

ABSTRACT
Plasma membrane proteins that enter cells by clathrin-independent endocytosis (CIE) are sorted either to lysosomes for degradation or recycled back to the plasma membrane. Expression of some MARCH E3 ubiquitin ligases promotes trafficking of CIE cargo proteins to lysosomes by ubiquitylating the proteins. Here, we show that co-expression of the ubiquitin-specific protease TRE17/USP6 counteracts the MARCH-dependent targeting of CIE cargo proteins, but not that of transferrin receptor, to lysosomes, leading to recovery of the stability and cell surface level of the proteins. The ubiquitylation of CIE cargo proteins by MARCH8 was reversed by TRE17, suggesting that TRE17 leads to deubiquitylation of CIE cargo proteins. The effects of TRE17 were dependent on its deubiquitylating activity and expression of TRE17 alone led to a stabilization of surface major histocompatibility complex class I (MHCI) molecules, a CIE cargo, suggesting that deubiquitylation of endogenous CIE cargo proteins promotes their stability. This study demonstrates that cycles of ubiquitylation and deubiquitylation can determine whether CIE cargo proteins are degraded or recycled.
KEY WORDS: TRE17, USP6, Ubiquitin, Cargo sorting, Ubiquitin-specific protease, Lysosome
INTRODUCTION
Protein levels are carefully monitored in cells through the regulated balance of protein synthesis and degradation. In general, soluble and membrane proteins are tagged for destruction by ubiquitylation, which leads to degradation by the proteasome and lysosome, respectively. For cell surface proteins, ubiquitylation can lead to post-endocytic sorting into intraluminal vesicles that form the multi-vesicular body (MVB), which later fuses with the lysosome (Piper and Lehner, 2011; Clague et al., 2012a). Sorting into the MVB requires ubiquitylation of the cargo protein, and endosomal sorting complex required for transport (ESCRT) complexes. Ubiquitylation involves the covalent addition of ubiquitin, typically onto lysine residues, and this process involves the activity of E3 ligases. There are over 600 human E3 ligases, which are thought to exhibit specificity towards their substrates (Li et al., 2008). Conversely, deubiquitylating enzymes (DUBs) catalyze the removal of ubiquitin. There are ∼90 DUBs, but the extent to which DUBs have differing specificities is not clear (Clague et al., 2012b). Prior to degradation, the ubiquitin moiety on ubiquitylated proteins is removed by DUBs; this process allows for the recycling of the ubiquitin protein so it can be used for the next target. Recent evidence has emerged that in addition to providing a pathway for ubiquitin salvage, other DUBs can counteract or undo the effects of ubiquitylation, thus rescuing it from degradation.
We have been studying the trafficking and turnover of plasma membrane proteins that enter cells by clathrin-independent endocytosis (CIE). This mode of endocytosis occurs independently of clathrin and adaptor proteins and is used by a wide range of plasma membrane proteins (Naslavsky et al., 2004; Eyster et al., 2009). Some CIE cargo proteins, such as major histocompatibility complex class I (MHCI) molecules, can be either recycled back to the plasma membrane or routed to late endosomes and lysosomes for degradation (Naslavsky et al., 2004). Other CIE cargo proteins, such as CD98 and CD44, are mostly recycled back to the plasma membrane with little routing to degradation (Eyster et al., 2009; 2011). Members of the MARCH family of E3 ligases ubiquitylate CIE cargo proteins, causing them to be routed directly to late endosomes and lysosomes (Eyster et al., 2011). This change in trafficking, induced by expression of MARCH8, is observed for MHCI, but is most dramatic for CD98 and CD44, which normally avoid degradative compartments (Eyster et al., 2011).
TRE17, also known as USP6, is a member of the family of ubiquitin-specific proteases (USPs), the most abundant group of DUBs (>50 members). TRE17 was originally identified as an oncogene (Nakamura et al., 1992). Later studies revealed that translocation of the TRE17 locus leads to overexpression of the wild-type protein and is associated with two neoplasms, aneurysmal bone cyst (Oliveira et al., 2004a; Oliveira et al., 2004b; Oliveira et al., 2005; Panagopoulos et al., 2008) and nodular fasciitis (Erickson-Johnson et al., 2011). The USP domain of TRE17 is required for tumorigenesis (Ye et al., 2010; Pringle et al., 2012). However, relevant substrates have not been identified to date. TRE17 has another characteristic domain, the TBC (Tre-2, Bub2, Cdc16) domain, through which it binds to Arf6, a G protein associated with the CIE endosomal membrane system (Martinu et al., 2004). TRE17 colocalizes with Arf6 and CIE cargo proteins. TRE17 associates with GDP-bound Arf6 and promotes activation of Arf6 in vivo in a manner requiring its TBC domain (Martinu et al., 2004; Lau et al., 2010), and has been proposed to promote recycling of CIE cargo proteins. However, the role of the USP domain in the trafficking function of TRE17 has not been explored. In the current study, we re-examine the role of TRE17 in influencing CIE cargo protein trafficking. In particular, we investigate whether TRE17, through its USP activity, can counter the increased degradation of CIE cargo proteins triggered by MARCH expression.
RESULTS
TRE17 counteracts MARCH-dependent targeting of CIE cargo to late endosomes in a DUB-activity-dependent manner
In our previous work, we demonstrated that trafficking of CIE cargo proteins is altered by expression of MARCH proteins through ubiquitylation (Eyster et al., 2011). We hypothesized that expression of TRE17 might affect ubiquitylation-dependent CIE cargo protein trafficking through its DUB activity. To examine the effect of TRE17 on trafficking of CIE cargo proteins, we co-expressed TRE17 with the MARCH8 ubiquitin ligase in HeLa cells and followed the fate of internalized MHCI, a CIE cargo protein that is targeted by MARCH8 (Eyster et al., 2011). To track MHCI endocytosis and its intracellular trafficking, HeLa cells were incubated with monoclonal antibodies directed to the extracellular portion of the protein for 1 h to allow antibody-bound MHCI to enter the cells. Then, HeLa cells were treated with the proton ionophore NH4Cl for 2 h to neutralize the pH of the late endosome and block degradation, in order to visualize cargo delivery to late endosomes. As we reported previously, overexpression of MARCH8 caused downregulation of MHCI from the cell surface, with concomitant accumulation of the proteins in an enlarged juxtanuclear compartment (Fig. 1A, top panels). This compartment was co-stained with the late endosome/lysosome marker Lamp1 (Eyster et al., 2011) (data not shown), suggesting that MARCH8 targets MHCI to late endosomes for degradation. In clear contrast, most of cells co-expressing GFP–TRE17 and MARCH8 did not exhibit juxtanuclear accumulation of MHCI and instead MHCI was maintained at the cell surface (Fig. 1A, middle, outlined with dashed lines), suggesting that TRE17 can suppress the function of MARCH8. In contrast, expression of a TRE17 point mutant that lacks DUB activity (TRE17/USP−) (Shen et al., 2005) failed to suppress the effect of MARCH8. Cells co-expressing TRE17/USP− and MARCH8 were indistinguishable from those expressing MARCH8 alone (Fig. 1A, bottom). Quantification revealed that more than 90% of cells co-expressing MARCH8 with GFP or GFP–TRE17/USP− exhibited reduced surface labeling and increased juxtanuclear accumulation of MHCI (Fig. 1B). In contrast, only 15% of cells co-expressing MARCH8 and GFP–TRE17 exhibited reduced surface labeling and increased juxtanuclear accumulation of MHCI, as surface MHCI was once again apparent. These results suggest that TRE17 can counteract the effect of MARCH8 in a DUB-dependent manner.
Fig. 1.

TRE17 counteracts the MARCH8-mediated targeting of CIE cargo proteins to late endosomes. HeLa cells were transfected with MARCH8–FLAG and GFP, GFP–TRE17 wild type (WT) or GFP–TRE17/USP− (DUB mutant). (A,C) After 24 h, anti-MHCI (A) or anti-CD98 (C) antibodies were added to cells and allowed to bind to and be internalized for 1 h. Cells were washed and transferred to medium containing 25 mM NH4Cl for 2 h. Cells were then fixed, and the internalized anti-MHCI (A) and anti-CD98 (C) antibodies were detected with anti-mouse-IgG2a or anti-mouse-IgG antibody conjugated to Alexa Fluor 594, respectively (red). GFP-tagged proteins were immunolabeled in A with mouse anti-GFP and anti-mouse-IgG1 antibody conjugated to Alexa Fluor 488 (green). In C, GFP-tagged proteins were directly detected by their fluorescence. MARCH8–FLAG was detected by rabbit anti-FLAG and anti-rabbit-IgG antibody conjugated to Alexa Fluor 633 (blue). Cells co-expressing MARCH8 and GFP-tagged proteins are outlined by dashed lines. Arrows indicate the cells expressing MARCH8 alone. Scale bars: 10 µm. (B,D) Quantification of experiments shown in A and C are shown in B and D, respectively. 50–60 cells were randomly selected from transfected cells and the number of cells showing the effect of MARCH8 (reduced surface and perinuclear accumulation) was counted and plotted as the percentage of transfected cells. Shown are mean±s.e.m. from three independent experiments.
