

Abstract
Alcoholic liver disease encompasses a wide spectrum of pathogenesis including steatosis, fibrosis, cirrhosis, and alcoholic steatohepatitis. Autophagy is a lysosomal degradation process that degrades cellular proteins and damaged/excess organelles, and serves as a protective mechanism in response to various stresses. Acute alcohol treatment induces autophagy via FoxO3a-mediated autophagy gene expression and protects against alcohol-induced steatosis and liver injury in mice. Farnesoid X Receptor (FXR) is a nuclear receptor that regulates cellular bile acid homeostasis. In the present study, wild type and FXR knockout (KO) mice were treated with acute ethanol for 16 h. We found that ethanol treated-FXR KO mice had exacerbated hepatotoxicity and steatosis compared to wild type mice. Furthermore, we found that ethanol treatment had decreased expression of various essential autophagy genes and several other FoxO3 target genes in FXR KO mice compared with wild type mice. Mechanistically, we did not find a direct interaction between FXR and FoxO3. Ethanol-treated FXR KO mice had increased Akt activation, increased phosphorylation of FoxO3 resulting in decreased FoxO3a nuclear retention and DNA binding. Furthermore, ethanol treatment induced hepatic mitochondrial spheroid formation in FXR KO mice but not in wild type mice, which may serve as a compensatory alternative pathway to remove ethanol-induced damaged mitochondria in FXR KO mice. These results suggest that lack of FXR impaired FoxO3a-mediated autophagy and in turn exacerbated alcohol-induced liver injury.
Keywords: Autophagy, Farnesoid X receptor, FoxO3a, Alcohol, Liver injury
Abbreviations: ALT, alanine aminotransferase; ALD, alcoholic liver disease; CYP, cytochrome P450; 6ECDCA, 6α-ethyl-chenodeoxycholic acid; FoxO3a, forkhead Box O3a; FXR, farnesoid X receptor; GSK3β, glycogen synthase kinase beta; HEK, human embryonic kidney; LC3, microtubule associated protein light chain 3; KO, knockout; MnSOD, manganese superoxide dismutase; mTOR, mammalian target of rapamycin; PE, phosphatidylethanolamine; PI3-K, phosphoinositide 3-kinase complex; Shp, small heterodimer partner; Sqstm1, sequestosome-1; WT, wild type
Graphical abstract
Highlights
-
•
FXR knockout mice are more susceptible to acute ethanol-induced steatosis and liver injury due to defective hepatic autophagy.
-
•
FXR knockout mice had decreased FoxO3a activation and reduced expression of autophagy related genes in the liver after acute ethanol treatment.
-
•
FXR knockout mice had increased mitochondrial spheroid formation after acute ethanol treatment.
Introduction
Alcohol is widely consumed in the United States and worldwide, and could be beneficial in moderate amount. Excessive alcohol consumption and abuse may result in alcoholic liver disease (ALD), a major contributor of liver diseases and deaths [1,2]. Pathogenesis of ALD initiates with simple steatosis in a majority of drinkers and progresses to more severe pathologies including fibrosis, alcoholic hepatitis, and cirrhosis in a fraction of patients. In exceptional cases, ALD may progress to hepatocellular carcinoma [2]. Binge drinking is a form of alcohol abuse defined by consuming more than 5 drinks (males) or 4 drinks (females) in 2 h setting [3]. Around one out of three adults display high risk drinking pattern including binge drinking, but less attention has been given to liver injury induced by acute alcohol exposure despite the fact binge drinking is more common than chronic alcohol abuse [1]. At the physiological level, binge drinking induces glycogen depletion, acidosis, and hypoglycemia [4]. At the cellular level, binge drinking results in mitochondria damage, ablated insulin signaling, steatosis, and free radical generation [5–9]. Paradoxically, binge drinking induces autophagy as a cellular protective mechanism to selectively degrade damaged mitochondria (mitophagy) and lipid droplets (lipophagy), whereas suppression of autophagy by pharmacological inhibitors or small interfering RNAs exacerbates alcohol-induced hepatotoxicity and steatosis [10,11].
Autophagy is a cellular lysosomal degradation pathway responsible for degradation of cellular protein and damaged organelles to promote cell survival [12]. The autophagy process is characterized by formation of circular double membrane autophagosomes containing containing cargo. Autophagosomes then fuse with lysosomes to form autolysosomes to complete the degradation of autophagic cargo [12]. More than 30 Atg genes have been identified to participate in autophagy in yeast, and most of them have mammalian homologs [13]. Among them, two ubiquitin conjugation systems include Atg7 (E1-like protein), Atg3 (E2-like) and Atg5-Atg12-Atg16 complex (E3 ligase) play an essential role to mediate the conjugation of phosphatidylethanolamine (PE) to microtubule associated protein light chain 3 (LC3) [14,15]. This conjugated form of LC3 is known as LC3-II, and the unconjugated form of LC3 is referred to as LC3-I [16]. LC3-II is targeted to the autophagosomal membrane. After an autophagosome fuse with a lysosome, the inner membrane LC3-II on an autolysosome is degraded and the outer membrane LC3-II is de-conjugated by Atg4B and recycled [17]. Induction of autophagy or inhibition of autophagy degradation causes LC3-II accumulation; therefore, LC3-II is used as a marker to monitor autophagic flux [18]. Sequestome-1 (Sqstm1)/p62 is another specific autophagy substrate that also can be used to monitor autophagic flux, and is accumulated in autophagy deficient cells or mouse liver [19–21].
FoxO3a is a member of the FoxO transcription factor family, and regulates the transcription of genes involved in apoptosis, oxidative stress, cell-cycle transition and DNA repair [22,23]. FoxO3a also regulates the expression of autophagy genes in skeletal muscles [24,25], cardiomyocytes [26], and liver [27]. Multiple post-translational modifications including phosphorylation, ubiquitination, acetylation and methylation regulate the FoxO3a cellular localization and DNA-binding affinity [22,23]. Akt is the canonical regulator of FoxO3a by phosphorylating FoxO3a on serine 253 and sequestering FoxO3a in the cytosol, which renders FoxO3a unable to bind with DNA and induce gene transcription [22]. We have recently demonstrated that acute ethanol treatment inhibits Akt-mediated phosphorylation of FoxO3 resulting in nuclear FoxO3a retention and increased transcription of autophagy genes in mouse livers and primary mouse and human hepatocytes [27].
Farnesoid X receptor (FXR), the master regulator of bile acid homeostasis, is a member of the nuclear hormone receptor superfamily that is highly expressed in liver and intestines [28–30]. Bile acids are identified as the endogenous ligands for FXR, and bile acid-mediated FXR activation increases the expression of SHP, which serves as a negative regulator of bile acid synthesis. As a result, FXR knockout (KO) mice display elevated hepatic bile acid level due to the lack of SHP-mediated inhibition on bile acid synthesis [29,30]. We recently demonstrated that FXR KO mice display impaired hepatic autophagy due to increased hepatic bile acid level. Furthermore, bile acids suppress autophagosomal–lysosomal fusion [31]. Recent studies showed that activation of FXR protected against ethanol-induced hepatotoxicity and steatosis [32,33]. However, whether FXR would play a role in ethanol-induced autophagy is not known.
In the present study, we found that FXR was critical for protecting against acute ethanol-induced hepatotoxicity and steatosis by promoting ethanol-induced nuclear FoxO3a retention and activation. In response to acute ethanol treatment, there was an increased Akt activation resulting in decreased nuclear FoxO3a retention and FoxO3a-mediated expression of autophagy genes in FXR KO mice. Acute ethanol treatment also promoted mitochondrial spheroid formation in FXR KO mouse livers.
Materials and methods
Reagents
The following antibodies were used: p62 (H00008878-M01) from Abnova (Taipei, Taiwan), Beclin-1 (sc11427), FXR (sc13063), and HA (sc805) from Santa Cruz Biotechnology (Santa Cruz, CA), and Flag (F3165) from Sigma (St. Louis, MO), serine 473 phosphorylated Akt (4058), Akt (9272), serine 9 phosphorylated GSK3β (5558S), GSK3β (5676S), serine 253 phosphorylated FoxO3a, FoxO3a (2497), GAPDH (2118), and Lamin A/C (2032) from Cell Signaling Technology (Beverly, MA), and CYP2E1 (ab28146) from Abcam (Cambridge, MA). The rabbit polyclonal anti-LC3B antibody was described previously [34]. The secondary antibodies used for immunoblotting analysis were HRP-conjugated goat anti-mouse (115-035-062), rabbit (111-035-045), and rat (111-035-143) and a Dylight 549 conjugated goat anti-rabbit (111-505-144) antibody from Jackson ImmunoResearch (West Grove, PA) or an HRP-conjugated goat anti-rabbit antibody (31460) from Thermo Fisher Scientific (Waltham, MA). Ethanol was from Pharmaco (Brookfield, CT). All other chemicals were from Sigma-Aldrich, (St. Louis, MO), Thermo, Fisher Scientific (Waltham, MA), Invitrogen (Carlsbad, CA), or EMD Millipore (Billerica, MA).
