

Acute kidney injury (AKI) has remained a significant health care concern with high mortality rates for several decades. The overall mortality rate of AKI is estimated to range between 45-70% [1-7]. AKI may be observed in up to 5% of hospitalized patients, and a diagnosis of AKI increases the risk of mortality 5.5 to 6.5-fold as compared to a similarly ill patient without AKI [3, 8]. In addition, AKI is now recognized as a potential cause of chronic kidney disease (CKD), as a significant number of patients may require continued dialytic therapy [3, 5-15], or may show a decline of function following discharge [7, 10, 13, 16]. Recent estimates indicate that up to 3% of the yearly incidence of End-Stage Renal Disease in North America may be attributable, at least in part, to AKI [7, 17].
AKI can be classified clinically based on the genesis of the disorder as pre-renal, renal or post-renal. Ischemia due to hypotension or sepsis is the most common cause of human AKI [18] associated with frank renal injury. The activation of factors associated with vasoconstriction represents an important component of the renal injury process [19, 20]. It stands to reason that potential treatment modalities targeting vascular function in the setting of AKI may positively impact the dismal mortality rates currently achieved by supportive care alone.
This brief review will summarize recent advances on the understanding of renal endothelial function in the setting of AKI. We will consider primary roles of the endothelium in maintaining vascular tone and in influencing inflammation during progression of ischemia injury and highlight pathways for which interventional therapies have been shown efficacious in pre-clinical studies. Finally, we will consider the possible connection between AKI and CKD as a continuum, and reflect on the concept that promotion of vascular regeneration may represent a means to improve long term function following AKI.
Hemodynamic changes
The hallmark feature of AKI is a reduction in GFR, which implies an underlying impairment in hemodynamic regulation [21-25]. Indeed, this disorder was originally termed “vasomotor” nephropathy [21] and was characterized by a sustained increase in renal vascular resistance (RVR) [19, 26-28].
Renal hemodynamic responses have been studied in animal models in response to renal ischemia reperfusion injury. After release of renal artery occlusion, total renal blood flow (RBF) is restored to baseline levels within minutes followed by a subsequent decline in RBF, which takes place over several hours [29, 30] [31-33]. Methods that discriminate regional blood flow in the kidney, suggest that outer medullary RBF undergoes an earlier and more significant impairment relative to whole kidney RBF [32-35]. The outer medulla is normally hypoxic under physiological conditions, and sustained reductions in outer medullary flow are considered to exacerbate hypoxia and contribute to the more profound degree of morphological damage observed in this region [34, 36].
The increased RVR can be viewed as a vascular response to cellular events triggered by the initial ischemia. Increased RVR may manifest as the activation of vasoactive compounds, reactive oxygen species and/or inflammatory pathways which can affect perfusion. Renal endothelial cells may be the target or the culprit of these responses.
When viewed from a clinical perspective, an increase in RVR triggered during reperfusion may represent a critical shift in the pathophysiological process driving AKI, in which systemic complications initiating a reduction in perfusion activate renal-intrinsic responses sustaining reduced perfusion and fueling parenchymal tissue injury. Such a shift may represent what has been referred to as the extension phase of AKI by Molitoris and Sutton and has been suggested as promising clinical window for therapeutic intervention, since the restoration of blood flow at this time would mitigate subsequent hypoxic damage [37]. However, because a number of different factors influence RVR and their contribution may change during injury progression, some therapies may be only effective in early stages of injury may have reduced impact later in the injury process. In practice, the clinical window of interventional opportunity may be short, and missed due to a lack of accurate and timely assessment of GFR [38]. Therefore, the utility of potential novel therapies will require coordination with newer methods in biomarker discovery to more accurately assess the phases of AKI [39].
Mediators of vasoconstriction
No single factor is responsible for reduced RBF, however vasoconstriction, tubular congestion, edema and inflammation are all likely to contribute to the increased RVR following ischemia reperfusion, with vasoconstriction representing the most immediate of these responses.
Several factors have been proposed to modulate renal vascular tone following I/R. For example, evidence indicates impaired proximal Na reabsorption due to energy depletion activates tubuloglomerular feedback and adenosine-mediated vasoconstriction following I/R [40]. A host of other potential vasoconstrictors may be activated and contribute to reduced RBF following I/R injury, including the systemic activation of the sympathetic nervous system, renin-angiotensin II system, endothelin A, prostaglandins, and platelet activating factors. Several studies have been undertaken in which inhibition of these factors provides a partial preservation of RBF and/or GFR and diminishes the severity of AKI [41-52]. However, because vasoconstriction is mediated by a number of redundant pathways, the blockade of any single pathway is not likely to completely protect against injury. Moreover, such studies are almost always conducted by administration of an antagonist near the time of experimentally-induced reperfusion, while studies are rarely conducted to determine if delayed administration can reverse the course of injury after GFR becomes compromised. In some cases when treatments are delayed, there is a reduced efficacy in ameliorating AKI [53] [54] [55].
Endothelial involvement
Significant evidence exists that altered renal endothelial function contributes to a reduction in RBF following I/R. Sutton et al., demonstrated that I/R injury in rats alters cytoskeletal organization of small arterioles and endothelial cells [19, 56, 57]. These alterations contribute to the breakdown of VE-cadherin-mediated intercellular adhesion and the integrity of the tight junctions of peritubular capillaries as evidenced by leakiness of high molecular weight dextrans (>300,000 Da) into the interstitial space [57]. It has been suggested that increased leakiness may compromise renal perfusion by compressing peritubular capillaries and exacerbating erythrocyte trapping [19, 23, 58, 59].
Alterations in EC function may influence vasodilation, coagulation cascades and/or inflammatory processes [19, 60, 61]. Endothelial dysfunction is characterized, in part, by an impaired dilator capacity, which is often attributed to reduced production of nitric oxide. NOS3, (endothelial NOS) is produced at high levels in endothelial cells, particularly in the renal medulla [62], where it preserves medullary blood flow in response to renal vasoconstrictors such as Ang II [63]. However, following renal injury, there is impaired endothelial NOS (NOS3) function, demonstrated by a loss of vasodilator responses to acetylcholine and bradykinin [64]. This reduction in normal endothelial function does not appear to be the result of reduced NOS3 protein levels, but may result from inhibition of enzyme activity since bradykinin failed to produce measurable levels of NO in post-ischemic kidneys measured in real time by electrochemical methods [64].
A role for NOS3 in preservation of blood flow is suggested by the greater degree of renal damage that occurs in NOS3 knockout mice in response to endotoxin [65] or by studies in which inhibition of compensatory vasodilation in AKI with L-NAME or cyclooxygenase inhibitors, exacerbates the degree of renal injury in response to ischemia reperfusion[30]. Interestingly, renal autoregulatory responses are impaired for at least 7 days post-ischemia endothelial responses, well into the recovery phases when total RBF has returned to baseline levels, suggesting prolonged endothelial dysfunction continues despite an overall recovery response [66, 67] [68]. Such impaired responses have been suggested to prolong injury or predispose further damage, as evidence of fresh ischemic injury has been observed in biopsies from in AKI patients treated with hemodialysis during the recovery process. [69]
Impaired NOS3 activity may result from endothelial damage or from extrinsic free radical activity altering NO activity. Reactive oxygen species may influence the effects of vasocontrictors and vasodilators and lead to an increase in renal vascular resistance. Increased renal superoxide production augments renal vasoconstrictor responses particularly in the renal medulla. Superoxide promotes vasoconstriction and enhances the reactivity of Ang II in the renal cortex and medulla [70]. Oxidative stress in the kidney also enhances the vasoconstrictor effects of adenosine [71]. Superoxide, by virtue of its known activity to convert NO to peroxynitrite, may block the normal homeostatic mechanism maintaining medullary perfusion and these responses may be normalized with the use of antioxidants [70].
Several studies with the goal of increasing/preserving renal NO activity have demonstrated protective effects in AKI. For example, administration of L-arginine, the NO donor molsidomine, or the eNOS cofactor tetrohydroboipterin can preserve renal vascular perfusion and attenuate AKI induced by I/R [72-78]. Pharmacological approaches to impact NOS3 signaling by administration of PDE5 inhibitors have also been shown to ameliorate the severity of I/R induced AKI in rats[79] [80] or in pigs following cardiopulmonary bypass surgery [81].
Using videomicroscopy of blood vessels in the kidney, Yamamoto et al., reported that renal perfusion in peritubular vessels was compromised within minutes of unclamping, characterized by sluggish and occasionally retrograde blood flow [61]. Infusion of human umbilical vein endothelial cells to athymic nude rats, or other non-endothelial cells harboring the NOS3 gene expression construct, protected against this early compromise in blood flow [60]. Similar results were observed in endothelial-like cells were generated from mesenchymal cells in vitro and administered to rats in the setting of renal I/R [82]. Taken together, these data suggest that endothelial function, particularly via NOS3 activity, has protective effects on the severity of AKI.
