

Abstract
The mitochondrial respiratory chain is a major generator of cellular oxidative stress, thought to be an underlying cause of the carcinogenic and ageing process in many tissues including skin. Previous studies of the relative contributions of the respiratory chain (RC) complexes I, II and III towards production of reactive oxygen species (ROS) have focussed on rat tissues and certainly not on human skin which is surprising as this tissue is regularly exposed to UVA in sunlight, a potent generator of cellular oxidative stress. In a novel approach we have used an array of established specific metabolic inhibitors and DHR123 fluorescence to study the relative roles of the mitochondrial RC complexes in cellular ROS production in 2 types of human skin cells. These include additional enhancement of ROS production by exposure to physiological levels of UVA. The effects within epidermal and dermal derived skin cells are compared to other tissue cell types as well as those harbouring a compromised mitochondrial status (Rho-zero A549). The results show that the complex II inhibitor, TTFA, was the only RC inhibitor to significantly increase UVA-induced ROS production in both skin cell types (P<0.05) suggesting that the role of human skin complex II in terms of influencing ROS production is more important than previously thought particularly in comparison to liver cells. Interestingly, two-fold greater maximal activity of complex II enzyme was observed in both skin cell types compared to liver (P<0.001). The activities of RC enzymes appear to decrease with increasing age and telomere length is correlated with ageing. Our study showed that the level of maximal complex II activity was higher in the MRC5/hTERT (human lung fibroblasts transfected with telomerase) cells than the corresponding wild type cells (P=0.0012) which can be considered (in terms of telomerase activity) as models of younger and older cells respectively.
Keywords: Ageing, Mitochondria, Respiratory chain, Reactive oxygen species (ROS), Skin
Graphical abstract
Highlights
-
•
We examined the influence of mitochondrial complex II on ROS production in human skin.
-
•
Past studies have focussed on ROS production from mitochondrial complexes I and III.
-
•
DHR123 fluorescence was used following individual complex inhibition and UVA exposure.
-
•
Only complex II inhibition significantly increased ROS levels in both skin cell types.
-
•
Complex II had a two-fold greater activity in skin cells compared to liver cells.
Introduction
The cellular and molecular effects of UVA exposure in skin have been well documented and its relationship with increased reactive oxygen species (ROS) production and associated damage to lipids, proteins and nuclear DNA [1–3]. The mitochondrial respiratory chain (RC) is the major cellular generator of superoxide and associated ROS as electrons leak from sites at or within the RC complexes [4,5]. Mitochondrial DNA (mtDNA) is close to the site of superoxide production making it highly vulnerable to oxidative damage. As the integrity of mtDNA is essential for mitochondrial function, the generation of ROS from mitochondria and the accumulation of mutations and dysfunction in a vicious cycle of events is considered a contributor to ageing, cancer, neurodegeneration, and cell death in many tissues as well as skin [1,4,6]. We and others have previously shown that mtDNA mutations and ROS increase in skin due to UVA irradiation which has led to pioneering the use of mtDNA damage as biomarker of UV exposure in human skin [7–9].
Due to an increase of ROS being associated with many human diseases, there has been a considerable amount of interest and controversy in the literature regarding the most important sites of ROS production within the mitochondrial RC [5,10–12]. Many of the studies have been carried out in brain, muscle or liver mitochondria from humans and rats and this has led to the general opinion that complexes I (NADH–ubiquinone oxidoreductase) and III (ubiquinol–cytochrome c oxidoreductase) of the RC are the major sources of cellular ROS [5,10,13,14]. However, complex II (succinate–ubiquinone oxidoreductase) has been suggested as an under-studied source of cellular ROS [12,15,16]. To date there have been few investigations of the relative roles of these RC complexes in ROS production (even within the same experimental conditions); furthermore these have predominantly focussed on rat as opposed to human tissues. The few human studies conducted have not featured skin which is surprising as this tissue is regularly exposed to the harmful UVA rays in sunlight which are a potent generator of cellular oxidative stress [1,4].
Specific inhibitors of the RC complexes are commonly used as tools for assessing the role of individual complexes in ROS production. If a specific site of a complex is blocked, ROS is produced at or upstream of that site (usually via a free ubisemiquinone radical). The inhibitors rotenone (and to a lesser extent diphenyleneiodonium (DPI) and 1-trichloromethyl-1,2,3,4-tetrahydro-β-carboline (TaClo)), 2-trithenoylacetone (TTFA) (and to a lesser extent 3-nitropropionic acid (3NP)) and antimycin are frequently used as they are specific inhibitors of complexes I, II and III respectively [17–21].
In this novel approach we have used an array of commonly used specific metabolic inhibitors and DHR123 fluorescence to study the relative roles of the mitochondrial RC complexes in cellular ROS production in human skin cells under the same experimental conditions. These include additional enhancement of ROS production by exposure to physiological levels of UVA. The effects within skin cells (epidermal and dermal derived) were compared to other tissue cell types (e.g. liver) where the RC complexes have been extensively studied as well as those harbouring a compromised mitochondrial status (i.e. Rho-zero). The results show that the role of mitochondrial complex II in terms of influencing ROS production in human skin is more important than previously thought based upon previous studies in other tissues predominantly in non-human cell types (e.g. rat). It has been proposed that the activity of RC enzymes decrease with increasing age and that telomere length is correlated with ageing. Our investigations showed that the level of complex II activity was much higher in the MRC5/hTERT (human lung fibroblasts transfected with telomerase) cells than the corresponding wild type MRC5 cells which, in terms of considering telomerase activity as an ageing biomarker, can be considered as models of younger and older cells respectively.