We previously identified a new group of CIE cargo proteins (CD44, CD98, and CD147) that follow a different intracellular itinerary from MHCI (Eyster et al., 2009; Eyster et al., 2011). These cargoes largely avoid transport to degradative compartments and are instead recycled directly to the plasma membrane after internalization. We demonstrated that CD98 and CD44 are diverted to late endosome upon expression of MARCH8 (Eyster et al., 2011). Therefore, we predicted that TRE17 might also rescue them from a degradative fate. Similar to MHCI, CD98 delivery to late endosomes was increased and its surface level was decreased by the expression of MARCH8 (Fig. 1C, top; Fig. 1D). This effect was suppressed by co-expression of TRE17 but not by TRE17/USP− (Fig. 1C, middle and bottom; Fig. 1D). Similar results were obtained for CD44 (supplementary material Fig. S1). The downregulation of MHCI and CD98 and increased delivery to late endosomes by MARCH8 expression were also observed in the human bronchial epithelial cell line Beas2B, and these effects were suppressed by expression of TRE17 (supplementary material Fig. S2). Thus, TRE17 can alter the trafficking of two different types of CIE cargo proteins in two cell types that are targeted to late endosomes by MARCH8.
We next examined whether TRE17 could reverse the effects of other MARCH family members on trafficking of CIE cargo proteins. Interestingly, different CIE cargoes exhibit distinct sensitivities to MARCH members: MHCI is downregulated by MARCH1, MARCH4 and MARCH8, CD98 is downregulated by MARCH1 and MARCH8, and CD44 is targeted only by MARCH8 (Eyster et al., 2011). Therefore, we tested whether TRE17 can also suppress the effects of MARCH1 and MARCH4. MARCH1 promoted targeting of CD98 to late endosomes, coincident with its diminished surface expression. This was suppressed by the co-expression of TRE17 but not by the TRE17/USP− mutant (supplementary material Fig. S3A). Similarly, TRE17 reversed the late endosomal targeting of MHCI induced by both MARCH1 (data not shown) and MARCH4 (supplementary material Fig. S3B). Thus, TRE17 is capable of counteracting the function of multiple MARCH proteins that function in the CIE pathway.
Unique features of TRE17 determine its effects on CIE cargo trafficking
We next examined whether other USPs could suppress the effect of MARCH on CIE trafficking, or whether this was a specific function for TRE17. USP8 is an endosomal DUB that has been shown to deubiquitylate and regulate the trafficking of several plasma membrane proteins, including EGF receptor, the chemokine receptor CXCR4, the Ca2+-activated K+ channel and the epithelial Na+ channel ENaC (Berlin et al., 2010a; Berlin et al., 2010b; Balut et al., 2011; Zhou et al., 2013). Given that USP8 is suggested to function at the plasma membrane and also at endosomal compartments, it was possible that USP8 might also regulate CIE cargo trafficking as TRE17 does. However, in contrast to TRE17, co-expression of USP8 did not suppress MARCH8-induced surface downregulation and late endosomal accumulation of MHCI (Fig. 2A, middle) or CD98 (Fig. 2C, middle).
Fig. 2.

USP8 and USP32 cannot counteract the effect of MARCH8 on CIE cargo trafficking. HeLa cells were transfected with MARCH8–FLAG and wild-type GFP–TRE17, GFP–USP8 or GFP–USP32. (A,C) After 24 h, anti-MHCI (A) or anti-CD98 (C) antibodies were added to cells and allowed to internalize for 1 h. Cells were washed and transferred to medium containing 25 mM NH4Cl for 2 h. Antibodies for MHCI (A) and CD98 (C), GFP-tagged proteins and MARCH8–FLAG were used for detection as in Fig. 1. Cells co-expressing MARCH8 and GFP-tagged proteins are outlined by dashed lines. Arrows indicate the cells expressing MARCH8 alone. Scale bars: 10 µm. (B,D) Cells were counted as in Fig. 1 and the results are shown in B and D. Shown are means±s.e.m. from three independent experiments.
As an additional specificity control, we also tested USP32. The TRE17 gene is derived from the segmental duplication of USP32 and TBC1D3, followed by their fusion (Paulding et al., 2003). Although USP32 possesses a distinct N-terminus and lacks the TBC domain, its USP domain is 98% identical to that of TRE17. Strikingly, USP32 failed to rescue late endosomal targeting of MHCI and CD98 induced by MARCH8 (Fig. 2A,C). Quantification demonstrates that more than 90% of cells co-expressing USP8 or USP32 with MARCH8 still exhibit surface downregulation and late endosomal accumulation of both MHCI and CD98 (Fig. 2B,D). These results indicate that the ability of TRE17 to counteract the effects of MARCH proteins on CIE cargo trafficking is highly specific, and not merely a consequence of overexpression. Furthermore, our data indicate that the N-terminal region of TRE17, containing the TBC domain, confers specificity to TRE17 in regulating trafficking of cargo through this pathway.
TRE17 does not affect trafficking of TfR
In addition to CIE cargo proteins, we (Eyster et al., 2011) and others (Bartee et al., 2004; Nakamura et al., 2005; Fujita et al., 2013) have previously shown that the transferrin receptor (TfR), a plasma membrane protein that enters cells by clathrin-mediated endocytosis (CME), is downregulated by expression of some MARCH proteins. Therefore, we examined whether TRE17 would rescue the downregulation of TfR mediated by expression of MARCH. When we examined the steady-state level of TfR, we observed a significant downregulation of TfR in MARCH8-expressing cells (Fig. 3A, middle row) as reported previously (Eyster et al., 2011; Fujita et al., 2013). In contrast to CIE cargo proteins, however, expression of TRE17 failed to rescue this phenotype, with ∼95% of cells co-expressing MARCH8 and TRE17 exhibiting downregulation of TfR (Fig. 3A,B). These results suggest that TRE17 cannot counteract the effect of MARCH8 on TfR degradation and indicate that TRE17 has substrate specificity: TRE17 can rescue CIE cargoes that are targeted by MARCH proteins but not TfR, a CME cargo.
Fig. 3.
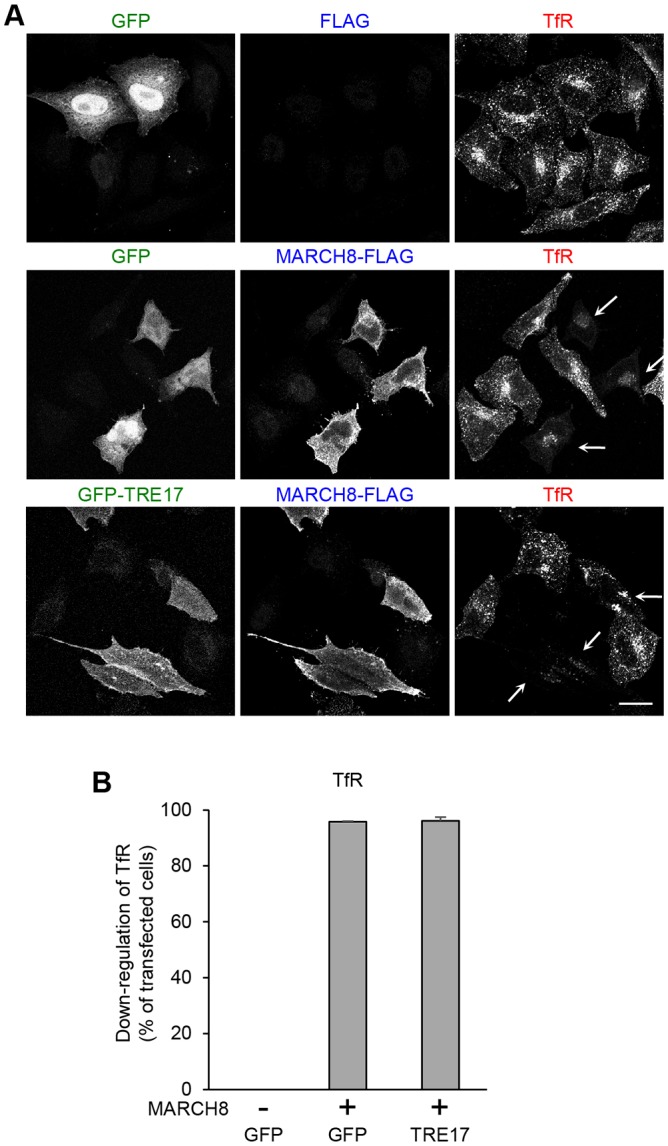
TRE17 does not counteract the MARCH8-mediated downregulation of TfR. (A) HeLa cells were transfected with either GFP or GFP–TRE17 with or without MARCH8–FLAG. After 24 h, cells were fixed and labeled with rabbit anti-FLAG and mouse anti-transferrin receptor antibodies, followed by immunofluorescence with anti-rabbit-IgG antibody conjugated to Alexa Fluor 633 (blue) and anti-mouse-IgG antibody conjugated to Alexa Fluor 594 (red). Arrows indicate the cells co-expressing MARCH8 and GFP-tagged proteins. Scale bar: 20 µm. (B) 50–60 cells were randomly selected from transfected cells and the number of cells showing the effect of MARCH8 (downregulation TfR) was counted and plotted as a percentage of transfected cells. Shown are mean±s.e.m. from four independent experiments.
