

The colon cancer tumor suppressor adenomatous polyposis coli (APC) negatively regulates Wnt signaling in the β-catenin destruction complex by binding to β-catenin and facilitating its phosphorylation and degradation. APC self-association plays an integral role in the assembly, stability, and activity of the destruction complex.
Abstract
The tumor suppressor adenomatous polyposis coli (APC) is an essential negative regulator of Wnt signaling through its activity in the destruction complex with Axin, GSK3β, and CK1 that targets β-catenin/Armadillo (β-cat/Arm) for proteosomal degradation. The destruction complex forms macromolecular particles we termed the destructosome. Whereas APC functions in the complex through its ability to bind both β-cat and Axin, we hypothesize that APC proteins play an additional role in destructosome assembly through self-association. Here we show that a novel N-terminal coil, the APC self-association domain (ASAD), found in vertebrate and invertebrate APCs, directly mediates self-association of Drosophila APC2 and plays an essential role in the assembly and stability of the destructosome that regulates β-cat degradation in Drosophila and human cells. Consistent with this, removal of the ASAD from the Drosophila embryo results in β-cat/Arm accumulation and aberrant Wnt pathway activation. These results suggest that APC proteins are required not only for the activity of the destructosome, but also for the assembly and stability of this macromolecular machine.
INTRODUCTION
Canonical Wnt signal transduction is an evolutionarily conserved pathway from hydra to humans that plays essential roles in embryonic development and adult tissue maintenance by regulating cellular differentiation, proliferation, and morphogenesis (Logan and Nusse, 2004; Guder et al., 2006). Loss or constitutive activation of the pathway is lethal during embryogenesis due to defects in proliferation and differentiation (Logan and Nusse, 2004). In humans, inappropriate activation of Wnt signaling is associated not only with various types of cancer (colon, breast, and ovarian) but also with a myriad of other diseases, including diabetes, Alzheimer's disease, and osteoporosis (Logan and Nusse, 2004; Welters and Kulkarni, 2008; Clevers and Nusse, 2012; Kim et al., 2013; Oliva et al., 2013). Thus the tight control of Wnt signaling is critical for both normal development and tissue homeostasis.
In the absence of a Wnt ligand, the pathway is negatively regulated by a complex of proteins called the destruction complex, which phosphorylates the key effector of the pathway, β-catenin (Armadillo in Drosophila; Cadigan and Peifer, 2009). Phosphorylated β-catenin (β-cat) is ubiquitinated by the β-TrCP ubiquitin E3 ligase to be degraded by the proteosome. Binding of the Wnt ligand to the coreceptor complex of Frizzled and LRP5/6 inactivates the destruction complex, allowing the accumulation and nuclear translocation of β-cat. Together with TCF/LEF-family transcription factors, β-cat activates transcription of Wnt target genes. Loss-of-function mutations in components of the destruction complex lead to ligand-independent accumulation of β-cat and the constitutive activation of Wnt target genes that play roles in proliferation, cell survival, and differentiation (Fodde, 2002; van de Wetering et al., 2002; Chen et al., 2003; Komori et al., 2014).
Adenomatous polyposis coli (APC) is a colon cancer tumor suppressor and an essential component of the destruction complex (McCartney and Näthke, 2008). Approximately 80% of all inherited and sporadic forms of colon cancer are associated with APC mutation (Polakis, 2012). The initiation of APC-dependent colorectal cancer is primarily due to the loss of destruction complex activity and the inappropriate activation of Wnt targets (Polakis, 2007), but APC's roles in cytoskeletal regulation may also contribute. APC is a core component of the destruction complex together with Axin and the kinases GSK3β and CK1. The cytoplasmic destruction complex appears to form macromolecular particles, or puncta, we termed the “destructosome” (Kunttas-Tatli et al., 2012; also known as the degradasome, Mendoza-Topaz et al., 2011; or Axin complex, Li et al., 2012). Despite its functional significance and abundant study, the inner workings of the destructosome and the precise role of APC in this molecular machine are enigmatic. Several hypotheses for APC's destruction complex function have been proposed. Owing to its large size and the presence of many putative protein–protein interaction domains, APC was initially believed to act as a scaffold. However, because Axin can bind directly to all of the core components of the destruction complex, including APC, β-cat, GSK3β, and CK1, as well as Dishevelled and the LRP5/6 coreceptor (Hart et al., 1998; Ikeda et al., 1998; Kishida et al., 1999; Mao et al., 2001; Ha et al., 2004), it is a stronger candidate for scaffolding function. Later studies proposed that APC–β-cat interactions are required 1) for phosphorylated β-cat to be recognized by the ubiquitination complex as a part of a catalytic cycle (Kimelman and Xu, 2006), 2) to protect β-cat from rapid dephosphorylation by PP2A upon β-cat release from the destruction complex (Su et al., 2008), and 3) to increase the activity of the destruction complex when cellular levels of β-cat are high (Ha et al., 2004). However, recent work has also called into question the importance of a direct APC–β-cat interaction for destruction complex function altogether (Yamulla et al., 2014). It has also been suggested that APC functions downstream of β-cat phosphorylation by mediating β-cat's ubiquitination by β-TRCP (Yang et al., 2006; Li et al., 2012).
In addition, APC self-association may contribute to both destruction complex function and dysfunction (Kunttas-Tatli et al., 2012). Vertebrate APC (vAPC) can self-associate via multiple mechanisms and domains. However, the precise role of APC self-association in normal destruction complex function and the affects this has on cancer initiation and progression are unclear. Two self-association domains C-terminal to the Armadillo (Arm) repeats in vAPC have been clearly implicated in APC's normal cytoskeletal functions. The dimerization coil ANS2 (Figure 1A) within the basic domain is required for APC's actin nucleation function (Okada et al., 2010), whereas a second oligomerization domain (OD-2; Figure 1A) can modulate the clustering of APC at microtubule plus ends at the tips of membrane protrusions (Li et al., 2008). N-terminal to the Arm repeats, vAPC can form coiled-coil–based dimers through an N-terminal coil (OD-1; Figure 1A), but the precise role of OD-1 in normal APC function is not well understood. The presence of multiple self-association sites within vAPC suggests that the protein may have the ability to form large oligomers in addition to dimers, although it is not clear whether this occurs in vivo.
FIGURE 1:
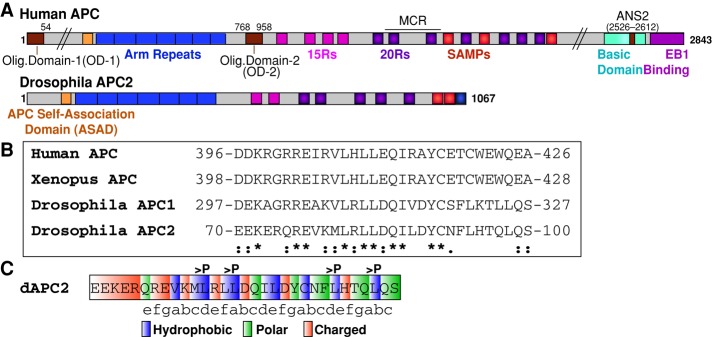
The ASAD is a conserved N-terminal coil. (A) Schematic representation of human APC and Drosophila APC2. ANS2, actin nucleation sequence 2; Arm repeats, Armadillo repeats (blue); ASAD, APC self-association domain (orange); MCR, mutation cluster region; 15Rs, 15–amino acid repeats (pink); 20Rs, 20–amino acid repeats (purple). (B) Sequence alignment of ASAD between human, Xenopus, and Drosophila APC proteins (*identical; [:], conserved substitution; [.], semiconserved substitution). (C) The ASAD coil fits into the classic heptad repeat (abcdefg) motif, where a and d are hydrophobic, e and g are charged, and b, c, and f tend to be polar amino acids (Gruber and Lupas, 2003). The four residues changed to proline in the APC2-ASADPro mutant are indicated.
The complexity of vAPC self-association prompted us to investigate the role of APC self-association in the destructosome using the simpler and more tractable Drosophila APC2 as a model. Although neither Drosophila APC1 nor APC2 contains sequence homology to any of vAPC's self-association domains, we and others have shown that Drosophila APC proteins do self-associate through an N-terminal domain (Mattie et al., 2010; Zhou et al., 2011; Roberts et al., 2012). Consistent with this, high levels of APC2 mutants lacking the central β-cat interaction domains (the 15– and 20–amino acid repeats) act as dominant negatives in Wnt signaling in the embryo (Roberts et al., 2011; Kunttas-Tatli et al., 2012). We predicted that this is because these mutants could associate with wild-type APC2 through the N-terminal domain and compete for Axin binding through their intact SAMP repeats. Finally, our data suggested an unanticipated cooperativity between APC2 and APC1 in the destruction complex, which may be mediated through hetero-oligomerization (Kunttas-Tatli et al., 2012).
To test the role of APC self-association in destruction complex function, we identified a novel N-terminal self-association domain in Drosophila APC proteins that appears to be conserved in all other APC proteins examined. Here we demonstrate that this APC self-association domain (ASAD) is necessary for the assembly and stability of the destructosome both in Drosophila S2 cells and in human SW480 colorectal cancer cells, and which in turn is essential for β-cat/Arm degradation. Furthermore, we show that loss of APC2 self-association in the Drosophila embryo leads to inappropriate activation of the Wnt signaling pathway due to loss of destructosome activity. These results suggest a novel role for APC proteins in the assembly and stability of the destructosome, in addition to their more established role in destructosome activity.