Animal experiments
Wild type C57BL and FXR KO C57BL were purchased from Jackson Laboratory (Bar Harbor, ME). 2–4 months age matched male wild type and FXR KO mice were used in this study. All mice were provided with humane care according to the NIH guidelines, and the Institutional Animal Care and Use Committee at the University of Kansas Medical Center approved all procedures.
Mouse ethanol binge treatment
The ethanol binge model was modified from the model Carson and Pruett established, which had been shown to closely mimic human binge ethanol consumption including blood alcohol levels and behavioral effects [35]. The mice were fasted for 6 h, and then administered with 33% (v/v) ethanol at a cumulative dose of 4.5 g/kg body weight by four equally divided gavages in 15 min intervals. Control mice received same volume of double-distilled water. Mice were sacrificed 16 h later after the treatment, and serum and liver samples were harvested. Serum alanine aminotransferase (ALT) levels and H&E staining were used to assess ethanol-induced liver injury. Total liver lysates were obtained by using radio-immunoprecipitation assay buffer (1% NP40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl (lauryl) sulfate).
Cell culture and transfection
Human embryonic kidney 293A cells were cultured in DMEM medium with FBS and l-glutamate and transiently transfected with HA tagged FoxO3, Flag tagged FXR, HA or Flag plasmid constructs using TurboFect Transfection reagent (Thermo, Fisher Scientific) for 24 h. Total cell lysates then were extracted using HA cell lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 2 mM EDTA pH 7.5, 1 mM EGTA pH 7.5, 1% Triton X-100, protease inhibitors).
Hepatic triglyceride analysis
Frozen liver tissues (50–100 mg) were grounded to fine powder using mortar and pestle. The powdered tissue was incubated in 1 ml of chloroform:methanol (2:1) mix with vigorous shaking for 1 h at room temperature. 200 µl of double-distilled water was added, and the mixture was centrifuged for 5 min at 3000g at 4 °C. The aqueous upper phase layer then was collected and air-dried at room temperature. The dried pellet was dissolved in tert-butanol and Triton X-114:methanol (2:1) solution. Hepatic triglyceride analysis was performed with a colormetric assay kit according to manufacturer’s instruction (Pointe Scientific, Ann Arbor, MI).
Immunoblot assay
Equal amount of nuclear fraction (15 µg), cytosol fraction (30 µg), or total liver lysates (50 µg) were separated by SDS-PAGE gel and transferred to PVDF membranes. The membranes were immunoblotted with primary antibodies followed by HRP-conjugated secondary antibodies. The membranes then were developed with either Pierce Supersignal West Pico chemiluminescent substrate (Thermo, Fisher Scientific, Rockford, IL) or Millipore Immobilon Western chemiluminescent HRP substrate (Billerica, MA). Densitometry was performed using ImageJ software and further normalized using beta-actin or GAPDH, and expressed as means±SEM.
qPCR
RNA was isolated from liver tissues using TRIzol (Life Technologies, Waltham, MA) and reversed transcribed into cDNA by RevertAid reverse transcriptase (Thermo, Fisher Scientific, Waltham, MA). Real-time PCR was performed on a Bio-Rad CFX384™ real-time PCR detection system using iTaq™ Universal SYBR® Green Supermix (Bio-Rad). The following genes were probed with quantitative PCR using β-actin gene as loading control: Atg5, Becn-1, Map1lc3b, MnSOD, p21, FoxO3a, Sqstm-1, and Shp. Primer sequences were as follows: β-actin, 5′-TGTTACCAACTGGGACGACA-3′ and 5′-GGGGTGTTGAAGGTCTCAAA-3′; Atg5, 5′-GACCACAAGCAGCTCTGGAT-3′ and 5′-GGTTTCCAGCATTGGCTCTA-3′; Becn-1 (Atg6), 5′-TGATCCAGGAGCTGGAAGAT-3′ and 5′-CAAGCGACCCAGTCTGAAAT-3′; FoxO3a, 5′-AGCCGTGTACTGTGGAGCTT-3′ and 5′-TCTTGGCGGTATATGGGAAG-3′; Map1lc3, 5′-CCGAGAAGACCTTCAAGCAG-3′ and 5′-ACACTTCGGAGATGGGAGTG-3′; MnSod, 5′-GGCCAAGGGAGATGTTACAA-3′ and 5′-AGACACGGCTGTCAGCTTCT-3′; p21, 5′-CGGTGGAACTTTGACTTCGT-3′ and 5′-CAGGGCAGAGGAAGTACTGG-3′; Sqstm1/p62, 5′-AGAATGTGGGGGAGAGTGTG-3′ and 5′-TCGTCTCCTCCTGAGCAGTT-3′; and Shp, 5′-CTGCAGGTCGTCCGACTATT-3′ and 5′-ACCTCGAAGGTCACAGCATC-3′.
Immunoprecipitation
Total cell lysates (200 µg) or total liver lysates (500 µg) were incubated with a FoxO3a antibody (sc11351, Santa Cruz) overnight at 4 °C rotating and pulled down with protein A/G agarose beads (sc2003, Santa Cruz) or EZview™ Red ANTI-FLAG® M2 Affinity Gel (F2425, Sigma). Immunoblot analysis was performed with immunoprecipitated and input proteins.
Chromatin immunoprecipitation
Fresh liver sections were minced or frozen liver sections were homogenized and cross-linked with 1% formaldehyde in PBS. Following incubation, the livers were quenched with 250 mM glycine. Crude nuclear extracts were obtained using hypotonic buffer (10 mM Tris–HCl pH 8.0, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40) and nuclear lysis buffer (1% SDS, 5 mM EDTA, 50 mM Tris–HCl pH 8.0). The chromatin extracts then were sheared using Active Motif Q800R sonicator to 200–500 bp fragments. Chromatin proteins (600 µg) were immunoprecipitated with a FoxO3a antibody (sc11351X) from Santa Cruz Biotechnology (Santa Cruz, CA) binded with Dynabead Protein A (10001D) from Invitrogen/Dynal (Oslo, Norway). DNA then was extracted from immunoprecipiated and input chromatin using GeneJet PCR Purificiation Kit (K0701) from Thermo Scientific (Waltham, MA). qPCR was performed and three FoxO3a binding sites in the Map1LC3b promoter site were probed. Primer sequences were as follows: 1379–1608 bp upstream, 5′-CATGCCTTGGGACACCAGAT-3′ and 5′-ACCTTCTTCAAGTGCTGTTTGT-3′; 3397–3595 bp upstream, 5′-TTTGACCAAACAGGGTTTCC-3′ and 5′-CCCTCCAGGTGTTTGTGATAA-3′; and 4673–4801 bp upstream, 5′-CCTCAGCTGGCTAAGAGCAT-3′ and 5′-CCC AAG GAT CTC AAC CAA AC-3′. The primer sequences were obtained from a previous report [24].
Fluorescence and electron microscopy
Liver sections were fixed with 4% paraformaldehyde then incubated in 20% sucrose in PBS and embedded in optimal cutting temperature (OCT) solution at −20 °C. Liver cyrosections were immunostained with FoxO3a antibody followed by Dylight 549 conjugated secondary antibody and Hoechst 33342 staining. The images of sections were acquired under a Nikon Eclipse 200 fluorescence microscope with MetaMorph software. For electron microscopy (EM), liver sections were fixed with 2.5% glutaraldehyde in 0.1 mol/l sodium cacodylate buffer (pH 7.4), followed by 1% OsO4. After dehydration, thin sections were cut and stained with uranyl acetate and lead citrate. Digital images were obtained using a JEM 1016CX electron microscope.
Nuclear fractionation
Mouse liver cytosol and nuclear proteins were extracted using NE-PER nuclear and cytoplasmic extraction reagents (Thermo, Fisher Scientific) according to manufacturer's instructions. In brief, 50–100 mg of liver was homogenized in cytoplasmic extraction reagent I followed by addition of cytoplasmic extraction reagent II and centrifuged. The supernatant was collected as cytoplasmic fraction, and the insoluble pellet was suspended in nuclear extraction reagent and centrifuged. The resulting supernatant contains nuclear fraction.