Compartmentalization of DUBs contributes to recognition of CIE cargo proteins
To further investigate the substrate specificity of TRE17, we analyzed the subcellular localization of TRE17 and cargo proteins MHCI, CD98 and TfR, by immunofluorescence staining. As reported previously (Masuda-Robens et al., 2003; Martinu et al., 2004), TRE17 localized at the plasma membrane and tubular endosomes (Fig. 4). TRE17 exhibited significant colocalization with CIE cargo proteins, MHCI and CD98, at both of these locations. However, we did not observe colocalization of TRE17 with TfR (Fig. 4).
Fig. 4.
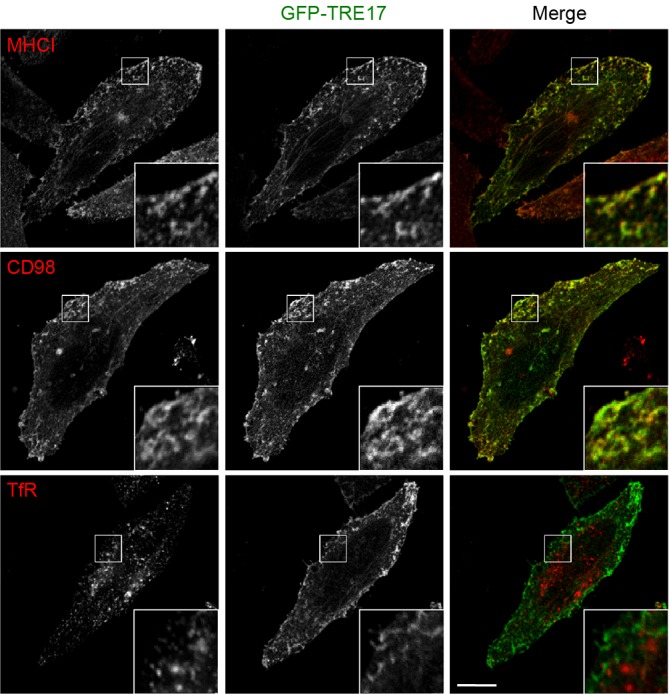
TRE17 colocalizes with MHCI and CD98 but not with TfR. HeLa cells were transfected with GFP–TRE17 and cultured for 24 h. Cells were fixed and incubated with rabbit anti-GFP and anti-MHCI, CD98 or TfR antibodies as indicated, followed by secondary antibodies, anti-rabbit-IgG antibody conjugated to Alexa Fluor 488 (green) and anti-mouse IgG antibody conjugated to Alexa Fluor 594 (red). Enlargements of the boxed region are shown in the insets. Scale bar: 10 µm.
These observations raise the possibility that the specificity of TRE17 on CIE cargo trafficking is achieved through its co-compartmentalization with CIE cargo. To test this, we re-directed USP8, which failed to alter trafficking of CIE cargo (Fig. 2), to the CIE pathway by appending the C-terminal 20 amino acids of H-Ras (denoted, USP8–Ras-C20). This region of H-Ras contains a farnesylation signal and mediates localization of H-Ras to the plasma membrane and clathrin-independent tubular endosomes and vesicles (Hancock et al., 1991; Porat-Shliom, et al., 2008; McKay et al., 2011). Furthermore, this peptide is sufficient to drive localization of GFP to the plasma membrane and clathrin-independent-derived endosomes and tubules. When expressed in HeLa cells, wild-type USP8 localized at cytosolic punctate structures and did not colocalize with the CIE cargoes MHCI and CD98 (Fig. 5A). In contrast, the USP8–Ras-C20 chimera was highly colocalized with both MHCI and CD98 at the plasma membrane and tubular endosomes, but not with TfR, a CME cargo (Fig. 5B), confirming that USP8–Ras-C20 is targeted to the CIE endocytic pathway.
Fig. 5.
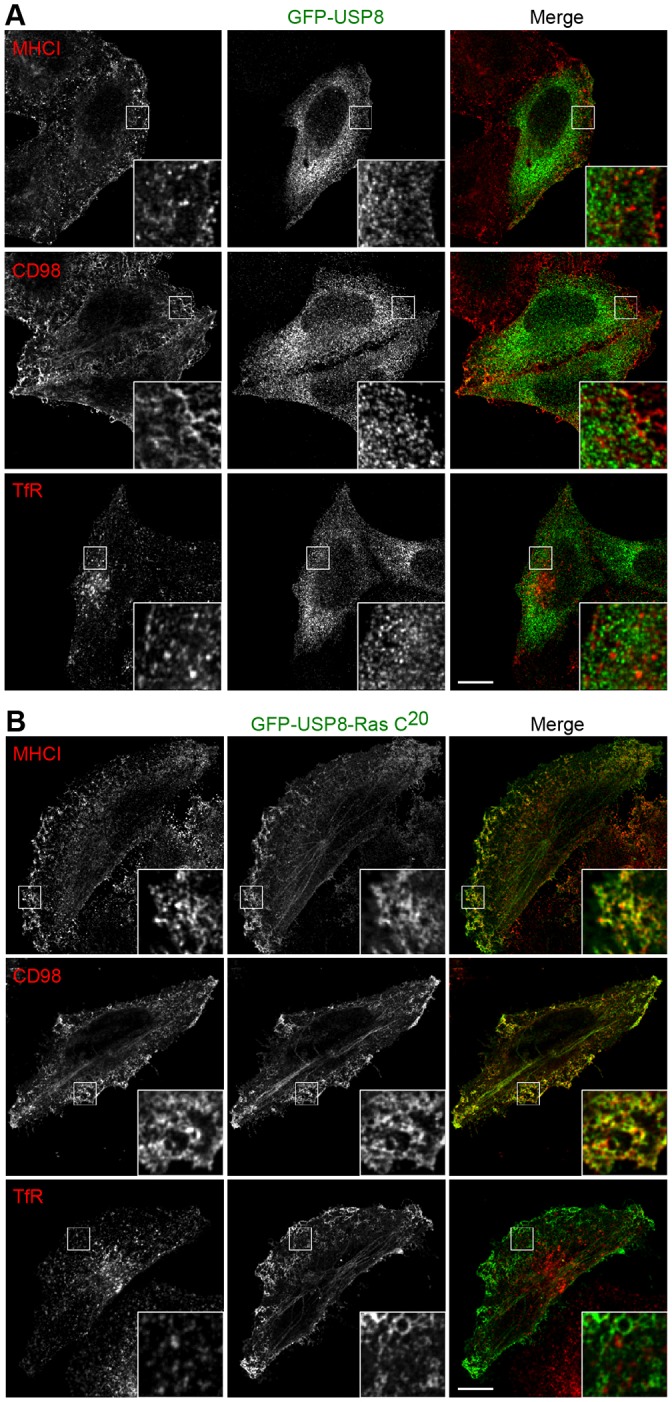
The USP8–Ras-C20 chimera colocalizes with CIE cargo proteins but not with TfR. HeLa cells were transfected with GFP–USP8 (A) or GFP–USP8–Ras-C20 (B) and cultured for 24 h. Cells were fixed and incubated with chicken anti-GFP and anti-MHCI, CD98 or TfR antibodies, followed by secondary antibodies, anti-chicken-IgG antibody conjugated to Alexa Fluor 488 (green) and anti-mouse IgG antibody conjugated to Alexa Fluor 594 (red). Enlargements of the boxed region are shown in the insets. Scale bars: 10 µm.