RESULTS
An N-terminal coil mediates the self-association of Drosophila APC proteins
To dissect the role of APC self-association in destructosome structure and function, we identified a novel self-association domain in the N-terminal half of Drosophila APC2. Previously we demonstrated that the N-terminal region of APC2 containing the Arm repeats (amino acids [aa] 1–490) mediates self-association (Zhou et al., 2011; Roberts et al., 2012), although it does not share sequence conservation with either OD-1 or OD-2 of human APC (Figure 1A). OD-1 mediates the formation of homodimers through a parallel coiled-coil (Joslyn et al., 1993). On this basis, we predicted that the Drosophila APC proteins would contain an N-terminal coil to promote self-association. We scanned the region of APC2 N-terminal to the Arm repeats (aa 1–112) using COILS to identify sequences likely to adopt a coiled-coil conformation (Lupas et al., 1991). Using these predictions and the recently solved crystal structure of the region in vAPC (Morishita et al., 2011; Zhang et al., 2012), we identified a putative coil fitting the classic heptad repeat model (Figure 1C; Gruber and Lupas, 2003) residing immediately N-terminal to the Arm-repeats (aa 70–100; Figure 1, A and B). Of interest, this N-terminal coil appears to be conserved in all bilateria APC proteins examined, whereas OD-1 was primarily present in the deuterostome lineage (Figure 1B and Supplemental Figure S1). Thus we designated this novel N-terminal coil the ASAD and hypothesized that it could mediate self-association of Drosophila APC proteins.
To test this hypothesis, we generated a mutant version of Drosophila APC2 lacking this region (APC2-∆ASAD; Figure 2A). In addition, we disrupted potential coiled-coil formation by changing four key hydrophobic leucine residues to proline (APC2-ASADPro; Figures 1C and 2A). To determine whether this domain is necessary to mediate APC2 self-association, we performed immunoprecipitation assays from transiently transfected Drosophila S2 cells. Previously we showed that mCherry-tagged (mCh) APC2-FL (full length) and APC2-N (aa 1–490) could coprecipitate untagged APC2-FL, unlike mCh-APC2-C (aa 491–1067; Zhou et al., 2011; Figure 2B). Neither mCh-APC2-∆ASAD nor mCh-APC2-ASADPro was able to coprecipitate untagged APC2-FL, demonstrating that the N-terminal coil is necessary for self-association (Figure 2B). Because human OD-1 mediates dimer formation through a direct protein–protein interaction (Joslyn et al., 1993), we asked whether ASAD mediates direct APC2–APC2 binding. Consistent with that model, APC2-N (Figure 2A) self-associated in a yeast two-hybrid (Y2H) assay, and this interaction was disrupted in both ASAD mutants (APC2-N-ΔASAD and APC2-N-ASADPro; Figure 2C).
FIGURE 2:

Removal of ASAD disrupts APC self-association. (A) Schematic representation of Drosophila APC2 and APC1 constructs used in the study. (B) mCherry (mCh)-tagged full-length APC2 protein (red arrow) coimmunoprecipitates untagged full-length protein (black arrow). mCh-APC2 ASAD mutants (both deletion and point mutant) and mCh-APC2-C (red arrows) fail to coimmunoprecipitate untagged APC2-FL (black arrows in 2–4). (C) Yeast two-hybrid experiments demonstrated that APC2-N can interact directly with APC2-N. Deletion of the ASAD (APC2-N-ΔASAD) or disruption of the potential coiled coil (APC2-N-ASADPro) abolishes this interaction. (D) mCh-APC1-N coimmunoprecipitates untagged APC2-FL protein but fails to coimmunoprecipitate the APC2-ΔASAD mutant (black arrow). APC2-N175K contains a mutation in the Arm repeats and retains the mCh-APC1-N interaction. (E) mCh-KAP3 coimmunoprecipitates both full-length APC2 and the APC2-ΔASAD mutant (black arrows). CL, cell lysate; DL, depleted lysate; P, pull down.
Previous work from our lab and others indicated that both human and Drosophila APCs (APC&APCL and APC1&APC2, respectively) can heteroassociate through an N-terminal domain and that this complex may collaborate in the destructosome (Mattie et al., 2010; Kunttas-Tatli et al., 2012; Schneikert et al., 2013). Because Drosophila APC1 also lacks OD-1 but contains the ASAD (Figure 1B), we asked whether the ASAD could mediate the association between Drosophila APC1 and APC2. Consistent with this hypothesis, mCh-APC1-N (aa 1–904) coprecipitated APC2-FL but not APC2-∆ASAD (Figure 2D). In contrast, a point mutation in the second Arm repeat of APC2 (N175K) that disrupts protein binding to the Arm repeats in human APC (hAPC; Watanabe et al., 2004), did not interfere with APC1–APC2 complex formation (Figure 2D).
Given the close proximity of the ASAD to the highly structured Arm repeats (Figure 1A), we asked whether deleting the ASAD domain disrupts the folding of these repeats. The crystal structure of the human APC Arm repeats was unaffected by the absence of sequences containing the ASAD (Zhang et al., 2012), suggesting that deletion of the ASAD alone is unlikely to disrupt Arm-repeat binding interactions. To test this directly, we examined the interaction between the Drosophila homologue of a known human APC Arm-repeat-binding protein, kinesin-associated protein 3 (KAP3; Jimbo et al., 2002), and APC2-∆ASAD. Deletion of the ASAD did not interfere with the ability of KAP3 to coprecipitate with APC2 (Figure 2E). In fact, KAP3 appeared to coprecipitate better in the absence of APC2 self-association, suggesting that APC2 self-association may negatively regulate Arm-repeat–mediated protein–protein interactions.
Disruption of APC2 self-association leads to defects in the assembly of destructosome puncta in both Drosophila and human cells
The destructosome is typically visualized as cytoplasmic Axin puncta that are observed both endogenously and when Axin is overexpressed in cell culture and intact tissues (Fagotto et al., 1999; Schwarz-Romond et al., 2007b; Faux et al., 2008; Fiedler et al., 2011). Overexpressed Axin tagged with green fluorescent protein (GFP), FLAG, red fluorescent protein, myc, or hemagglutinin (HA) localizes to cytoplasmic puncta in a variety of vertebrate and fly cultured cells, including S2, SW480, HeLa, MDCK, and Cos-7. Overexpressed Axin has been shown to rescue β-cat destruction in colorectal cancer cell lines (Behrens et al., 1998; Hart et al., 1998; Nakamura et al., 1998; Roberts et al., 2011), and in Drosophila embryos, cytoplasmic Axin-GFP puncta become cortical when cells activate the Wnt pathway, suggesting that these overexpression puncta are responsive to Wnt pathway activation (Mendoza-Topaz et al., 2011). Axin can self-associate via its C-terminal DIX domain (also called DAX), which is essential for its function in β-cat destruction and for its ability to form puncta (Schwarz-Romond et al., 2007a, b). It was recently shown that APC is essential for destructosome assembly, as in the absence of APC, Axin failed to form functional destructosomes (Mendoza-Topaz et al., 2011). Like Axin, we predicted that APC2 contributes to the formation of the destructosome through its ability to self-associate and form larger macromolecular assemblages (Kunttas-Tatli et al., 2012). To test this hypothesis, we coexpressed Axin and APC2 in Drosophila S2 cells. When expressed alone, both Axin-GFP (Figure 3A1) and Axin-HA (Supplemental Figure S2A) formed cytoplasmic puncta, albeit smaller in the case of Axin-HA. Thus the GFP tag may have a slight effect on puncta size. On the other hand, mCh-APC2-FL localized primarily to the cell cortex (Figure 3A2; Zhou et al., 2011). When coexpressed with Axin-GFP, mCh-APC2-FL redistributed and localized primarily in the cytoplasmic Axin puncta (Figure 3A3). Deletion of the Axin-binding SAMP repeats from APC2 (APC2-∆SAMP) restored cortical localization of APC2 (Figure 3A4), indicating that the primary mechanism for APC2's incorporation into Axin puncta is its direct association with Axin (Roberts et al., 2011).
FIGURE 3:

Disruption of APC2 self-association leads to defects in destructosome assembly in live Drosophila S2 cells. In all cases, Axin is GFP tagged and APC2 is mCherry tagged. Dotted lines indicate cell boundaries. (A) When expressed alone in S2 cells, Axin-GFP oligomers can be visualized as cytoplasmic puncta (1), and mCh-APC2-FL (2) is primarily cortical. When coexpressed, mCh-APC2-FL colocalizes in cytoplasmic puncta with Axin-GFP (3). Removal of APC2's Axin interaction domains (APC2-∆SAMP) disrupts this colocalization (4). ASAD mutants (both ΔASAD and ASADPro) colocalize with Axin-GFP, but cells coexpressing these proteins exhibit defects in puncta assembly and morphology (5, 6). (B) Quantification of puncta size in S2 cells expressing Axin alone and coexpressing Axin and APC2 in cells sorted into three expression level categories (high, medium, and low) by FACS using Axin-GFP. Images were taken under the same imaging conditions, and puncta size was determined using Imaris. Puncta were then divided into three classes based on area (micrometers squared). Coexpression with APC2-FL is associated with fewer, larger puncta at all three expression levels (see puncta/cell ratios). Coexpression with the ASAD mutants showed increase in the number of small puncta only in the low category. Owing to the disrupted puncta morphology in ASAD mutants, we were only able to assess the puncta size in this category. Two-tailed z test demonstrates significant differences between different groups. Scale bar, 10 μm.