Materials and methods
Cell culture
HaCaT cells are a spontaneously immortalised human skin keratinocyte cell line. Human dermal fibroblasts (HDFn) cells are neonatal human dermal fibroblasts. HepG2 cells are a hepatocellular carcinoma cell line. A549 Parental cells are adenocarcinomic human alveolar basal epithelial cells whereas A549 Rho-zero cells are treated with ethidium bromide to deplete mtDNA, producing cells lacking a functional RC (gift from Prof. Ian Holt, University of Cambridge). MRC5 cells are human foetal lung fibroblast cells, and MRC5/hTERT cells are lung fibroblast cells which overexpress a subunit of telomerase. All cell lines were grown in Dulbecco's Modified Eagles' Medium (DMEM) supplemented with 10% Foetal Calf Serum (FCS) and 5% penicillin/streptomycin with the exception of a549 Rho-zero cells which were supplemented with 50 µM uridine.
UVA treatment and fluorimetric analysis of DHR123 (dihydrorhodamine 123) fluorescence intensity (FI) in cultured cells
UVA irradiation used a glass filtered TL-09 lamp (315–400 nm, peak emission at 360 nm). Cells grown in the presence or absence (controls) of respiratory inhibitor (18 h) were loaded with 25 µM DHR123 (Sigma-Aldrich, Poole, UK) for 20 min. Excess DHR123 was removed (centrifugation and washing with PBS) then fluorescence intensity measurement was measured using a Varian, Cary Eclipse fluorimeter (Varian, UK) (λex=488 nm and λem=520 nm respectively). Controls included cells loaded with DHR123 and no UVA; cells without DHR123 but exposed to UVA, to ensure that the increase in fluorescence intensity (FI) was due to increased DHR123 fluorescence and not autofluorescence of the cells. Cell viability was determined by the MTS assay (Promega, UK). Absorbance was measured at 490 nm with a 96-well plate reader (SpectraMax 250, Molecular Devices) using SOFTmax pro 3.1.1. An optimal sub-lethal UVA dose was derived from these detailed dose response experiments which provided a significant induction of FI without compromising cell viability. The sub-lethal optimal dose was 14 J/cm2 for the skin cells (Figs. 1 and 2) and the same UVA dose was found to provide the same profile of response in the other cell types that were tested (results not shown).
Fig. 1.
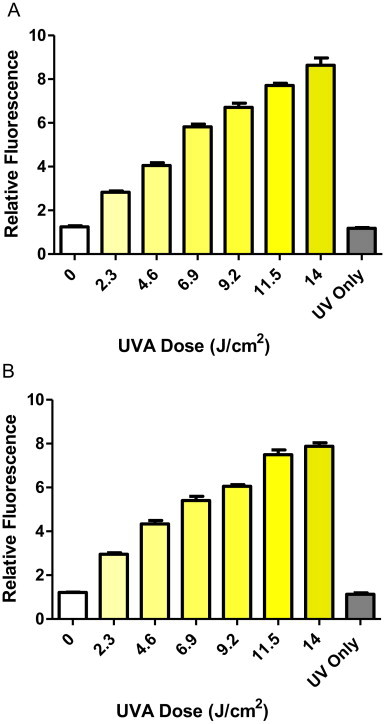
UVA dose response of DHR123 fluorescence in (A) HaCaT and (B) HDFn cells. Cells loaded with DHR123 were shown to exhibit significantly increased fluorescence intensity over controls at all UVA irradiances (one-way ANOVA including Bonferroni's post-hoc test, P<0.001, n=8). Data representative of 2 repeats, n=8 replicates for each experimental dose, bars represent means±SEM.
Fig. 2.
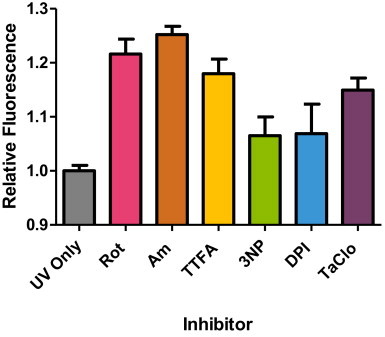
Summary of relative fluorescence intensity of HaCaT cells treated with respiratory chain inhibitors for 18 h prior to exposure of 14 J/cm2 UVA. Significant increases in DHR123 fluorescence intensity over control (i.e. UVA exposure in the absence of inhibitor) were found in rotenone (Rot), antimycin (Am), TTFA and TaClo for 18 h compared to UVA alone (control) (*P<0.05, n=8, as analysed by a one-way ANOVA with Dunnett's post-hoc test). No significant difference in fluorescence intensity was found for 3NP and DPI treatment 18 h, (P>0.05). Data representative of 2 repeats, n=8 replicates for each inhibitor treatment, bars represent means±SEM.
Measurement of complex II and citrate synthase activity
Enzyme activity was determined by previously described spectrophotometric methods [22].
Statistical analyses
For each data set, an analysis of variance (ANOVA) was performed with post-hoc test for multiple groups was performed using commercially available software (GraphPad, Prism 5).
Results
The effect of different RC inhibitors on ROS production in UVA-irradiated skin cells
The initial aim of the study was to perform a detailed investigation of the effect of established inhibitors of the RC complexes on UVA-induced ROS production in different human cultured skin cells, namely HaCaTs and HDFns (epidermal and dermal skin fibroblasts respectively). Firstly, it was necessary to derive a maximal but sub-lethal concentration of the inhibitors for the RC complexs in the skin cells (and the other cell types used in the study) by performing an extensive range of cell viability dose–response experiments. Cell viability was determined using the MTS assay over a nanomolar to millimolar concentration range of each RC inhibitor to derive a sub-lethal experimental concentration in all of the cultured cells (Table 1).