Statistic analysis
All experimental data were expressed as means±SE and subjected to Student t-test or one-way analysis of variance with Holm–Sidak post hoc test or one-way analysis of variance on ranks with Dunn post hoc test where appropriate. *p<0.05 was considered significant.
Results
Acute ethanol treatment exacerbated liver injury and steatosis in FXR KO mice compared to WT mice
Acute ethanol treatment increased serum alanine aminotransferase (ALT) levels and hepatic triglyceride levels in wild type (WT) mice, which is consistent with our previous report [10]. Compared to WT mice, acute ethanol-treated FXR KO mice had higher serum ALT and hepatic triglyceride levels, suggesting ethanol treatment exacerbated liver injury and steatosis in FXR KO mice (Fig. 1A and B). Notably, control FXR KO mice already had a mild increase of serum ALT level, which is consistent with previous reports suggesting the lack of FXR causes mild liver injury [36] (Fig. 1A). However, there was no difference in hepatic triglycerides level between age matched WT and FXR KO control mice (Fig. 1B). Hematoxylin and eosin staining revealed that ethanol induced mild hepatic steatosis in WT mice, which was further exacerbated in FXR KO mice (Fig. 1C). In line with these findings, oil red o staining for lipids showed that ethanol-treatment increased oil red o staining in WT mouse livers compared to control mice. However, oil red o staining was much more severe in ethanol-treated FXR KO mouse livers compared to ethanol-treated WT mouse livers, suggesting that lack of FXR exacerbates ethanol-induced hepatic steatosis (Fig. 1D). Furthermore, EM analysis also revealed that ethanol treatment increased the number of lipid droplets in WT mouse livers. The number of lipid droplets was further increased in ethanol-treated FXR KO mouse livers compared to WT mouse livers (Fig. 1E and F). Taken together these data indicate that FXR deficiency exacerbates acute ethanol-induced liver injury and steatosis.
Fig. 1.
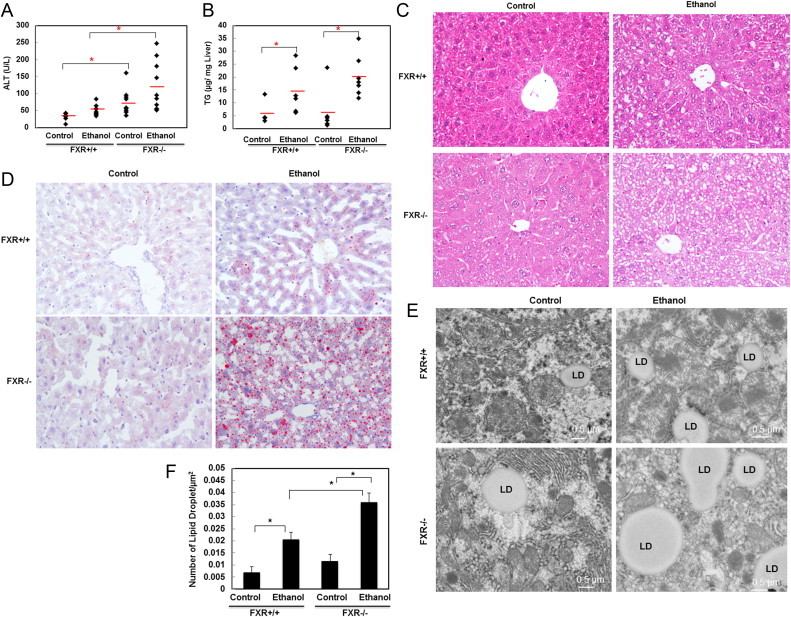
FXR KO mice exhibited increased ethanol-induced liver injury and steatosis. Age matched WT and FXR KO mice were treated with 4.5 g/kg ethanol or water by gavage for 16 h. Serum alanine aminotransferase (ALT) (A) and hepatic triglycerides (TG) (B) were measured (n=4–9). Representative H&E stain images are shown in (C) and representative oil red o images are shown in (D). Representative EM images are shown in (E) and lipid droplets (LD) are quantified from at least 24 different liver sections. * p<0.05. One-way ANOVA analysis. FXR KO control vs WT control, FXR KO ethanol vs WT ethanol (A). WT ethanol vs control, and FXT KO ethanol vs control (B).
Impaired autophagy in acute ethanol-treated FXR KO mouse livers
We previously demonstrated that FXR KO mice have impaired hepatic autophagy [31]. In accordance with previous findings, we showed that control FXR KO mice had higher basal LC3-II and p62 protein levels compared to WT mice. Ethanol treatment increased LC3-II protein levels in WT but not in FXR KO mouse livers (Fig. 2A). However, ethanol treatment increased p62 protein levels in WT mouse livers, which was further increased in FXR KO mouse livers (Fig. 2A). Ethanol treatment also increased CYP2E1 protein levels in both WT and FXR KO mouse livers (Fig. 2B), consistent with previous reports [37]. Ethanol treatment increased the mRNA levels of p62 in WT mouse livers but not in FXR KO mouse livers (Fig. 2C), suggesting that ethanol treatment-increased p62 protein levels could be mediated by the increased transcription for p62 in WT mice whereas increased p62 levels in FXR KO mice were mainly regulated at the protein level. EM analysis revealed that ethanol treatment increased the number of autolysosomes in WT mouse livers although it did not reach the statistical significance. Consistent with our previous report [31], control FXR KO mice already had increased number of autophagosomes and autolysosomes likely due to impaired fusion of autophagosomes with lysosomes. Treatment with acute ethanol further increased the number of autophagosomes and autolysosomes (Fig. 2D and E). These results indicate that autophagy was impaired in FXR KO mouse livers, which might contribute to exacerbated liver injury and steatosis following ethanol treatment.
Fig. 2.
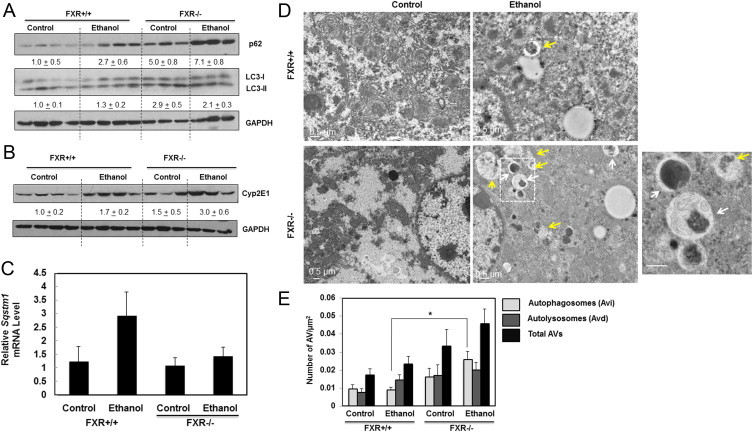
Ethanol induced p62 and LC3-II protein expression was enhanced in FXR KO mice. Age matched WT and FXR KO mice were treated with 4.5 g/kg ethanol or water by gavage for 16 h. Total liver lysates were subjected to immunoblot analysis for p62 and LC3 (A) and CYP2E1 (B). Densitometry analysis data are presented as a ratio of control (n=3–4). Hepatic mRNA was isolated and qRT-PCR was performed for p62 (C). The gene expression levels were normalized to β-actin and shown as fold increase over wild type mice (n=4–7). Representative EM images are shown in (D). Right image is the enlarged image from the box area. White arrows: early autophagosomes (Avi) and yellow arrows: autolysosomes (Avd) (D). Autophagosomes (Avi) and autolysosomes (Avd) were quantified (E) (<24 different liver sections). *p<0.05. One-way ANOVA analysis. FXR ethanol vs WT ethanol (E).
Lack of FXR impaired acute ethanol-induced FoxO3a activation.