Consistent with the hypothesis that APC proteins promote the assembly of the destructosome, coexpression of Axin-GFP with mCh-APC2-FL resulted in the formation of fewer, larger Axin-GFP puncta (Figure 3B). Because expression levels could influence this effect, we used FACS to sort the cells into three different groups based on expression of Axin-GFP (high, medium, and low) and assessed puncta size and number. Consistent with our observations, Axin formed fewer, larger puncta in the presence of APC2-FL at all three expression levels (Figure 3B). To test the hypothesis that APC2-FL promotes the formation of larger puncta by increasing the rate of puncta growth, we examined puncta from cells expressing Axin-GFP alone or coexpressed with APC2-FL over time (Supplemental Figure S2B). Axin-GFP expressed alone formed puncta even at the lowest detectable expression level a few hours after induction, suggesting that puncta formation is not the result of significant overexpression. Cells expressing Axin-GFP alone contained puncta that reached their maximum size by 24–48 h postinduction, and at 96 h, these cells contained many smaller puncta. In contrast, cells coexpressing Axin and APC2 displayed large, and often single, misshapen puncta by 96 h postinduction. This suggests that APC2 primarily promotes puncta assembly rather than accelerates their growth rate.
To determine whether the APC2 ASAD mediates puncta assembly, we coexpressed the APC2 self-association mutants (APC2-∆ASAD and APC2-ASADPro) with Axin-GFP (Figure 3A, 5 and 6). Expression of these self-association mutants produced a dramatic change in destructosome morphology. Puncta that incorporated the APC2 self-association mutants appeared smaller, fragmented, and dispersed throughout the cytoplasm (Figure 3A, 5 and 6, and B). The striking alteration in destructosome morphology precluded quantification of their size at medium and high expression levels (Supplemental Figure S3).
To rule out the possibility that this is a cell type–specific effect, we examined the role of APC2 in destructosome assembly in SW480 human colon cancer cells. Similar to Drosophila S2 cells, expression of Axin-GFP in SW480 cells led to the formation of discrete cytoplasmic puncta (Figure 4A1; Fiedler et al., 2011). In the presence of Drosophila APC2-FL, Axin-GFP puncta decreased in number and increased in size (Figure 4A, 1 and 2, and B), suggesting that the role of APC proteins in destructosome assembly is conserved in human cells. Consistent with this hypothesis, expression of APC2-∆ASAD resulted in fragmented, dispersed Axin-GFP puncta (Figure 4A3), suggesting that APC2 self-association is also required for destructosome assembly in human cells.
FIGURE 4:

APC2 self-association is necessary to degrade β-cat and regulate Wnt target gene expression in SW480 cells. (A) Similar to S2 cells, Axin-GFP forms cytoplasmic puncta (1) and mCh-APC2 colocalizes with Axin-GFP in SW480 cells (2). Coexpression of APC2-ΔASAD with Axin-GFP does not disrupt colocalization (3) but is also associated with defects in puncta assembly and morphology. (B) Similar to S2 cells, coexpression of Axin with APC2-FL in SW480 cells leads to fewer, larger puncta. Two-tailed z test demonstrates significant differences between the two conditions. (C, D) Expression of full-length Drosophila APC2 was sufficient to suppress the elevated levels of β-cat (2) (compare to the empty vector control [1]) in SW480 cells. The APC2-ΔASAD mutant moderately suppressed the elevated β-cat levels (3). (E) In SW480 cells, expression of APC2-FL strongly suppressed activation of Wnt targets as assessed by TOP/Flash activity compared with the empty vector control. Expression of the APC2-ΔASAD mutant suppressed target gene activation compared with the empty vector control but exhibited significantly less activity than APC2-FL. Student's t test revealed significant differences between the conditions in D and E. (F) mCh-tagged APC2-FL (2) and APC2-ΔASAD (3) were expressed at equal levels in SW480 cells used in the TOP/Flash assays. Scale bar, 10 μm.
APC2 self-association is necessary for destructosome activity in SW480 cells
Next we asked whether the defects in destructosome assembly and morphology affect destructosome function. SW480 cells express a truncated version of human APC (Nishisho et al., 1991), resulting in elevated levels of β-cat due to loss of destruction complex activity (Munemitsu et al., 1995). This has made SW480 cells a useful tool to investigate the mechanisms of destructosome function. Expression of Drosophila APC2-FL can compensate for the loss of hAPC function and suppress the elevated levels of β-cat (Figure 4, C and D; Roberts et al., 2011). Although APC2-∆ASAD still contains all other domains required for APC's destructosome function, including the β-cat and Axin interaction domains (20Rs and SAMP repeats; Roberts et al., 2011), it only moderately suppressed the high levels of β-cat protein (Figure 4, C3 and D). This suggests that the fragmented destructosomes do not effectively target β-cat for destruction, and APC self-association is required for proper destruction complex activity. In SW480 cells expressing APC2-∆ASAD, β-cat levels appear to decrease in both the cytoplasm and the nucleus (Figure 4C3). Although APC2-∆ASAD–mediated destruction is likely decreasing the overall level of β-catenin protein, APC2-∆ASAD may also be reducing nuclear β-cat by tethering it in the cytoplasm (Roberts et al., 2011).
Owing to elevated β-cat levels, SW480 cells also display high levels of Wnt target gene expression, which can be detected using the well-established TOP-Flash luciferase assay (Korinek et al., 1997). Expression of Drosophila APC2-FL significantly reduced the high level of reporter gene expression in SW480 cells (Figure 4E; Roberts et al., 2011), consistent with the strong reduction in β-cat levels (Figure 4, C and D). APC2-∆ASAD and APC2-ASADPro were significantly impaired in their ability to suppress β-cat–mediated transcription (Figure 4E and unpublished data). Collectively these results suggest that APC2 self-association plays a functionally significant role in destructosome activity in SW480 cells.
APC2 self-association stabilizes the destructosome
One simple model for the role of APC self-association in destructosome assembly and morphology is that APC–APC interactions, together with APC–Axin interactions, provide stability to the complex. When APC self-association is blocked but APC retains its interaction with Axin and the complex, destructosome stability is reduced, leading to both fragmentation and loss of activity. To test this hypothesis, we used fluorescence recovery after photobleaching (FRAP) to assess the turnover of Axin-GFP within the puncta in S2 cells. If this hypothesis is correct, we predicted that cells expressing both Axin and APC2-FL would have a relatively large immobile fraction and a relatively small free mobile pool of Axin-GFP within the puncta. We expected that cells expressing Axin-GFP and only endogenous APC2 would exhibit Axin-GFP dynamics similar to that of cells overexpressing APC2-FL, although we may observe a larger immobile fraction of Axin-GFP in cells with additional APC2-FL. Conversely, we predicted that cells expressing Axin and APC2-∆ASAD would have a larger mobile fraction, as the rate of turnover of Axin-GFP is higher with a smaller immobile fraction of Axin-GFP.
For these FRAP experiments, we chose similarly sized puncta for each condition and kept the bleached area constant. For the ∆ASAD mutant, where the puncta can be interconnected at higher expression levels, we chose cells with relatively low expression, for which we could see isolated, individual puncta. To compare the Axin-GFP fluorescence recovery among the three conditions, we first normalized for the starting postbleach fluorescence by calculating ∆F/F for each time point in each condition and plotted this over time (Figure 5, A–C). To compare the rates of recovery, we compared the mean slope of the regression lines for each condition (Figure 5D). Axin-GFP/APC2-∆ASAD puncta displayed a significantly greater slope than either Axin-GFP alone or Axin-GFP/APC2-FL puncta. Conversely, Axin-GFP/APC2-FL puncta exhibited a significantly reduced slope compared with the other conditions (Figure 5D). Axin-alone puncta exhibited the greatest variation in rate of recovery; some puncta displayed Axin-GFP/APC2-FL–like properties, whereas others exhibited Axin-GFP/APC2-∆ASAD–like properties (Figure 5A). Cells expressing Axin-GFP alone express significantly more Axin than the low level of endogenous APC2 in these cells (Zhou and McCartney, unpublished data). This suggests that at a high Axin:APC2 ratio, Axin turnover rates are not well controlled and fluctuate as a consequence. When the Axin:APC2 ratio is closer to 1, as in the case of Axin-GFP/APC2-FL puncta, Axin-GFP is stabilized, and the overall rates of recovery decrease significantly (Figure 5D). On the other hand, disrupting APC2 self-association appears to drive Axin-GFP toward the opposite end of its dynamic spectrum (Figure 5D).