Table 1.
The maximal sub-lethal doses of the respiratory chain inhibitors (µM) for all cell types. Cultured cells were pre-treated with a range of concentrations of inhibitors (rotenone, DPI and TaClo inhibit complex I; TTFA and 3NP inhibit complex II; and antimycin inhibits complex III) and cell viability was assessed using the MTS assay. The numbers refer to the maximum sub-lethal concentrations (µM) for each inhibitor. Statistical significance of cell death was assessed by performing a one-way ANOVA with Dunnett’s post-hoc test compared to control (untreated).
| Inhibitor |
Cell line |
||||
|---|---|---|---|---|---|
| HaCaT | HDFn | HepG2 | A549Par | A549Rho-zero | |
| Rotenone | 2.5 | 25 | 15 | 2.5 | 2.5 |
| Antimycin | 1.8 | 1800 | 9 | 1.8 | 4.5 |
| TTFA | 220 | 220 | 440 | 88 | 220 |
| 3NP | 10 | 10 | 100 | 100 | 10 |
| DPI | 0.1 | 0.1 | 0.1 | 0.5 | 1 |
| TaClo | 100 | 100 | 400 | 100 | 200 |
UVA in sunlight is a well-known physiological inducer of oxidative stress in human skin. Using electrochemical studies we have previously shown that the mechanism of the UVA-induced ROS production involves species such as singlet oxygen [23] and superoxide in skin cells [24] and isolated mitochondria from those skin cells [25]. UVA was used as an inducer of ROS rather than hydrogen peroxide as, apart from UVA exposure being more physiologically relevant to skin, we have also previously shown that both inducers produce the same profile of ROS production in human skin cells [26]. It was therefore necessary to derive a sub-lethal dose of UVA for the generation of increased ROS in the cultured cells by means of detailed UVA dose response experiments. As non-fluorescent DHR123 is oxidised to fluorescent rhodamine-123 by cellular peroxides, fluorimetry was used to assess cellular peroxide production in the cultured skin cells loaded with DHR123 following irradiation with increasing UVA doses. In all cases, an MTS assay was performed to detect cell viability. An optimal sub-lethal UVA dose of 14 J/cm2 was derived from these detailed dose response experiments. This dose provided a significant induction of fluorescent intensity (FI) without compromising cell viability. An example of the UVA dose curves for both HaCaT and HDFn skin cells is shown in Fig. 1 (parts A and B respectively). The UVA dose of 14 J/cm2 is a physiologically relevant dose being consistent with previous studies such as those described by Gniadecki et al. [13] and Aitken et al. [24].
Following the derivation of the sub-lethal/maximal effect of both the RC inhibitor concentrations and the UVA doses, the combined effect of RC inhibitor concentrations on UVA-induced ROS production (DHR123 fluorescence) was determined in HaCaT and HDFn cells (Figs. 2 and 3 respectively). The effect profiles of the different RC inhibitors (in terms of enhancing the UVA-induced ROS production) was different between the two skin cell cultures as shown by a different hierarchy of effect following individual inhibitor treatment. This is not surprising as the two skin cell cultures reflect or are derived from different parts of the skin where the cellular bioenergy demands from mitochondria are different [27]. However, a closer examination of the data shows quite clearly that the complex II inhibitor, TTFA, was the only RC inhibitor to significantly increase UVA-induced ROS production in both skin cell types (P<0.05, one-way ANOVA with Dunnett's correction). This consistent effect of TTFA treatment is important as it suggests that the influence of complex II in skin cells may be more important than previously thought based upon the prevailing literature view in other cell types where the dominant influence centres upon complexes I and III. In detail, a high proportion of these latter studies have been initially performed in whole cells and/or isolated mitochondria particularly from liver (but also from other rat and animal tissue (e.g. muscle) rather than human).
Fig. 3.
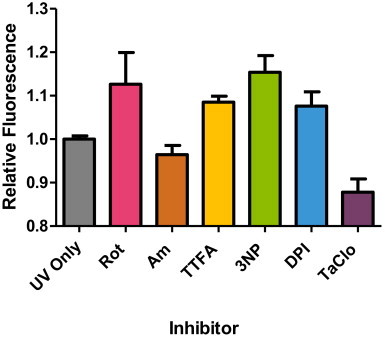
Summary of relative fluorescence intensity of HDFn cells treated with respiratory chain inhibitors for 18 h before exposure to 14 J/cm2 UVA. Significant increases in DHR123 fluorescence intensity over control (i.e. UVA exposure in the absence of inhibitor) were found in TTFA and 3NP at 18 h treatment compared to UVA alone (control) (*P<0.05, n=8, as analysed by a one-way ANOVA with Dunnett's post-hoc test). No significant difference in fluorescence intensity was found for all other inhibitor treatment 18 h, (P>0.05). Data representative of 2 repeats, n=8 replicates for each inhibitor treatment, bars represent means±SEM.
This issue was addressed by repeating the experimental protocol utilised to generate data shown in Figs. 2 and 3 but using HepG2 cells derived from human liver. In order to ensure a direct comparison, the exact protocol used on the skin cells was performed on the liver cells. Hydrogen peroxide is often used as an inducer of ROS production in cultured cells, however we have observed that UVA and hydrogen peroxide produce the same profile of ROS production response in vitro [4]. Unsurprisingly, the data from this series of experiments described in Fig. 4 confirms the predominant view in the liver (and muscle/brain) literature that complex I (inhibited by rotenone) and complex III (inhibited by antimycin) are indeed the major sites of influence on ROS generation in the RC. However, it should be noted that the role of complex II appears to be greater than previously assumed even in liver as portrayed by the 3NP results. The addition of this other commonly used complex II inhibitor shows a degree of inhibition which is comparable to that observed using the other complex I inhibitor TaClo [28]. Interestingly, although 3NP is a well-documented RC inhibitor, the emphasis in the literature in terms of numbers of studies remains biased towards the complex I and III inhibitors, rotenone and antimycin, with an additional focus on (rat) liver and muscle studies as opposed to skin.