We previously reported that acute ethanol treatment induces FoxO3a-mediated hepatic expression of autophagy related genes [27]. Consistent with our previous report, ethanol treatment increased the mRNA levels of Atg5, Becn-1 and Microtubule-associated protein 1 light chain 3 beta (Map1lc3b) in WT mouse livers (Fig. 3A). In contrast, ethanol treatment did not cause such an increase, and actually showed a slight decrease of hepatic expression of these autophagy related genes in FXR KO mice. In addition to autophagy related genes, ethanol treatment also increased expression of MnSod, p21 and FoxO3a, which are known FoxO3a target genes, in WT but not FXR KO mouse livers (Fig. 3B). Small heterodimer partner (Shp) is a well-known target gene of FXR, and ethanol treatment had no effect on the expression of hepatic Shp mRNA level in WT mice, suggesting that acute ethanol treatment may not activate FXR. In contrast, FXR KO mice had dramatically decreased expression of hepatic Shp, which was further decreased with ethanol treatment (Fig. 3C). These results suggest that lack of FXR impairs ethanol-induced FoxO3a activation in mouse livers.
Fig. 3.
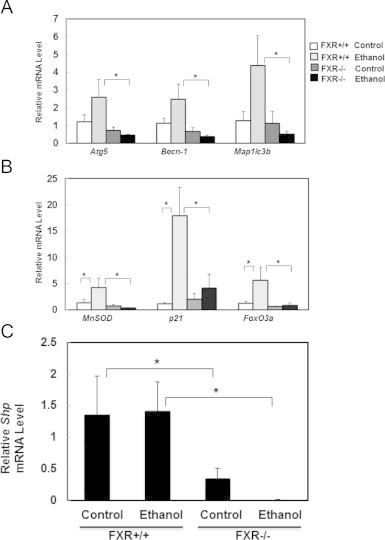
Ethanol-induced FoxO3a-mediated transcription of autophagy related and FoxO3a target genes were inhibited in FXR KO mouse livers. Age matched WT and FXR KO mice were treated with 4.5 g/kg ethanol or water by gavage for 16 h. Hepatic mRNA was isolated and qRT-PCR was performed for autophagy related genes, Atg5, Becn-1, and MAP1LC3B (A) and FoxO3a target genes, MnSOD, p21, and FoxO3a (B). qRT-PCR was also performed for FXR target gene, Shp (C). The gene expression levels were normalized to β-actin and shown as fold increase over wild type mice (n=4–7). *p<0.05. One-way ANOVA analysis. WT control vs FXR KO control, WT ethanol vs FXO KO ethanol.
No direct interaction between FoxO3a and FXR both in vitro and in vivo
To determine whether FXR would directly interact with FoxO3a, we transiently transfected HA-tagged FoxO3a, Flag-tagged FXR, or both in human embryonic kidney (HEK) 293A cells followed by co-immunoprecipitation assay. We found that there was a weak interaction between Flag-FoxO3a and HA-FXR. However, Flag tag only also showed similar weak interaction with HA-FXR, suggesting that the interaction between Flag-FoxO3a and HA-FXR is none-specific. Immunoblotting analysis of the total lysate revealed that the transfection was efficient in HEK293 cells (Fig. 4A). We further found that FoxO3a was present in both nuclear and cytosolic fractions, whereas FXR was dominantly expressed in nuclear fraction, suggesting that the lack of interaction between FXR and FoxO3a was not due to the alterations of cellular localization as a result of the overexpression of these two proteins (Fig. 4A). To determine whether endogenous FXR in mouse livers would interact with endogenous FoxO3a, and whether this interaction would be altered by ethanol treatment, immunoprecipitation was performed using endogenous liver proteins from mouse livers with or without ethanol treatment. Similar to the results from the overexpression of FoxO3a and FXR in HEK293 experiment, no interaction between FoxO3a and FXR was found in both control and ethanol-treated mouse livers (Fig. 4B). These results suggest that FXR may not directly interact with FoxO3a in ethanol-treated mouse livers.
Fig. 4.
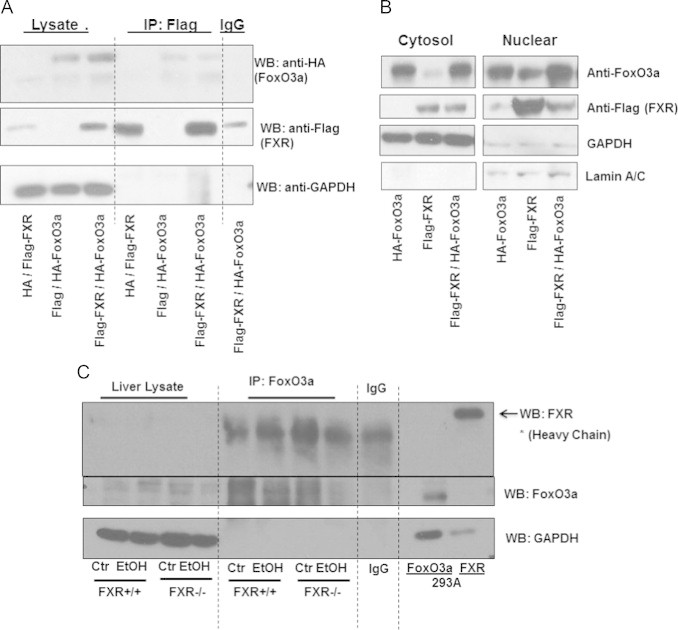
FXR and FoxO3a did not interact in vitro or in vivo. HEK 293A cells were transfected with plasmids containing flag-FXR, HA-FoxO3a, HA or flag. Total cell lysates were isolated, and flag was pulled down by immunoprecipitation and subjected to immunoblot analysis for flag and HA. Input total lysates from transfected HEK 293A were used as positive controls for FoxO3a and FXR (A). Cytosol and nuclear fractions were obtained from transfected HEK 293A cells, and immunoblot analysis was performed for flag and FoxO3 (B). Age matched WT and FXR KO mice were treated with 4.5 g/kg ethanol or water by gavage for 16 h. FoxO3a in total liver lysates was pulled down by immunopreciptation and subjected to immunoblot analysis for FXR and FoxO3a (C).
Ethanol-induced FoxO3a nuclear translocation was inhibited in FXR KO mouse livers
We next determined whether lack of FXR would affect the cellular localizations of FoxO3a in ethanol treated mouse livers. Immunostaining analysis for endogenous FoxO3a revealed that acute ethanol treatment increased the number of cells with nuclear FoxO3a signals in WT but not in FXR KO mouse livers (Fig. 5A and B). To confirm the findings from the immunostaining analysis, cytosolic and nuclear fractions were prepared from WT and FXR KO mouse livers followed by immunoblotting analysis. We found that there was a decrease of cytosolic but increase of nuclear FoxO3a in WT mouse livers after ethanol treatment. However, acute ethanol treatment failed to increase nuclear FoxO3a levels in FXR KO mice (Fig. 5C). These data suggest that acute ethanol-induced nuclear retention of FoxO3a is inhibited in FXR KO mouse livers.
Fig. 5.
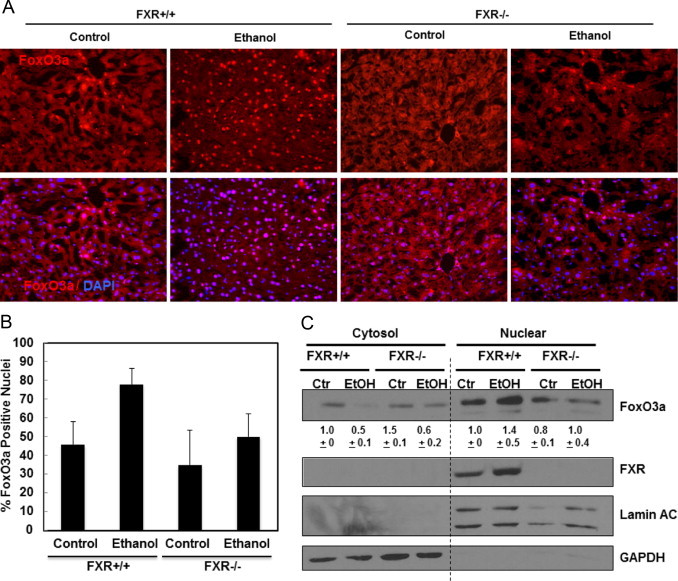
Ethanol-induced FoxO3a nuclear translocation was diminished in FXR KO mouse livers. Age matched WT and FXR KO mice were treated with 4.5 g/kg ethanol or water by gavage for 16 h. Liver cyrosections were immunostained for FoxO3a and Hoechst 33342 for nuclei, and representative images are shown in (A). Nuclei positive for FoxO3a were quantified from at least 3 images (B). Cytosolic and nuclear fractions were isolated from liver and subjected to immunoblot analysis for FoxO3a and FXR (C) densitometry analysis data are presented as a ratio of control (n=3).