Next, we examined whether USP8–Ras-C20 alters the trafficking of CIE cargo proteins. In contrast to USP8, which did not suppress MARCH8-dependent downregulation and accumulation of MHCI, co-expression of USP8–Ras-C20 markedly suppressed the effect of MARCH8: nearly 70% of cells co-expressing USP8–Ras-C20 recovered MHCI at the cell surface (Fig. 6A,B). Similar results were obtained for CD98 (nearly 60% were recovered) (Fig. 6C,D). By contrast, USP8–Ras-C20 could not counteract the downregulation of TfR caused by expression of MARCH8 (Fig. 6E,F), indicating that USP8–Ras-C20 has similar substrate specificity to TRE17. Therefore, localization of the DUBs TRE17, USP8 and USP8–Ras-C20, clearly correlated with the ability to alter the itinerary of cargo proteins. These results suggest that the substrate specificity against CIE cargo proteins is determined in part by the subcellular localization of the DUB.
Fig. 6.

The USP8–Ras-C20 chimera counteracts the MARCH8-mediated targeting of CIE cargo proteins to late endosomes but not the downregulation of TfR. (A–D) HeLa cells were transfected with MARCH8–FLAG and GFP–TRE17 (not shown in A and C), and GFP–USP8 or GFP–USP8–Ras-C20. After 24 h, anti-MHCI (A,B) or anti-CD98 (C,D) antibodies were added to cells and treated as in Fig. 1. Cells were then fixed, and the internalized anti-MHCI (A,B) and anti-CD98 (C,D) antibodies were detected with anti-mouse-IgG antibody conjugated to Alexa Fluor 546 (red). GFP-tagged proteins were immunolabeled with chicken anti-GFP and anti-chicken-IgG antibody conjugated to Alexa Fluor 488 (green). MARCH8–FLAG was detected by rabbit anti-FLAG and anti-rabbit-IgG antibody conjugated to Alexa Fluor 633 (blue). The cell number was counted and plotted in B and D as in Fig. 1. Shown are mean±s.e.m. from three independent experiments. (E,F) HeLa cells were transfected with MARCH8–FLAG and GFP–TRE17 (not shown in E), GFP–USP8 or GFP–USP8–Ras-C20. After 24 h, cells were fixed and labeled with chicken anti-GFP, rabbit anti-FLAG and mouse anti-transferrin receptor, followed by immunofluorescence with anti-chicken-IgG antibody conjugated to Alexa Fluor 488 (green), anti-rabbit-IgG antibody conjugated to Alexa Fluor 633 (blue) and anti-mouse-IgG antibody conjugated to Alexa Fluor 546 (red). The cell number was counted and plotted in F as in Fig. 3. Shown are mean±s.e.m. from three independent experiments. In A, C and E, cells co-expressing MARCH8 and GFP-tagged proteins are outlined by dashed lines. Arrows indicate the cells expressing MARCH8 alone. Scale bars: 20 µm.
CIE cargo proteins are deubiquitylated in TRE17-expressing cells
To test whether the function of TRE17 is mediated through deubiquitylation of target cargo proteins, we examined whether ubiquitylation of CIE cargo proteins by MARCH is reversed by TRE17. To facilitate detection of ubiquitylation of CIE cargoes, we used C-terminally SNAP-tagged CD98. The SNAP tag is an engineered form of a 20-kDa DNA repair enzyme (O6-alkylguanine-DNA alkyltransferase) that binds covalently to O6-benzylguanine (Gautier et al., 2008), which can be attached to Alexa Fluor dyes and biotin.
We first sought to confirm that trafficking of CD98–SNAP recapitulates that of the endogenous protein. HeLa cells expressing CD98–SNAP were exposed to O6-benzylguanine–Alexa-Fluor-594 for 1 h at 37°C and chased for 2 h in medium containing 25 mM NH4Cl to follow the trafficking of CD98–SNAP. Similar to endogenous CD98, CD98–SNAP was lost from the cell surface and accumulated at late endosomes upon MARCH8 overexpression (supplementary material Fig. S4A, second row). This effect was counteracted by TRE17, but not TRE17/USP− (supplementary material Fig. S4A, third and bottom rows). Thus, both MARCH8 and TRE17 regulate the trafficking of CD98–SNAP in a similar manner to endogenous CD98.
Next, we investigated the ubiquitylation status of CD98–SNAP in cells expressing MARCH8 and TRE17. We previously demonstrated that MARCH8 induces the ubiquitylation and degradation of both endogenous CD98 and CD98–SNAP (Eyster et al., 2011). To test whether TRE17 can reverse this, HeLa cells expressing CD98–SNAP were labeled with O6-benzylguanine–biotin for 4 h, and biotin-labeled CD98–SNAP was isolated on streptavidin beads and detected by anti-ubiquitin blotting. To ensure that anti-ubiquitin signals were derived from CD98–SNAP and not co-purifying proteins, cell lysates were denatured by SDS and boiled before the pull down. CD98–SNAP exhibited a low basal level of ubiquitylation that was strongly enhanced by co-expressing MARCH8 (Fig. 7A, top panel, compare lanes 1 and 2). The smeared bands observed in the anti-ubiquitin blot were not detected in control cells (no CD98–SNAP) confirming that the band corresponds to ubiquitylated CD98–SNAP (data not shown). Concomitant with this increased ubiquitylation, total levels of CD98–SNAP were dramatically reduced upon co-expression with MARCH8, consistent with its targeting for lysosomal degradation (Fig. 7A, second and third panels). When TRE17 was co-expressed with MARCH8, the smeared band observed in cells expressing MARCH8 alone was remarkably decreased (Fig. 7A, top panel, compare lanes 2 and 3). In contrast, expression of the TRE17/USP− mutant did not reduce MARCH8-dependent ubiquitylation of CD98-SNAP (Fig. 7A, top panel, lane 4). Co-expression of TRE17, but not the TRE17/USP− mutant, partially restored levels of CD98–SNAP protein (Fig. 7A, compare lanes 1–4 in the anti-SNAP blot), correlating with the level of ubiquitylation. These results suggest that TRE17 counteracts the function of MARCH8 by promoting deubiquitylation of CD98–SNAP. Furthermore, expression of the other DUBs USP8 and USP32, did not affect MARCH8-dependent ubiquitylation substantially (Fig. 7A, lanes 5 and 6), suggesting that the effects of TRE17 on CD98–SNAP ubiquitylation and protein levels are specific. Quantification of these results further supports the above observation. We measured band intensities of the anti-ubiquitin blot and normalized to the amount of isolated CD98–SNAP to calculate the level of ubiquitylation of CD98–SNAP (Fig. 7B). Expression of MARCH8 increased ubiquitylation of CD98–SNAP more than 35-fold compared to the control (Fig. 7B, columns 1 and 2). Co-expression of TRE17 markedly decreased MARCH8-dependent ubiquitylation (Fig. 7B, columns 2 and 3). By contrast, the amount of ubiquitylation of CD98–SNAP in cells co-expressing the TRE17/USP−, USP8 or USP32 was not significantly different from that of cells expressing MARCH8 alone (Fig. 7B, columns 2, 4, 5 and 6).
Fig. 7.

