition losses, in addition to fitness costs and the breadth of the CTL response, dramatically affect the rate and the order of escape mutations during an HIV infection. These findings help to interpret the positive correlation between fitness costs and recognition losses observed in the Pol gene [26] and make the testable prediction that the strength of the correlation should decrease with the time post-infection. Our results call for direct measurements of recognition losses for different escape mutations. Combined with the proposed trajectory approach (Figure 2), these data will serve as a basis for improved prediction of conserved epitopes for use in vaccines.
Materials and Methods
Model of HIV dynamics
We model SIV/HIV evolution in the presence of multiple CTL clones (Figure 1A). Each CTL clone recognizes a distinct viral epitope that is presented by an infected cell. Mutations occur in the proviral genomes of infected cell that allow an infected cell to partially evade CTL recognition, but come at a cost in terms of viral replication. The model is given by the following equations:
| (6) |
| (7) |
| (8) |
where 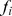 and
and 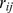 are the relative viral replication rate of viral genome i and the relative CTL recognition of sequence i by clone j, as compared to the transmitted sequence, respectively:
are the relative viral replication rate of viral genome i and the relative CTL recognition of sequence i by clone j, as compared to the transmitted sequence, respectively:
 |
(9) |
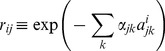 |
(10) |
The processes described are, as follows. Highly infectable target cells (T) are replenished at a rate 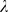 cells per day, leave the highly infectable phase with a rate
cells per day, leave the highly infectable phase with a rate 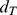 and are infected by virus at a rate that depends on the fitness on the infecting strain, with maximum rate per cell β, and the number of productively infected cells
and are infected by virus at a rate that depends on the fitness on the infecting strain, with maximum rate per cell β, and the number of productively infected cells 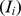 in the system. We consider n epitopes, each consists of m amino acid positions (sites), giving a total of
in the system. We consider n epitopes, each consists of m amino acid positions (sites), giving a total of  possible strains. An cell infected is labeled by proviral genome i with n epitopes denoted
possible strains. An cell infected is labeled by proviral genome i with n epitopes denoted 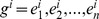 , where each epitope,
, where each epitope, 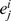 , has m sites
, has m sites 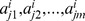 , and
, and 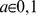 indicates the presence or absence of a mutation at an epitope site. The fitness of genome i is reduced by mutations. A mutation in epitope j at site k contributes cost
indicates the presence or absence of a mutation at an epitope site. The fitness of genome i is reduced by mutations. A mutation in epitope j at site k contributes cost  to the logarithm of the reduction in replication rate. The strain with all 0 is the transmitted strain, which has fitness 1.
to the logarithm of the reduction in replication rate. The strain with all 0 is the transmitted strain, which has fitness 1.
Effector CTL (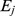 ) are replenished with a constant rate
) are replenished with a constant rate  cells per day, divide with a rate dependent on the number of infected cells that they recognize and their avidity,
cells per day, divide with a rate dependent on the number of infected cells that they recognize and their avidity, 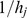 , with maximum rate c, and die with rate
, with maximum rate c, and die with rate 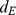 . CTL clones each respond to a distinct viral epitope that is presented by an infected cell and kill infected cells at a maximum rate
. CTL clones each respond to a distinct viral epitope that is presented by an infected cell and kill infected cells at a maximum rate  . The recognition ability of the CTL clone to epitope j in strain i is reduced by mutations, as given by Equation 10. A mutation in epitope j at site k contributes
. The recognition ability of the CTL clone to epitope j in strain i is reduced by mutations, as given by Equation 10. A mutation in epitope j at site k contributes 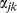 to the logarithm reduction in CTL recognition. In the present work, we assume CTL of equal avidities,
to the logarithm reduction in CTL recognition. In the present work, we assume CTL of equal avidities, 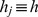 . Throughout the text, we describe the relative loss in fitness and recognition due to escape mutations in terms of notation
. Throughout the text, we describe the relative loss in fitness and recognition due to escape mutations in terms of notation  and
and 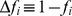 .
.
Mutations are generated randomly with rate  per site per generation of infected cells (i.e., the average lifespan of an infected cell,
per site per generation of infected cells (i.e., the average lifespan of an infected cell, 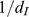 ) between strains that differ by one site. Strains with average copy number above one infected cell are simulated deterministically according to the above equations; below this threshold a strain is considered extinct. The basic model with one escape-conferring site per epitope (
) between strains that differ by one site. Strains with average copy number above one infected cell are simulated deterministically according to the above equations; below this threshold a strain is considered extinct. The basic model with one escape-conferring site per epitope (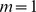 ) has been introduced by Althaus and De Boer [14]. In this work, we adapt the model to focus on the effect of partial recognition and fitness losses during a narrow time interval after acute infection (see
Discussion
for a detailed comparison of the two models). Further information on model parameters and the estimated range of parameter values are listed in Table 1. All simulations were performed in Matlab (Mathworks) and the source code can be obtained freely upon request from the authors.