While vasoconstriction contributes to the initial loss of GFR, it remains unclear whether therapeutic targeting can be used effectively to treat AKI, in part because tissue injury, once established, prevents vasodilatory therapy to generate desired effects. Perhaps the most well-documented example of this principle is represented by the use of “renal-dose” dopamine. At low doses (i.e., 3 μg/kg/min) dopamine is vasodilatory and promotes diuresis primarily through the D-1 receptor and has protective effects in preclinical models of AKI [83]. Despite this, multiple studies have consistently demonstrated that renal-dose dopamine or the dopamine agonist fenoldapam, do not influence outcome of AKI in patients despite producing diuresis [84-88].
Endothelial cell involvement in inflammation
Despite some uncertainty about the role of specific leukocyte populations in human AKI, an infiltration of leukocytes, primarily neutrophils occurs fairly rapidly following ischemia/reperfusion injury in rodents [89-91]. For more thorough discussion on the role of inflammation in AKI, the reader is referred to other articles on this topic [92, 93]. Leukocyte adhesion to activated endothelial cells following I/R may exacerbate injury by contributing to peritubular capillary congestion, by producing molecules that affect vascular tone (e.g., ROS or vasoactive lipids), or by liberating cytokines which contribute to parenchymal cell injury [36, 92].
Reductions in medullary blood flow occur prior to changes in total RBF. Whether leukocyte adhesion is associated with this decreased MBF in unclear since there have been no studies attempting to correlate medullary leukocyte adhesion and medullary perfusion. Despite this, we advocate the proposed view that leukocyte adhesion represents a critical step in the switch to the extension phase of AKI and therefore may partially explain why vasodilator therapies alone are ineffective in established AKI. For this reason, there has been increased interest in understanding endothelial/leukocyte interactions and their potential targeting to mitigate the severity of I/R induced AKI.
There is an increase surface expression of the leukocyte adhesion molecules ICAM-1 and P and E-selectin on endothelial cells in response to injury [94-96]. In vivo imaging studies have shown leukocyte adhesiveness to the peritubular capillary wall occurs within hours of reperfusion [97, 98]. The endothelium may also be the source of chemoattractant factors, such as fractalkine (CX3CL1), which is expressed following renal injury and may promote infiltration of macrophages.
Treatments geared toward reducing endothelial/leukocyte interactions by targeting these endothelial adhesion molecules preserve blood flow and protect against renal damage in I/R induced AKI [94-96]. Blocking P selectin increases renal blood flow following I/R [29]. A landmark study in this area was published by Kelly et al., demonstrating that inhibition of ICAM or genetic deletion of ICAM resulted in a significant protection of ischemia reperfusion injury [99]. In human AKI, ICAM antibody pretreatment protected against delayed graft function in transplant recipients, relative to a group of recipients receiving the contralateral kidneys from the same donors. Interestingly, immunoneutralization following transplant did not affect the development of DGF [100-102].
Microthrombus formation has been described in renal I/R as well as in transplant biopsies[19, 103]. Although these pathways have not received prominent attention, the activation of prothrombotic events by activated endothelial cells represents a potentially important step in I/R injury. For example, tissue factor, a membranous glycoprotein that functions as a receptor for the coagulation system and is normally not present on quiescent endothelial cells, is expressed on peritubular capillary endothelial cells following renal I/R injury. Inhibition of endothelial tissue factor expression in rats following I/R injury results in significant improvement in mortality and preservation of overall morphology [104]. In addition, there is evidence that thrombomodulin, a thrombin inhibitor, is reduced on the surface of endothelial cells following renal ischemia reperfusion [19]. It was recently shown that administration of soluble thrombomodulin inhibitor to rats attenuated I/R induced AKI by preserving perfusion within renal capillaries and inhibiting leukocyte adhesion [98].
Additional therapies with potential anti thrombotic activity have received consideration in the setting of acute kidney injury. For example, activated protein C binds to receptors on the surface of endothelial cells where is has antithrombotic and direct cytoprotective effects on endothelial cells mediated by the protease activated receptor-1 (PAR-1) [105]. In a model of kidney ischemia reperfusion injury, APC administration preserved renal blood flow and protected against renal damage and inflammation [105].
It has been suggested that low protein C levels may predict morality in sepsis induced AKI. Administration of APC to septic rats improves renal function and lowers mortality. In a clinical trial of human sepsis patients, Drotrecogin (a recombinant human activated protein C) significantly decreased 28 d mortality over the placebo group [106]. However, a more recent study of sepsis patients, recombinant APC provided no significant difference in mortality at 28 or 90 days [107]. Whether APC might provide beneficial effects in AKI in settings other than sepsis has not been evaluated.
Regeneration
The kidney is well known for its impressive regenerative capacity. In patients surviving AKI, recovery of renal function is generally expected. In many patients, recovery may not be complete, while some patients may develop chronic renal insufficiency and/or become susceptible to future episodes of AKI [10, 36]. The processes of renal repair and recovery after I/R injury have been studied extensively in animal models. In most studies, GFR and RBF return to basal levels rapidly (i.e., within days), while repair of damaged proximal tubules leads to the restoration of renal histological structure over several weeks. Despite this, even in otherwise healthy animals, there are persistent alterations in renal structure and function, and post AKI animals become predisposed to chronic disease.
Several theories have been proposed to describe the mechanisms by which predisposition to CKD may ensue following I/R injury [36, 108-111]. One possible mechanism relates to the reduction in peritubular capillary density that persists following the otherwise uneventful recovery from AKI [112]. Our laboratory demonstrated that there was a 30-50% reduction of PTC density which was present following the resolution of GFR, RBF and tubular morphology, at 4 weeks post I/R in rats [112, 113]. Similar results have been reported in other species such as mice [114], and in human kidneys following recovery from delayed graft function [115, 116].
The potential functional relationship between peritubular capillary loss and progressive fibrosis following I/R injury has been summarized in previous reviews [117-119]. In general, 3 possible mechanisms may contribute to the progressive development of renal fibrosis following injury, these include: 1) the exacerbation of hypoxia as a well-known physiological trigger promoting renal fibrosis [113], 2) an alteration in renal hemodynamic control, particularly in the renal medulla, which accentuates salt-sensitive hypertension and fuels progression of CKD [120, 121], and 3) the proliferation of new fibroblasts secondary to endothelial-mesenchymal transition [122]. As described below, treatments which ameliorate capillary loss post AKI reduce the development of secondary CKD [123]. Therefore strategies to preserve or restore renal vascular structure following injury represent an important potential area for therapeutic development.
The cellular basis of PTC rarefaction is incompletely understood. It is reasonable to suggest that apoptotic cell death underlies vascular (i.e., endothelial) cell loss following the induction of acute injury. Lee et al., demonstrated peritubular capillary endothelial cell apoptosis following severe liver I/R in mice which results in a subsequent AKI [124]. Recent studies have also reported apoptotic cells following acute injury in the region of the peritubular vasculature, but positive identification that EC are primarily affected is not clear [125]. In contrast, Hörbelt et al., used Tie-GFP mice to label renal endothelial cells and were not able to identify apoptotic endothelial cells in response to renal ischemia reperfusion, but did identify apoptotic endothelial cells in response to CD95 ligand infusion [114]. Therefore, it is not clear whether apoptosis represents a prominent modality for the reduction of kidney endothelial cells post AKI.
We recently proposed an alternative mechanism for PTC rarefaction in which endothelial cells loss is manifested by a phenotypic transition via endothelial-mesenchymal transition (i.e., EndoMT). This suggestion is based on the co-localization of endothelial markers (CD31 or cablin) with mesenchymal markers (S100A4) as early as 6 hours and for up to 7 days post AKI. Moreover, fate tracing studies using Cre-Lox targeting of EC, indicated that a population of interstitial cells present at 2 weeks post I/R may have derived from the endothelium [122].
Impaired mechanisms of capillary regeneration
Regardless of the mechanisms of dropout, PTC rarefaction post-AKI appears to be permanent [112] suggesting that the intrinsic regenerative capacity of renal capillaries is profoundly limited. This impaired regenerative response may stem in part from a lack of trophic support following AKI. In general, there is shift in the balance of paracrine factors toward an environment which likely inhibits endothelial stability [36, 117]. For example, TGF-beta is prominently but transiently induced post-AKI[126]. TGF-beta is a negative regulator of endothelial proliferation and may induce endoMT. Inhibition of TGF beta by administration of neutralizing antibodies to rats post AKI for 5 weeks preserved vascular density and attenuated the deposition of peritubular fibroblasts [127]. In addition, other inhibitors of endothelial cell growth are expressed following AKI including angiostatin, sFLT, ADAMTS-1, endostatin, arrestin and canstatin [114, 128, 129].
Renal tubular expression of VEGF is thought to be the primary source of local vascular trophic support, and the loss of this trophic activity may also shift the balance to impaired recovery. Despite hypoxia post AKI, there is no increase, and in some reports, a reduction in VEGF expression following AKI [129, 130]. Interestingly, other models of CKD are also characterized by a counter-intuitive reduction of VEGF expression which persists with the loss of peritubular and/or glomerular vessels [119, 131].