TRE17 deubiquitylates CD98 and Tac. (A) HeLa cells were transfected with CD98–SNAP and GFP, GFP–TRE17 wild-type (WT), GFP–TRE17/USP−, GFP–USP8 or GFP–USP32 with or without MARCH8–FLAG as indicated. After 24 h, CD98–SNAP was labeled with O6-benzylguanine–biotin and pulled down by streptavidin–agarose from denatured cell lysates as described in the Materials and Methods. Cell lysates and precipitates were separated by SDS-PAGE and immunoblotted with antibodies as indicated. (B) Quantification of ubiquitylated proteins shown in A. The intensities of ubiquitylated CD98–SNAP that are indicated by the bracket in anti-ubiquitin blot were measured and normalized to the respective precipitated CD98–SNAP level. The values are plotted as the fold increase from control cells (GFP without MARCH8). Shown are mean±s.e.m. from four independent experiments. *P<0.05; n.s., not significant (compared with cells transfected with GFP and MARCH8 by one-way ANOVA with post hoc Dunnett's test). (C) HeLa cells were transfected with SNAP–Tac and GFP, GFP–TRE17 WT, GFP–TRE17/USP−, GFP–USP8 or GFP–USP32 with or without MARCH8–FLAG as indicated. After 24 h, SNAP–Tac was labeled, pulled down and immunoblotted as in A. (D) Ubiquitylated SNAP–Tac from C was quantified and plotted as in B. Shown are mean±s.e.m. from three independent experiments. **P<0.005, n.s., not significant (compared with cells transfected with GFP and MARCH8 by one-way ANOVA with post hoc Dunnett's test).
To determine whether the effects of TRE17 on ubiquitylation and degradation could be extrapolated to other CIE cargoes, we performed similar analysis of Tac, the alpha subunit of the interleukin 2 receptor. Tac is a CIE cargo protein that follows a similar intracellular trafficking itinerary to MHCI (Naslavsky et al., 2003). We appended the SNAP tag to the N-terminus of Tac, and examined the trafficking and ubiquitylation of SNAP–Tac. Similar to the observations for CD98–SNAP, SNAP–Tac was routed to late endosomes and the cell surface level was decreased by the expression of MARCH8 (supplementary material Fig. S4B). These phenotypes were suppressed by the co-expression of TRE17 but not by the TRE17/USP− mutant. When we examined ubiquitylation of SNAP–Tac, two discrete bands and a faint smeared band that ran slightly higher than SNAP–Tac were observed in cells expressing MARCH8 in anti-ubiquitin blots (Fig. 7C, top panel, compare lanes 1 and 2). These bands were not observed in control cells, confirming that the bands are ubiquitylated SNAP–Tac (data not shown). When TRE17 was co-expressed with MARCH8, ubiquitylation of SNAP–Tac was strongly suppressed (Fig. 7C, compare lanes 2 and 3). However, expression of TRE17/USP−, USP8 and USP32 did not affect the ubiquitylation of SNAP–Tac by MARCH8. Coupled to the ubiquitylation status, the expression level of SNAP–Tac was markedly decreased in cells expressing MARCH8 alone and recovered in cells specifically upon co-expression of TRE17. Quantification of ubiquitylated SNAP–Tac gave similar results to that of CD98–SNAP. Expression of TRE17 decreased MARCH8-dependent ubiquitylation of SNAP–Tac, whereas expression of either TRE17/USP-, USP8 or USP32 had no significant effect (Fig. 7D). Taken together, these findings demonstrate that TRE17 promotes the deubiquitylation of two different classes of CIE cargo proteins that are targeted by MARCH, leading to their enhanced stability through inhibition of delivery to late endosomes.
MHCI is stabilized in TRE17-expressing cells
Having determined that TRE17 can counteract the effect of MARCH targeting cargo proteins for degradation, we next examined whether expression of TRE17 alone could alter the stability of endogenous cargo proteins. We previously demonstrated that pulse-labeled MHCI proteins are routed to degradative compartments resulting in almost complete disappearance of the labeled proteins from the cell surface after 24 h (Naslavsky et al., 2003). We followed the long-term fate of surface MHCI in cells expressing TRE17 in order to examine its effect on the stability of CIE cargo. Cells expressing TRE17 were incubated with anti-MHCI antibody on ice for 1 h to label the surface MHCI. Antibody was washed out and cells were either fixed immediately (0 h) or further incubated for 20 h at 37°C. After incubation, cells were fixed and antibody remaining at the cell surface was detected by using secondary antibodies without permeabilizing the cells (Fig. 8A). By measuring intensities of the cell surface MHCI as described in the Materials and Methods, we compared the surface level of MHCI among cells expressing GFP control, wild-type TRE17 and TRE17/USP− (Fig. 8B). At the 0 h time point, the surface level of MHCI was slightly, but significantly, increased in cells expressing TRE17 but not in cells expressing TRE17/USP−. This indicates that TRE17 modestly upregulates steady-state levels of MHCI in a DUB-dependent manner. After 20 h of incubation, the cell surface MHCI labeled with the antibodies was dim in control (GFP) cells (Fig. 8A, right panels). However, in cells expressing wild-type TRE17, the signal intensities of surface MHCI were higher than those of non-expressing cells. In contrast, cells expressing TRE17/USP− did not show significant differences in the amount of surface MHCI compared to non-expressing control cells. Quantification of cell surface MHCI levels demonstrated that cells expressing wild-type TRE17 but not the TRE17/USP− mutant had ∼60% more surface MHCI compared to GFP control cells (Fig. 8B, right). These results indicate that expression of TRE17 by itself can stabilize endogenous MHCI resulting in increased levels of the protein at the cell surface. Taken together, our results suggest that TRE17 not only counteracts the effects of overexpressed MARCH E3 ligases, but also regulates the trafficking of endogenous CIE cargo proteins by promoting deubiquitylation and thus blocking their degradation.
Fig. 8.

TRE17 expression alone increases cell surface MHCI. (A) HeLa cells were transfected with GFP, GFP–TRE17 wild-type (WT) or GFP–TRE17/USP−. After 24 h, cells were pre-incubated with anti-MHCI antibodies at 4°C for 1 h to label the cell surface MHCI. Cells were washed and further incubated at 37°C for 0 h or 20 h. Cells were then fixed and incubated with anti-mouse-IgG2a antibody conjugated to Alexa Fluor 594 without saponin to label the cell surface anti-MHCI antibodies. GFP-tagged proteins were detected by using mouse anti-GFP and anti-mouse-IgG1 antibody conjugated to Alexa Fluor 488. Images of 0 h and 20 h were obtained with individual settings of a confocal microscope. Scale bars: 20 µm. (B) Quantification of cell surface MHCI in A. 40–50 cells were randomly selected from transfected cells and the intensity of fluorescence signals of the surface MHCI were measured and normalized to each cell area. The values were plotted as the fold increase from GFP-expressing cells in each time point (0 h and 20 h). Shown are mean±s.d. for one representative experiment, repeated one additional time. **P<0.005; n.s., not significant (compared with GFP-expressing cells by one-way ANOVA with post hoc Dunnett's test).
DISCUSSION
In this study, we show that TRE17 counteracts the MARCH-dependent ubiquitylation and downregulation of CIE cargo proteins in a DUB activity-dependent manner. Among several USPs localized to the periphery, TRE17 was specifically able to regulate the trafficking of CIE cargo proteins upon overexpression. Taken together with our previous studies, our work indicates that trafficking of CIE cargoes can be regulated by reversible ubiquitylation mediated by the coordinated activities of TRE17 and MARCH proteins.
We had previously shown that MARCH isoforms exhibit substrate specificity for the downregulation of CIE cargo proteins: CD44 is downregulated by overexpression of MARCH8, CD98 by MARCH1 and MARCH8, and MHCI by MARCH4 and MARCH8 (Eyster et al., 2011). In contrast, TRE17 suppressed the effects of all these MARCH proteins, and promoted recovery of CD44, CD98 and MHCI at the cell surface. Therefore, it is likely that TRE17 can widely recognize CIE cargo proteins that are ubiquitylated. By contrast, TRE17 could not counteract the MARCH-dependent downregulation of TfR, a CME cargo. Thus, TRE17 appears to selectively regulate CIE but not CME pathway cargoes. In support of this view, TRE17 was highly colocalized with CIE cargo proteins, MHCI and CD98, but not with TfR, at the plasma membrane and tubular recycling endosomes, characteristic structures for CIE trafficking pathways (Naslavsky et al., 2004; Martinu et al., 2004) (Fig. 4). Furthermore, we did not observe colocalization of TRE17 with AP-2, an adaptor protein subunit for CME (our unpublished observations).