) has been introduced by Althaus and De Boer [14]. In this work, we adapt the model to focus on the effect of partial recognition and fitness losses during a narrow time interval after acute infection (see
Discussion
for a detailed comparison of the two models). Further information on model parameters and the estimated range of parameter values are listed in Table 1. All simulations were performed in Matlab (Mathworks) and the source code can be obtained freely upon request from the authors.
Killing of infected cells by CTL
Following the original work [14], our model assumes that the majority of infected cells are killed by CTL rather than by viral cytopathicity. It has also been proposed that CTL decrease the viral production in infected cells in a non-lytic fashion, by suppressing viral replication, and that cells die within a fixed period of time (∼1 d) due to viral cytopathicity [50]. A specific choice of the mechanism of viral control does not affect the competition between CTL escape variants (although the long-range nature of the cytokine interaction may slow down the process of escape [51]). However, because there has been considerable controversy on the matter, it deserves a brief discussion.
Two variations of the lytic model have been studied: a simple model in which infected cells are described by a single compartment and a slightly more complex model with two linked compartments of infected cells (cells in the eclipse phase of virion production and cells within a shorter, virus-producing phase). Several authors have argued against single-compartment models for the following reasons: (i) Although viremia and CTL levels vary strongly among patients, the decay rate of viremia under anti-retroviral therapy (ART) (interpreted as the lifespan of infected cells [52], [53]) is approximately 1 d−1 with less than 50% variation between patients [54]. (ii) The rate of viremia growth under CD8 T cell depletion is 2–3 fold faster than the rate of viremia decay under ART, whereas in the simplest lytic model, the two rates must be the same [2]. (iii) Finally, CD8 depletion does not affect the rate viremia decay rate under ART [55], [56]. These observations are, however, compatible with two-compartment lytic models [14], [57]–[60]. In these models the decay rate of viremia under ART is determined by the length of the eclipse phase (lasting ∼1 d), which does not depend on CTL.
Elemans et al [61] compared the ability of two-compartment lytic models with non-lytic models to explain data in Refs. [55], [56] and reported that Akaike information criterion generally favors the non-lytic models. However, they conceded that the accuracy of the cited experiments may be insufficient to detect the effect of CD8 T cells. On the other hand, the short duration of the virus-producing phase measured by Wick et al [62], [63] and a faster decay under ART including an integrase inhibitor compared to therapies including only protease and reverse transcriptase inhibitors [59] support the lytic models with short-lived virus-expressing cells. For these reasons, we choose a lytic model of HIV control (though since we do not aim to predict accurately the viremia decay rate under ART we do not include the eclipse phase). The matter is open to further investigation.
The total CTL killing rate in our model is given by 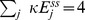 d−1, which is higher than assumed in the original work. Elemans et al [50] reviewed some estimates of the CTL killing rate in a steady state HIV infection. Using antigenic escape data (assuming no mutation cost and full recognition loss), the killing rate was estimated to be 0.1–0.2 d−1 per CTL response [64]. Somewhat larger estimates,
d−1, which is higher than assumed in the original work. Elemans et al [50] reviewed some estimates of the CTL killing rate in a steady state HIV infection. Using antigenic escape data (assuming no mutation cost and full recognition loss), the killing rate was estimated to be 0.1–0.2 d−1 per CTL response [64]. Somewhat larger estimates,  d−1 for the total CTL response, were obtained by another indirect method, which compared the death rate of SIV infected cells in animals treated with antiretroviral therapy (ART) between CD8+ depleted and control groups [61]. The most direct (and highest) estimate, 4–10 d−1 for the total CTL response, was obtained by Wick et al [62], [63] who re-infused into patients autologous, invitro expanded, CD8+ T cells to 2.5% of total CD8 count. Wick et al then directly quantified the decay rate of virus-expressing cells. The estimate they obtained is 4–10 fold higher than the rate of viremia decay under ART, indicating a short virus producing phase of 2–8 hours. The estimate agrees with those obtained by modeling data from acute SIV infection and HIV dynamics during ART [47], [65] and with the killing rate per CTL in acute LCMV infection [66] once the difference in CTL level between the two systems is accounted for.
d−1 for the total CTL response, were obtained by another indirect method, which compared the death rate of SIV infected cells in animals treated with antiretroviral therapy (ART) between CD8+ depleted and control groups [61]. The most direct (and highest) estimate, 4–10 d−1 for the total CTL response, was obtained by Wick et al [62], [63] who re-infused into patients autologous, invitro expanded, CD8+ T cells to 2.5% of total CD8 count. Wick et al then directly quantified the decay rate of virus-expressing cells. The estimate they obtained is 4–10 fold higher than the rate of viremia decay under ART, indicating a short virus producing phase of 2–8 hours. The estimate agrees with those obtained by modeling data from acute SIV infection and HIV dynamics during ART [47], [65] and with the killing rate per CTL in acute LCMV infection [66] once the difference in CTL level between the two systems is accounted for.