In contrast to CKD models, the reduction in VEGF expression is transient post-AKI, while the loss of vessels is permanent. We investigated whether the transient reduction in VEGF expression may influence vascular dropout. Exogenous VEGF-121 attenuated PTC rarefaction only when administration was initiated within the first few days of ischemia [123], but not when provided at a later time point, suggesting that replenishment of trophic support is only efficacious within a critical window of time during recovery.
In recent years, the question of how to replenish renal capillaries has received increased interest. The literature contains various examples of treatments that preserve renal PTC loss in response to injury using treatments such as VEGF infusion [123, 132, 133], comp-angiopoiten-1 [134] or ramapril [135]. However, delayed administration of VEGF does not restore lost vessels [123, 136]. To the best of our knowledge, there have no reports in rodents have shown that renal vascular loss can be reversed once it has been firmly established. It may be interesting to note that at least one study using a swine model of renal artery stenosis has demonstrated improvement of vascular structure with VEGF treatment [137].
Because endogenous renal expression of pro- and anti-angiogenic are restored to basal levels following recovery from AKI without evidence of vascular repair, and delayed VEGF administration has no apparent effect on capillary regeneration, we propose that sustained reduction in peritubular capillary density is the result of impaired growth responsiveness by the resident kidney endothelial cells. Indeed, there is a paucity of studies indicating that resident kidney endothelial cells have significant proliferative potential following completion of embryonic development [138]. Although a small number of studies using disease models such as diabetes and immune complex disease have reported evidence for capillary sprouting and endothelial cell proliferation in the glomerulus [139-143], most studies indicate peritubular capillary (PTC) endothelial cell proliferation is minimal in response to injury. Hoenstein et al., demonstrated a small degree of PTC endothelial proliferation in response to an antibody-mediated injury. The proliferative response was enhanced by erythropoietin, but erythropoietin did not provide a sustained benefit on PTC density [144]. In a recent study by our group, BrdU was dosed over a 7-day period to maximize detection of proliferating cells; we observed that PTC-EC proliferation was an extremely rare event following I/R; no BrdU positive endothelial cells were detected within 2 days of I/R, while fewer than 1% of BrdU cells were endothelial after 7 days of continuous administration post I/R [145]. The administration of VEGF-121 preserved PTC density post AKI, but it did not increase the number of BrdU-positive ECs [145] suggesting that VEGF-mediated protection of the renal vasculature occurs via a non-proliferative mechanism.
It is possible that a small degree of EC proliferation may mediate vascular repair responses in the injured kidney. Interestingly, Lin et al, have posited that capillary rarefaction may be mediated by a loss of the critical interaction of the endothelial cells with their neighboring pericytes in response to injury [146]. Renal derived pericytes stabilize human umbilical vein ECs in in vitro co-culture assays, but pericytes which have been activated by injury lose this stabilizing feature. The molecular pathways of this stabilizing effect have yet to be elucidated but may relate to the expression of TIMPs or ADAMTS-1 [147]. These studies suggest that if endothelial cell proliferation might be sufficient to induce a repair, activated renal pericytes may contribute to an environment incapable of maintaining repaired vessels. A better elucidation of these events will likely represent an important component in an overall strategy to revascularize the kidney.
Intrinsic and extrinsic endothelial progenitor cells in the kidney
There has been considerable interest in the role and the potential utility of endothelial progenitor cells (EPCs) in the setting of renal injury. The term endothelial progenitor cell was originally coined in a manuscript by Asahara et al., when a population of cells isolated from blood appeared to incorporate into sites of active angiogenesis [148]. EPCs are isolated when either low-density MNCs or isolated CD34+ or CD133+ enriched cells are plated on fibronectin-coated tissue culture surfaces, allowing distinct adherent colonies emerge after 5–9 days. These are referred to as colony-forming unit-Hill (CFU-Hill) endothelial colonies, which express cell surface antigens CD31, CD105, CD144, CD146, vWF and KDR [149]. However, it is increasingly recognized that these cells do not represent true EPCs, since they are rarely found in the endothelial layer of a repaired vessel [150-152]. In addition, these cells express markers of hematopoetic origin that are not present on endothelial cells, such as CD45. Therefore, they have more recently been referred to as “angiogenic macrophages”, “pro-angiogenic hematopoietic stem/progenitor cells” or proangiogenic cells (PAC) [151, 152].
In kidney, understanding of PAC activity has been facilitated by studies in which labeled BM-derived cells have been found in the glomerulus following Thy 1.1 injury [153] or in the peritubular capillaries following adriamycin [154] or ischemic injury [155]. These studies report that these BM-derived cells differentiate into renal ECs on the basis of their expression of “endothelial” markers. However, it is well known that the expression of surface endothelial markers in bone marrow cells is not sufficient to define ECs [156]. Moreover, in a study by Li et al [154], BM-derived cells expressing endothelial markers peak within 14 days and decline by 28 days in kidney following adriamycin injury. Such a result is consistent with the transient homing of PACs to support local vascular remodeling, rather than stable incorporation and differentiation into more long-lived vascular endothelial cells. Despite significant interest in elucidating the trafficking of PACs to injured kidney, rarefaction of both glomerular and peritubular capillaries remains a prominent feature of these models, indicating that the endogenous activity of these cells is not sufficient to preserve or restore renal vascular injury.
Exogenous supplementation of PACs has shown great promise for multiple therapeutic applications by providing an enriched paracrine environment suitable for angiogenic/repair activity [157-160]. Within the kidney, studies from Goligorsky's group have shown that bone marrow derived PACs protect the loss of function following ischemia or adriamycin induced injury, by influencing hemodynamic or anti-inflammatory processes [150, 161, 162]. Strategies which increase the activity or mobilization of pro-angiogenic bone marrow derived cells increase these protective effects [150, 161, 162]. In one study, the early administration of BM-PAC reduced the degree of PTC rarefaction following adriamycin- induced injury [161]. However, this study did not evaluate whether PACs were stably incorporated into PTCs. In addition, to the best of our knowledge, no studies in rodent models of acute injury have evaluated if PACs might restore PTC after rarefaction.
A second type of EPC has been described by Yoder and colleagues that can be isolated from peripheral blood and are not of hematopoietic origin. These cells are referred to as endothelial colony forming cells (ECFCs); they are clonally distinct from PACs and they have robust proliferative potential. ECFCs can form perfused vessels in vivo through a process of de novo vasculogenesis, while PACs do not display this activity [151]. ECFCs have also been shown to incorporate into and stimulate neovascularization in a hindlimb ischemia model [163].
A novel hypothesis has been proposed by Ingram et al., regarding cooperative interaction of ECFCs and BM-derived PACs in vascular repair/vasculogenesis [164]. Simply stated, it is suggested that hematopoietic cells (BM-PACs) home to sites of injury/remodeling to create a pro-angiogenic environment. The secretion of paracrine factors by BM-PACs recruits ECFCs from either the circulation or the local vascular wall. Migration and proliferation of ECFCs are guided by BM-PAC activity leading to vascular repair and/or angiogenesis [164] In support of this hypothesis, when cultured in Matrigel plugs, these two cell types rapidly form capillary-like structures where the ECFC form the lumenized structures and the BM-PACs migrate into the lumen of the formed vessels. In addition, co-adoptive transfer of both cell types has resulted in superior in vivo neo-angiogenesis when compared to a single cell transfers alone [165].
The isolation of ECFC illustrates that there is a hierarchy of ECFC proliferative potential which can be assessed by single-cell colony forming assays. Some ECFCs form large colonies (>10,000 over two weeks), which are referred to as high proliferative potential (HPP-ECFCs) and are enriched in human cord blood. ECFC are reduced in the blood of patients characterized by high cardiovascular risk status. ECFCs can also be isolated from the vessel walls of a variety of tissue sources in multiple species. This has led to the suggestion that the source of ECFC resides within the local vasculature [166].
We attempted to isolate rat kidney endothelial cells (KEC) and attempted to identify ECFC content within the isolated KEC. Consistent with reports from other investigators, kidney ECs proved difficult to maintain in long-term culture without transformation into mesenchymal cells. Compared to other EC cell types under optimal culture conditions, KEC displayed extremely slow growth kinetics. No ECFCs were identified when KEC were plated at a clonal level which were capable of forming large colonies (i.e., > 2000 cells/2 weeks). Moreover, rat kidney ECs failed to proliferate in response to VEGF addition to the culture media, despite showing prominent expression of numerous VEGF receptors. These cells do not express markers typical of senescence and we propose that these cells may be growth restricted by a post-receptor signaling pathway [167].
In consideration of how kidney endothelial cell growth potential and PACs may operate in vascular repair, we recall that the paradigm outlined by Ingram et al., indicates that that vascular repair mediated by PACs is contingent upon the activation of a local ECFC, capable of participating in repair. Based on the fairly-common observation that PTC capillaries lack regenerative potential we hypothesize that limited renal ECFC potential is the basis for persistent vascular loss despite the apparent deposition of a population of cells that would be classified as PACs following injury.