The localization of TRE17 appears to be a key determinant of how it recognizes its substrates. The results of experiments using the USP8–Ras-C20 chimera (Figs 5, 6) further support this possibility. Although USP8 itself does not affect CIE cargo trafficking, when it is targeted to CIE trafficking pathways by connecting H-Ras C-terminal farnesylation signal, the chimeric protein colocalized with CIE cargoes and altered their itinerary. These results suggest that a DUB relocalized to CIE trafficking pathways obtains substrate specificity against CIE cargoes. It is likely that recruitment of the DUB to the CIE-cargo-specific compartment is important for it to achieve substrate specificity against CIE cargoes. TRE17 is recruited to the plasma membrane from which CIE cargoes enter, and to endosomal compartments that are related to CIE trafficking pathways. Some factors of the CIE trafficking machinery might recruit TRE17 to the sites where cargo proteins are deubiquitylated. Interestingly, TRE17 is known to bind Arf6, a key regulatory factor for the CIE trafficking pathway. It has been shown that Arf6 and TRE17 colocalize at the plasma membrane and tubular recycling endosomes (Masuda-Robens et al., 2003; Martinu et al., 2004). In addition, Rueckert and Haucke (Rueckert and Haucke, 2012) have shown that subcellular localization of TRE17 depends on Arf6. Thus, Arf6 might serve to recruit TRE17 to target CIE cargo proteins for deubiquitylation.
We found that expression of TRE17 by itself can stabilize MHCI and increase the surface level of the protein. Although it has not been clarified whether endogenous MARCH proteins contribute to the turnover of MHCI proteins in HeLa cells, this result indicates that the trafficking of MHCI is regulated by endogenous ubiquitylation to some extent. Expression of TRE17 likely deubiquitylates endogenously ubiquitylated MHCI and leads to its stabilization. Thus, the effect of TRE17 is not limited to suppression of overexpressed MARCH proteins.
Whether endogenous TRE17 contributes to stabilization of CIE cargo proteins remains to be determined. Although TRE17 expression in normal tissues appears to be limited to the testis (Paulding et al., 2003), low levels have been reported in HeLa cells. We examined whether knockdown of TRE17 could affect trafficking of CIE cargo proteins in HeLa cells, but unfortunately we could not consistently detect substantial levels of endogenous TRE17 (unpublished observations). Nevertheless, we believe that the effects of TRE17 on CIE cargo trafficking are very likely physiologically relevant because, even upon overexpression, other USPs were incapable of regulating CIE cargo as TRE17 does. Most notably, USP32, which shares 98% identity with TRE17 in the catalytic domain, failed to reverse the juxtanuclear accumulation of CIE cargo. Furthermore, the overexpression phenotype of TRE17 is highly relevant to oncogenesis, as TRE17 is translocated and vastly overexpressed in two human tumors.
TRE17 was originally identified as an oncogene from human Ewing's sarcoma (Nakamura et al., 1992) and is genetically linked to aneurysmal bone cyst (ABC) and nodular fasciitis, tumors in which TRE17 is overexpressed because of a chromosomal translocation (Oliveira et al., 2004a; Oliveira et al., 2004b; Oliveira et al., 2005; Panagopoulos et al., 2008; Erickson-Johnson et al., 2011). In addition, it has been shown that injection of cells overexpressing TRE17 into nude mice induces formation of ABC-like tumors in a DUB-activity-dependent manner (Ye et al., 2010). The overexpression of TRE17 induced expression of the matrix metalloproteinases (MMPs) MMP-9 and MMP-10 (Ye et al., 2010), MMPs known to be enriched in ABC tumors (Kumta et al., 2003). The induction of MMP-9 was mediated by NF-κB, and inhibition of NF-κB in TRE17-overexpressing cells dramatically reduced tumorigenesis (Ye et al., 2010; Pringle et al., 2012). Ruekert and Haucke (Ruekert and Haucke, 2012) have also shown that TRE17 is involved in the regulation of cell migration and cytokinesis through the Arf6-dependent pathway, indicating that TRE17 contributes to oncogenesis through the activation of Arf6. In addition to these previous studies, our findings might provide another mechanism by which overexpression of TRE17 contributes to tumorigenesis. The expression levels of CD44, CD98 and CD147, cell surface proteins that we previously identified as CIE cargo (Eyster et al., 2009), are known to be elevated in various types of cancers. CD44 is a hyaluronan receptor that mediates cell–matrix interactions and CD98 influences integrin trafficking and its signaling, both of which are related to growth, survival, motility and invasion of cancer cells (Zöller, 2011; Louderbough and Schroeder, 2011; Cantor and Ginsberg, 2012). CD147 also associates with integrins (Berditchevski et al., 1997) and elevated levels of cell surface CD147 on cancer cells induces production of MMPs in neighboring fibroblasts (Iacono et al., 2007). By preventing the lysosomal degradation of these CIE cargo proteins, TRE17 would increase the cell surface level of these proteins, promoting tumor formation and progression. An examination of whether levels of these CIE cargo proteins are elevated in TRE17-expressing tumors and investigation of how they are upregulated in the tumors will reveal the pathogenesis of TRE17-dependent tumors and also help to elucidate the physiological relevance of TRE17.
To our knowledge, TRE17 is the first identified DUB that regulates trafficking of multiple CIE cargoes, including MHCI, CD98, CD44 and Tac. CIE is in general a non-selective process, however, once internalized, CIE cargo proteins follow different itineraries in the cell (Eyster et al., 2011). We recently identified a mechanism that facilitates recycling of specific CIE cargo proteins (Maldonado-Báez et al., 2013). Reversible ubiquitylation mediated by MARCH and TRE17 might play an additional role in regulating the trafficking of CIE cargo proteins, providing a quality control check at the level of the endosome. Elucidating how their activities are regulated will contribute to our understanding of regulatory mechanisms of CIE pathways.
MATERIALS AND METHODS
Antibodies and plasmids
Mouse monoclonal antibodies to MHCI (clone W6/32; IgG2a), CD44 (clone BJ18; IgG1) and CD98 (clone MEM-108; IgG1) were purchased from Biolegend (San Diego, CA) and used for antibody internalization. Mouse monoclonal antibodies to transferrin receptor (clone 236-15375; IgG1) were purchased from Invitrogen Molecular Probes (Grand Island, NY). Mouse monoclonal antibodies to GFP (clone B34; IgG1) and chicken anti-GFP antibodies were purchased from Covance Research Products (Princeton, NJ) and Merk Millipore (Billerica, MA), respectively, and used for immunofluorescence. Rabbit anti-GFP antibodies were purchased from Invitrogen Molecular Probes and used for immunoblotting and immunofluorescence. Rabbit anti-FLAG antibodies were purchased from Sigma-Aldrich (St Louis, MO) and used for immunofluorescence and immunoblotting. Mouse monoclonal antibodies to ubiquitin (clone P4D1; IgG1) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antibodies to SNAP (clone MGMT-214; IgG1) were purchased from Sigma-Aldrich. All Alexa-Fluor-conjugated secondary antibodies were purchased from Invitrogen Molecular Probes (Grand Island, NY).
MARCH1–FLAG, MARCH4–FLAG and MARCH8–FLAG were as described previously (Eyster et al., 2011). GFP–TRE17 and GFP–TRE17/USP− are as described previously (Martinu et al., 2004; Shen et al., 2005). cDNA of USP8 was generously provided by Yihong Ye (NIDDK, Bethesda, MD). USP8 and USP32 were amplified by PCR and subcloned into pEGFP-C1 (Clontech, Mountain View, CA) to generate GFP–USP8 and GFP–USP32. GFP–USP8–Ras-C20 was generated by inserting the farnesylation signal from pEGFP-F (Clontech), which contains 20 amino acids from the C-terminus of c-Ha-Ras, and the USP8 gene into pEGFP-C1. CD98–SNAP was as described previously (Eyster et al., 2011). SNAP–Tac was generated in pcDNA3.1 using the signal peptide of hen egg lysozyme (MRSLLILVLCFLPLAALG) introduced before the second amino acid of the SNAP tag (DKDCEMKR) sequence followed by a (GGGGS)2 linker and then extracellular, transmembrane and cytoplasmic tail domains of Tac.
Cell culture and transfection
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Lonza) with 4.5 g/l glucose, 10% fetal bovine serum, 2 mM glutamine, 100 units/ml penicillin and 100 µg/ml streptomycin and grown under a 5% CO2 atmosphere at 37°C. Beas2B cells were cultured as HeLa cells except for a glucose concentration of 1.0 g/l. For transfections, HeLa or Beas2B cells were grown on coverslips (for immunofluorescence) or six-well dishes (for pull-down) and transfected using X-tremeGENE, Fugene 6 (both from Roche Diagnostics, Indianapolis, IN), or Lipofetamine 2000 (Life Technologies, Grand Island, NY) according to the manufacturers' instructions. Transfections with multiple plasmids were performed with an equal amount of each plasmid. Experiments were performed 20–24 h after transfection.