We also introduce the virus-induced infected cell death rate,  d−1, which is several-fold smaller than the CTL killing rate in steady state [55]. Parameter
d−1, which is several-fold smaller than the CTL killing rate in steady state [55]. Parameter  is defined as the inverse length of the highly infectable period of the activated cell cycle, End G1-S-G2-M, which lasts less than 1 day. During that period, a virion entry leads to a successful infection, because the concentrations of nucleotides are maximal, and reverse transcription is completed quickly before HIV RNA is degraded by vigorous innate responses [67]. The choice
is defined as the inverse length of the highly infectable period of the activated cell cycle, End G1-S-G2-M, which lasts less than 1 day. During that period, a virion entry leads to a successful infection, because the concentrations of nucleotides are maximal, and reverse transcription is completed quickly before HIV RNA is degraded by vigorous innate responses [67]. The choice 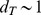 over, e.g.,
over, e.g., 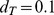 d−1 (often used as an estimate of the total activated cell lifespan) does not change the dynamics of escape.
d−1 (often used as an estimate of the total activated cell lifespan) does not change the dynamics of escape.
Simplified model to study the order of escape mutations
We introduce a simplified model, which does not explicitly consider dynamics, in order to study the sequence of escape mutations for a realistic size genome. Fractional fitness costs (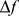 ) and recognition losses (
) and recognition losses (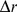 ) are randomly generated from a uniform distribution
) are randomly generated from a uniform distribution 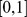 for 100 sites (10 epitopes with 10 sites per epitope) in order to study the sequence of escaped sites. All sites are ranked in order of
for 100 sites (10 epitopes with 10 sites per epitope) in order to study the sequence of escaped sites. All sites are ranked in order of 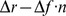 and escape sites in similar ranking over many runs are considered together. CTL decay is introduced for all additional sites in an epitope once a site in the epitope escapes: after each round of escape,
and escape sites in similar ranking over many runs are considered together. CTL decay is introduced for all additional sites in an epitope once a site in the epitope escapes: after each round of escape, 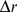 is reduced for all sites in the epitope by:
is reduced for all sites in the epitope by: 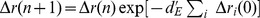 for all i in the epitope that have escaped. Here parameter
for all i in the epitope that have escaped. Here parameter 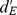 is defined as the decay rate per escape, in contrast to
is defined as the decay rate per escape, in contrast to  in the main model which is defined per day.
in the main model which is defined per day.
Supporting Information
Escape rate and
 are negatively correlated in two experimental studies. The frequency of a mutated epitope for epitopes over time is fit to the curve
are negatively correlated in two experimental studies. The frequency of a mutated epitope for epitopes over time is fit to the curve 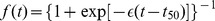 , which describes deterministic selection on a single site with selection coefficient
, which describes deterministic selection on a single site with selection coefficient  , in order to determine parameters
, in order to determine parameters  and
and  . Colored dots show data from a single patient studied in [11] (blue) and multiple patients studied in [30] CH40 (red), CH58 (green), CH77 (cyan). Inset: Simulation example showing the correlation between escape rate,
. Colored dots show data from a single patient studied in [11] (blue) and multiple patients studied in [30] CH40 (red), CH58 (green), CH77 (cyan). Inset: Simulation example showing the correlation between escape rate,  , and the time that the mutation spreads to 50% of the population of infected cells, denoted
, and the time that the mutation spreads to 50% of the population of infected cells, denoted 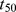 . Parameters
. Parameters  and
and 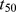 are found for the three escape mutations shown in Figure 1B that occur in the first 200 days post infection. Thus, variation in recognition and fitness losses across many epitopes successfully reproduces this feature of escape dynamics.
are found for the three escape mutations shown in Figure 1B that occur in the first 200 days post infection. Thus, variation in recognition and fitness losses across many epitopes successfully reproduces this feature of escape dynamics.
(TIF)
Estimating the relationship between
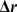 and
and
 approximately from three published experiments. Our model contains one parameter for CTL recognition loss caused by a mutation,
approximately from three published experiments. Our model contains one parameter for CTL recognition loss caused by a mutation,  . In order to compare model predictions with data from Mostowy et al [26], where HLA binding impairment caused by mutations was considered rather than overall CTL recognition loss, we sought to compare HLA binding impairment with overall CTL recognition loss. By combine data from three references, Schneidewind et al [27], Kawashima et al [28] and Matthews et al [29], we were able to demonstrate a strong correlation between the two parameters. This justifies our comparison of our model predictions with data from [26].