FIGURE 5:

APC2 self-association stabilizes the destructosome. (A–C) Plots of ΔF/F for each condition. Similar-sized Axin-GFP puncta were selected, and the recovery of individual bleached spots is shown in unique colors for each condition. Black lines are regression lines. Regression analysis indicates that the relationship between time and fluorescence varies by condition. ANOVA for regression lines, p < 0.0001; Tukey–Kramer HSD posthoc test for each pair, p < 0.05. (D) To compare the rate of recovery, we calculated the slope of the regression line for each individual sample and compared the means of these slopes for each condition. Means are plotted with SEM whiskers; Tukey–Kramer HSD posthoc test for each pair, p < 0.05. (E) To compare the difference in mobile and immobile fractions at the end of the experiment, time-zero normalized degree of recovery at our last time point (384.12 s) for the three conditions was determined. Means are plotted with SEM whiskers; Tukey–Kramer HSD posthoc test for each pair, p < 0.05. (F) We observed a significant difference in the number of puncta merging and separating events (a measure of puncta dynamics) between Axin-GFP/APC2-ΔASAD puncta compared with Axin-GFP or Axin-GFP/APC2-FL puncta. This behavior is rarely observed in Axin-GFP and Axin-GFP/APC2-FL puncta. Scale bar, 2 μm. Student's t test, p < 0.001 between the mutant and either of the other conditions. (G) Axin-GFP/APC2-ΔASAD puncta are highly dynamic. The white arrow tracks the merging of two puncta. Time stamp in minutes:seconds.
After ∼400 s postbleach, Axin-GFP in Axin-GFP/APC2-∆ASAD puncta had recovered the greatest fluorescence, reflecting a relatively large mobile pool (Figure 5E). although the degree of fluorescence recovery was more similar between Axin-alone puncta and Axin-GFP/APC2-FL puncta, they were significantly different, with Axin-GFP/APC2-FL puncta displaying the weakest recovery and therefore the smallest mobile fraction. Taken together, these data suggest that APC2 promotes stability of the destructosome through its ability to self-associate.
In addition to the increased mobility of Axin-GFP in cells coexpressing APC2-∆ASAD, we observed that the fragmented puncta themselves were remarkably dynamic (Supplemental Movies S1–S3). Furthermore, the fragmented puncta frequently split and merged with neighboring puncta (Figure 5, F and G), behavior rarely observed in cells expressing Axin alone or Axin and APC2-FL (Figure 5F).
APC2 self-association is required for destructosome activity in the Drosophila embryo
Because APC2 self-association is necessary for proper β-cat regulation in cultured cells, we asked whether APC2 self-association is also necessary for destructosome activity and the negative regulation of Wnt signaling in the more physiologically relevant context of the Drosophila embryo. We expressed GFP-tagged APC2-FL or APC2-∆ASAD in the embryo under the native APC2 promoter (McCartney et al., 2006) and found that the two tagged proteins are expressed at levels comparable to that of the endogenous wild-type protein (Figure 6A). As previously shown, APC2-FL protein expressed in APC2-null (APC2g10) embryos is enriched at the cell cortex of embryonic epithelia similar to endogenous APC2 (McCartney et al., 1999; Zhou et al., 2011). However, APC2-∆ASAD exhibited limited enrichment at the cortex (Figure 6B), consistent with our observations in S2 cells (Supplemental Figure S4). We previously demonstrated that the localization of APC2 to the cell cortex requires both the N-terminal region (aa 1–490) and the most-C-terminal 30 amino acids (C30; Figure 1A; Zhou et al., 2011). Because we have now demonstrated that the function of C30 requires APC2 dimerization (McCartney and Molinar, unpublished data), it is not surprising that the ASAD is necessary for cortical localization. We previously showed that cortical localization of APC2 is not required to regulate Wnt signaling (Zhou et al., 2011); therefore, lack of APC2-∆ASAD cortical localization will not affect its destructosome activity.
FIGURE 6:

APC2 self-association is required to negatively regulate Wnt signaling in the Drosophila embryo. (A) Immunoblot of 0- to 6-h embryonic lysates demonstrates that the level of expression of GFP-APC2-FL and GFP-APC2-ΔASAD is comparable to that of endogenous APC2. (B) GFP-APC2-FL is enriched at the cell cortex with Arm in embryonic epithelia, whereas GFP-APC2-ΔASAD is primarily cytoplasmic. Scale bar, 10 μm. (C, D) Expression of GFP-APC2-FL rescued the lethality of APC2-null (APC2g10) embryos and restored the wild- type cuticle phenotype, whereas the APC2-ΔASAD mutant only moderately rescued the lethality and cuticle phenotype. The numbers in D indicate the phenotypic average for each genotype (scoring criteria as in McCartney et al., 2006). Cuticle images are shown at the same scale. (E) Representative embryos showing Arm and En protein expression in wild-type (1) and APC2-null (2) embryos. APC2-FL restored wild-type Arm levels and the En expression domain of APC2-null (APC2g10) embryos. APC2-ΔASAD weakly suppressed Arm accumulation and restored a weak Arm stripe pattern in the epidermis. The En expression domain remains expanded in APC2-null embryos expressing APC2-ΔASAD. Scale bar, 25 μm.
Between 4 and 6 h after egg laying, Wnt signaling is activated in a subset of ectodermal cells within each developing segment. Cells receiving Wnt produce smooth cuticle, whereas cells not receiving Wnt produce microtubule- and actin-based apical projections that result in the formation of cuticular outgrowths called denticles. Thus Wnt signaling results in a segmentally repeated pattern of denticles and smooth cuticle on the ventral surface of the embryo (like Figure 6D, APC2-FL; APC2g10). This segmentally repeated pattern is reflected in the accumulation of Arm in “stripes” of cells receiving Wnt (Figure 6E) and in the patterned expression of the Wnt target gene engrailed (en; Figure 6E). Embryos activating Wnt signaling uniformly throughout the ectoderm, as in the null APC2g10, were embryonic lethal (0% hatch rate to the larval stage; Figure 6C), produced excess smooth cuticle at the expense of denticles (Figure 6D), accumulated Arm uniformly across the ectoderm (Figure 6E2), and exhibited an expanded expression domain of en (Figure 6E2). Addition of APC2-FL into the null background rescued all of these defects to a virtually wild-type phenotype (Figure 6, C–E). In contrast, expression of APC2-∆ASAD in the APC2 null suppressed, but failed to fully rescue, these defects. Whereas 98% of APC2-FL; APC2g10 embryos hatched (Figure 6C), only 39% of APC2-∆ASAD; APC2g10 embryos hatched to the larval stage (Figure 6C). The 61% of APC2-∆ASAD; APC2g10 embryos that failed to hatch exhibited a suppressed phenotype compared with the null alone (Figure 6D); denticle bands were restored but were frequently incomplete, and the overall size of the embryo increased but not to the level of APC2-FL rescue (Figure 6D). Consistent with the incomplete rescue, cells not receiving the Wnt signal in the APC2-∆ASAD; APC2g10 embryos still exhibited elevated Arm (Figure 6E4), and the en expression domain remained expanded (Figure 6E4). In conclusion, the APC2-∆ASAD protein supports only weak destructosome activity in the Drosophila embryo, consistent with our results in SW480 cells (Figure 4).
Drosophila expresses a splice form of APC2 lacking the ASAD
Although we showed that self-association is necessary for APC2's destructosome function, self-association may interfere with other aspects of APC function and may be regulated. Although little is known about the regulation of self-association via OD-1 in human APC, splice variants of APC that alter that domain have been reported (Santoro and Groden, 1997; Carson et al., 2004). In addition, exon 9, containing the human ASAD, can be alternatively spliced (Groden et al., 1991; Joslyn et al., 1991) Remarkably, we identified a splice variant of APC2 in the modENCODE database that selectively removes a region within the ASAD. We confirmed at the mRNA level that both isoforms are found in 4- to 8-h Drosophila embryos, albeit at dramatically different levels (Supplemental Figure S5). These data suggest that a monomeric form of APC2 may have a functional role during development.
DISCUSSION
As core members of the β-cat destruction complex, APC proteins are indispensable negative regulators of Wnt signaling. APC loss leads to unregulated accumulation of β-cat and Wnt pathway activation. Previous studies focusing on APC's binding to β-cat and Axin demonstrated the importance of these interactions for APC's destructosome activity (Roberts et al., 2011; Kunttas-Tatli et al., 2012). Here we reveal a novel role for APC proteins in destructosome function by promoting destructosome assembly and stability through self-association. Although it has been known for almost 20 yr that APC proteins are essential negative regulators of Wnt signaling (Munemitsu et al., 1995), novel roles for APC in this process are still being uncovered.