We propose that future studies seek to determine whether proliferation competent ECFCs and PACs may work to minimize vascular rarefaction and/or regenerate renal vessels post rarefaction. Since rodents have no circulating ECFCs, studies will have to determine how adoptive transfer of competent ECFCs may occur and home to the kidney to work with PAC. In contrast, larger animals, including humans, may have a circulating ECFC population but may require strategies to target these to the kidney and participate in repair. Regardless, since PTC rarefaction is a feature of nearly all forms of CKD, it will be important to further investigate potential therapies seeking to revascularize the kidney.
Summary
Impaired endothelial function plays a central role in the pathogenesis of acute kidney injury. The potential to treat endothelial dysfunction and alter the course of acute kidney injury requires an understanding of the fundamental processes of injury an progression in response to ischemia. Figure 1 summarizes how alterations in endothelial function may relate the pathogenesis of acute kidney injury. During the initiation phase, vasodilator therapies may by effective in animal models, however the lack of efficacy in clinical trials may relate to fact that AKI diagnosis is delayed until at least the extension phase, when alterations in endothelial leukocyte interactions may be a prominent activity. Long term alterations may relate to peritubular capillary loss, which can be preserved by experimental treatments during the early phases of injury and repair, but ultimately become refractory to pro-angiogenic therapies.
Figure 1.
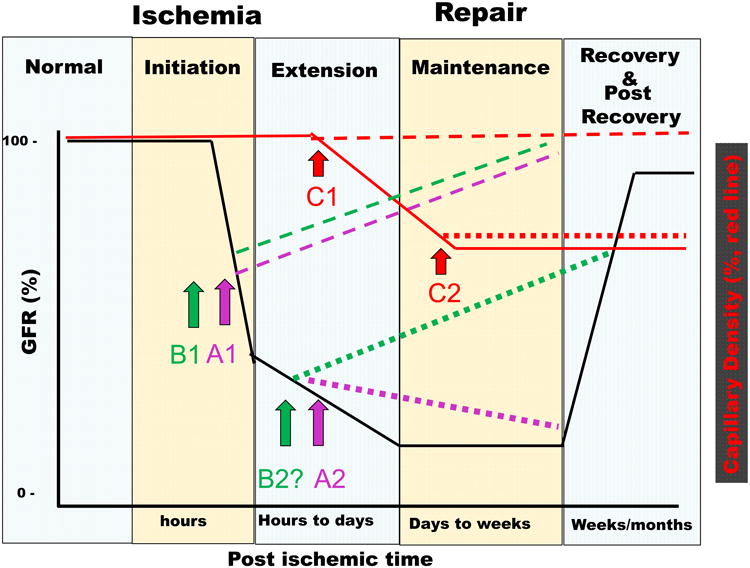
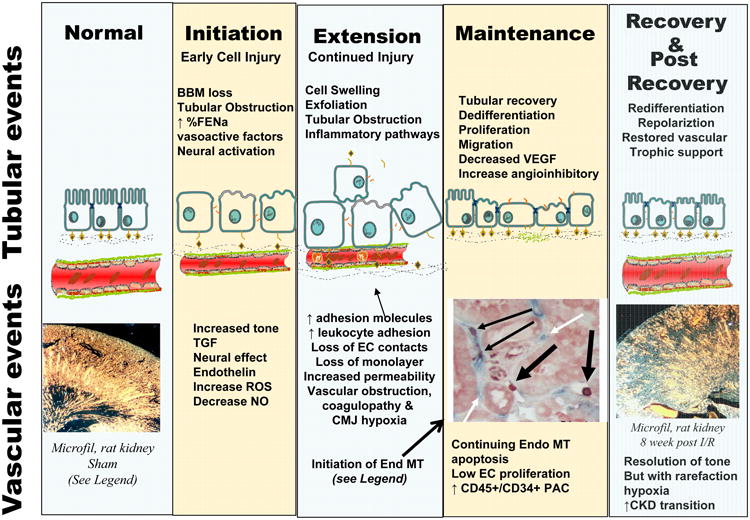
Summary of tubular and vascular events in the initiation and recovery from ischemic AKI. Top Panel, Time line of loss of GFR in response to renal ischemia shown in relation to the corresponding phases (Initiation, Extension, Maintenance, and Recovery) described by Molitoris and Sutton [37]. Early reduction of GFR in the initiation phase is mediated initially be changes in vascular tone. Arrows and lines show the anticipated effects of endothelial targeted treatments. Treatment A1 (purple dashed line) indicates preservation of function when treatments effecting tone, (e.g., supplementation of NO or vasoconstrictor antagonists) are provided during initiation, while Treatment A2 (purple dotted line) shows a lack of efficacy of these treatments when provided during the extension phase. Similarly, treatments that disrupt endothelial leukocyte interactions are effective when provided during initiation (B1, green dashed line). The effect of anti-leukocyte during the extension phase is not yet clear, but is predicted to have potential beneficial effects (B2?, green dotted line). Capillary density is indicated by the red lines: rarefaction progresses in the extension and maintenance phases and does not resolve; pro-vasculogenic treatments preserve capillary density when provided during the extension phase (C1), but lack effect at later times (C2). Bottom Panel summarizes altered cellular and molecular events associated with the phases of injury in both the tubular and vascular compartments, with an emphasis on events linked with vascular perfusion. The photomicrograph in “normal” state illustrates intact vascular density in control rats, while capillary rarefaction is observed in the post-recovery phase (photomicrographs from [112]). Loss of microvasculature occurs, in part, via endothelial mesenchymal transition (EndoMT) which can be observed in the extension and maintenance phase. EndoMT is illustrated by co-localizing S100-A4 (red) in capillary EC (blue) (highlighted by thin black arrow, from citation [122].
Acknowledgments
DPB and MY are supported by NIH grant DK063114.
Citations
- 1.Liangos O, Wald R, O'Bell JW, Price L, Pereira BJ, Jaber BL. Epidemiology and Outcomes of Acute Renal Failure in Hospitalized Patients: A National Survey. Clinical Journal of the American Society of Nephrology. 2006;1(1):43–51. doi: 10.2215/CJN.00220605. [DOI] [PubMed] [Google Scholar]
- 2.Bagshaw SM, Laupland KB, Doig CJ, Mortis G, Fick GH, Mucenski M, Godinez-Luna T, Svenson LW, Rosenal T. Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: a population-based study. Crit Care. 2005;9(6):R700–9. doi: 10.1186/cc3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levy EM, Viscoli CM, Horwitz RI. The effect of acute renal failure on mortality. A cohort analysis. JAMA. 1996;275(19):1489–94. [PubMed] [Google Scholar]
- 4.Mehta RL, Chertow GM. Acute renal failure definitions and classification: time for change? J Am Soc Nephrol. 2003;14(8):2178–87. doi: 10.1097/01.asn.0000079042.13465.1a. [DOI] [PubMed] [Google Scholar]
- 5.Nash K, Hafeez A, Hou S. Hospital-acquired renal insufficiency. Am J Kidney Dis. 2002;39(5):930–6. doi: 10.1053/ajkd.2002.32766. [DOI] [PubMed] [Google Scholar]
- 6.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Ronco C. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294(7):813–8. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- 7.Wald R, Quinn RR, Luo J, Li P, Scales DC, Mamdani MM, Ray JG. Chronic dialysis and death among survivors of acute kidney injury requiring dialysis. JAMA. 2009;302(11):1179–85. doi: 10.1001/jama.2009.1322. [DOI] [PubMed] [Google Scholar]
- 8.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute Kidney Injury, Mortality, Length of Stay, and Costs in Hospitalized Patients. Journal of the American Society of Nephrology. 2005;6(11):3365–3370. doi: 10.1681/ASN.2004090740. [DOI] [PubMed] [Google Scholar]
- 9.Chertow GM, Christiansen CL, Cleary PD, Munro C, Lazarus JM. Prognostic stratification in critically ill patients with acute renal failure requiring dialysis. Arch Intern Med. 1995;155(14):1505–11. [PubMed] [Google Scholar]
- 10.Ishani A, Xue JL, Himmelfarb J, Eggers PW, Kimmel PL, Molitoris BA, Collins AJ. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol. 2009;20(1):223–8. doi: 10.1681/ASN.2007080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liano F, Junco E, Pascual J, Madero R, Verde E. The spectrum of acute renal failure in the intensive care unit compared with that seen in other settings. The Madrid Acute Renal Failure Study Group. Kidney Int Suppl. 1998;66:S16–24. [PubMed] [Google Scholar]
- 12.Liano F, Pascual J. Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Madrid Acute Renal Failure Study Group. Kidney Int. 1996;50(3):811–8. doi: 10.1038/ki.1996.380. [DOI] [PubMed] [Google Scholar]
- 13.Lo LJ, Go AS, Chertow GM, McCulloch CE, Fan D, Ordonez JD, Hsu CY. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int. 2009;76(8):893–9. doi: 10.1038/ki.2009.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgera S, Kraft AK, Siebert G, Luft FC, Neumayer HH. Long-term outcomes in acute renal failure patients treated with continuous renal replacement therapies. Am J Kidney Dis. 2002;40(2):275–9. doi: 10.1053/ajkd.2002.34505. [DOI] [PubMed] [Google Scholar]
- 15.Schiffl H. Renal recovery from acute tubular necrosis requiring renal replacement therapy: a prospective study in critically ill patients. Nephrol Dial Transplant. 2006;21(5):1248–52. doi: 10.1093/ndt/gfk069. [DOI] [PubMed] [Google Scholar]
- 16.Amdur RL, Chawla LS, Amodeo S, Kimmel PL, Palant CE. Outcomes following diagnosis of acute renal failure in U.S. veterans: focus on acute tubular necrosis. Kidney Int. 2009;76(10):1089–97. doi: 10.1038/ki.2009.332. [DOI] [PubMed] [Google Scholar]