. In order to compare model predictions with data from Mostowy et al [26], where HLA binding impairment caused by mutations was considered rather than overall CTL recognition loss, we sought to compare HLA binding impairment with overall CTL recognition loss. By combine data from three references, Schneidewind et al [27], Kawashima et al [28] and Matthews et al [29], we were able to demonstrate a strong correlation between the two parameters. This justifies our comparison of our model predictions with data from [26].
(TIF)
CTL lifetime changes the frequency with which intra-epitope escape patterns are observed. Here we show a modified version of Figure 5 for the case when CTL are long-lived, 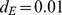 . Using simulation of the model (Figure 1A, Equations 6 to 8) with two sites per epitope,
. Using simulation of the model (Figure 1A, Equations 6 to 8) with two sites per epitope,  , the pattern of escape is calculated for a range of recognition and fitness losses. The pattern that is obtained is plotted as a function of the parameters of recognition loss at the first and second site (
, the pattern of escape is calculated for a range of recognition and fitness losses. The pattern that is obtained is plotted as a function of the parameters of recognition loss at the first and second site (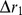 and
and 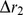 , respectively). In each panel, certain parameters are fixed in order to focus on the effect of recognition loss. Fixed parameters are: the escape rate of the first haplotype and the number of targeted epitopes:
, respectively). In each panel, certain parameters are fixed in order to focus on the effect of recognition loss. Fixed parameters are: the escape rate of the first haplotype and the number of targeted epitopes: 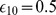 ,
, 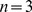 (A,C) which correspond to early infection and
(A,C) which correspond to early infection and 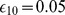 ,
, 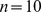 (B,D) which correspond to chronic infection. Fitness costs are chosen such that the second site is less costly than the first:
(B,D) which correspond to chronic infection. Fitness costs are chosen such that the second site is less costly than the first: 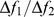 equal to 3 (A,B) or much less costly than the first,
equal to 3 (A,B) or much less costly than the first,  (C,D). Other parameters given in Table 1. Equations S6 (red line) and S9 (blue line) determine the region where the leapfrog pattern can be observed. Regions that require
(C,D). Other parameters given in Table 1. Equations S6 (red line) and S9 (blue line) determine the region where the leapfrog pattern can be observed. Regions that require 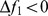 are not allowed by definition (magenta line). The shaded regions between these three lines correspond to regions of parameter space where both sites escape. The corresponding patterns are: “leapfrog” (
are not allowed by definition (magenta line). The shaded regions between these three lines correspond to regions of parameter space where both sites escape. The corresponding patterns are: “leapfrog” (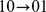 , Figure 4C), “nested” (
, Figure 4C), “nested” (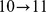 , Figure 4E), “nested leapfrog” (
, Figure 4E), “nested leapfrog” ( ). Observation of the leapfrog pattern in an epitope tightly constrains the fraction of CTL recognition loss conferred by sites in an epitope. The inset shows the length of time during which haplotype 01 is dominant in the escaping epitope. When CTLs are long-lived, the leapfrog pattern (Figure 4C) is observed more often, since the haplotype 01 has time to grow to dominate the population. The death rate of CTL is
). Observation of the leapfrog pattern in an epitope tightly constrains the fraction of CTL recognition loss conferred by sites in an epitope. The inset shows the length of time during which haplotype 01 is dominant in the escaping epitope. When CTLs are long-lived, the leapfrog pattern (Figure 4C) is observed more often, since the haplotype 01 has time to grow to dominate the population. The death rate of CTL is 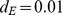 , ten times smaller than the value used in Figure 5. The difference between the predicted leapfrog region as determined by Equations S6 (red line) and S9 (blue line) and the shaded region where leapfrog is actually observed is lessened for long-lived CTL. This is the case because each time that haplotype 01 gains the advantage over haplotype 10, it grows to dominate the population before CTL decay to the level where haplotype 01 also reverts.
, ten times smaller than the value used in Figure 5. The difference between the predicted leapfrog region as determined by Equations S6 (red line) and S9 (blue line) and the shaded region where leapfrog is actually observed is lessened for long-lived CTL. This is the case because each time that haplotype 01 gains the advantage over haplotype 10, it grows to dominate the population before CTL decay to the level where haplotype 01 also reverts.
(TIF)
Steady state and the escape rate.
(PDF)
Escape causes contraction of CTL clones.
(PDF)
General conditions for observing the leapfrog pattern of escape.
(PDF)
Simulation: Leapfrog pattern can occur in a broad range of fitness and recognition losses.
(PDF)
Finding the relationship between
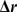 and HLA binding loss from three different experiments.
and HLA binding loss from three different experiments.
(PDF)
Acknowledgments
We thank Todd Allen, Christian Boutwell, John Coffin, Richard Neher, Damien Tully, Alan Perelson, and Leor Weinberger for helpful comments and discussion.