Fig. 4.
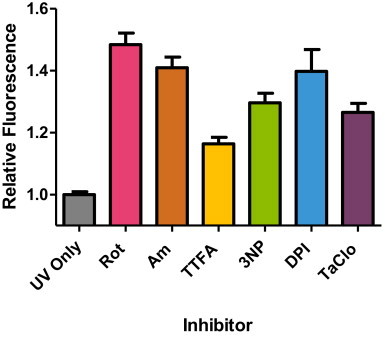
Summary of relative fluorescence intensity of HepG2 cells treated with respiratory chain inhibitors for 18 h before exposure to 14 J/cm2 UVA. Significant increases in DHR123 fluorescence intensity over control (i.e. UVA exposure in the absence of inhibitor) were observed for all inhibitors at 18 h treatment compared to UVA alone (control) (*P<0.01, n=8, analysed by a one-way ANOVA with Dunnett's post-hoc test). Data representative of 2 repeats, n=8 replicates for each inhibitor treatment, bars represent means±SEM.
Comparison of complex II activity in skin cells with human liver, Rho-zero and Parental cells
This apparent importance of human skin complex II in ROS production compared to the scenario observed in liver was investigated further by direct measurement of the maximal activity of complex II in these and other cell types to provide context. This was performed using methods previously established in our laboratory and used for many years in other laboratories to investigate many cell types [29]. The maximum capacity of complex II activity values were normalised to citrate synthase activity which is a standard procedure in mitochondrial research [29]. Citrate synthase is a mitochondrial matrix enzyme not involved in the RC and is frequently used a marker of mitochondrial content as opposed to simply normalising to total cellular protein where it is not possible to discern non-mitochondrial protein [22]. The results in Table 2 show that the citrate synthase normalised complex II activity in both types of skin cells (HaCaT and HDFn) is approximately two-fold greater than that observed in liver cells (HepG2). This represents a statistically significant difference (HepG2 vs. HaCaT, P<0.001 and HepG2 vs. HDFn, P<0.001, unpaired t-test) whereas there was no statistical difference in complex II activity between the two types of skin cells (HaCa T vs. HDFn, P=0.51, unpaired t-test).
Table 2.
Complex II activity in the different cell types. The activity of complex II in different cell types was normalised against the activity of the mitochondrial housekeeping enzyme citrate synthase to obtain the ratio of mitochondrial content to complex II activity (nmols DCPIP reduced min−1/unit citrate synthase). The degree of complex II inhibition by the specific inhibitors, TTFA and 3NP is expressed as a percentage of the total activity.
| Cell type | Mean CII/CS activity | % Inhibition by TTFA | % Inhibition by 3-NP |
|---|---|---|---|
| HaCaT | 0.27±0.016, N=8 | 42.40±1.72 | 51.85±6.89 |
| HDFn | 0.25±0.016, N=7 | 30.22±7.33 | 32.1±1.11 |
| HepG2 | 0.13±0.005, N=6 | 49.21±6.59 | 57.74±0.72 |
| Parental | 0.15±0.009, N=6 | 26.30±0.06 | 49.88±1.84 |
| Rho-zero | 0.29±0.020, N=4 | 0.00 | 48.77±3.90 |
| MRC5 | 0.27±0.014, N=7 | 35±5.46 | 36±4.43 |
| MRC5/hTERT | 0.37±0.011, N=4 | 38±0.48 | 42±0.94 |
The two-fold difference in complex II activity coupled with the associated difference in the inhibitor profiles (i.e. particularly complex II, Figs. 2–4) between the liver and the skin cells (Table 2) prompted further investigations of other cell types to provide context. This entailed the determination of complex II activity in the A549 Rho-0 cell line. These cells are depleted of the mtDNA genome encoding for specific parts of the RC complexes I–V except for complex II which is uniquely and exclusively nuclear encoded and therefore contrasts with the other RC complexes which are under dual genetic control (i.e. both mitochondrial and nuclear DNA) [30]. Although Rho-zero cells are effectively absent of mtDNA they do retain mitochondria and carry out some electron transport activities which allows them to be used as a model for ROS production from the RC complexes [31]. The enzyme activity of complex II was determined in both the Parental and Rho-zero A549 cells (Table 2) and the activity measurements were again normalised to citrate synthase activity. In addition the degree of inhibition by the specific complex II inhibitors, TTFA and 3NP, was also determined. The data shows that the maximum specific activity of complex II in the Rho-zero cells was approximately two-fold greater than the Parental cells (P<0.0001, unpaired t-test). In terms of sensitivity to inhibition of complex II activity, it is clear that the Rho-zero cells are completely resistant to inhibition by TTFA in contrast to the Parental cells which exhibit a significantly different inhibition of 26% (P<0.0004, unpaired t-test). Interestingly, 3NP inhibits complex II activity in both the Parental and Rho-zero cells to the same degree at approximately 50% and 49% respectively (i.e. no significant difference in the 3NP inhibition between the 2 cell types (P=0.85, unpaired t-test)) which is also within the range of 3NP inhibition observed in the other cell types (Table 2)). Furthermore, the levels of complex II activity in the Rho zero and Parental cells were very similar to those activity levels observed in the skin cells and liver cells respectively.