Antibody internalization and immunofluorescence
HeLa cells or Beas2B cells were plated on glass coverslips 2 days prior to use. The next day, cells were transfected and further cultured for 20–24 h. For antibody internalization experiments, cells were preincubated with primary antibodies to MHCI (5 µg/ml), CD44 (2.5 µg/ml) or CD98 (2.5 µg/ml) for 1 h at 37°C and then transferred to fresh medium containing 25 mM NH4Cl for 2 h. Cells were fixed for 10 min in 2% formaldehyde in PBS, rinsed with PBS, and incubated with primary antibodies in PBS containing 10% fetal bovine serum with 0.2% saponin. Alexa-Fluor-conjugated secondary antibodies were used to detect the primary antibodies. All images were obtained using 510 LSM (Zeiss, Thornwood, NY), FV10i-LIV (Olympus, Tokyo, Japan), and TCS-SP5 (Leica Microsystems, Wetzlar, Germany) confocal microscopes. To quantify the effect of DUB proteins, 50–60 cells expressing both MARCH8 and GFP-tagged proteins were selected and the number of cells where cargo proteins were downregulated and/or accumulated at a juxtanuclear compartment were counted. In experiments that followed the long-term fate of surface proteins, HeLa cells were preincubated with anti-MHCI antibodies at 4°C for 1 h to label the surface MHCI, washed with medium, and further incubated at 37°C for 20 h. Cells were fixed and incubated with anti-mouse-IgG2a antibody conjugated to Alexa Fluor 594 without saponin to detect the surface MHCI–antibody complex. Then transfected GFP-tagged proteins were detected by anti-GFP antibody as described above. Images of the surface anti-MHCI antibodies were obtained using a 510 LSM confocal microscope with the pinhole completely opened. To quantify the surface level of MHCI, 40–50 transfected cells were selected and the fluorescence signals were quantified with MetaMorph (Molecular Devices, Sunnyvale, CA). The value of each cell was normalized to the cell area. To calculate the fold increase from the control (GFP-transfected cells), the mean value of the GFP control was calculated and then intensity of each cell was normalized to the mean value of GFP control and plotted.
Pulldown and immunofluorescence of SNAP-tagged proteins
For pulldown experiments, HeLa cells (six-well plate) were transfected with CD98–SNAP or SNAP–Tac, and GFP-tagged proteins with or without MARCH8–FLAG. After 24 h, cells were labeled with O6-benzylguanine-PEG4–biotin substrate (3 mM) for 4 h at 37°C. Cells were rinsed three times with PBS, lifted, and pelleted at 300 g. Cell pellets were solubilized in 50 µl of lysis buffer [50 mM Tris-HCl, pH 7.4, 0.25 M NaCl, 0.1% Triton X-100, 1 mM EDTA, 1 mM dithiothreitol and complete protease inhibitor tablets (Roche Diagnostics, Indianapolis, IN)] containing 400 µM O6-benzylguanine-NH2 and incubated on ice for 15 min. The cell extract was denatured by adding SDS to the final concentration of 1% and boiled for 15 min. The SDS was quenched by adding 50 µl of 10% Triton X-100 and 450 µl of lysis buffer without protease inhibitors and placed on ice for 30 min. The lysate was centrifuged at 13,000 g for 5 min, and 12 µl of a 1∶1 slury of streptavidin–agarose (Sigma-Aldrich) was added to the supernatant. The lysate was rocked at room temperature for 1 h, and the beads were washed three times with lysis buffer without protease inhibitors. The beads were boiled for 10 min with 2× sample buffer to elute the bound proteins. The eluate was separated by SDS-PAGE (4–12% Tris-glycine gel; Invitrogen), transferred onto nitrocellulose membrane and immunoblotted with the specified antibodies. The intensities of anti-ubiquitin blot and anti-SNAP blot of the pulled-down fraction were quantified with Odyssey Imaging System (LI-COR, Lincoln, NE).
For immunofluorescence, HeLa cells were incubated with DMEM containing O6-benzylguanine conjugated to Alexa Fluor 594 to label the SNAP-tagged proteins at 37°C for 1 h. Cells were than washed and transferred to fresh medium containing 25 mM NH4Cl for 2 h. Cells were stained as describe above.
Supplementary Material
Acknowledgments
The authors would like to thank Chad Williamson and Darya Karabacheva (NHLBI, Bethesda, MD) for comments on the manuscript and the Donaldson laboratory group for discussions. We also thank Nelson Cole (NHLBI, Bethesda, MD) for the SNAP tag constructs and Yihong Yi (NIDDK, Bethesda, MD) for USP8 construct. Microscopes used were in the NHLBI Light Microscopy Core Facility.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
Y.F., M.M.C., Y.K. and J.G.D. planned the experiments; Y.F. executed the experiments; Y.F. and J.G.D. interpreted the data and prepared the draft manuscript; Y.F., M.M.C., Y.K. and J.G.D. edited the manuscript.
Funding
This work was supported by the Intramural Research Program in the National Heart, Lung and Blood Institute at the National Institutes of Health [grant number HL006060] to J.G.D.; and by a grant from the National Cancer Institute [grant number CA126452] to M.M.C. This work was also supported by the Institutional Program for Young Researcher Overseas Visits and the Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation from the Ministry of Education, Science, Sports and Culture of Japan to Y.F. and a research grant [grant number 26440044] from the Ministry of Education, Science, Sports and Culture of Japan to Y.K. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.156786/-/DC1
References
- Balut C. M., Loch C. M., Devor D. C. (2011). Role of ubiquitylation and USP8-dependent deubiquitylation in the endocytosis and lysosomal targeting of plasma membrane KCa3.1. FASEB J. 25, 3938–3948. 10.1096/fj.11-187005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartee E., Mansouri M., Hovey Nerenberg B. T., Gouveia K., Früh K. (2004). Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J. Virol. 78, 1109–1120. 10.1128/JVI.78.3.1109-1120.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berditchevski F., Chang S., Bodorova J., Hemler M. E. (1997). Generation of monoclonal antibodies to integrin-associated proteins. Evidence that alpha3beta1 complexes with EMMPRIN/basigin/OX47/M6. J. Biol. Chem. 272, 29174–29180. 10.1074/jbc.272.46.29174 [DOI] [PubMed] [Google Scholar]
- Berlin I., Higginbotham K. M., Dise R. S., Sierra M. I., Nash P. D. (2010a). The deubiquitinating enzyme USP8 promotes trafficking and degradation of the chemokine receptor 4 at the sorting endosome. J. Biol. Chem. 285, 37895–37908. 10.1074/jbc.M110.129411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin I., Schwartz H., Nash P. D. (2010b). Regulation of epidermal growth factor receptor ubiquitination and trafficking by the USP8·STAM complex. J. Biol. Chem. 285, 34909–34921. 10.1074/jbc.M109.016287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor J. M., Ginsberg M. H. (2012). CD98 at the crossroads of adaptive immunity and cancer. J. Cell Sci. 125, 1373–1382. 10.1242/jcs.096040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague M. J., Liu H., Urbé S. (2012a). Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev. Cell 23, 457–467. 10.1016/j.devcel.2012.08.011 [DOI] [PubMed] [Google Scholar]
- Clague M. J., Coulson J. M., Urbé S. (2012b). Cellular functions of the DUBs. J. Cell Sci. 125, 277–286. 10.1242/jcs.090985 [DOI] [PubMed] [Google Scholar]
- Erickson-Johnson M. R., Chou M. M., Evers B. R., Roth C. W., Seys A. R., Jin L., Ye Y., Lau A. W., Wang X., Oliveira A. M. (2011). Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab. Invest. 91, 1427–1433. 10.1038/labinvest.2011.118 [DOI] [PubMed] [Google Scholar]