Acute ethanol treatment increased Akt-mediated FoxO3a phosphorylation and inhibited ethanol-induced FoxO3 binding in Map1lc3b promoter sites in FXR KO mouse livers
We found that acute ethanol treatment slightly decreased the phosphorylation of Akt at serine 473 in WT mouse livers, which is consistent with our previous report [27]. Conversely, we found that acute ethanol treatment dramatically increased the phosphorylation of Akt in FXR KO mouse livers by five-fold (Fig. 6A). Glycogen synthase kinase beta (GSK3β) can be phosphorylated by Akt and thus serves as an indirect marker for Akt kinase activity. We found that the phosphorylation of GSK3β at serine 9 was reduced in ethanol treated WT mouse livers, but it was enhanced by ethanol treatment in FXR KO mouse livers by approximately 2.5-fold (Fig. 6A). These data suggest that acute ethanol treatment inhibits Akt activity in WT mouse livers but increases Akt activity in FXR KO mouse livers. As a result, we found that ethanol treatment attenuated the phosphorylation of FoxO3a at serine 253 in WT mouse livers but increased the phosphorylation of FoxO3a in FXR KO mouse livers (Fig. 6B). Moreover, ethanol increased total hepatic FoxO3a protein expression in WT mouse livers by two-fold but decreased the hepatic FoxO3a protein level in FXR KO mice. Since FoxO3a itself is a target gene of FoxO3a, these data thus may also imply that there is a decrease of FoxO3a transcriptional activity in FXR KO mouse livers after ethanol treatment compared to WT mice. We next performed ChIP analysis to determine whether there would be any changes for FoxO3a binding on its target genes between WT and FXR KO mouse livers after ethanol treatment. It has previously been reported that FoxO3a binds to mouse Map1lc3b transcription promoter region in three different sites at approximately 1.5, 3.5 and 4.5 kilo-base pairs upstream of the Map1lc3b transcription start site [24]. Indeed, ethanol increased FoxO3a binding to all three binding sites in WT mouse livers, but these bindings were significantly diminished in FXR KO mouse livers (Fig. 6C). Collectively, these data suggest that ethanol treatment activates Akt and increased phosphorylation of FoxO3a resulting in decreased FoxO3a transcriptional activity in FXR KO mouse livers.
Fig. 6.
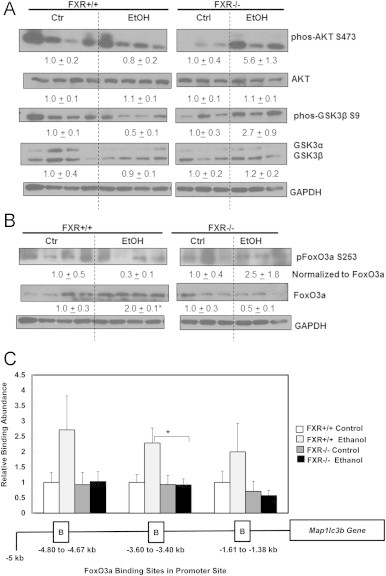
Increased Akt activity and reduced FoxO3a binding in Map1lc3b promoter sites in acute ethanol-treated FXR KO mouse livers. Age matched WT and FXR KO mice were treated with 4.5 g/kg ethanol or water by gavage for 16 h. Total liver lysates were subjected to immunoblot analysis for serine 473 phosphorylated and total AKT, serine 9 phosphorylated and total GSK3β (A), and serine 253 phosphorylated and total FoxO3a (B). Densitometry analysis data are presented as a ratio of control (n=3–4). Chromatin immunoprecipitation using FoxO3a antibody was performed to probe the three putative FoxO3a binding sites in Map1lc3b promoter site. DNA was isolated from chromatin pull-down and qPCR analysis was performed, and data was normalized to input chromatin. Relative binding abundance in comparison to untreated WT control was presented in (C). *p<0.05. One-way ANOVA analysis. WT ethanol vs FXR KO ethanol.
Acute ethanol treatment increased the number of mitochondrial spheroid in ethanol-treated FXR KO mouse livers
We previously reported that mitochondrial spheroids are induced to serve as an alternative mitochondrial quality control pathway in response to stresses that induce mitochondrial damage as a result of impaired conventional mitophagy [38]. In ethanol treated FXR KO mouse livers, we found that some mitochondria underwent a dramatic structural remodeling that form a vesicular-like structure with enveloped contents. Some part of the mitochondria formed a four-membrane structure with squeezed matrix (Fig. 7A, arrows), which is consistent with the formation of mitochondrial spheroids that we previously reported [38]. We did not detect any mitochondrial spheroids in either control or ethanol treated WT mouse livers but quantitative EM analysis showed increased number of mitochondrial spheroid in control FXR KO mouse livers, which was further increased after ethanol treatment (Fig. 7B). These results suggest that acute ethanol treatment may induce the formation of mitochondrial spheroid as an alternatively mitochondrial quality control to compensate for the impaired autophagy in FXR KO mouse livers. We previously demonstrated that Parkin negatively regulates the formation of mitochondrial spheroids by promoting the degradation of mitofusin 1 (Mfn1) and Mfn2, two important proteins that regulate mitochondrial fusion [38,39]. Interestingly, we found that acute ethanol treatment caused almost 50% loss of hepatic Parkin. While the mechanisms of how ethanol decreased Parkin protein levels in the mouse livers were not clear, we have found that chronic plus binge alcohol treatment (Gao-binge) led to the Parkin mitochondrial translocation (Williams J. et al., unpublished observations). Thus it is possible that the decreased Parkin in acute ethanol-treated mouse livers was due to the increased autophagic degradation of Parkin positive mitochondria (mitophagy). Interestingly, we also found that increased degradation of Mfn1 and Mfn2 in ethanol-treated wild type mouse livers but the levels of Mfn1 and Mfn2 were less affected in FXR KO mouse livers (Fig. 7C). The relatively higher levels of Mfn1 and Mfn2 in ethanol-treated FXO KO mouse livers than that of wild type mice may account for the increased mitochondrial spheroids formation in FXO KO mouse livers. We also found that acute ethanol treatment led to approximately 30% decrease on hepatic Lamp1 and Lamp2 proteins in both wild type and FXR KO mice (Fig. 7C). Alcohol consumption has been implicated in impaired lysosomal functions but whether the decreased Lamp1 and Lamp2 would contribute to the impaired lysosomal functions need to be further studied. The proposed cellular mechanisms of how FXR regulates FoxO3 and mitochondria homeostasis are shown in Fig. 8.
Fig. 7.
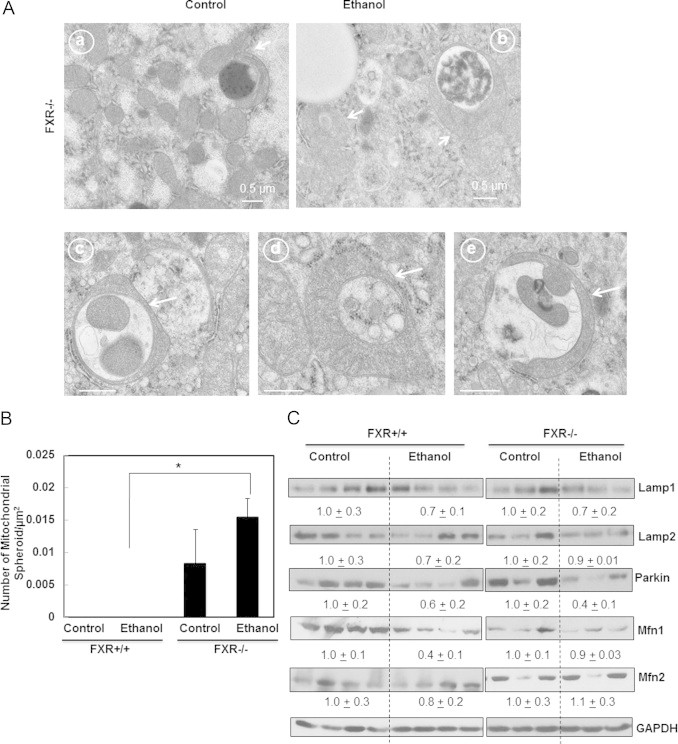
Ethanol induced mitochondrial spheroid formation in FXR KO mouse livers. Age matched WT and FXR KO mice were treated with 4.5 g/kg ethanol or water by gavage for 16 h. Representative EM images of mitochondrial spheroids from FXR KO control (a) and ethanol-treated (b–e) liver sections were shown (A). Arrows denote mitochondrial spheroids. Mitochondrial spheroid formation was quantified (>24 liver sections) (B). *p<0.05. One-way ANOVA analysis. WT ethanol vs FXR KO ethanol. (C) Mice were treated as in (A), total liver lysates were subjected to western blot analysis followed by densitometry analysis. Densitometry analysis data are presented as a ratio of control (n=3–4).