Decline of complex II activity associated with ageing biomarkers
It has been proposed that the activity of RC enzymes decrease with increasing age [32–34] and furthermore that telomere length is correlated with ageing [35]. Based upon these observations it was decided to compare the level of complex II activity in human lung fibroblasts transfected with a subunit of telomerase (enzyme involved in telomere repair) (i.e. MRC5/hTERT) with cells lacking additional telomerase (i.e. MRC5), as a model for younger and older cells respectively. It was found that the level of complex II activity was much higher in the MRC5/hTERT cells than the MRC5 cells (Table 2) (MRC5 vs. MRC5/hTERT, P=0.0012, unpaired t-test).
Discussion
Increased cellular oxidative stress is thought to be an underlying cause of the carcinogenic and ageing processes in many tissues as well as skin [4]. This is important because it is well established that the mitochondrial RC is the major cellular generator of superoxide as a result of leakage of single electrons which reduce O2 to form [5]. The effect of UVA in our study involves singlet oxygen [23] and superoxide and our previous electrochemical studies have shown that skin cells and their isolated mitochondria produce superoxide following electron leakage from the RC in response to UVR [24,25]. DHR123 is an established fluorescent probe for cellular ROS measurements and used previously in skin cells [26] although one should be mindful of its limitations versus other cellular probes and include appropriate controls [36,37].
Using specific established inhibitors of the RC, our study shows that the complex II inhibitor, TTFA, was the only inhibitor to significantly increase UVA-induced ROS generation in both skin cell types against a background of differential effects of the other inhibitors observed in the skin cells. This suggests that the effect of complex II in skin cells may be more important than previously thought based upon the prevailing literature view in other cell types where historically the research tends to focus on complexes I and III. Indeed this latter scenario was confirmed in liver cells (the present study) in which much of the previous work in the literature has been performed as well as to muscle and brain.
The increased importance of complex II in UVA-induced ROS production in skin cells compared to liver cells (Figs. 2–4) is associated with an approximately two-fold greater activity of complex II enzyme (Table 1) in skin cells compared to liver. Interestingly, a two-fold greater complex II activity is also seen on comparison of Rho-zero A549 with the Parental A549 cells (Table 2). The greater reliance and emphasis on the nuclear encoded complex II in the mtDNA depleted Rho-zero cells may explain this observation and may also further support the suggestion of a greater influence or emphasis of complex II in skin cells compared to liver cells. The complex II inhibitors TTFA and 3NP, were used to enhance ROS production at or upstream of the site of complex II. TTFA is an incomplete inhibitor binding at the (distal) site of UQ cycling whereas 3NP is a competitive inhibitor at the succinate binding (proximal) site [18,19]. The relative similarities in the degree of complex II inhibition by TTFA and 3NP in both skin cells and liver cells compared to the marked differences observed between the Rho-zero and Parental A549 cells suggests unsurprisingly that the stochastic arrangement of mitochondrial and nuclear encoded enzyme subunits immediately proximal and distal to complex II is similar in both skin and liver cells.
The findings in our study of increased importance of complex II in terms of influencing ROS production is strongly supported by a recent investigation showing that complex II under certain conditions can generate ROS at high rates comparable to the other ROS producing sites at complexes I and III [16]. Although this study was performed in rat muscle the authors suggest that complex II may be an important contributor to physiological and pathological ROS production. In this respect, it is interesting to note that the increase in 14 J/cm2 UVA-induced DHR123 fluorescence compared to un-irradiated controls is two-fold greater in skin cells than in liver cells (i.e. 6.5±1.00 and 6.9±0.15 in HDFns and HaCaTs respectively vs. 3.3±0.63 in HepG2s P<0.001; one way ANOVA with Bonferroni's post-hoc test for both HDFns and HaCaTs vs. HepG2s). Indeed, increased ROS production from complex II has been found in human diseases such as heart failure [38] and complex II inhibition has a key role in the development of Huntington's disease [39] as well as tumourigenesis in a number of tissues [40].
Respiratory chain enzyme activity and/or mitochondrial derived cellular bioenergy are known to decline with age in many tissues [32–34] and telomere length has been correlated with ageing. In keeping with this we have observed that the activity of complex II was higher in the MRC5/hTERT cells compared to the same cells without additional telomerase (MRC5), as a model for younger and older cells respectively [41,42].
In terms of mechanism, our study shows that UVA irradiation enhanced the increase in ROS production caused by the presence of the RC inhibitors. In this context, it has been postulated that iron may play a critical role in the modulation of UVA-induced oxidative damage. An increased presence of free iron further exacerbates oxidative damage through interaction with reactive oxygen intermediates via Fenton reactions. Studies have reported an increased level of iron subsequent to UVA irradiation, which has been shown to potentiate irreversible oxidative damage [43–45]. Enzymes with iron–sulphur (Fe–S) centres have been shown to be sensitive to modification by UVA rendering the enzyme inactive [46] and consequently resulting in the release and accumulation of free intracellular iron [47]. Interestingly, Fe–S centres are integral to the RC complexes I, II and III [48] which may make them vulnerable targets for photosensitisation and a potential hotspot for iron-mediated oxidative damage. Indeed our group has recently shown that tiron (4,5-dihydroxy-1,3-benzenedisulfonic) which chelates iron (and other metals), exhibits ROS scavenging properties and enters mitochondria, is able to prevent UVA and hydrogen peroxide induced mitochondrial DNA damage in human skin cells [49].