Role of APC in the formation of the destructosome
Axin is believed to drive destructosome assembly through polymerization via its C-terminal DAX domain (Fiedler et al., 2011). Furthermore, Axin's direct binding to all other core members of the complex suggests that its primary function is scaffolding (Luo and Lin, 2004). Surprisingly, Mendoza-Topaz et al. (2011) demonstrated that APC is essential for Axin complex assembly in vivo; without APC, Axin failed to form functional destructosomes in Drosophila embryos. They suggested two scenarios to explain this. First, Axin is unstable without APC, resulting in the observed reduction in Axin protein that may be below the minimum concentration needed for polymerization. Second, APC is a cofactor clustering Axin via its multiple Axin interaction domains (SAMPs), APC self-association, or both. Our data support the model that APC promotes destructosome assembly by stimulating Axin polymerization via APC self-association (Figure 7). With APC2, Axin formed larger and more stable structures in cultured cells (Figures 3 and 4). Disrupting APC2 self-association by removing the ASAD had no effect on APC2–Axin interactions but resulted in fragmented and significantly more dynamic Axin puncta (Figures 3–5). One might expect that loss of APC2 self-association would result in Axin-GFP puncta similar to those in cells lacking additional APC2. We predict that APC2-∆ASAD fragments the Axin puncta because monomeric APC2 (APC2m) retains its interaction with Axin and interferes with Axin polymerization (Figure 7). This might be due to the stronger APC–Axin interaction (KD = 50 nM in vitro) versus the weaker Axin–Axin interaction (KD = 5–20 μM in vitro; Lee et al., 2003; Schwarz-Romond et al., 2007b). This dramatic affinity difference further supports the idea that a cofactor like APC is required to efficiently polymerize the less abundant Axin (Figure 7; Lee et al., 2003).
FIGURE 7:

Model for the role of APC self-association in promoting Axin puncta formation. (A) Normal Axin polymers form via the weak DAX interactions (thin lines) in the absence of APC. (B) Larger Axin polymers form via the stronger interaction between APC's SAMP repeats and Axin's RGS domains (thick lines) due in part to APC's ability to self-associate (unknown binding affinity; wavy lines). (C) Smaller Axin polymers form in the absence of APC self-association.
Our data also suggest that once APC drives Axin polymerization, it stabilizes Axin in the complex (Figure 5). Because of Axin's ability to bind the other core components of the complex, stabilized Axin may in turn stabilize the presence of GSK3β and CK1, leading to more efficient β-cat phosphorylation and degradation. This is consistent with our functional data in cultured cells and during Drosophila embryogenesis; the expression of APC2m resulted in significant reduction, but not complete loss, of destructosome activity (Figures 4 and 6). The low level of APC1 in the embryo does support destructosome function (Ahmed et al., 2002; Akong et al., 2002; Kunttas-Tatli et al., 2012), and we predict that this activity is due in part to APC1's association with the more abundant APC2 through the ASAD and through APC1's self-association. Taken together, we conclude that whereas APC self-association is not strictly essential for destructosome activity, it is necessary for normal function. Because even slight elevations in Wnt signaling due to the reduction of negative regulation can lead to dramatic defects (McCartney et al., 2006; Komori et al., 2014), we predict that maximally efficient destructosome activity is essential for both normal development and to prevent Wnt signaling–mediated cancers.
Our findings appear to contrast with some previous work about the role of APC's N-terminal domains in destructosome function. Overexpression of hAPC internal fragments containing at least three 20Rs but lacking OD1, OD2, and the Arm repeats rescues β-cat destruction in SW480 cells (Rubinfeld et al., 1997; Roberts et al., 2011; Li et al., 2012). This appears to suggest that APC's N-terminal domains are dispensable. In contrast, overexpression of analogous dAPC2 fragments or dAPC2 N-terminal deletions failed to rescue destructosome function in SW480 cells (Roberts et al., 2011; unpublished data). Consistent with these results and our findings here, we previously demonstrated that the N-terminus of APC2 is essential in vivo (Roberts et al., 2011, 2012). Moreover, hypomorphic point mutations in the Arm repeats of dAPC2 (McCartney et al., 1999, 2006) and mouse studies of colon cancer (Crist et al., 2010) suggest that the N-terminus is functionally important. It is unclear how internal fragments of hAPC rescue destruction, whereas the analogous Drosophila fragments do not; however, this collection of in vivo data provides a compelling argument that the N-terminus of APC is essential for β-cat destruction in Drosophila and mammals.
The self-association of APC proteins significantly affects their functions
Previously the only functions ascribed to APC dimerization were in cytoskeletal regulation. ANS2 in the basic domain is necessary for dimerization and APC's actin nucleation activity (Okada et al., 2010). Phosphorylation enhances OD-2 dimer formation, which in turn enhances the assembly of microtubule-associated APC clusters at the cell periphery (Li et al., 2008). Dispersing these APC clusters by disrupting OD-2 reduced cell migration. The potential parallels in APC function in these clusters and in destructosomes are intriguing; in both cases, blocking self-association disrupts the assembly of these prominent macromolecular assemblages.
It is unclear what role OD1 plays in normal APC function. Clinically relevant APC C-terminal truncations forming dimers with wild-type APC through OD1 in heterozygous cells may promote chromosomal instability and aneuploidy by interfering with microtubule functions (Green and Kaplan, 2003; Green et al., 2005). Of interest, a splice isoform of APC that skips exon-1 encoding OD1 is enriched in mouse and human brain and heart (Thliveris et al., 1994). A splice variant of hAPC that deletes a portion of the ASAD has also been observed (Groden et al., 1991; Joslyn et al., 1991), and there are some reports of colorectal cancer–associated mutations in this region that may result in increased production of the ASAD-lacking isoform (van der Luijt et al., 1995). However, the data are limited and the functional consequences unclear. Our intriguing observation that Drosophila express an alternate splice form of APC2 lacking the ASAD (Supplemental Figure S5) suggests that Drosophila may be a relevant and simple system in which to examine the functional consequences of these alternative APC isoforms.
Although the function of APCm is unknown, our observations suggest that it may complex more efficiently with Arm repeat–binding proteins such as KAP3 (Figure 2E). Similarly, the binding of OD2 and the C-terminal domains of vAPC (aa 2545–2843) decreased Kap3 association with the Arm repeats (Li and Näthke, 2005). This suggests that APCm may exhibit enhanced binding to a broad array of Arm repeat partners, including KAP3, Asef, IQGAP, and the PP2A regulatory subunit (Seeling et al., 1999; Kawasaki, 2000; Jimbo et al., 2002; Watanabe et al., 2004). The cytoskeletal functions of the first three suggest that APCm may have enhanced cytoskeletal roles. APC's association with PP2A is believed to be in the context of the destructosome (Seeling et al., 1999), suggesting that APCm may also have destructosome function.
MATERIALS AND METHODS
Constructs and molecular biology
Site-directed mutagenesis primers were designed, and a standard PCR-based mutagenesis protocol was followed. The resulting APC2 mutants were cloned into the pGEM-T Easy (Promega, Madison, WI) shuttle vector and then into the EcoRI site in pRmHa-3 (metallothionein promoter vector for S2 cells), pCS2(+) (cytomegalovirus promoter vector for SW480 cells), and pCaSpeR-2 modified to contain the native APC2 promoter and GFP for expression in whole Drosophila (McCartney et al., 2006). The mutant constructs were confirmed by sequencing. The specific amino acid positions of the Drosophila APC2 (FlyBase annotation symbol, CG6193) fragments are as follows: APC2-ΔASAD, 1–69 plus 100–1067; APC2-ASADPro, Pro81Leu, Pro84Leu, Pro94Leu, Pro98Leu; APC2-N175K, Asn175Lys APC2-N, 1–490; APC2-C, 491–1067; APC2-ΔSAMP, 1–930 plus 1037–1067; APC1-N, 1–904. Full-length Kap3 (aa 1–945) was PCR amplified from DGRC cDNA clone LD13052 and shuttled through pGEM-T Easy to EcoR1 of pRmHa-3. For the Axin-GFP construct, GFP-Gateway-3X STOP cassette was inserted downstream of the pMT promoter in pMT V5/His (Invitrogen, Carlsbad, CA). Full-length Axin was then cloned into the pCR8 Gateway entry vector and Gateway cloned into the pMT GFP-W destination vector. For the Axin-HA construct, 3XHA-Gateway-3X STOP cassette was inserted downstream of the pMT promoter in pMT V5/His (Invitrogen), and full-length Axin was then cloned into the EcoRI and Xho sites of the pMT HA destination vector.
For validation of the newly identified APC2 isoform, forward (5′-GCACAACATCGTCCACAATAATCC-3′) and reverse (5′-GCTCCCAGTTCGCACATAGTCTG-3′) primers were used to amplify the region of APC2 containing the putative intron encompassing the ASAD. We used 4- to 8-h embryonic cDNA to PCR amplify the region, and the unspliced isoform (231 base pairs) and the spliced form (168 base pairs) were identified on a GelStar (Lonza, Walkersville, MD) agarose gel. The less abundant spliced isoform was then isolated from the agarose gel (with some contamination from the unspliced form) and reamplified using the same primers. Both isoforms were directly sequenced for validation.