- 17.Waikar SS, Winkelmayer WC. Chronic on acute renal failure: long-term implications of severe acute kidney injury. JAMA. 2009;302(11):1227–9. doi: 10.1001/jama.2009.1364. [DOI] [PubMed] [Google Scholar]
- 18.Xue JL, Daniels F, Star RA, Kimmel PL, Eggers PW, Molitoris BA, Himmelfarb J, Collins AJ. Incidence and mortality of acute renal failure in medicare beneficiaries, 1992 to 2001. J Am Soc Nephrol. 2006;17:1135–1142. doi: 10.1681/ASN.2005060668. [DOI] [PubMed] [Google Scholar]
- 19.Sutton TA, Fisher CJ, Molitoris BA. Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney International. 2002;62(5):1539–49. doi: 10.1046/j.1523-1755.2002.00631.x. [DOI] [PubMed] [Google Scholar]
- 20.Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol. 2003;14:2199–2210. doi: 10.1097/01.asn.0000079785.13922.f6. [DOI] [PubMed] [Google Scholar]
- 21.Hollenberg NK, Epstein FH, Rosen S, Basch R, Oken D. Merrill, Acute oliguric renal failure in man: evidence for preferential renal cortical ischemia. Medicne (Balt) 1968;47(6):455–74. doi: 10.1097/00005792-196811000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Karlberg L, Norlen BJ, Ojteg G, Wolgast M. Impaired medullary circulation in postischemic acute renal failure. Acta Physiologica Scandinavica. 1983;118(1):11–7. doi: 10.1111/j.1748-1716.1983.tb07234.x. [DOI] [PubMed] [Google Scholar]
- 23.Hellberg POA, Kallskog OT, Ojteg G, Wolgast M. Peritubular capillary permeability and intravascular RBC aggregation after ischemia: effects of neutrophils. American Journal of Physiology. 1990;258:F1018–F1025. doi: 10.1152/ajprenal.1990.258.4.F1018. Renal Fluid Electrolyte Physiology. [DOI] [PubMed] [Google Scholar]
- 24.Mason J, Torhorst J, Welsch J. Role of the medullary perfusion defect in the pathogenesis of ischemic renal failure. Kidney Int. 1984;26:283–293. doi: 10.1038/ki.1984.171. [DOI] [PubMed] [Google Scholar]
- 25.Dagher PC, Herget-Rosenthal S, Ruehm SG, Jo SK, Star RA, Agarwal R, Molitoris BA. Newly developed techniques to study and diagnose acute renal failure. Journal of the American Society of Nephrology. 2003;14(8):2188–98. doi: 10.1097/01.asn.0000079790.91292.4a. [DOI] [PubMed] [Google Scholar]
- 26.Sheridan AM, Bonventre JV. Cell biology and molecular mechanisms of injury in ischemic acute renal failure. Current Opinion in Nephrology & Hypertension. 2000;9(4):427–34. doi: 10.1097/00041552-200007000-00015. [DOI] [PubMed] [Google Scholar]
- 27.Park K, Chen A, Bonventre JV. Prevention of kidney ischemia/reperfusion induced functional injury and JNK, p38 and MAPK kinase activation by remote ischemic pretreatment. Journal of Biological Chemistry. 2001;276(15):11870–6. doi: 10.1074/jbc.M007518200. [DOI] [PubMed] [Google Scholar]
- 28.Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. Journal of the American Society of Nephrology. 2003;14(8):2199–210. doi: 10.1097/01.asn.0000079785.13922.f6. [DOI] [PubMed] [Google Scholar]
- 29.Bojakowski K, Abramczyk P, Bojakowska M, Zwolinska A, Przybylski J, Gaciong Z. Fucoidan improves the renal blood flow in the early stage of renal ischemia/reperfusion in the rat. Journal of Physiology and Pharmacology. 2001;52(1):137–143. [PubMed] [Google Scholar]
- 30.Cristol JP, Thiemermann C, Mitchell JA, Walder C, Vane JR. Support of renal blood flow after ischaemic-reperfusion injury by endogenous formation of nitric oxide and of cyclo-oxygenase vasodilator metabolites. British Journal of Pharmacology. 1993;109(1):188–194. doi: 10.1111/j.1476-5381.1993.tb13552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Versteilen AMG, Korstjens IJM, Musters RJP, Groeneveld ABJ, Sipkema P. Rho kinase regulates renal blood flow by modulating eNOS activity in ischemia-reperfusion of the rat kidney. American Journal of Physiology - Renal Physiology. 2006;291(3):F606–F611. doi: 10.1152/ajprenal.00434.2005. [DOI] [PubMed] [Google Scholar]
- 32.Regner KR, Zuk A, Van Why SK, Shames BD, Ryan RP, Falck JR, Manthati VL, McMullen ME, Ledbetter SR, Roman RJ. Protective effect of 20-HETE analogues in experimental renal ischemia reperfusion injury. Kidney Int. 2008;75(5):511–517. doi: 10.1038/ki.2008.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi H, Patschan D, Epstein T, Goligorsky MS, Winaver J. Delayed recovery of renal regional blood flow in diabetic mice subjected to acute ischemic kidney injury. American Journal of Physiology - Renal Physiology. 2007;293(5):F1512–F1517. doi: 10.1152/ajprenal.00215.2007. [DOI] [PubMed] [Google Scholar]
- 34.Regner K, Roman R. Role of medullary blood flow in the pathogenesis of renal ischemia-reperfusion injury. Current Opinion in Nephrology and Hypertension. 2012;21(1):33–38. doi: 10.1097/MNH.0b013e32834d085a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saito M, Miyagawa I. Real-time monitoring of nitric oxide in ischemia-reperfusion rat kidney. Urological Research. 2000;28(2):141–146. doi: 10.1007/s002400050153. [DOI] [PubMed] [Google Scholar]
- 36.Basile DP, Anderson M, Sutton TA. Pathophysiology of acute kidney injury. Comprehensive Physiology. 2012;2:1303–1353. doi: 10.1002/cphy.c110041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molitoris BA, Sutton TA. Endothelial injury and dysfunction: role in the extension phase of acute renal failure. Kidney International. 2004;66(2):496–9. doi: 10.1111/j.1523-1755.2004.761_5.x. [DOI] [PubMed] [Google Scholar]
- 38.Jo SK, Rosner MH, Okusa MD. Pharmacologic Treatment of Acute Kidney Injury: Why Drugs Haven't Worked and What Is on the Horizon. Clinical Journal of the American Society of Nephrology. 2007;2(2):356–365. doi: 10.2215/CJN.03280906. [DOI] [PubMed] [Google Scholar]
- 39.Honore P, Jacobs R, Joannes-Boyau O, Verfaillie L, De Regt J, Van Gorp V, De Waele E, Boer W, Collin V, Spapen H. Biomarkers for early diagnosis of AKI in the ICU: ready for prime time use at the bedside? Annals of Intensive Care. 2012;2(1):24. doi: 10.1186/2110-5820-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blantz RC, Pelayo JC. A functional role for the tubuloglomerular feedback mechanism. Kidney Int. 1984;25(5):739–746. doi: 10.1038/ki.1984.84. [DOI] [PubMed] [Google Scholar]
- 41.Fujii T, Kurata H, Takaoka M, Muraoka T, Fujisawa Y, Shokoji T, Nishiyama A, Abe Y, Matsumura Y. The role of renal sympathetic nervous system in the pathogenesis of ischemic acute renal failure. European Journal of Pharmacology. 2003;481(2-3):241–248. doi: 10.1016/j.ejphar.2003.09.036. [DOI] [PubMed] [Google Scholar]
- 42.Salman IM, Ameer OZ, Sattar MA, Abdullah NA, Yam MF, Najim HS, Khan AH, Johns EJ. Role of the renal sympathetic nervous system in mediating renal ischaemic injury-induced reductions in renal haemodynamic and excretory functions. Pathology. 42(3):259–266. doi: 10.3109/00313021003631304. [DOI] [PubMed] [Google Scholar]
- 43.Wang W, Falk SA, Jittikanont S, Gengaro PE, Edelstein CL, Schrier RW. Protective effect of renal denervation on normotensive endotoxemia-induced acute renal failure in mice. American Journal of Physiology - Renal Physiology. 2002;283(3):F583–F587. doi: 10.1152/ajprenal.00270.2001. [DOI] [PubMed] [Google Scholar]
- 44.Sreedharan R, Devarajan P, Van Why S. Pathogenesis of acute renal failure. In. In: Avner E, Harmon W, Niaudet P, Yoshikawa N, editors. Pediatric Nephrology. 6th. Springer-Verlag; Heidleberg: 2009. pp. 1579–1602. [Google Scholar]
- 45.Nishiyama A, Miyatake A, Aki Y, Fukui T, Rahman M, Kimura S, Abe Y. Adenosine A1 Receptor Antagonist KW-3902 Prevents Hypoxia-Induced Renal Vasoconstriction. Journal of Pharmacology and Experimental Therapeutics. 1999;291(3):988–993. [PubMed] [Google Scholar]