In summary, we have used an array of established specific metabolic inhibitors and DHR123 fluorescence as a novel approach to study the relative roles of the mitochondrial RC complexes in cellular ROS production in human skin cells under the same experimental conditions. These include additional enhancement of ROS production by exposure to physiological levels of UVA which is a potent inducer of oxidative stress to sunlight exposed skin tissue. The effects within epidermal and dermal derived skin cells are compared to other tissue cell types (e.g. liver) where the RC complexes have been extensively studies as well as those harbouring a compromised mitochondrial status (i.e. Rho-zero). The results show that the role of human skin mitochondrial complex II in terms of influencing ROS production is more important than previously thought based upon previous studies in other tissues predominantly in non-human cell types. Further investigation of complex II showed a decline in enzyme activity with ageing biomarkers (telomerase). Additionally, our observations may help to partly explain the comparatively recent clinical success of a complex II metabolic substrate (i.e. fumarate) which has been used in the clinical treatment of patients suffering from the proliferating and energy demanding skin disease psoriasis [50].
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Acknowledgements
The work was supported by the Medical Research Council (MRC), the Institute of Cellular Medicine, the Faculty of Medical Sciences (Newcastle University (C0241N3010)), the North Eastern Skin Research Fund (NESRF) (RES/0241/7536) and the National Institute for Health Research Newcastle Biomedical Research Centre based at Newcastle Hospitals Foundation Trust and Newcastle University (BH120752) . The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
References
- 1.Tulah A.S., Birch-Machin M.A. Stressed out mitochondria: the role of mitochondria in ageing and cancer focussing on strategies and opportunities in human skin. Mitochondrion. 2013;13:444–453. doi: 10.1016/j.mito.2012.11.007. 23195682 [DOI] [PubMed] [Google Scholar]
- 2.Huang X.X., Bernerd F., Halliday G.M. Ultraviolet A within sunlight induces mutations in the epidermal basal layer of engineered human skin. American Journal of Pathology. 2009;174:1534–1543. doi: 10.2353/ajpath.2009.080318. 19264911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mouret S., Forestier A., Douki T. The specificity of UVA-induced DNA damage in human melanocytes. Photochemical & Photobiological Sciences: Official Journal of the European Photochemistry Association and the European Society for Photobiology. 2012;11:155–162. doi: 10.1039/c1pp05185g. 21986862 [DOI] [PubMed] [Google Scholar]
- 4.Birch-Machin M.A., Swalwell H. How mitochondria record the effects of UV exposure and oxidative stress using human skin as a model tissue. Mutagenesis. 2010;25:101–107. doi: 10.1093/mutage/gep061. 19955330 [DOI] [PubMed] [Google Scholar]
- 5.Murphy M.P. How mitochondria produce reactive oxygen species. Journal of Biological Chemistry. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Moura M.B., Dos Santos L.S., Van Houten B. Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environmental and Molecular Mutagenesis. 2010;51:391–405. doi: 10.1002/em.20575. 20544881 [DOI] [PubMed] [Google Scholar]
- 7.Berneburg M., Grether-Beck S., Kürten V., Ruzicka T., Briviba K., Sies H., Krutmann J. Singlet oxygen mediates the UVA-induced generation of the photoaging-associated mitochondrial common deletion. Journal of Biological Chemistry. 1999;274:15345–15349. doi: 10.1074/jbc.274.22.15345. 10336420 [DOI] [PubMed] [Google Scholar]
- 8.Birch-Machin M.A., Tindall M., Turner R., Haldane F., Rees J.L. Mitochondrial DNA deletions in human skin reflect photo-rather than chronologic aging. Journal of Investigative Dermatology. 1998;110:149–152. doi: 10.1046/j.1523-1747.1998.00099.x. 9457910 [DOI] [PubMed] [Google Scholar]
- 9.Krishnan K.J., Harbottle A., Birch-Machin M.A. The use of a 3895 bp mitochondrial DNA deletion as a marker for sunlight exposure in human skin. Journal of Investigative Dermatology. 2004;123:1020–1024. doi: 10.1111/j.0022-202X.2004.23457.x. 15610508 [DOI] [PubMed] [Google Scholar]
- 10.Brand M.D. The sites and topology of mitochondrial superoxide production. Experimental Gerontology. 2010;45:466–472. doi: 10.1016/j.exger.2010.01.003. 20064600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kowaltowski A.J., de Souza-Pinto N.C., Castilho R.F., Vercesi A.E. Mitochondria and reactive oxygen species. Free Radical Biology & Medicine. 2009;47:333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. 19427899 [DOI] [PubMed] [Google Scholar]
- 12.Lemarie A., Grimm S. Mitochondrial respiratory chain complexes: apoptosis sensors mutated in cancer? Oncogene. 2011;30:3985–4003. doi: 10.1038/onc.2011.167. 21625217 [DOI] [PubMed] [Google Scholar]
- 13.Gniadecki R., Thorn T., Vicanova J., Petersen A., Wulf H.C. Role of mitochondria in ultraviolet-induced oxidative stress. Journal of Cellular Biochemistry. 2000;80:216–222. doi: 10.1002/1097-4644(20010201)80:2<216::aid-jcb100>3.0.co;2-h. 11074592 [DOI] [PubMed] [Google Scholar]