Yeast two-hybrid analysis
Yeast two-hybrid (Y2H) analysis was performed using the Matchmaker System (Clontech, Mountain View, CA). Briefly, the pGBKT7 and pGADT7 yeast vectors were engineered to be Gateway compatible by inserting a Gateway-3X STOP cassette downstream of the Gal4 DNA-binding domain or Gal4 transcriptional activation domain, respectively. APC2 constructs containing the Arm repeats were TOPO-TA cloned into the pCR8/GW/TOPO vector (Life Technologies, Grand Island NY) and Gateway cloned into pGBKT7-W and pGADT7-W. Resulting constructs were transformed into the Y2HGold and Y187 yeast strains, respectively, using the SC Easy Transformation kit (Life Technologies). After transformation and selection, appropriate yeast colonies were mated in 2× Yeast extract-peptone-dextrose medium + adenine (YPAD) for 24 h and plated on double-selection –Leu –Trp plates. Resulting yeast colonies were inoculated in liquid –Leu –Trp medium, and β-galactosidase assays performed using the Yeast β-galactosidase Assay Kit (Thermo Scientific, Rockford, IL). Several different colonies were tested per experiment, and each experiment was conducted independently three times. β-Galactosidase activity was calculated using the equation, activity = (1000 × OD420)/(TV × OD660), where T is the duration of the reaction in minutes and V is the volume of the reaction in milliliters.
S2 cell culture, transfections, and coimmunoprecipitation experiments
S2 cells were cultured at 25°C in Schneider's Drosophila Medium (Lonza) with 10% heat-inactivated fetal bovine serum (FBS) and 1× penicillin-streptomycin (Pen/Strep). DNA constructs were transfected into S2 cells using Effectene (Qiagen, Valencia, CA) and standard protocols at a cell density of 2.5 × 105 cells in six-well plates. Expression of constructs was induced 24 h posttransfection with CuSO4 (40 mM final concentration) for 14–16 h. Coimmunoprecipitation experiments were performed as described in Zhou et al. (2011). Briefly, cells were lysed and preincubated with Rec-G beads (Invitrogen) for 30 min at 4°C. mCherry antibody (632496; Clontech) was used to pull down tagged proteins from the precleared lysate. Rec-G beads were then added and incubated for 1 h at 4°C. After washing the beads several times, SDS–PAGE and immunoblotting were performed using standard procedures. Anti-APC2 antibody (McCartney et al., 1999) was used to visualize the various APC2 constructs. To visualize Axin-HA localization, the same transfection procedure was applied, and S2 cells were fixed with 4% paraformaldehyde 14–16 h after induction and labeled with phalloidin (to label cortical actin) and anti-HA (mouse 1:200; gift from Adam Linstedt's lab, Carnegie Mellon University).
Cell sorting
The BD FacsVantage Diva option (laser 488) was used to sort the high (33%)-, medium (33%)-, and low (33%)-expressing S2 cells 24 h after induction for the indicated constructs. Cells were sorted into phosphate-buffered saline (PBS), and images of live cells were taken immediately under identical imaging conditions for puncta size measurements. Imaris (Bitplane, Zurich, Switzerland) was then used to measure the area of the Axin puncta in micrometers squared.
SW480 cell culture, transfections, and immunofluorescence
SW480 cells were cultured in DMEM with high glucose (DMEM-H) supplemented with 10% heat-inactivated and 1× Pen/Strep/glutamine. Cells were maintained at 37°C and 5% CO2. For transfections, SW480 cells were plated at a density of 2.5 × 105 cells in six-well plates and grown overnight. pCS2(+)-APC2 DNA constructs were transfected using TurboFect (Thermo Fisher) according to the manufacturer's instructions. For immunofluorescence, cells were plated on glass coverslips, transfected with 4 μg of the relevant mCherry-tagged APC2 DNA construct, and fixed 24 h posttransfection with 4% formaldehyde in 1× PBS for 10 min. Cells were washed three times with 1× PBS, blocked for 15 min in 1× PBTN (1× PBS containing 1% normal goat serum and 0.1% Triton-100), and then antibody stained as previously described (Roberts et al., 2011). The primary antibody was mouse anti–β-cat (1:1000; 610153; BD Transduction Laboratories), and the secondary was goat anti-mouse Alexa 488 (1:1000; Life Technologies). For quantification of β-cat levels, 30 cell images were taken for each condition using identical settings on the spinning disk confocal microscope (see later description). The fluorescence intensities for three circular regions of interest (1264 square pixels) were measured for each cell (using ImageJ) and averaged for each condition.
TOP/FOP luciferase reporter assay
The TOP/FOPFlash luciferase constructs and the pRL Renilla transfection control were provided by Hans Clevers (Hubrecht Institute, Utrecht, Netherlands). Luciferase assays were performed using the Dual Glo Luciferase System (Promega) according to the manufacturer's protocol. Briefly, SW480 cells were transiently cotransfected with 1 μg of either the TOP or FOPFlash luciferase reporter, 1 μg of pRL, and 2 μg of the appropriate APC2 construct. At 24 h posttransfection, cells were lysed in a hypotonic 0.1× PBS solution and subjected to a 5-min freeze-thaw at −80°C. Cells were scraped and cellular debris pelleted at 3000 × g in a microcentrifuge. Lysates were mixed with the provided luciferase substrate, and luciferase activity was measured using a PerkinElmer EnSpire plate reader. Luciferase signal was normalized to Renilla activity and overall values normalized to the mCherry-only control. All samples were measured in triplicate per experiment, and three independent experiments were performed. None of the constructs displayed significant FOPFlash activity.
Fly genetics, hatch rate, and cuticle analysis
Transgenic flies expressing P[endoP-EGFP-APC2-FL] (Zhou et al., 2011) and P[endoPEGFP-APC2-ΔASAD] were generated using P-element–mediated germline transformation (Model System Genomics; Duke University, Durham, NC). Two independent second chromosome insertions for each transgene were crossed into the APC2g10 (APC2 null) background using standard methods. Embryonic cuticles were prepared and hatch rate analysis was performed as previously described (Wieschaus and Nusslein-Volhard, 1998). Scoring criteria for the cuticle phenotype was previously described (McCartney et al., 2006). Cuticle images were taken with darkfield illumination at 20× zoom with a Spot RT Color Model 2.2.0 camera from Diagnostic Instruments.
Immunohistochemistry in the Drosophila embryo
Embryos were collected 4-6 h at 27ºC and fixed and stained as previously described (McCartney et al., 1999). Anti-Armadillo (Arm; ms, N27A1, 1:250) and anti-Engrailed (En; ms, 4D9, 1:50) were obtained from the Developmental Studies Hybridoma Bank at the University of Iowa (Iowa City, IA). Anti-GFP (1:5000; Abcam) was preabsorbed against w1118 embryos before using for immunohistochemistry. Anti-APC2 (rt, 1:1000) was used as previously described (McCartney et al., 1999). Secondary antibodies were conjugated with various Alexa dyes (1:1000; Invitrogen).
Imaging and image analysis
Images were acquired with a spinning disk confocal microscope with a Yokagawa scan head (Solamere Technology Group) and a QICAM-IR camera (Qimaging) on a Zeiss Axiovert 200M, using QED InVivo software. For images of whole embryos stained for Arm and En, multiple images acquired at 25× were merged using Photoshop (Adobe) to generate whole-embryo images.
Sequence alignments of oligomerization domain 1 and ASAD
Sequences for various species used in the study were acquired from the NCBI (National Center for Biotechnology Information) pblast database: Drosophila melanogaster (AAF56249.1), Nasonia vitripennis (XP001602839.2), Capitella teleta (ELU12449.1), Lottia gigantea (ESO95067.1), Strongylocentrotus purpuratus (XP783363.3), Saccoglossus kowalevskii (XP002738523.1), Ciona intestinalis (XP002124987.2), and Homo sapiens (AAA03586.1). The Nasonia, Capitella, and Lottia genomes have not been annotated, and therefore we relied on manual confirmation of the APC sequences based on conservation of the protein. ClustalW was used for generating sequence alignments. We considered a minimum of 50% conservation to decide the presence of OD-1 or ASAD.
Live imaging and FRAP analysis
FRAP experiments were carried out using a Zeiss LSM510 confocal microscope with ZEN software. After two prebleach scans at 3× zoom, we performed 10 bleaching scans with 100% intensity of 488 nm over the region of interest (15 by 15–pixel circle). This area was kept constant, and similar-sized puncta were bleached for the analysis. After photobleaching, the fluorescence recovery was monitored every 3.96 s for 100 frames. The recoveries of the fluorescence intensities of each image series were quantified with ImageJ and processed using Excel. We calculated ∆F/F by taking the difference between the time-zero fluorescence measurement (arbitrary units) and the fluorescence measurement at each time point divided by the time-zero fluorescence measurement. This showed the degree of recovery postbleaching normalized by the time-zero fluorescence. The three groups were compared with each other in a regression analysis model. The analysis was protected overall by analysis of variance (ANOVA), and individual pairwise comparisons were assessed by the Tukey–Kramer honest significant difference (HSD) posthoc test. To assess that rate of fluorescence recovery, we calculated the sum of least squares best-fit regression line for each site and compared it with ANOVA and the Tukey–Kramer HSD posthoc test. Time-zero normalized degree of recovery at our last time point (384.12 s) for each condition was compared using ANOVA and the Tukey–Kramer HSD posthoc test. All analyses were conducted using JMP 11.