- 46.Wilhelm SM, Simonson MS, Robinson AV, Stowe NT, Schulak JA. Endothelin up-regulation and localization following renal ischemia and reperfusion. Kidney International. 1999;55(3):1011–8. doi: 10.1046/j.1523-1755.1999.0550031011.x. [DOI] [PubMed] [Google Scholar]
- 47.Huang C, Huang C, Hestin D, Dent PC, Barclay P, Collis M, Johns EJ. The effect of endothelin antagonists on renal ischaemia-reperfusion injury and the development of acute renal failure in the rat. Nephrology Dialysis Transplantation. 2002;17(9):1578–1585. doi: 10.1093/ndt/17.9.1578. [DOI] [PubMed] [Google Scholar]
- 48.Knoll T, Schult S, Birck R, Braun C, Michel MS, Bross S, Juenemann KP, Kirchengast M, Rohmeiss P. Therapeutic administration of an endothelin-A receptor antagonist after acute ischemic renal failure dose-dependently improves recovery of renal function. Journal of Cardiovascular Pharmacology. 2001;37:483–8. doi: 10.1097/00005344-200104000-00015. [DOI] [PubMed] [Google Scholar]
- 49.Büyükgebiz O, Aktan AÖ, Haklar G, Yalçin AS, Yeğen C, Yalin R, Ercan ZS. BQ-123, a specific endothelin (ET1) receptor antagonist, prevents ischemia- reperfusion injury in kidney transplantation. Transplant International. 1996;9(3):201–207. doi: 10.1007/BF00335386. [DOI] [PubMed] [Google Scholar]
- 50.Lopez-Novoa JM. Potential role of platelet activating factor in acute renal failure. Kidney Int. 1999;55(5):1672–1682. doi: 10.1046/j.1523-1755.1999.00450.x. [DOI] [PubMed] [Google Scholar]
- 51.Wang W, Zolty E, Falk S, Summer S, Stearman R, Geraci M, Schrier R. Prostacyclin in endotoxemia-induced acute kidney injury: cyclooxygenase inhibition and renal prostacyclin synthase transgenic mice. American Journal of Physiology – Renal Physiology. 2007;293(4):F1131–F1136. doi: 10.1152/ajprenal.00212.2007. [DOI] [PubMed] [Google Scholar]
- 52.Tobimatsu M, Euda Y, Saito S, Tsumagari T, Konomi K. Effects of a stable prostocyclin analog on experimental ischemic acute renal failure. Ann Surg. 1988;208:65–70. doi: 10.1097/00000658-198807000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vargas A, Krishnamurthi V, Masih R, Robinson A, Shchulak J. Prostaglandin E1 attenuation of ischemic renal reperfusion in the rat. J Am Coll Surg. 1995;180:713–717. [PubMed] [Google Scholar]
- 54.Gellai M, Jugus M, Fletcher T, DeWolf R, Nambi P. Reversal of postischemic acute renal failure with a selective endothlin-A receptor antagonist. The Journal of Clinical Investigation. 1994;93:900–906. doi: 10.1172/JCI117046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boffa JJ, Just A, Coffman TM, Arendshorst WJ. Thromboxane Receptor Mediates Renal Vasoconstriction and Contributes to Acute Renal Failure in Endotoxemic Mice. Journal of the American Society of Nephrology. 2004;15(9):2358–2365. doi: 10.1097/01.ASN.0000136300.72480.86. [DOI] [PubMed] [Google Scholar]
- 56.Kwon O, Phillips CL, Molitoris BA. Ischemia induces alterations in actin filaments in renal vascular smooth muscle cells. American Journal of Physiology - Renal Fluid & Electrolyte Physiology. 2002;282:F1012–9. doi: 10.1152/ajprenal.00294.2001. [DOI] [PubMed] [Google Scholar]
- 57.Sutton TA, Mang HE, Campos SB, Sandoval RM, Yoder MC, Molitoris BA. Injury of the renal microvascular endothelium alters barrier function after ischemia. American Journal of Physiology - Renal Physiology. 2003;285(2):F191–8. doi: 10.1152/ajprenal.00042.2003. [DOI] [PubMed] [Google Scholar]
- 58.Hellberg PO, Kallskog O, Wolgast M. Nephron function in the early phase of ischemic renal failure. Significance of erythrocyte trapping. Kidney International. 1990;38:432–9. doi: 10.1038/ki.1990.223. [DOI] [PubMed] [Google Scholar]
- 59.Hellberg POA, Kallskog O, Wolgast M. Red cell trapping and postischemic renal blood flow. Differences between the cortex, outer and inner medulla. Kidney Int. 1991;40:625–631. doi: 10.1038/ki.1991.254. [DOI] [PubMed] [Google Scholar]
- 60.Brodsky SV, Yamamoto T, Tada T, Kim B, Chen J, Kajiya F, Goligorsky MS. Endothelial dysfunction in ischemic acute renal failure: rescue by transplanted endothelial cells. American Journal of Physiology - Renal Fluid & Electrolyte Physiology. 2002;282(6):F1140–9. doi: 10.1152/ajprenal.00329.2001. [DOI] [PubMed] [Google Scholar]
- 61.Yamamoto T, Tada T, Brodsky SV, Tanaka H, Noiri E, Kajiya F, Goligorsky MS. Intravital videomicroscopy of peritubular capillaries in renal ischemia. American Journal of Physiology - Renal Fluid & Electrolyte Physiology. 2002;282(6):F1150–5. doi: 10.1152/ajprenal.00310.2001. [DOI] [PubMed] [Google Scholar]
- 62.Mattson DL, Lu S, Cowley AW., Jr Role of nitric oxide in the control of the renal medullary circulation. Clinical & Experimental Pharmacology & Physiology. 1997;24(8):587–90. doi: 10.1111/j.1440-1681.1997.tb02096.x. [DOI] [PubMed] [Google Scholar]
- 63.Zou AP, Wu F, Cowley AW., Jr Protective effect of angiotensin II-induced increase in nitric oxide in the renal medullary circulation. Hypertension. 1998;31:271–6. doi: 10.1161/01.hyp.31.1.271. [DOI] [PubMed] [Google Scholar]
- 64.Conger JD, Robinette JB, Schrier RW. Smooth muscle calcium and endothelium derived relaxing factor in the abnormal vascular responses of acute renal failure. Journal of Clinical Investigation. 1988;82:532–537. doi: 10.1172/JCI113628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang W, Mitra A, Poole B, Falk S, Lucia MS, Tayal S, Schrier R. Endothelial nitric oxide synthase-deficient mice exhibit increased susceptibility to endotoxin-induced acute renal failure. American Journal of Physiology - Renal Physiology. 2004;287(5):F1044–F1048. doi: 10.1152/ajprenal.00136.2004. [DOI] [PubMed] [Google Scholar]
- 66.Conger JD, Schultz MF, Miller F, Robinette JB. Responses to hemorrhagic arterial pressure reduction in different ischemic renal failure models. Kidney International. 1994;46(2):318–23. doi: 10.1038/ki.1994.277. [DOI] [PubMed] [Google Scholar]
- 67.Conger JD, Robinette JB, Villar A, Raij L, Shultz P. Increased nitric oxide synthase activity despite lack of response to endothelium-dependent vasodilators in post-ischemic acute renal failure in rats. Journal of Clinical Investigation. 1995;96:631–638. doi: 10.1172/JCI118078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Adams PL, Adams FF, Bell PD, Navar LG. Impaired renal blood flow autoregulation in ischemic acute renal failure. Kidney Int. 1980;18(1):68–76. doi: 10.1038/ki.1980.111. [DOI] [PubMed] [Google Scholar]
- 69.Solez K, Morel-Maroger L, Sraer JD. The Morphology of “Acute Tubular Necrosis” in Man: Analysis of 57 Renal Biopsies and a Comparison with the Glycerol Model. Medicine. 1979;58(5):362–376. [PubMed] [Google Scholar]
- 70.Zou AP, Li N, Cowley AW., Jr Production and actions of superoxide in the renal medulla. Hypertension. 2001;37:547–53. doi: 10.1161/01.hyp.37.2.547. [DOI] [PubMed] [Google Scholar]
- 71.Chen YF, Li PL, Zou AP. Oxidative stress enhances the production and actions of adenosine in the kidney. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2001;281(6):R1808–R1816. doi: 10.1152/ajpregu.2001.281.6.R1808. [DOI] [PubMed] [Google Scholar]
- 72.Schneider R, Raff U, Vornberger N, Schmidt M, Freund R, Reber M, Schramm L, Gambaryan S, Wanner C, Schmidt HHHW, Galle J. L-Arginine counteracts nitric oxide deficiency and improves the recovery phase of ischemic acute renal failure in rats. Kidney Int. 2003;64(1):216–225. doi: 10.1046/j.1523-1755.2003.00063.x. [DOI] [PubMed] [Google Scholar]
- 73.Garcia-Criado FJ, Eleno N, Santos-Benito F, Valdunciel JJ, Reverte M, Lozano-Sanchez FS, Ludena MD, Gomez-Alonso A, Lopez-Novoa JM. Protective effect of exogenous nitric oxide on the renal function and inflammatory response in a model of ischemia-reperfusion. Transplantation. 1998;66(8):982–90. doi: 10.1097/00007890-199810270-00003. [DOI] [PubMed] [Google Scholar]