Funding Statement
This work was funded by NIH grants R01AI 063926 to IMR and R37CA 089441 to John Coffin. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Schmitz JE (1999) Control of Viremia in Simian Immunodeficiency Virus Infection by CD8+ Lymphocytes. Science 283: 857–860. [DOI] [PubMed] [Google Scholar]
- 2. Jin X, Bauer D, Tuttleton S, Lewin S, Gettie A, et al. (1999) Dramatic rise in plasma viremia after CD8+ T cell depletion in simian immunodeficiency virus–infected macaques. The Journal of experimental medicine 189: 991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Finlay BB, McFadden G (2006) Anti-Immunology: Evasion of the Host Immune System by Bacterial and Viral Pathogens. Cell 124: 767–782. [DOI] [PubMed] [Google Scholar]
- 4. Rouzine IM, Coffin JM (1999) Linkage disequilibrium test implies a large effective population number for HIV in vivo. Proceedings of the National Academy of Sciences of the United States of America 96: 10758–10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Turnbull EL, Wong M, Wang S, Wei X, Jones NA, et al. (2009) Kinetics of Expansion of Epitope-Specific T Cell Responses during Primary HIV-1 Infection. The Journal of Immunology 182: 7131–7145. [DOI] [PubMed] [Google Scholar]
- 6. Karlsson AC, Iversen AKN, Chapman JM, de Oliviera T, Spotts G, et al. (2007) Sequential broadening of CTL responses in early HIV-1 infection is associated with viral escape. PLoS ONE 2: e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu MKP, Hawkins N, Ritchie AJ, Ganusov VV, Whale V, et al. (2012) Vertical T cell immunodominance and epitope entropy determine HIV-1 escape. J Clin Invest 123: 380–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leslie A (2005) Transmission and accumulation of ctl escape variants drive negative associations between hiv polymorphisms and hla. Journal of Experimental Medicine 201: 891–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Friedrich TC, Dodds EJ, Yant LJ, Vojnov L, Rudersdorf R, et al. (2004) Reversion of ctl escape–variant immunodeficiency viruses in vivo. Nat Med 10: 275–281. [DOI] [PubMed] [Google Scholar]
- 10. Troyer RM, McNevin J, Liu Y, Zhang SC, Krizan RW, et al. (2009) Variable Fitness Impact of HIV-1 Escape Mutations to Cytotoxic T Lymphocyte (CTL) Response. PLoS Pathogens 5: e1000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu Y, Mcnevin JP, Holte S, Mcelrath MJ, Mullins JI (2011) Dynamics of viral evolution and ctl responses in hiv-1 infection. PLoS ONE 6: e15639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ganusov VV, Goonetilleke N, Liu MKP, Ferrari G, Shaw GM, et al. (2011) Fitness Costs and Diversity of the Cytotoxic T Lymphocyte (CTL) Response Determine the Rate of CTL Escape during Acute and Chronic Phases of HIV Infection. Journal of Virology 85: 10518–10528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, et al. (2012) Whole Genome Deep Sequencing of HIV-1 Reveals the Impact of Early Minor Variants Upon Immune Recognition During Acute Infection. PLoS Pathogens 8: e1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Althaus CL, Boer RJD (2008) Dynamics of immune escape during hiv/siv infection. PLoS Computational Biology 4: e1000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mostowy R, Kouyos RD, Fouchet D, Bonhoeffer S (2011) The Role of Recombination for the Coevolutionary Dynamics of HIV and the Immune Response. PLoS ONE 6: e16052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Deutekom HWM, Wijnker G, De Boer RJ (2013) The Rate of Immune Escape Vanishes When Multiple Immune Responses Control a HIV Infection. J Immunol 191: 3277–86. [DOI] [PubMed] [Google Scholar]
- 17. Kearney M, Maldarelli F, Shao W, Margolick JB, Daar ES, et al. (2009) Human Immunodeficiency Virus Type 1 Population Genetics and Adaptation in Newly Infected Individuals. Journal of Virology 83: 2715–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fryer HR, Frater J, Duda A, Roberts MG, Phillips RE, et al. (2010) Modelling the evolution and spread of HIV immune escape mutants. PLoS Pathogens 6: e1001196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kelleher AD, Long C, Holmes EC, Allen RL, Wilson J, et al. (2001) Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. The Journal of experimental medicine 193: 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Crawford H, Matthews PC, Schaefer M, Carlson JM, Leslie A, et al. (2011) The Hypervariable HIV-1 Capsid Protein Residues Comprise HLA-Driven CD8+ T-Cell Escape Mutations and Covarying HLA-Independent Polymorphisms. Journal of Virology 85: 1384–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Song H, Pavlicek JW, Cai F, Bhattacharya T, Li H, et al. (2012) Impact of immune escape mutations on HIV-1 fitness in the context of the cognate transmitted/founder genome. Retrovirology 9: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boutwell CL, Carlson JM, Lin TH, Seese A, Power KA, et al. (2013) Frequent and variable cytotoxic-T-lymphocyte escape-associated fitness costs in