- Eyster C. A., Higginson J. D., Huebner R., Porat-Shliom N., Weigert R., Wu W. W., Shen R. F., Donaldson J. G. (2009). Discovery of new cargo proteins that enter cells through clathrin-independent endocytosis. Traffic 10, 590–599. 10.1111/j.1600-0854.2009.00894.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyster C. A., Cole N. B., Petersen S., Viswanathan K., Früh K., Donaldson J. G. (2011). MARCH ubiquitin ligases alter the itinerary of clathrin-independent cargo from recycling to degradation. Mol. Biol. Cell 22, 3218–3230. 10.1091/mbc.E10-11-0874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H., Iwabu Y., Tokunaga K., Tanaka Y. (2013). Membrane-associated RING-CH (MARCH) 8 mediates the ubiquitination and lysosomal degradation of the transferrin receptor. J. Cell Sci. 126, 2798–2809. 10.1242/jcs.119909 [DOI] [PubMed] [Google Scholar]
- Gautier A., Juillerat A., Heinis C., Corrêa I. R., Jr, Kindermann M., Beaufils F., Johnsson K. (2008). An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 15, 128–136. 10.1016/j.chembiol.2008.01.007 [DOI] [PubMed] [Google Scholar]
- Hancock J. F., Cadwallader K., Paterson H., Marshall C. J. (1991). A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 10, 4033–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacono K. T., Brown A. L., Greene M. I., Saouaf S. J. (2007). CD147 immunoglobulin superfamily receptor function and role in pathology. Exp. Mol. Pathol. 83, 283–295. 10.1016/j.yexmp.2007.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumta S. M., Huang L., Cheng Y. Y., Chow L. T. C., Lee K. M., Zheng M. H. (2003). Expression of VEGF and MMP-9 in giant cell tumor of bone and other osteolytic lesions. Life Sci. 73, 1427–1436. 10.1016/S0024-3205(03)00434-X [DOI] [PubMed] [Google Scholar]
- Lau A. W., Pringle L. M., Quick L., Riquelme D. N., Ye Y., Oliveira A. M., Chou M. M. (2010). TRE17/ubiquitin-specific protease 6 (USP6) oncogene translocated in aneurysmal bone cyst blocks osteoblastic maturation via an autocrine mechanism involving bone morphogenetic protein dysregulation. J. Biol. Chem. 285, 37111–37120. 10.1074/jbc.M110.175133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Bengtson M. H., Ulbrich A., Matsuda A., Reddy V. A., Orth A., Chanda S. K., Batalov S., Joazeiro C. A. P. (2008). Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS ONE 3, e1487. 10.1371/journal.pone.0001487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louderbough J. M. V., Schroeder J. A. (2011). Understanding the dual nature of CD44 in breast cancer progression. Mol. Cancer Res. 9, 1573–1586. 10.1158/1541-7786.MCR-11-0156 [DOI] [PubMed] [Google Scholar]
- Maldonado-Báez L., Cole N. B., Krämer H., Donaldson J. G. (2013). Microtubule-dependent endosomal sorting of clathrin-independent cargo by Hook1. J. Cell Biol. 201, 233–247. 10.1083/jcb.201208172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinu L., Masuda-Robens J. M., Robertson S. E., Santy L. C., Casanova J. E., Chou M. M. (2004). The TBC (Tre-2/Bub2/Cdc16) domain protein TRE17 regulates plasma membrane-endosomal trafficking through activation of Arf6. Mol. Cell. Biol. 24, 9752–9762. 10.1128/MCB.24.22.9752-9762.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda-Robens J. M., Kutney S. N., Qi H., Chou M. M. (2003). The TRE17 oncogene encodes a component of a novel effector pathway for Rho GTPases Cdc42 and Rac1 and stimulates actin remodeling. Mol. Cell. Biol. 23, 2151–2161. 10.1128/MCB.23.6.2151-2161.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay J., Wang X., Ding J. A., Buss J. E., Ambrosio L. (2011). H-ras resides on clathrin-independent ARF6 vesicles that harbor little RAF-1, but not on clathrin-dependent endosomes. Biochim. Biophys. Acta 1813, 298–307 [DOI] [PubMed] [Google Scholar]
- Nakamura T., Hillova J., Mariage-Samson R., Onno M., Huebner K., Cannizzaro L. A., Boghosian-Sell L., Croce C. M., Hill M. (1992). A novel transcriptional unit of the tre oncogene widely expressed in human cancer cells. Oncogene 7, 733–741 [PubMed] [Google Scholar]
- Nakamura N., Fukuda H., Kato A., Hirose S. (2005). MARCH-II is a syntaxin-6-binding protein involved in endosomal trafficking. Mol. Biol. Cell 16, 1696–1710. 10.1091/mbc.E04-03-0216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky N., Weigert R., Donaldson J. G. (2003). Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol. Biol. Cell 14, 417–431. 10.1091/mbc.02-04-0053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky N., Weigert R., Donaldson J. G. (2004). Characterization of a nonclathrin endocytic pathway: membrane cargo and lipid requirements. Mol. Biol. Cell 15, 3542–3552. 10.1091/mbc.E04-02-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira A. M., Hsi B. L., Weremowicz S., Rosenberg A. E., Dal Cin P., Joseph N., Bridge J. A., Perez-Atayde A. R., Fletcher J. A. (2004a). USP6 (Tre2) fusion oncogenes in aneurysmal bone cyst. Cancer Res. 64, 1920–1923. 10.1158/0008-5472.CAN-03-2827 [DOI] [PubMed] [Google Scholar]
- Oliveira A. M., Perez-Atayde A. R., Inwards C. Y., Medeiros F., Derr V., Hsi B. L., Gebhardt M. C., Rosenberg A. E., Fletcher J. A. (2004b). USP6 and CDH11 oncogenes identify the neoplastic cell in primary aneurysmal bone cysts and are absent in so-called secondary aneurysmal bone cysts. Am. J. Pathol. 165, 1773–1780. 10.1016/S0002-9440(10)63432-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira A. M., Perez-Atayde A. R., Dal Cin P., Gebhardt M. C., Chen C. J., Neff J. R., Demetri G. D., Rosenberg A. E., Bridge J. A., Fletcher J. A. (2005). Aneurysmal bone cyst variant translocations upregulate USP6 transcription by promoter swapping with the ZNF9, COL1A1, TRAP150, and OMD genes. Oncogene 24, 3419–3426. 10.1038/sj.onc.1208506 [DOI] [PubMed] [Google Scholar]
- Panagopoulos I., Mertens F., Löfvenberg R., Mandahl N. (2008). Fusion of the COL1A1 and USP6 genes in a benign bone tumor. Cancer Genet. Cytogenet. 180, 70–73. 10.1016/j.cancergencyto.2007.09.017 [DOI] [PubMed] [Google Scholar]
- Paulding C. A., Ruvolo M., Haber D. A. (2003). The Tre2 (USP6) oncogene is a hominoid-specific gene. Proc. Natl. Acad. Sci. USA 100, 2507–2511. 10.1073/pnas.0437015100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper R. C., Lehner P. J. (2011). Endosomal transport via ubiquitination. Trends Cell Biol. 21, 647–655. 10.1016/j.tcb.2011.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porat-Shliom N., Kloog Y., Donaldson J. G. (2008). A unique platform for H-Ras signaling involving clathrin-independent endocytosis. Mol. Biol. Cell 19, 765–775. 10.1091/mbc.E07-08-0841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle L. M., Young R., Quick L., Riquelme D. N., Oliveira A. M., May M. J., Chou M. M. (2012). Atypical mechanism of NF-κB activation by TRE17/ubiquitin-specific protease 6 (USP6) oncogene and its requirement in tumorigenesis. Oncogene 31, 3525–3535. 10.1038/onc.2011.520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueckert C., Haucke V. (2012). The oncogenic TBC domain protein USP6/TRE17 regulates cell migration and cytokinesis. Biol. Cell 104, 22–33. 10.1111/boc.201100108 [DOI] [PubMed] [Google Scholar]
- Shen C., Ye Y., Robertson S. E., Lau A. W., Mak D. O. D., Chou M. M. (2005). Calcium/calmodulin regulates ubiquitination of the ubiquitin-specific protease TRE17/USP6. J. Biol. Chem. 280, 35967–35973. 10.1074/jbc.M505220200 [DOI] [PubMed] [Google Scholar]
- Ye Y., Pringle L. M., Lau A. W., Riquelme D. N., Wang H., Jiang T., Lev D., Welman A., Blobel G. A., Oliveira A. M. et al. (2010). TRE17/USP6 oncogene translocated in aneurysmal bone cyst induces matrix metalloproteinase production via activation of NF-kappaB. Oncogene 29, 3619–3629. 10.1038/onc.2010.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R., Tomkovicz V. R., Butler P. L., Ochoa L. A., Peterson Z. J., Snyder P. M. (2013). Ubiquitin-specific peptidase 8 (USP8) regulates endosomal trafficking of the epithelial Na+ channel. J. Biol. Chem. 288, 5389–5397. 10.1074/jbc.M112.425272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zöller M. (2011). CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 11, 254–267. 10.1038/nrc3023 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.