Fig. 8.
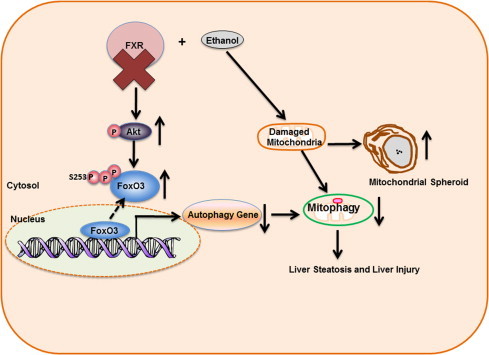
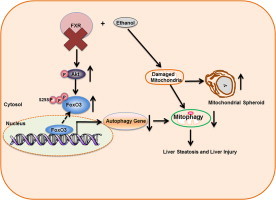
Possible molecular and cellular events following acute ethanol treatment in FXR KO mouse livers. Acute ethanol exposure induces FoxO3a activation and mitophagy as a protective mechanism. FXR deficiency promotes Akt activation and subsequent serine 253 phosphorylation of FoxO3a, resulting in decreased nuclear FoxO3a retention and abolished transcription of autophagy related genes in response to ethanol. The defective hepatic autophagy in FXR KO mouse livers results in impaired mitophagy and compensatory induction of mitochondrial spheroid formation. Altogether, impaired FoxO3a-mediated autophagy and mitophagy in ethanol-treated FXR KO mouse livers lead to increased liver injury and steatosis.
Discussion
In the present study, using an acute ethanol binge model, we showed that FXR KO mice had exacerbated steatosis and liver injury in comparison with WT mice. Mechanistically, following acute ethanol treatment, FXR KO mice had impaired hepatic autophagy due to the failure of activating FoxO3a-mediated up-regulation of autophagy related genes and likely reduced fusion of autophagosomes with lysosomes.
It is well known that the nuclear receptor FXR is the master regulator of bile acid homeostasis. FXR deficiency caused an increased concentration of bile acid in liver and serum due to activation of Cyp7A1 [29,30]. We previously reported that FXR KO mice had reduced hepatic autophagy likely due to impaired fusion of autophagosomes with lysosomes as a result of increased hepatic bile acid levels [31]. In addition to removing cytosolic proteins and damaged/excess organelles, autophagy has been known to remove excess lipid droplets, a process named lipophagy [40]. Impaired autophagy has been reported in many experimental NAFLD and NASH models, which was associated with endoplasmic reticulum (ER) stress and post-translational modifications of several autophagy proteins [1,41]. We also showed that fatty acids differentially regulated autophagy in which saturated fatty acids suppressed whereas unsaturated fatty acids induced autophagy in cultured hepatoma cells [42]. In the context of alcoholic liver diseases, we also showed that pharmacological inhibition of autophagy exacerbated acute ethanol-induced steatosis [10]. Therefore, the impaired autophagy in FXR KO mice is likely one of the major contributors for the exacerbated hepatic steatosis in acute ethanol-treated FXR KO mice.
Interestingly, it has been reported that chronic ethanol exposure inhibited FXR activation, resulting in up-regulation of bile acid synthesis enzymes and down-regulation of bile acid transporter and in turn elevated hepatic bile acid pool [33,43]. Moreover, pharmacological activation of FXR using WAY-362450 and 6α-ethyl-chenodeoxycholic acid (6ECDCA) attenuated chronic ethanol exposure-induced liver injury and steatosis [32,43]. While it is still controversial regarding the autophagy status in the mouse livers after chronic ethanol exposure, it has been suggested that chronic ethanol exposure may lead to impaired hepatic autophagy [44,45]. This notion is supported by the observations that chronic ethanol exposure leads to hepatomegaly [46], accumulated ubiquitin positive protein aggregates [47,48] and decreased protein degradation [49]. In the future, it will be interesting to determine whether increased bile acid after chronic ethanol exposure would contribute to impaired hepatic autophagy and alcohol-induced liver injury.
Emerging evidence supports that transcriptional regulation of the expression of autophagy related genes is critical in induction of autophagy in yeast [50,51], Caenorhabditis elegans [52] and mammals [53,54]. Among the several known transcriptional factors that regulate autophagy, the FoxO family members play critical roles in regulating expression of autophagy related genes in skeletal muscle [24,25], cardiomyocyte [26] and liver [55]. In addition, cytosolic FoxO proteins were also showed to regulate autophagy independent of their transcriptional activities [56,57]. We recently demonstrated that FoxO3a is critical for acute ethanol-induced hepatic autophagy, and plays a protective role against ethanol-induced liver injury [27]. In agreement with our previous findings, we found that ethanol induced FoxO3a-mediated expression of autophagy related genes in WT mouse livers. The activation of FoxO3a is mainly regulated by its post-translational modifications including phosphorylation, acetylation, ubiquitination and methylation. It has been well-documented that FoxOs are phosphorylated by the serine/threonine protein kinase AKT and become sequestered in the cytoplasm, where they are unable to regulate gene expression [22]. In WT mice, we found that acute ethanol treatment decreased the level of phosphorylated AKT and FoxO3a and in turn increased nuclear retention of FoxO3a. In contrast, acute ethanol treatment increased AKT activity resulting in the increased phosphorylation of FoxO3a and decreased nuclear retention FoxO3a in FXR KO mice. Currently it is not clear how AKT was activated in FXR KO mouse livers. However, FXR KO mice develop spontaneous hepatocellular carcinoma [36], and increased AKT activity can promote tumorigenesis [58]. In addition to post-translational modifications, FoxO family members bind to a variety of nuclear hormone receptors to regulate either their own or nuclear hormone receptors' DNA binding affinity and transcriptional activities [59]. FoxO3a contains LxxLL motif in the C-terminal of forkhead DNA binding region, which may enable FoxO3a to interact with nuclear hormone receptors [59,60]. Thus it is possible that the decreased FoxO3a-mediated expression of autophagy related genes is due to the lack of a direct interaction between FoxO3a and FXR. However, the failure to detect the direct interaction between FXR and FoxO3a both in vitro and in vivo excludes this possibility. In addition, we also found that acute ethanol treatment increased the binding of FoxO3a to the promoter regions of Map1lc3b in WT but not in FXR KO mouse livers by the ChIP assay. While the increased nuclear retention of FoxO3a in ethanol-treated WT mouse livers could contribute to these observations, it is also likely that other FoxO3a co-factors/suppressors may influence the binding of FoxO3a to the promoter regions of Maplc3b. It is known that FoxO3a also interacts with other co-factors such as PGC1-α [61–63] and p300/CBP [64]. Future studies will be needed to determine the changes of the interactions of FoxO3a with PGC1-α or p300/CBP after ethanol treatment in WT and FXR KO mice.
In addition to the defect of FoxO3a-mediated transcriptional regulation of autophagy in FXR KO mouse livers, increased bile acid levels may also contribute to the impaired autophagy in FXR KO mouse livers. As we previously demonstrated that bile acids themselves did not have any effects on the expression of autophagy related genes regardless of the conjugated or unconjugated bile acids [31]. However, bile acids impaired the fusion of autophagosomes with lysosomes through the alteration of intracellular Rab7, a key protein that regulates the fusion of autophagosomes with lysosomes [31]. The increased number of autophagosomes in ethanol-treated FXR KO mouse livers may reflect the impaired fusion of autophagosomes with lysosomes. Thus it is likely that the combination of impaired FoxO3a-mediated expression of autophagy related genes and bile acid-mediated defect on the fusion of autophagosomes with lysosomes contribute to the exacerbated steatosis and liver injury in ethanol-treated FXR KO mice.