Supplementary Material
Acknowledgments
We thank Javier Lopez and Deborah Makin for providing 4- to 8-h cDNAs and assistance with the analysis of the second APC isoform; Marcel Bruchez and Manoj Puthenveedu for assistance with FRAP; Justin Crowley for statistical analysis; Dannie Durand and Charles Ettensohn for reviewing Supplemental Figure S1; Gerard Campbell for use of his microscope to acquire cuticle images; Adam Linstedt for providing the HA antibody; Haibing Teng for microscope assistance; Yehuda Creeger for FACS; Mengning Zhou for contributions to cloning; Malachi Blundon for contributions to the S2 cell studies; Olivia Molinar for sharing her unpublished results on APC2's cortical localization; and all the members of the McCartney laboratory for their input. This work was supported by National Institutes of Health Grants R01 GM073891 (to B.M.M.) and R15 GM107796 (to D.M.R.), start-up funds from Franklin and Marshall College (to D.M.R.), and Carnegie Mellon University Graduate Small Project Help (GuSH) funding (to E.K.-T.).
Abbreviations used:
- 15/20R
15/20 amino acid repeat
- ANS2
actin nucleation sequence 2
- APC
adenomatous polyposis coli
- APCm
monomeric APC
- Arm
Armadillo
- ASAD
APC self-association domain
- β-cat
β-catenin
- CK1
casein kinase 1
- dAPC
Drosophila APC
- En
engrailed
- FL
full length
- FRAP
fluorescent recovery after photobleaching
- GFP
green fluorescent protein
- GSK3
glycogen synthase kinase 3
- KAP3
kinesin-associated protein 3
- LRP5/6
LDL receptor-related proteins 5 and 6
- mCh
mcherry
- MCR
mutation cluster region
- modENCODE
Model Organism Encyclopedia of DNA Elements
- NCBI
National Center for Biotechnology Information
- NGS
normal goat serum
- OD
optical density
- OD-1
oligomerization domain 1
- PBS
phosphate-buffered saline
- TCF
T-cell factor
- vAPC
vertebrate APC
- Wg
Wingless
- Y2H
yeast two-hybrid
Footnotes
This article was publised online ahead of print in MBoC in Press (http://www .molbiolcell.org/cgi/doi/10.1091/mbc.E14-04-0885) on September 10, 2014.
The authors declare that they have no competing interests.
REFERENCES
- Ahmed Y, Nouri A, Wieschaus E. Drosophila Apc1 and Apc2 regulate Wingless transduction throughout development. Development. 2002;129:1751–1762. doi: 10.1242/dev.129.7.1751. [DOI] [PubMed] [Google Scholar]
- Akong K, Grevengoed EE, Price MH, McCartney BM, Hayden MA, DeNofrio JC, Peifer M. Drosophila APC2 and APC1 play overlapping roles in wingless signaling in the embryo and imaginal discs. Dev Biol. 2002;250:91–100. doi: 10.1006/dbio.2002.0776. [DOI] [PubMed] [Google Scholar]
- Behrens J, Jerchow BA, Würtele M, Grimm J, Asbrand C, Wirtz R, Kühl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280:596–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- Cadigan KM, Peifer M. Wnt signaling from development to disease: insights from model systems. Cold Spring Harb Perspect Biol. 2009;1:a002881. doi: 10.1101/cshperspect.a002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson DJ, Santoro IM, Groden J. Isoforms of the APC tumor suppressor and their ability to inhibit cell growth and tumorigenicity. Oncogene. 2004;23:7144–7148. doi: 10.1038/sj.onc.1207954. [DOI] [PubMed] [Google Scholar]
- Chen T, Yang I, Irby R, Shain KH, Wang HG, Quackenbush J, Coppola D, Cheng JQ, Yeatman TJ. Regulation of caspase expression and apoptosis by adenomatous polyposis coli. Cancer Res. 2003;63:4368–4374. [PubMed] [Google Scholar]
- Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Crist RC, Roth JJ, Baran AA, McEntee BJ, Siracusa LD, Buchberg AM. The armadillo repeat domain of Apc suppresses intestinal tumorigenesis. Mamm Genome. 2010;21:450–457. doi: 10.1007/s00335-010-9288-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagotto F, Jho Eh, Zeng L, Kurth T, Joos T, Kaufmann C, Costantini F. Domains of axin involved in protein-protein interactions, Wnt pathway inhibition, and intracellular localization. J Cell Biol. 1999;145:741–756. doi: 10.1083/jcb.145.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faux MC, Coates JL, Catimel B, Cody S, Clayton AH, Layton MJ, Burgess AW. Recruitment of adenomatous polyposis coli and beta-catenin to axin-puncta. Oncogene. 2008;27:5808–5820. doi: 10.1038/onc.2008.205. [DOI] [PubMed] [Google Scholar]
- Fiedler M, Mendoza-Topaz C, Rutherford TJ, Mieszczanek J, Bienz M. Dishevelled interacts with the DIX domain polymerization interface of Axin to interfere with its function in down-regulating β-catenin. Proc Natl Acad Sci USA. 2011;108:1937–1942. doi: 10.1073/pnas.1017063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodde R. The APC gene in colorectal cancer. Eur J Cancer. 2002;38:867–871. doi: 10.1016/s0959-8049(02)00040-0. [DOI] [PubMed] [Google Scholar]
- Green RA, Kaplan KB. Chromosome instability in colorectal tumor cells is associated with defects in microtubule plus-end attachments caused by a dominant mutation in APC. J Cell Biol. 2003;163:949–961. doi: 10.1083/jcb.200307070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RA, Wollman R, Kaplan KB. APC and EB1 function together in mitosis to regulate spindle dynamics and chromosome alignment. Mol Biol Cell. 2005;16:4609–4622. doi: 10.1091/mbc.E05-03-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- Gruber M, Lupas AN. Historical review: another 50th anniversary—new periodicities in coiled coils. Trends Biochem Sci. 2003;28:679–685. doi: 10.1016/j.tibs.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Guder C, Philipp I, Lengfeld T, Watanabe H, Hobmayer B, Holstein TW. The Wnt code: cnidarians signal the way. Oncogene. 2006;25:7450–7460. doi: 10.1038/sj.onc.1210052. [DOI] [PubMed] [Google Scholar]
- Ha N-C, Tonozuka T, Stamos JL, Choi H-J, Weis WI. Mechanism of phosphorylation-dependent binding of APC to beta-catenin and its role in beta-catenin degradation. Mol Cell. 2004;15:511–521. doi: 10.1016/j.molcel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Hart MJ, de los Santos R, Albert IN, Rubinfeld B, Polakis P. Downregulation of beta-catenin by human Axin and its association with the APC tumor suppressor, beta-catenin and GSK3 beta. Curr Biol. 1998;8:573–581. doi: 10.1016/s0960-9822(98)70226-x. [DOI] [PubMed] [Google Scholar]
- Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998;17:1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimbo T, Kawasaki Y, Koyama R, Sato R, Takada S, Haraguchi K, Akiyama T. Identification of a link between the tumour suppressor APC and the kinesin superfamily. Nat Cell Biol. 2002;4:323–327. doi: 10.1038/ncb779. [DOI] [PubMed] [Google Scholar]
- Joslyn G, Carlson M, Thliveris A, Albertsen H, Gelbert L, Samowitz W, Groden J, Stevens J, Spirio L, Robertson M. Identification of deletion mutations and three new genes at the familial polyposis locus. Cell. 1991;66:601–613. doi: 10.1016/0092-8674(81)90022-2. [DOI] [PubMed] [Google Scholar]
- Joslyn G, Richardson DS, White R, Alber T. Dimer formation by an N-terminal coiled coil in the APC protein. Proc Natl Acad Sci USA. 1993;90:11109–11113. doi: 10.1073/pnas.90.23.11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y. Asef, a link between the tumor suppressor APC and G-protein signaling. Science. 2000;289:1194–1197. doi: 10.1126/science.289.5482.1194. [DOI] [PubMed] [Google Scholar]
- Kim JH, Liu X, Wang J, Chen X, Zhang H, Kim SH, Cui J, Li R, Zhang W, Kong Y, et al. Wnt signaling in bone formation and its therapeutic potential for bone diseases. Ther Adv Musculoskelet Dis. 2013;5:13–31. doi: 10.1177/1759720X12466608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelman D, Xu W. beta-Catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006;25:7482–7491. doi: 10.1038/sj.onc.1210055. [DOI] [PubMed] [Google Scholar]
- Kishida S, Yamamoto H, Hino S, Ikeda S, Kishida M, Kikuchi A. DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate beta-catenin stability. Mol Cell Biol. 1999;19:4414–4422. doi: 10.1128/mcb.19.6.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori H, Xiao Q, McCartney BM, Lee C-Y. Brain tumor specifies intermediate progenitor cell identity by attenuating β-catenin/Armadillo activity. Development. 2014;141:51–62. doi: 10.1242/dev.099382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Kunttas-Tatli E, Zhou M-N, Zimmerman S, Molinar O, Zhouzheng F, Carter K, Kapur M, Cheatle A, Decal R, McCartney BM. Destruction complex function in the Wnt signaling pathway of Drosophila requires multiple interactions between Adenomatous polyposis coli 2 and Armadillo. Genetics. 2012;190:1059–1075. doi: 10.1534/genetics.111.133280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Salic A, Krüger R, Heinrich R, Kirschner MW. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 2003;1:E10. doi: 10.1371/journal.pbio.0000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, Mohammed S, Heck AJ, Maurice MM, Mahmoudi T, et al. Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell. 2012;149:1245–1256. doi: 10.1016/j.cell.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Li Z, Kroboth K, Newton IP, Näthke IS. Novel self-association of the APC molecule affects APC clusters and cell migration. J. Cell Sci. 2008;121:1916–1925. doi: 10.1242/jcs.029470. [DOI] [PubMed] [Google Scholar]