the human immunodeficiency virus type 1 subtype B Gag proteins. Journal of Virology 87: 3952–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu Y, Mcnevin JP, Holte S, Mcelrath MJ, Mullins JI (2011) Dynamics of Viral Evolution and CTL Responses in HIV-1 Infection. PLoS ONE 6: e15639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cale EM, Hraber P, Giorgi EE, Fischer W, Bhattacharya T, et al. (2011) Epitope-specific cd8+ t lymphocytes cross-recognize mutant simian immunodeficiency virus (siv) sequences but fail to contain very early evolution and eventual fixation of epitope escape mutations during siv infection. Journal of Virology 85: 3746–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ganusov VV, De Boer RJ (2005) Estimating Costs and Benefits of CTL Escape Mutations in SIV/HIV Infection. PLoS Computational Biology preprint: e24. [DOI] [PMC free article] [PubMed]
- 26. Mostowy R, Kouyos RD, Hoof I, Hinkley T, Haddad M, et al. (2012) Estimating the fitness cost of escape from HLA presentation in HIV-1 protease and reverse transcriptase. PLoS Computational Biology 8: e1002525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schneidewind A, Brockman MA, Sidney J, Wang YE, Chen H, et al. (2008) Structural and functional constraints limit options for cytotoxic T-lymphocyte escape in the immunodominant HLA-B27-restricted epitope in human immunodeficiency virus type 1 capsid. Journal of Virology 82: 5594–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, et al. (2009) Adaptation of HIV-1 to human leukocyte antigen class I. Nature 458: 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matthews PC, Koyanagi M, Kloverpris HN, Harndahl M, Stryhn A, et al. (2012) Differential Clade-Specific HLA-B*3501 Association with HIV-1 Disease Outcome Is Linked to Immunogenicity of a Single Gag Epitope. Journal of Virology 86: 12643–12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goonetilleke N, Liu MK, Salazar-Gonzalez JF, Ferrari G, Giorgi E, et al. (2009) The first t cell response to transmitted/founder virus contributes to the control of acute viremia in hiv-1 infection. Journal of Experimental Medicine 206: 1253–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fischer W, Ganusov VV, Giorgi EE, Hraber PT, Keele BF, et al. (2010) Transmission of Single HIV-1 Genomes and Dynamics of Early Immune Escape Revealed by Ultra-Deep Sequencing. PLoS ONE 5: e12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rouzine IM, Weinberger LS (2013) The quantitative theory of within-host viral evolution. Journal of Statistical Mechanics: Theory and Experiment 2013: P01009. [Google Scholar]
- 33. Turnbull EL, Wong M, Wang S, Wei X, Jones NA, et al. (2009) Kinetics of expansion of epitope-specific t cell responses during primary hiv-1 infection. The Journal of Immunology 182: 7131–7145. [DOI] [PubMed] [Google Scholar]
- 34. Kouyos RD, Althaus CL, Bonhoeffer S (2006) Stochastic or deterministic: what is the effective population size of HIV-1? Trends in microbiology 14: 507–511. [DOI] [PubMed] [Google Scholar]
- 35.Pennings P, Kryazhimskiy S, Wakeley J (2014) PLOS Genetics: Loss and Recovery of Genetic Diversity in Adapting Populations of HIV. PLoS Genetics. [DOI] [PMC free article] [PubMed]
- 36. Richman DD, Havlir D, Corbeil J, Looney D, Ignacio C, et al. (1994) Nevirapine resistance mutations of human immunodeficiency virus type 1 selected during therapy. Journal of Virology 68: 1660–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu Y, Mullins JI, Mittler JE (2006) Waiting times for the appearance of cytotoxic T-lymphocyte escape mutants in chronic HIV-1 infection. Virology 347: 140–146. [DOI] [PubMed] [Google Scholar]
- 38. Quiñones-Mateu ME, Arts EJ (2002) Fitness of drug resistant HIV-1: methodology and clinical implications. Drug Resistance Updates 5: 224–233. [DOI] [PubMed] [Google Scholar]
- 39. Bonhoeffer S (2004) Evidence for Positive Epistasis in HIV-1. Science 306: 1547–1550. [DOI] [PubMed] [Google Scholar]
- 40. Hinkley T, Martins J, Chappey C, Haddad M, Stawiski E, et al. (2011) A systems analysis of mutational effects in HIV-1 protease and reverse transcriptase. Nature Genetics 43: 487–489. [DOI] [PubMed] [Google Scholar]
- 41. Poon AFY, Swenson LC, Dong WWY, Deng W, Kosakovsky Pond SL, et al. (2010) Phylogenetic Analysis of Population-Based and Deep Sequencing Data to Identify Coevolving Sites in the nef Gene of HIV-1. Molecular Biology and Evolution 27: 819–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rouzine IM, Coffin JM (1999) Search for the mechanism of genetic variation in the pro gene of human immunodeficiency virus. Journal of Virology 73: 8167–8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kadolsky UD, Asquith B (2010) Quantifying the Impact of Human Immunodeficiency Virus-1 Escape From Cytotoxic T-Lymphocytes. PLoS Computational Biology 6: e1000981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haase AT (2011) Early Events in Sexual Transmission of HIV and SIV and Opportunities for Interventions. Annual review of medicine 62: 127–139. [DOI] [PubMed] [Google Scholar]