It is known that alcohol consumption can lead to increased oxidative stress and mitochondrial damage [1,65]. Metabolism of alcohol may play a critical role in alcohol-induced oxidative stress and mitochondrial damage. Alcohol is mainly metabolized in the liver through cytosolic alcohol dehydrogenase (ADH) to acetaldehyde, a highly reactive molecule that is further metabolized through mitochondrial acetaldehyde dehydrogenase (ALDH) to acetate. Consequently, alcohol metabolism increases hepatic NADH/NAD+ ratio and oxidative stress. Moreover, alcohol can also induce hepatic Cyp2E1 expression to oxidize alcohol, which also leads to increased oxidative stress and mitochondrial damage [66]. Previous works including ours have shown that the metabolism of alcohol either via AHD or Cyp2E1 is important for alcohol-induced changes on hepatic autophagy [10,67,68]. Interestingly, we found that the levels of hepatic Cyp2E1 were much higher induced by acute ethanol treatment in FXR KO mice than wild type mice. It is likely that the higher levels of Cyp2E1 in FXR KO mouse livers may also contribute to the increased liver injury by acute ethanol treatment. Future studies will be needed to further determine the levels of hepatic oxidative stress after ethanol treatment in FXR KO mice.
We previously demonstrated that at least one purpose of acute ethanol-induced autophagy is to remove damaged mitochondrial through mitophagy [10,11]. However, increased evidence suggests that cells may use alternative pathways or activate other forms of autophagy (i.e. microautophagy or chaperone-mediated autophagy) to regulate cellular organelle homeostasis [69]. We recently reported that several mitochondrial damage stressors induce the formation of mitochondrial spheroid likely as an alternative pathway to regulate mitochondrial homeostasis in cultured cells or mouse livers in both WT and autophagy-deficient cells [38,70]. Interestingly, we also found that acute ethanol treatment increased the number of mitochondrial spheroid in FXR KO but not WT mouse livers. Moreover, mitochondrial spheroid structures were also found in the control FXR KO mouse livers. It is likely that increased bile acid levels might induce mitochondrial damage in control FXR KO mouse livers, which was further exacerbated after ethanol treatment. Damaged mitochondria can normally be removed via mitophagy but this process was impaired in FXR KO mouse livers. The increased mitochondrial spheroid in ethanol-treated FXR KO mouse livers thus may also serve as an alternative pathway to regulate mitochondrial homeostasis in FXR KO mouse livers.
In conclusion, we demonstrated that FXR is a protective factor against ethanol-induced hepatotoxicity and steatosis. Furthermore, we also showed that FXR is associated with ethanol-induced FoxO3a activation and FoxO3a-mediated autophagy. Modulating FXR activity may be a promising novel therapeutic target for ALD.
Acknowledgements
We want to thank Barbara Fegley from the electron microscopy core facility at the University of Kansas Medical Center for her assistance with processing the specimens for EM. Finally, we want to thank Hua Yang, Yuan Li, and Shaogui Wang for their assistance with the mouse experiments. The research work in Wen-Xing Ding's lab was supported in part by the NIAAA funds (R01 AA020518), National Center for Research Resources (5P20RR021940), the National Institute of General Medical Sciences (8P20 GM103549 and T32 ES007079) and an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (P20 GM103418).
References
- 1.Ding W.X., Manley S., Ni H.M. The emerging role of autophagy in alcoholic liver disease. Experimental Biology and Medicine. 2011;236(5):546–556. doi: 10.1258/ebm.2011.010360. 21478210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao B., Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141(5):1572–1585. doi: 10.1053/j.gastro.2011.09.002. 21920463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wechsler H., Austin S.B. Binge drinking: the five/four measure. Journal of Studies on Alcohol. 1998;59(1):122–124. doi: 10.15288/jsa.1998.59.122. 9498324 [DOI] [PubMed] [Google Scholar]
- 4.Zakhari S., Li T.K. Determinants of alcohol use and abuse: impact of quantity and frequency patterns on liver disease. Hepatology. 2007;46(6):2032–2039. doi: 10.1002/hep.22010. 18046720 [DOI] [PubMed] [Google Scholar]
- 5.Bailey S.M., Cunningham C.C. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radical Biology & Medicine. 2002;32(1):11–16. doi: 10.1016/s0891-5849(01)00769-9. 11755312 [DOI] [PubMed] [Google Scholar]
- 6.Carmiel-Haggai M., Cederbaum A.I., Nieto N. Binge ethanol exposure increases liver injury in obese rats. Gastroenterology. 2003;125(6):1818–1833. doi: 10.1053/j.gastro.2003.09.019. 14724834 [DOI] [PubMed] [Google Scholar]
- 7.Lieber C.S. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34(1):9–19. doi: 10.1016/j.alcohol.2004.07.008. 15670660 [DOI] [PubMed] [Google Scholar]
- 8.He J., de la Monte S., Wands J.R. Acute ethanol exposure inhibits insulin signaling in the liver. Hepatology. 2007;46(6):1791–1800. doi: 10.1002/hep.21904. 18027876 [DOI] [PubMed] [Google Scholar]
- 9.Lu Y., Cederbaum A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radical Biology and Medicine. 2008;44(5):723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding W.X. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139(5):1740–1752. doi: 10.1053/j.gastro.2010.07.041. 20659474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding W.X., Li M., Yin X.M. Selective taste of ethanol-induced autophagy for mitochondria and lipid droplets. Autophagy. 2011;7(2):248–249. doi: 10.4161/auto.7.2.14347. 21150309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizushima N., Yoshimori T., Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi: 10.1016/j.cell.2010.01.028. 20144757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klionsky D.J., Emr S.D. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. doi: 10.1126/science.290.5497.1717. 11099404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nature Reviews. Molecular Cell Biology. 2001;2(3):211–216. doi: 10.1038/35056522. 11265251 [DOI] [PubMed] [Google Scholar]
- 15.Kabeya Y. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO Journal. 2000;19(21):5720–5728. doi: 10.1093/emboj/19.21.5720. 11060023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamada Y., Sekito T., Ohsumi Y. Autophagy in yeast: a TOR-mediated response to nutrient starvation. Current Topics in Microbiology and Immunology. 2004;279:73–84. doi: 10.1007/978-3-642-18930-2_5. 14560952 [DOI] [PubMed] [Google Scholar]
- 17.Kirisako T. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. Journal of Cell Biology. 2000;151(2):263–276. doi: 10.1083/jcb.151.2.263. 11038174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klionsky D.J. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544. doi: 10.4161/auto.19496. 22966490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirkin V. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Molecular Cell. 2009;33(4):505–516. doi: 10.1016/j.molcel.2009.01.020. 19250911 [DOI] [PubMed] [Google Scholar]
- 20.Komatsu M. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163. doi: 10.1016/j.cell.2007.10.035. 18083104 [DOI] [PubMed] [Google Scholar]
- 21.Ni H.M. Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury. Toxicological Sciences: An Official Journal of the Society of Toxicology. 2012;127(2):438–450. doi: 10.1093/toxsci/kfs133. 22491424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tzivion G., Dobson M., Ramakrishnan G. FoxO transcription factors; regulation by AKT and 14-3-3 proteins. Biochimica et Biophysica Acta. 2011;1813(11):1938–1945. doi: 10.1016/j.bbamcr.2011.06.002. 21708191 [DOI] [PubMed] [Google Scholar]
- 23.Huang H., Tindall D.J. Dynamic FoxO transcription factors. Journal of Cell Science. 2007;120(15):2479–2487. doi: 10.1242/jcs.001222. 17646672 [DOI] [PubMed] [Google Scholar]
- 24.Zhao J. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metabolism. 2007;6(6):472–483. doi: 10.1016/j.cmet.2007.11.004. 18054316 [DOI] [PubMed] [Google Scholar]
- 25.Mammucari C. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metabolism. 2007;6(6):458–471. doi: 10.1016/j.cmet.2007.11.001. 18054315 [DOI] [PubMed] [Google Scholar]
- 26.Sengupta A. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. Journal of Biological Chemistry. 2011;286(9):7468–7478. doi: 10.1074/jbc.M110.179242. 21159781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ni H.M., Du K., You M., Ding W.X. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. American Journal of Pathology. 2013;183:1815–1825. doi: 10.1016/j.ajpath.2013.08.011. 24095927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forman B.M. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81(5):687–693. doi: 10.1016/0092-8674(95)90530-8. 7774010 [DOI] [PubMed] [Google Scholar]
- 29.Kim I. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis. 2007;28(5):940–946. doi: 10.1093/carcin/bgl249. 17183066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinal C.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102(6):731–744. doi: 10.1016/s0092-8674(00)00062-3. 11030617 [DOI] [PubMed] [Google Scholar]