- Li Z, Näthke IS. Tumor-associated NH2-terminal fragments are the most stable part of the adenomatous polyposis coli protein and can be regulated by interactions with COOH-terminal domains. Cancer Res. 2005;65:5195–5204. doi: 10.1158/0008-5472.CAN-04-4609. [DOI] [PubMed] [Google Scholar]
- Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- Luo W, Lin S-C. Axin: a master scaffold for multiple signaling pathways. Neurosignals. 2004;13:99–113. doi: 10.1159/000076563. [DOI] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Mao J, Wang J, Liu B, Pan W, Farr GH, Flynn C, Yuan H, Takada S, Kimelman D, Li L, Wu D. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell. 2001;7:801–809. doi: 10.1016/s1097-2765(01)00224-6. [DOI] [PubMed] [Google Scholar]
- Mattie FJ, Stackpole MM, Stone MC, Clippard JR, Rudnick DA, Qiu Y, Tao J, Allender DL, Parmar M, Rolls MM. Directed microtubule growth, +TIPs, and kinesin-2 are required for uniform microtubule polarity in dendrites. Curr Biol. 2010;20:2169–2177. doi: 10.1016/j.cub.2010.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney BM, Dierick HA, Kirkpatrick C, Moline MM, Baas A, Peifer M, Bejsovec A. Drosophila APC2 is a cytoskeletally-associated protein that regulates wingless signaling in the embryonic epidermis. J Cell Biol. 1999;146:1303–1318. doi: 10.1083/jcb.146.6.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney BM, Näthke IS. Cell regulation by the Apc protein Apc as master regulator of epithelia. Curr Opin Cell Biol. 2008;20:186–193. doi: 10.1016/j.ceb.2008.02.001. [DOI] [PubMed] [Google Scholar]
- McCartney BM, Price MH, Webb RL, Hayden MA, Holot LM, Zhou M, Bejsovec A, Peifer M. Testing hypotheses for the functions of APC family proteins using null and truncation alleles in Drosophila. Development. 2006;133:2407–2418. doi: 10.1242/dev.02398. [DOI] [PubMed] [Google Scholar]
- Mendoza-Topaz C, Mieszczanek J, Bienz M. The Adenomatous polyposis coli tumour suppressor is essential for Axin complex assembly and function and opposes Axin's interaction with Dishevelled. Open Biol. 2011;1:110013. doi: 10.1098/rsob.110013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita EC, Murayama K, Kato-Murayama M, Ishizuka-Katsura Y, Tomabechi Y, Hayashi T, Terada T, Handa N, Shirouzu M, Akiyama T, Yokoyama S. Crystal structures of the armadillo repeat domain of adenomatous polyposis coli and its complex with the tyrosine-rich domain of Sam68. Structure. 2011;19:1496–1508. doi: 10.1016/j.str.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Hamada F, Ishidate T, Anai K, Kawahara K, Toyoshima K, Akiyama T. Axin, an inhibitor of the Wnt signalling pathway, interacts with beta-catenin, GSK-3beta and APC and reduces the beta-catenin level. Genes Cells. 1998;3:395–403. doi: 10.1046/j.1365-2443.1998.00198.x. [DOI] [PubMed] [Google Scholar]
- Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- Okada K, Bartolini F, Deaconescu AM, Moseley JB, Dogic Z, Grigorieff N, Gundersen GG, Goode BL. Adenomatous polyposis coli protein nucleates actin assembly and synergizes with the formin mDia1. J Cell Biol. 2010;189:1087–1096. doi: 10.1083/jcb.201001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva CA, Vargas JY, Inestrosa NC. Wnts in adult brain: from synaptic plasticity to cognitive deficiencies. Front Cell Neurosci. 2013;7:224. doi: 10.3389/fncel.2013.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012;4:a008052. doi: 10.1101/cshperspect.a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DM, Pronobis MI, Poulton JS, Kane EG, Peifer M. Regulation of Wnt signaling by the tumor suppressor adenomatous polyposis coli does not require the ability to enter the nucleus or a particular cytoplasmic localization. Mol Biol Cell. 2012;23:2041–2056. doi: 10.1091/mbc.E11-11-0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DM, Pronobis MI, Poulton JS, Waldmann JD, Stephenson EM, Hanna S, Peifer M. Deconstructing the ßcatenin destruction complex: mechanistic roles for the tumor suppressor APC in regulating Wnt signaling. Mol Biol Cell. 2011;22:1845–1863. doi: 10.1091/mbc.E10-11-0871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Munemitsu S, Polakis P. Loss of beta-catenin regulation by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res. 1997;57:4624–4630. [PubMed] [Google Scholar]
- Santoro IM, Groden J. Alternative splicing of the APC gene and its association with terminal differentiation. Cancer Res. 1997;57:488–494. [PubMed] [Google Scholar]
- Schneikert J, Vijaya Chandra SH, Ruppert JG, Ray S, Wenzel EM, Behrens J. Functional comparison of human adenomatous polyposis coli (APC) and APC-like in targeting beta-catenin for degradation. PLoS One. 2013;8:e68072. doi: 10.1371/journal.pone.0068072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz-Romond T, Fiedler M, Shibata N, Butler PJG, Kikuchi A, Higuchi Y, Bienz M. The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nat Struct Mol Biol. 2007a;14:484–492. doi: 10.1038/nsmb1247. [DOI] [PubMed] [Google Scholar]
- Schwarz-Romond T, Metcalfe C, Bienz M. Dynamic recruitment of axin by Dishevelled protein assemblies. J Cell Sci. 2007b;120:2402–2412. doi: 10.1242/jcs.002956. [DOI] [PubMed] [Google Scholar]
- Seeling JM, Miller JR, Gil R, Moon RT, White R, Virshup DM. Regulation of beta-catenin signaling by the B56 subunit of protein phosphatase 2A. Science. 1999;283:2089–2091. doi: 10.1126/science.283.5410.2089. [DOI] [PubMed] [Google Scholar]
- Su Y, Fu C, Ishikawa S, Stella A, Kojima M, Shitoh K, Schreiber EM, Day BW, Liu B. APC is essential for targeting phosphorylated beta-catenin to the SCFbeta-TrCP ubiquitin ligase. Mol Cell. 2008;32:652–661. doi: 10.1016/j.molcel.2008.10.023. [DOI] [PubMed] [Google Scholar]
- Thliveris A, Samowitz W, Matsunami N, Groden J, White R. Demonstration of promoter activity and alternative splicing in the region 5' to exon 1 of the APC gene. Cancer Res. 1994;54:2991–2995. [PubMed] [Google Scholar]
- Van der Luijt RB, Vasen HF, Tops CM, Breukel C, Fodde R, Meera Khan P. APC mutation in the alternatively spliced region of exon 9 associated with late onset familial adenomatous polyposis. Hum Genet. 1995;96:705–710. doi: 10.1007/BF00210303. [DOI] [PubMed] [Google Scholar]
- Van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis A-P, et al. The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Wang S, Noritake J, Sato K, Fukata M, Takefuji M, Nakagawa M, Izumi N, Akiyama T, Kaibuchi K. Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev Cell. 2004;7:871–883. doi: 10.1016/j.devcel.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Welters HJ, Kulkarni RN. Wnt signaling: relevance to beta-cell biology and diabetes. Trends Endocrinol Metab. 2008;19:349–355. doi: 10.1016/j.tem.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Wieschaus E, Nusslein-Volhard C. Drosophila: A Practical Approach. Oxford, UK: Oxford University Press; 1998. [Google Scholar]
- Yamulla RJ, Kane EG, Moody AE, Politi KA, Lock NE, Foley AV, Roberts DM. Testing models of the APC tumor suppressor/β-catenin interaction reshapes our view of the destruction complex in Wnt signaling. Genetics. 2014;197:1285–1302. doi: 10.1534/genetics.114.166496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhang W, Evans PM, Chen X, He X, Liu C. Adenomatous polyposis coli (APC) differentially regulates beta-catenin phosphorylation and ubiquitination in colon cancer cells. J Biol Chem. 2006;281:17751–17757. doi: 10.1074/jbc.M600831200. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chen L, Gao L, Lin K, Zhu L, Lu Y, Shi X, Gao Y, Zhou J, Xu P, et al. Structural basis for the recognition of Asef by adenomatous polyposis coli. Cell Res. 2012;22:372–386. doi: 10.1038/cr.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M-N, Kunttas-Tatli E, Zimmerman S, Zhouzheng F, McCartney BM. Cortical localization of APC2 plays a role in actin organization but not in Wnt signaling in Drosophila. J Cell Sci. 2011;124:1589–1600. doi: 10.1242/jcs.073916. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.