- 45. Ogg GS (1998) Quantitation of HIV-1-Specific Cytotoxic T Lymphocytes and Plasma Load of Viral RNA. Science 279: 2103–2106. [DOI] [PubMed] [Google Scholar]
- 46. Kuroda MJ, Schmitz JE, Charini W, Nickerson C, Lifton M, et al. (1999) Emergence of CTL coincides with clearance of virus during primary simian immunodeficiency virus infection in rhesus monkeys. Journal of immunology (Baltimore, Md: 1950) 162: 5127–5133. [PubMed] [Google Scholar]
- 47. Sergeev RA, Batorsky RE, Rouzine IM (2010) Model with two types of CTL regulation and experiments on CTL dynamics. Journal of Theoretical Biology 263: 369–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Batorsky R, Kearney MF, Palmer SE, Maldarelli F, Rouzine IM, et al. (2011) Estimate of effective recombination rate and average selection coefficient for HIV in chronic infection. Proceedings of the National Academy of Sciences 108: 5661–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Neher RA, Shraiman BI, Fisher DS (2010) Rate of Adaptation in Large Sexual Populations. Genetics 184: 467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Elemans M, Florins A, Willems L, Asquith B (2014) Rates of CTL Killing in Persistent Viral Infection In Vivo. PLoS Computational Biology 10: e1003534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Seich Al Basatena NK, Chatzimichalis K, Graw F, Frost SDW, Regoes RR, et al. (2013) Can non-lytic CD8+ T cells drive HIV-1 escape? PLoS Pathogens 9: e1003656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ho D, Neumann AU, Perelson AS, Chen W, Leonard JM, et al. (1995) Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373: 123–126. [DOI] [PubMed] [Google Scholar]
- 53. Perelson A, Neumann A, Markowitz M, Leonard J, Ho D (1996) Hiv-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271: 1582–1586. [DOI] [PubMed] [Google Scholar]
- 54. Markowitz M, Louie M, Hurley A, Sun E, Di Mascio M, et al. (2003) A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-cell decay in vivo. Journal of Virology 77: 5037–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Klatt NR, Shudo E, Ortiz AM, Engram JC, Paiardini M, et al. (2010) CD8+ lymphocytes control viral replication in SIVmac239-infected rhesus macaques without decreasing the lifespan of productively infected cells. PLoS Pathogens 6: e1000747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wong JK, Strain MC, Porrata R, Reay E, Sankaran-Walters S, et al. (2010) In Vivo CD8+ T-Cell Suppression of SIV Viremia Is Not Mediated by CTL Clearance of Productively Infected Cells. PLoS Pathogens 6: e1000748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Klenerman P, Phillips RE, Rinaldo CR, Wahl LM, Ogg G, et al. (1996) Cytotoxic T lymphocytes and viral turnover in HIV type 1 infection. Proceedings of the National Academy of Sciences of the United States of America 93: 15323–15328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rouzine IM, Sergeev RA, Glushtsov AI (2006) Two types of cytotoxic lymphocyte regulation explain kinetics of immune response to human immunodeficiency virus. Proceedings of the National Academy of Sciences 103: 666–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sedaghat AR, Dinoso JB, Shen L, Wilke CO, Siliciano RF (2008) Decay dynamics of HIV-1 depend on the inhibited stages of the viral life cycle. Proceedings of the National Academy of Sciences 105: 4832–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Althaus C (2011) Implications of CTL-Mediated Killing of HIV-Infected Cells during the Non-Productive Stage of Infection. PLoS ONE 6: e16468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Elemans M, Basatena NKSA, Klatt NR, Gkekas C, Silvestri G, et al. (2011) Why Don't CD8+ T Cells Reduce the Lifespan of SIV-Infected Cells In Vivo? PLoS Computational Biology 7: e1002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Brodie SJ, Lewinsohn DA, Patterson BK, Jiyamapa D, Krieger J, et al. (1999) In vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nature Medicine 5: 34–41. [DOI] [PubMed] [Google Scholar]
- 63. Wick WD, Yang OO, Corey L, Self SG (2005) How many human immunodeficiency virus type 1-infected target cells can a cytotoxic T-lymphocyte kill? Journal of Virology 79: 13579–13586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Asquith B, Edwards CTT, Lipsitch M, McLean AR (2006) Inefficient Cytotoxic T Lymphocyte–Mediated Killing of HIV-1–Infected Cells In Vivo. PLoS Biology 4: e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sergeev RA, Batorsky RE, Coffin JM, Rouzine IM (2010) Interpreting the effect of vaccination on steady state infection in animals challenged with Simian immunodeficiency virus. Journal of Theoretical Biology 263: 385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ganusov VV, De Boer RJ (2008) Estimating In Vivo Death Rates of Targets due to CD8 T-Cell-Mediated Killing. Journal of Virology 82: 11749–11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pan X, Baldauf HM, Keppler OT, Fackler OT (2013) Restrictions to HIV-1 replication in resting CD4+ T lymphocytes. Cell research 23: 876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li Q, Duan L, Estes JD, Ma ZM, Rourke T, et al. (2005) Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 434: 1148–1152. [DOI] [PubMed] [Google Scholar]