- 74.Dagher F, Pollina RM, Rogers DM, Gennaro M, Ascer E. The value and limitations of L-arginine infusion on glomerular and tubular function in the ischemic/reperfused kidney. Journal of Vascular Surgery. 1995;21(3):453–8. doi: 10.1016/s0741-5214(95)70287-3. [DOI] [PubMed] [Google Scholar]
- 75.Schramm L, La M, Heidbreder E, Hecker M, Beckman JS, Lopau K, Zimmermann J, Rendl J, Reiners C, Winderl S, Wanner C, Schmidt HH. L-arginine deficiency and supplementation in experimental acute renal failure and in human kidney transplantation. Kidney International 2002. 2002 Apr;61(4):1423–32. doi: 10.1046/j.1523-1755.2002.00268.x. [DOI] [PubMed] [Google Scholar]
- 76.Schneider R, Raff U, Vornberger N, Schmidt M, Freund R, Reber M, Schramm L, Gambaryan S, Wanner C, Schmidt HH, Galle J. L-Arginine counteracts nitric oxide deficiency and improves the recovery phase of ischemic acute renal failure in rats. Kidney International. 2003;64:216–25. doi: 10.1046/j.1523-1755.2003.00063.x. [DOI] [PubMed] [Google Scholar]
- 77.Kakoki M, Hirata Y, Hayakawa H, Suzuki E, Nagata D, Tojo A, Nishimatsu H, Nakanishi N, Hattori Y, Kikuchi K, Nagano T, Omata M. Effects of tetrahydrobiopterin on endothelial dysfunction in rats with ischemic acute renal failure. J Am Soc Nephrol. 2000;11:301–309. doi: 10.1681/ASN.V112301. [DOI] [PubMed] [Google Scholar]
- 78.Sucher R, Gehwolf P, Oberhuber R, Hermann M, Margreiter C, Werner ER, Obrist P, Schneeberger S, Ollinger R, Margreiter R, Brandacher G. Tetrahydrobiopterin protects the kidney from ischemia-reperfusion injury. Kidney Int. 2010;77(8):681–689. doi: 10.1038/ki.2010.7. [DOI] [PubMed] [Google Scholar]
- 79.Sohotnik R, Nativ O, Abbasi A, Awad H, Frajewicki V, Bishara B, Sukhotnik I, Armaly Z, Aronson D, Heyman SN, Nativ O, Abassi ZA. Phosphodiesterase5 Inhibition Attenuates Early Renal Ischemia/Reperfusion-Induced Acute Kidney Injury: Assessment by measurement of urinary NGAL and KIM-1. American Journal of Physiology - Renal Physiology. 2013 doi: 10.1152/ajprenal.00649.2012. [DOI] [PubMed] [Google Scholar]
- 80.Choi DE, Jeong JY, Lim BJ, Chung S, Chang YK, Lee SJ, Na KR, Kim SY, Shin YT, Lee KW. Pretreatment of sildenafil attenuates ischemia-reperfusion renal injury in rats. American Journal of Physiology - Renal Physiology. 2009;297(2):F362–F370. doi: 10.1152/ajprenal.90609.2008. [DOI] [PubMed] [Google Scholar]
- 81.Patel NN, Lin H, Toth T, Jones C, Ray P, Welsh GI, Satchell SC, Sleeman P, Angelini GD, Murphy GJ. Phosphodiesterase-5 Inhibition Prevents Postcardiopulmonary Bypass Acute Kidney Injury in Swine. The Annals of Thoracic Surgery. 2012;92(6):2168–2176. doi: 10.1016/j.athoracsur.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 82.Arriero M, Brodsky SV, Gealekman O, Lucas PA, Goligorsky MS. Adult skeletal muscle stem cells differentiate into endothelial lineage and ameliorate renal dysfunction after acute ischemia. American Journal of Physiology - Renal Physiology. 2004;287(4):F621–7. doi: 10.1152/ajprenal.00126.2004. [DOI] [PubMed] [Google Scholar]
- 83.Denton Mark D, Chertow GM, Brady HR. [ldquo]Renal-dose[rdquo] dopamine for the treatment of acute renal failure: Scientific rationale, experimental studies and clinical trials. Kidney Int. 1996;50(1):4–14. doi: 10.1038/ki.1996.280. [DOI] [PubMed] [Google Scholar]
- 84.Kellum JA, M Decker J. Use of dopamine in acute renal faiulre: a meta analysis. Crit Care Med. 2001;29(8):1526–31. doi: 10.1097/00003246-200108000-00005. [DOI] [PubMed] [Google Scholar]
- 85.Kellum JA. Systematic review: The use of diuretics and dopamine in acute renal failure: a systematic review of the evidence. Critical Care. 1997;1(2):53–59. doi: 10.1186/cc103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bellomo R, Chapman M, Hickling K, Myburgh J. Low-dose dopamine in patients with early renal dysfunction: a placebo-controlled randomised trial. The Lancet. 2000;356(9248):2139–2143. doi: 10.1016/s0140-6736(00)03495-4. [DOI] [PubMed] [Google Scholar]
- 87.Lauschke A, Teichgraber UKM, Frei U, Eckardt KU. ‘Low-dose’ dopamine worsens renal perfusion in patients with acute renal failure. Kidney Int. 2006;69(9):1669–1674. doi: 10.1038/sj.ki.5000310. [DOI] [PubMed] [Google Scholar]
- 88.Tumlin JA, Finkel KW, Murray PT, Samuels J, Cotsonis G, Shaw AD. Fenoldopam Mesylate in Early Acute Tubular Necrosis: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2005;46(1):26–34. doi: 10.1053/j.ajkd.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 89.Furuichi K, Wada T, Iwata Y, Sakai N, Yoshimoto K, Kobayashi K, Mukaida N, Matsushima K, Yokoyama H. Administration of FR167653, a new anti-inflammatory compound, prevents renal ischaemia/reperfusion injury in mice. Nephrology Dialysis Transplantation. 2002;17(3):399–407. doi: 10.1093/ndt/17.3.399. [DOI] [PubMed] [Google Scholar]
- 90.Miyazawa S, Watanabe H, Miyaji C, Hotta O, Abo T. Leukocyte accumulation and changes in extra-renal organs during renal ischemia reperfusion in mice. Journal of Laboratory and Clinical Medicine. 2002;139(5):269–278. doi: 10.1067/mlc.2002.122832. [DOI] [PubMed] [Google Scholar]
- 91.Ysebaert DK, De Greef KE, Vercauteren SR, Ghielli M, Verpooten GA, Eyskens EJ, De Broe ME. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrology Dialysis Transplantation. 2000;15(10):1562–1574. doi: 10.1093/ndt/15.10.1562. [DOI] [PubMed] [Google Scholar]
- 92.Kinsey GR, Li L, Okusa MD. Inflammation in Acute Kidney Injury. Nephron Experimental Nephrology. 2008;109(4):e102–e107. doi: 10.1159/000142934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Friedewald JJ, Rabb H. Inflammatory cells in ischemic acute renal failure. Kidney Int. 2004;66(2):486–491. doi: 10.1111/j.1523-1755.2004.761_3.x. [DOI] [PubMed] [Google Scholar]
- 94.Molitoris BA, Sandoval R, Sutton TA. Endothelial injury and dysfunction in ischemic acute renal failure. Critical Care Medicine. 2002;30(5 Suppl):S235–40. doi: 10.1097/00003246-200205001-00011. [DOI] [PubMed] [Google Scholar]
- 95.Kelly KJ, Williams WW, Jr, Bonventre JV. Antibody to intercellular adhesion molecule 1 protects the kidney against ischemic injury. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(2):812–6. doi: 10.1073/pnas.91.2.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fuller TF, Sattler B, Binder L, Vetterlein F, Ringe B, Lorf T. Reduction of severe ischemia/reperfusion injury in rat idney grafts by a soluble P-Selectin glycprotein ligand. Tranplantation. 2001;72:216–222. doi: 10.1097/00007890-200107270-00008. [DOI] [PubMed] [Google Scholar]
- 97.Kelly KJ, Sutton TA, Weathered N, Ray N, Caldwell EJ, Plotkin Z, Dagher PC. Minocycline inhibits apoptosis and inflammation in a rat model of ischemic renal injury. Am J Physiol. 2004;287:F760–F766. doi: 10.1152/ajprenal.00050.2004. [DOI] [PubMed] [Google Scholar]
- 98.Sharfuddin AA, Sandoval RM, Berg DT, McDougal GE, Campos SB, Phillips CL, Jones BE, Gupta A, Grinnell BW, Molitoris BA. Soluble Thrombomodulin Protects Ischemic Kidneys. Journal of the American Society of Nephrology. 2009;20(3):524–534. doi: 10.1681/ASN.2008060593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kelly KJ, Williams WW, Williams WW, Jr, Colvin RB, Meehan SM, Springer TA, Gutierrez-Ramos WW, Bonventre JV. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. Journal of Clinical Investigation. 1996;97(4):1056–63. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Friedewald JJ, Rabb H. Inflammatory cells in ischemic acute renal failure. Kidney International. 2004;66(2):486–91. doi: 10.1111/j.1523-1755.2004.761_3.x. [DOI] [PubMed] [Google Scholar]