- 14.Kudin A.P., Debska-Vielhaber G., Kunz W.S. Characterization of superoxide production sites in isolated rat brain and skeletal muscle mitochondria. Biomedicine & Pharmacotherapy. 2005;59:163–168. doi: 10.1016/j.biopha.2005.03.012. 15862710 [DOI] [PubMed] [Google Scholar]
- 15.Guzy R.D., Sharma B., Bell E., Chandel N.S., Schumacker P.T. Loss of the SdhB, but not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Molecular and Cellular Biology. 2008;28:718–731. doi: 10.1128/MCB.01338-07. 17967865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quinlan C.L., Orr A.L., Perevoshchikova I.V., Treberg J.R., Ackrell B.A., Brand M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. Journal of Biological Chemistry. 2012;287:27255–27264. doi: 10.1074/jbc.M112.374629. 22689576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bringmann G., Feineis D., God R., Peters K., Peters E.M., Scholz J., Riederer F., Moser A. 1-Trichloromethyl-1,2,3,4-tetrahydro-β-carboline (TaClo) and related derivatives: chemistry and biochemical effects on catecholamine biosynthesis. Bioorganic & Medicinal Chemistry. 2002;10:2207–2214. doi: 10.1016/s0968-0896(02)00060-3. [DOI] [PubMed] [Google Scholar]
- 18.Huang L.S., Sun G., Cobessi D., Wang A.C., Shen J.T., Tung E.Y., Anderson V.E., Berry E.A. 3-Nitropropionic acid is a suicide inhibitor of mitochondrial respiration that, upon oxidation by complex II, forms a covalent adduct with a catalytic base arginine in the active site of the enzyme. Journal of Biological Chemistry. 2006;281:5965–5972. doi: 10.1074/jbc.M511270200. 16371358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandavilli B.S., Boldogh I., Van Houten B. 3-Nitropropionic acid-induced hydrogen peroxide, mitochondrial DNA damage, and cell death are attenuated by Bcl-2 overexpression in PC12 cells. Brain Research. Molecular Brain Research. 2005;133:215–223. doi: 10.1016/j.molbrainres.2004.10.033. 15710238 [DOI] [PubMed] [Google Scholar]
- 20.Nasr P., Delorme T. Age-dependent vulnerability of the striatal mitochondrial to 3-nitropropionic acid. Ohio Journal of Sciences. 2007;107:120–124. [Google Scholar]
- 21.Palfi S., Ferrante R.J., Brouillet E., Beal M.F., Dolan R., Guyot M.C., Peschanski M., Hantraye P. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of Huntington's disease. Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1996;16:3019–3025. doi: 10.1523/JNEUROSCI.16-09-03019.1996. 8622131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birch-Machin M.A., Briggs H.L., Saborido A.A., Bindoff L.A., Turnbull D.M. An evaluation of the measurement of the activities of complexes I–IV in the respiratory chain of human skeletal muscle mitochondria. Biochemical Medicine and Metabolic Biology. 1994;51:35–42. doi: 10.1006/bmmb.1994.1004. 8192914 [DOI] [PubMed] [Google Scholar]
- 23.Sakurai H., Yasui H., Yamada Y., Nishimura H., Shigemoto M. Detection of reactive oxygen species in the skin of live mice and rats exposed to UVA light: a research review on chemiluminescence and trials for UVA protection. Photochemical & Photobiological Sciences: Official Journal of the European Photochemistry Association and the European Society for Photobiology. 2005;4:715–720. doi: 10.1039/b417319h. 16121282 [DOI] [PubMed] [Google Scholar]
- 24.Aitken G.R., Henderson J.R., Chang S.C., McNeil C.J., Birch-Machin M.A. Direct monitoring of UV-induced free radical generation in HaCaT keratinocytes. Clinical and Experimental Dermatology. 2007;32:722–727. doi: 10.1111/j.1365-2230.2007.02474.x. 17953641 [DOI] [PubMed] [Google Scholar]
- 25.Henderson J.R., Swalwell H., Boulton S., Manning P., McNeil C.J., Birch-Machin M.A. Direct, real-time monitoring of superoxide generation in isolated mitochondria. Free Radical Research. 2009;43:796–802. doi: 10.1080/10715760903062895. 19562601 [DOI] [PubMed] [Google Scholar]
- 26.Swalwell H., Latimer J., Haywood R.M., Birch-Machin M.A. Investigating the role of melanin in UVA/UVB- and hydrogen peroxide-induced cellular and mitochondrial ROS production and mitochondrial DNA damage in human melanoma cells. Free Radical Biology & Medicine. 2012;52:626–634. doi: 10.1016/j.freeradbiomed.2011.11.019. 22178978 [DOI] [PubMed] [Google Scholar]
- 27.Birch-Machin M.A. The role of mitochondria in ageing and carcinogenesis. Clinical and Experimental Dermatology. 2006;31:548–552. doi: 10.1111/j.1365-2230.2006.02161.x. 16716161 [DOI] [PubMed] [Google Scholar]
- 28.Boulton S.J., Keane P.C., Morris C.M., McNeil C.J., Manning P. Real-time monitoring of superoxide generation and cytotoxicity in neuroblastoma mitochondria induced by 1-trichloromethyl-1,2,3,4-tetrahydro-beta-carboline. Redox Report: Communications in Free Radical Research. 2012;17:108–114. doi: 10.1179/1351000212Y.0000000011. 22664359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Birch-Machin M.A., Turnbull D.M. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods in Cell Biology. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. 11381612 [DOI] [PubMed] [Google Scholar]
- 30.Scheffler I. Mitochondria. second edition. John Wiley and Sons; Hoboken, NJ: 2008. [Google Scholar]