- 31.Manley S. Suppression of autophagic flux by bile acids in hepatocytes. Toxicological Sciences: An Official Journal of the Society of Toxicology. 2014;137(2):478–490. doi: 10.1093/toxsci/kft246. 24189133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lívero F.A. The FXR agonist 6ECDCA reduces hepatic steatosis and oxidative stress induced by ethanol and low-protein diet in mice. Chemico-Biological Interactions. 2014;217:19–27. doi: 10.1016/j.cbi.2014.03.014. 24713361 [DOI] [PubMed] [Google Scholar]
- 33.Xie G. Alteration of bile acid metabolism in the rat induced by chronic ethanol consumption. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2013;27(9):3583–3593. doi: 10.1096/fj.13-231860. 23709616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding W.X. Oncogenic transformation confers a selective susceptibility to the combined suppression of the proteasome and autophagy. Molecular Cancer Therapeutics. 2009;8(7):2036–2045. doi: 10.1158/1535-7163.MCT-08-1169. 19584239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carson E.J., Pruett S.B. Development and characterization of a binge drinking model in mice for evaluation of the immunological effects of ethanol. Alcoholism, Clinical and Experimental Research. 1996;20(1):132–138. doi: 10.1111/j.1530-0277.1996.tb01055.x. 8651442 [DOI] [PubMed] [Google Scholar]
- 36.Yang F. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Research. 2007;67(3):863–867. doi: 10.1158/0008-5472.CAN-06-1078. 17283114 [DOI] [PubMed] [Google Scholar]
- 37.Wu D., Cederbaum A.I. Ethanol consumption by the nursing mother induces cytochrome P-4502E1 in neonatal rat liver. Journal of Pharmacology and Experimental Therapeutics. 1993;267(1):560–566. 8229787 [PubMed] [Google Scholar]
- 38.Ding W.X. Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. Journal of Biological Chemistry. 2012;287(50):42379–42388. doi: 10.1074/jbc.M112.413682. 23095748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin X.M., Ding W.X. The reciprocal roles of PARK2 and mitofusins in mitophagy and mitochondrial spheroid formation. Autophagy. 2013;9(11):1687–1692. doi: 10.4161/auto.24871. 24162069 [DOI] [PubMed] [Google Scholar]
- 40.Singh R. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. doi: 10.1038/nature07976. 19339967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang L. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metabolism. 2010;11(6):467–478. doi: 10.1016/j.cmet.2010.04.005. 20519119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mei S. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. Journal of Pharmacology and Experimental Therapeutics. 2011;339(2):487–498. doi: 10.1124/jpet.111.184341. 21856859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu W. Activation of farnesoid X receptor attenuates hepatic injury in a murine model of alcoholic liver disease. Biochemical and Biophysical Research Communications. 2014;443(1):68–73. doi: 10.1016/j.bbrc.2013.11.057. 24269813 [DOI] [PubMed] [Google Scholar]
- 44.Dolganiuc A. Autophagy in alcohol-induced liver diseases. Alcoholism, Clinical and Experimental Research. 2012;36(8):1301–1308. doi: 10.1111/j.1530-0277.2012.01742.x. 22551004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Czaja M.J. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9(8):1131–1158. doi: 10.4161/auto.25063. 23774882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baraona E. Alcoholic hepatomegaly: accumulation of protein in the liver. Science. 1975;190(4216):794–795. doi: 10.1126/science.1198096. 1198096 [DOI] [PubMed] [Google Scholar]
- 47.Harada M. Autophagy is involved in the elimination of intracellular inclusions, Mallory–Denk bodies, in hepatocytes. Medical Molecular Morphology. 2010;43(1):13–18. doi: 10.1007/s00795-009-0476-5. 20340001 [DOI] [PubMed] [Google Scholar]
- 48.Zatloukal K. From Mallory to Mallory–Denk bodies: What, how and why? Experimental Cell Research. 2007;313(10):2033–2049. doi: 10.1016/j.yexcr.2007.04.024. 17531973 [DOI] [PubMed] [Google Scholar]
- 49.Donohue T.M., Jr., Zetterman R.K., Tuma D.J. Effect of chronic ethanol administration on protein catabolism in rat liver. Alcoholism, Clinical and Experimental Research. 1989;13(1):49–57. doi: 10.1111/j.1530-0277.1989.tb00283.x. 2646978 [DOI] [PubMed] [Google Scholar]
- 50.Williams R.M. The Ume6 regulon coordinates metabolic and meiotic gene expression in yeast. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(21):13431–13436. doi: 10.1073/pnas.202495299. 12370439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bartholomew C.R. Ume6 transcription factor is part of a signaling cascade that regulates autophagy. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(28):11206–11210. doi: 10.1073/pnas.1200313109. 22733735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang P., Zhang H. You are what you eat: Multifaceted functions of autophagy during C. elegans development. Cell Research. 2014;24(1):80–91. doi: 10.1038/cr.2013.154. 24296782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Füllgrabe J., Klionsky D.J., Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nature Reviews. Molecular Cell Biology. 2014;15(1):65–74. doi: 10.1038/nrm3716. 24326622 [DOI] [PubMed] [Google Scholar]
- 54.Pietrocola F. Regulation of autophagy by stress-responsive transcription factors. Seminars in Cancer Biology. 2013;23(5):310–322. doi: 10.1016/j.semcancer.2013.05.008. 23726895 [DOI] [PubMed] [Google Scholar]
- 55.Xiong X. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. Journal of Biological Chemistry. 2012;287(46):39107–39114. doi: 10.1074/jbc.M112.412569. 22992773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van der Vos K.E. Modulation of glutamine metabolism by the PI(3)K–PKB-FOXO network regulates autophagy. Nature Cell Biology. 2012;14(8):829–837. doi: 10.1038/ncb2536. 22820375 [DOI] [PubMed] [Google Scholar]
- 57.Zhao Y. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nature Cell Biology. 2010;12(7):665–675. doi: 10.1038/ncb2069. 20543840 [DOI] [PubMed] [Google Scholar]
- 58.Bellacosa A. Activation of AKT kinases in cancer: implications for therapeutic targeting. Advances in Cancer Research. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. 16095999 [DOI] [PubMed] [Google Scholar]
- 59.van der Vos K.E., Coffer P.J. FOXO-binding partners: it takes two to tango. Oncogene. 2008;27(16):2289–2299. doi: 10.1038/onc.2008.22. 18391971 [DOI] [PubMed] [Google Scholar]
- 60.Wang F. Structures of KIX domain of CBP in complex with two FOXO3a transactivation domains reveal promiscuity and plasticity in coactivator recruitment. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(16):6078–6083. doi: 10.1073/pnas.1119073109. 22474372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Borniquel S. Inactivation of Foxo3a and subsequent downregulation of PGC-1 alpha mediate nitric oxide-induced endothelial cell migration. Molecular and Cellular Biology. 2010;30(16):4035–4044. doi: 10.1128/MCB.00175-10. 20547753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sandri M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(44):16260–16265. doi: 10.1073/pnas.0607795103. 17053067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Olmos Y. Mutual dependence of Foxo3a and PGC-1alpha in the induction of oxidative stress genes. Journal of Biological Chemistry. 2009;284(21):14476–14484. doi: 10.1074/jbc.M807397200. 19324885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van der Heide L.P., Smidt M.P. Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends in Biochemical Sciences. 2005;30:81–86. doi: 10.1016/j.tibs.2004.12.002. 15691653 [DOI] [PubMed] [Google Scholar]
- 65.Hoek J.B., Cahill A., Pastorino J.G. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology. 2002;122(7):2049–2063. doi: 10.1053/gast.2002.33613. 12055609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu D., Cederbaum A.I. Ethanol cytotoxicity to a transfected HepG2 cell line expressing human cytochrome P4502E1. Journal of Biological Chemistry. 1996;271(39):23914–23919. doi: 10.1074/jbc.271.39.23914. 8798623 [DOI] [PubMed] [Google Scholar]
- 67.Thomes P.G. Multilevel regulation of autophagosome content by ethanol oxidation in HepG2 cells. Autophagy. 2013;9(1):63–73. doi: 10.4161/auto.22490. 23090141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu D. Alcohol steatosis and cytotoxicity: the role of cytochrome P4502E1 and autophagy. Free Radical Biology & Medicine. 2012;53(6):1346–1357. doi: 10.1016/j.freeradbiomed.2012.07.005. 22819980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cuervo A.M., Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Research. 2014;24(1):92–104. doi: 10.1038/cr.2013.153. 24281265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ni H.M. Zonated induction of autophagy and mitochondrial spheroids limits acetaminophen-induced necrosis in the liver. Redox Biology. 2013;1(1):427–432. doi: 10.1016/j.redox.2013.08.005. 24191236 [DOI] [PMC free article] [PubMed] [Google Scholar]