- 69. Kuroda MJ, Schmitz JE, Charini W, Nickerson C, Lifton M, et al. (1999) Emergence of CTL coincides with clearance of virus during primary simian immunodeficiency virus infection in rhesus monkeys. Journal of immunology (Baltimore, Md: 1950) 162: 5127–5133. [PubMed] [Google Scholar]
- 70. Ogg GS, Jin X, Bonhoeffer S, Moss P, Nowak MA, et al. (1999) Decay kinetics of human immunodeficiency virus-specific effector cytotoxic T lymphocytes after combination antiretroviral therapy. Journal of Virology 73: 797–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Escape rate and
are negatively correlated in two experimental studies. The frequency of a mutated epitope for epitopes over time is fit to the curve , which describes deterministic selection on a single site with selection coefficient , in order to determine parameters and . Colored dots show data from a single patient studied in [11] (blue) and multiple patients studied in [30] CH40 (red), CH58 (green), CH77 (cyan). Inset: Simulation example showing the correlation between escape rate, , and the time that the mutation spreads to 50% of the population of infected cells, denoted . Parameters and are found for the three escape mutations shown in Figure 1B that occur in the first 200 days post infection. Thus, variation in recognition and fitness losses across many epitopes successfully reproduces this feature of escape dynamics.
(TIF)
Estimating the relationship between
and
approximately from three published experiments. Our model contains one parameter for CTL recognition loss caused by a mutation, . In order to compare model predictions with data from Mostowy et al [26], where HLA binding impairment caused by mutations was considered rather than overall CTL recognition loss, we sought to compare HLA binding impairment with overall CTL recognition loss. By combine data from three references, Schneidewind et al [27], Kawashima et al [28] and Matthews et al [29], we were able to demonstrate a strong correlation between the two parameters. This justifies our comparison of our model predictions with data from [26].
(TIF)
CTL lifetime changes the frequency with which intra-epitope escape patterns are observed. Here we show a modified version of Figure 5 for the case when CTL are long-lived, . Using simulation of the model (Figure 1A, Equations 6 to 8) with two sites per epitope, , the pattern of escape is calculated for a range of recognition and fitness losses. The pattern that is obtained is plotted as a function of the parameters of recognition loss at the first and second site ( and , respectively). In each panel, certain parameters are fixed in order to focus on the effect of recognition loss. Fixed parameters are: the escape rate of the first haplotype and the number of targeted epitopes: , (A,C) which correspond to early infection and , (B,D) which correspond to chronic infection. Fitness costs are chosen such that the second site is less costly than the first: equal to 3 (A,B) or much less costly than the first, (C,D). Other parameters given in Table 1. Equations S6 (red line) and S9 (blue line) determine the region where the leapfrog pattern can be observed. Regions that require are not allowed by definition (magenta line). The shaded regions between these three lines correspond to regions of parameter space where both sites escape. The corresponding patterns are: “leapfrog” (, Figure 4C), “nested” (, Figure 4E), “nested leapfrog” (). Observation of the leapfrog pattern in an epitope tightly constrains the fraction of CTL recognition loss conferred by sites in an epitope. The inset shows the length of time during which haplotype 01 is dominant in the escaping epitope. When CTLs are long-lived, the leapfrog pattern (Figure 4C) is observed more often, since the haplotype 01 has time to grow to dominate the population. The death rate of CTL is , ten times smaller than the value used in Figure 5. The difference between the predicted leapfrog region as determined by Equations S6 (red line) and S9 (blue line) and the shaded region where leapfrog is actually observed is lessened for long-lived CTL. This is the case because each time that haplotype 01 gains the advantage over haplotype 10, it grows to dominate the population before CTL decay to the level where haplotype 01 also reverts.
(TIF)
Steady state and the escape rate.
(PDF)
Escape causes contraction of CTL clones.
(PDF)
General conditions for observing the leapfrog pattern of escape.
(PDF)
Simulation: Leapfrog pattern can occur in a broad range of fitness and recognition losses.
(PDF)
Finding the relationship between
and HLA binding loss from three different experiments.
(PDF)