- 31.Cuperus R., Leen R., Tytgat G.A., Caron H.N., van Kuilenburg A.B. Fenretinide induces mitochondrial ROS and inhibits the mitochondrial respiratory chain in neuroblastoma. Cellular and Molecular Life Sciences. 2010;67:807–816. doi: 10.1007/s00018-009-0212-2. 19941060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma Y.S., Wu S.B., Lee W.Y., Cheng J.S., Wei Y.H. Response to the increase of oxidative stress and mutation of mitochondrial DNA in aging. Biochimica et Biophysica Acta. 2009;1790:1021–1029. doi: 10.1016/j.bbagen.2009.04.012. 19397952 [DOI] [PubMed] [Google Scholar]
- 33.Short K.R., Bigelow M.L., Kahl J., Singh R., Coenen-Schimke J., Raghavakaimal S., Nair K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. 15800038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwong L.K., Sohal R.S. Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Archives of Biochemistry and Biophysics. 2000;373:16–22. doi: 10.1006/abbi.1999.1495. 10620319 [DOI] [PubMed] [Google Scholar]
- 35.Tsuji A., Ishiko A., Takasaki T., Ikeda N. Estimating age of humans based on telomere shortening. Forensic Science International. 2002;126:197–199. doi: 10.1016/s0379-0738(02)00086-5. 12062940 [DOI] [PubMed] [Google Scholar]
- 36.Boulton S.J., Anderson A., Swalwell H., Henderson J.R., Manning P., Birch-Machin M.A. Implications of using the fluorescent probes, dihydrorhodamine 123 and 2′,7′-dichlorodihydrofluorescein diacetate, for the detection of UVA-induced reactive oxygen species. Free Radical Research. 2011;45(2):139–146. doi: 10.3109/10715762.2010.517751. 20942573 [DOI] [PubMed] [Google Scholar]
- 37.Zmijewski J.W., Moellering D.R., Le Goffe C.L., Landar A., Ramachandran A., Darley-Usmar V.M. Oxidized LDL induces mitochondrially associated reactive oxygen/nitrogen species formation in endothelial cells. American Journal of Physiology. Heart and Circulatory Physiology. 2005;289:H852–H861. doi: 10.1152/ajpheart.00015.2005. 15805232 [DOI] [PubMed] [Google Scholar]
- 38.Redout E.M., Wagner M.J., Zuidwijk M.J., Boer C., Musters R.J.P., van Hardeveld C., Paulus W.J., Simonides W.S. Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovascular Research. 2007;75:770–781. doi: 10.1016/j.cardiores.2007.05.012. 17582388 [DOI] [PubMed] [Google Scholar]
- 39.Liot G., Bossy B., Lubitz S., Kushnareva Y., Sejbuk N., Bossy-Wetzel E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death and Differentiation. 2009;16:899–909. doi: 10.1038/cdd.2009.22. 19300456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoekstra A.S., Bayley J.P. The role of complex II in disease. Biochimica et Biophysica Acta. 2013;1827:543–551. doi: 10.1016/j.bbabio.2012.11.005. 23174333 [DOI] [PubMed] [Google Scholar]
- 41.Bodnar A.G., Ouellette M., Frolkis M., Holt S.E., Chiu C.P., Morin G.B., Harley C.B., Shay J.W., Lichtsteiner S., Wright W.E. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. 9454332 [DOI] [PubMed] [Google Scholar]
- 42.Vaziri H., Benchimol S. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Current Biology. 1998;8:279–282. doi: 10.1016/s0960-9822(98)70109-5. 9501072 [DOI] [PubMed] [Google Scholar]
- 43.Pourzand C., Watkin R.D., Brown J.E., Tyrrell R.M. Ultraviolet A radiation induces immediate release of iron in human primary skin fibroblasts: the role of ferritin. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6751–6756. doi: 10.1073/pnas.96.12.6751. 10359784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pygmalion M.J., Ruiz L., Popovic E., Gizard J., Portes P., Marat X., Lucet-Levannier K., Muller B., Galey J.B. Skin cell protection against UVA by Sideroxyl, a new antioxidant complementary to sunscreens. Free Radical Biology & Medicine. 2010;49:1629–1637. doi: 10.1016/j.freeradbiomed.2010.08.009. 20826208 [DOI] [PubMed] [Google Scholar]
- 45.Aroun A., Zhong J.L., Tyrrell R.M., Pourzand C. Iron, oxidative stress and the example of solar ultraviolet A radiation. Photochemical & Photobiological Sciences: Official Journal of the European Photochemistry Association and the European Society for Photobiology. 2012;11:118–134. doi: 10.1039/c1pp05204g. 21986918 [DOI] [PubMed] [Google Scholar]
- 46.Brazzolotto X., Gaillard J., Pantopoulos K., Hentze M.W., Moulis J.M. Human cytoplasmic aconitase (iron regulatory protein 1) is converted into its [3Fe–4S] form by hydrogen peroxide in vitro but is not activated for iron-responsive element binding. Journal of Biological Chemistry. 1999;274:21625–21630. doi: 10.1074/jbc.274.31.21625. 10419470 [DOI] [PubMed] [Google Scholar]
- 47.Kruszewski M. Labile iron pool: the main determinant of cellular response to oxidative stress. Mutation Research. 2003;531:81–92. doi: 10.1016/j.mrfmmm.2003.08.004. 14637247 [DOI] [PubMed] [Google Scholar]
- 48.Gerber J., Lill R. Biogenesis of iron–sulfur proteins in eukaryotes: components, mechanism and pathology. Mitochondrion. 2002;2(1–2):71–86. doi: 10.1016/s1567-7249(02)00041-7. 16120310 [DOI] [PubMed] [Google Scholar]
- 49.Oyewole A.O., Wilmot M.C., Fowler M., Birch-Machin M.A. Comparing the effects of mitochondrial targeted and localized antioxidants with cellular antioxidants in human skin cells exposed to UVA and hydrogen peroxide. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2014;28:485–494. doi: 10.1096/fj.13-237008. 24115050 [DOI] [PubMed] [Google Scholar]
- 50.Salgo R., Thaçi D. Treatment of moderate-to-severe plaque psoriasis. Giornale Italiano di Dermatologia e Venereologia: Organo Ufficiale, Società Italiana di Dermatologia e Sifilografia. 2009;144:701–711. 19907408 [PubMed] [Google Scholar]