- 101.Haug CE, Colvin RB, Delmonico FL, Auchincloss HJ, Tolkoff-Rubin N, Preffer FI, Rothlein R, Norris S, Scharschmidt L, Cosimi AB. A Phase I Trial Of Immunosuppression With Antiic Am-1 (Cd54) mAb In Renal Allograft Recipients. Transplantation. 1993;55(4):766–772. doi: 10.1097/00007890-199304000-00016. [DOI] [PubMed] [Google Scholar]
- 102.Salmela K, Wramner L, Ekberg H, Hauser I, Bentdal O, Lins LE, Isoniemi H, Bäckman L, Persson N, Neumayer HH, Jørgensen PF, Spieker C, Hendry B, Nicholls A, Kirste G, Hasche G. A Randomized Multicenter Trial of the Anti-Icam-1 Monoclonal Antibody (Enlimomab) for the Prevention of Acute Rejection and Delayed Onset of Graft Function in Cadaveric Renal Transplantation: A Report of the European Anti-ICAM-1 Renal Transplant Study Group. Transplantation. 1999;67(5):729–736. doi: 10.1097/00007890-199903150-00015. [DOI] [PubMed] [Google Scholar]
- 103.Enestrom S, Druid H, Rammer L. Fibrin deposition in the kidney in post-ischaemic renal damage. British Journal of Experimental Pathology. 1988;69:387–394. [PMC free article] [PubMed] [Google Scholar]
- 104.Matsuyama M, Yoshimura R, Akioka K, Okamoto M, Ushigome H, Kadotani Y, Nakatani T, Yoshimura M. Tissue factor antisense oligonucleotides prevent renal ischema reperfusion injury. Tranplantation. 2003;76:786–791. doi: 10.1097/01.TP.0000079630.68668.C2. [DOI] [PubMed] [Google Scholar]
- 105.Gupta A, Williams MD, Macias WL, Molitoris B, Grinnell B. Activated protein C and acute kidney injury: Selective Targeting of PAR-1. Current Drug Targets. 2009;10:1212–26. doi: 10.2174/138945009789753291. [DOI] [PubMed] [Google Scholar]
- 106.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ. Efficacy and Safety of Recombinant Human Activated Protein C for Severe Sepsis. New England Journal of Medicine. 2001;344(10):699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 107.Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, Gårdlund B, Marshall JC, Rhodes A, Artigas A, Payen D, Tenhunen J, Al-Khalidi HR, Thompson V, Janes J, Macias WL, Vangerow B, Williams MD. Drotrecogin Alfa (Activated) in Adults with Septic Shock. New England Journal of Medicine. 2012;366(22):2055–2064. doi: 10.1056/NEJMoa1202290. [DOI] [PubMed] [Google Scholar]
- 108.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16(5):535–543. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK. Acute kidney injury: a springboard for progression in chronic kidney disease. American Journal of Physiology - Renal Physiology. 2010;298(5):F1078–F1094. doi: 10.1152/ajprenal.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zager RA, Johnson ACM. Renal ischemia-reperfusion injury upregulates histone-modifying enzyme systems and alters histone expression at proinflammatory/profibrotic genes. American Journal of Physiology - Renal Physiology. 2009;296(5):F1032–F1041. doi: 10.1152/ajprenal.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim J, Seok YM, Jung KJ, Park KM. Reactive oxygen species/oxidative stress contributes to progression of kidney fibrosis following transient ischemic injury in mice. American Journal of Physiology - Renal Physiology. 2009;297(2):F461–F470. doi: 10.1152/ajprenal.90735.2008. [DOI] [PubMed] [Google Scholar]
- 112.Basile DP, Donohoe DL, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. American Journal of Physiology. 2001;281(10):F887–F899. doi: 10.1152/ajprenal.2001.281.5.F887. [DOI] [PubMed] [Google Scholar]
- 113.Basile DP, Donohoe DL, Roethe K, Mattson DL. Chronic renal hypoxia following ischemia/reperfusion injury: Effects of L-Arginine on hypoxia and secondary damage. Am J Physiol Renal Physiol. 2003;284:F338–348. doi: 10.1152/ajprenal.00169.2002. [DOI] [PubMed] [Google Scholar]
- 114.Horbelt M, Lee SY, Mang HE, Knipe NL, Sado Y, Kribben A, Sutton TA. Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2007;293(3):F688–695. doi: 10.1152/ajprenal.00452.2006. [DOI] [PubMed] [Google Scholar]
- 115.Kwon O, Hong SM, Sutton TA, Temm CJ. Preservation of peritubular capillary endothelial integrity and increasing pericytes may be critical to recovery from postischemic acute kidney injury. Am J Physiol Renal Physiol. 2008;295(2):F351–359. doi: 10.1152/ajprenal.90276.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Steegh FMEG, Gelens MACJ, Nieman FHM, van Hooff JP, Cleutjens JPM, van Suylen RJ, Daemen MJAP, van Heurn ELW, Christiaans MHL, Peutz-Kootstra CJ. Early Loss of Peritubular Capillaries after Kidney Transplantation. Journal of the American Society of Nephrology. 2011;22(6):1024–1029. doi: 10.1681/ASN.2010050531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney International. 2007;72:151–156. doi: 10.1038/sj.ki.5002312. [DOI] [PubMed] [Google Scholar]
- 118.Basile DP. Rarefaction of peritubular capillaries following ischemic acute renal failure: a potential factor predisposing progressive nephropathy. Current Opinion in Nephrology and Hypertension. 2004;13:1–13. doi: 10.1097/00041552-200401000-00001. [DOI] [PubMed] [Google Scholar]
- 119.Norman J, Fine LG. Intrarenal oxygenation in chronic renal failure. Clinical & Experimental Pharmacology & Physiology. 2006;133:989–996. doi: 10.1111/j.1440-1681.2006.04476.x. [DOI] [PubMed] [Google Scholar]
- 120.Pechman KR, De Miguel C, Lund H, Leonard EC, Basile DP, Mattson DL. Recovery from renal ischemia-reperfusion injury is associated with altered renal hemodynamics, blunted pressure natriuresis, and sodium-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol. 2009;297(5):R1358–1363. doi: 10.1152/ajpregu.91022.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Spurgeon-Pechman KR, Donohoe DL, Mattson DL, Lund H, James L, Basile DP. Recovery from acute renal failure predisposes hypertension and secondary renal disease in response to elevated sodium. Am J Physiol Renal Physiol. 2007;293:F269–278. doi: 10.1152/ajprenal.00279.2006. [DOI] [PubMed] [Google Scholar]
- 122.Basile DP, Friedrich JL, Spahic J, Knipe NL, Mang HE, Leonard EC, Ashtiyani SC, Bacallao RL, Molitoris BA, Sutton TA. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. American Journal of Physiology - Renal Physiology. 2011;300:F721–33. doi: 10.1152/ajprenal.00546.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Leonard EC, Friderich J, Basile DP. VEGF-121 preserves renal microvessel structure and ameliorates secondary renal disease following acute kidney injury. Am J Physiol-Renal Physiol. 2008;295:F1648–57. doi: 10.1152/ajprenal.00099.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee HT, Park SW, Kim M, D'Agati VD. Acute kidney injury after hepatic ischemia and reperfusion injury in mice. Lab Invest. 2008;89(2):196–208. doi: 10.1038/labinvest.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kida Y, Ieronimakis N, Schrimpf C, Reyes M, Duffield JS. EphrinB2 Reverse Signaling Protects against Capillary Rarefaction and Fibrosis after Kidney Injury. Journal of the American Society of Nephrology. 2013;24(4):559–572. doi: 10.1681/ASN.2012080871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Basile DP, Rovak JM, Martin DR, Hammerman MR. Increased transforming growth factor-β expression in regenerating rat renal tubules following ischemic injury. American Journal of Physiology. 1996;270:F500–F509. doi: 10.1152/ajprenal.1996.270.3.F500. Renal Fluid Electrolyte Physiol. 39. [DOI] [PubMed] [Google Scholar]
- 127.Spurgeon KS, Donohoe DL, Basile DP. Transforming growth factor-β in acute renal failure: receptor expression, influence in cell proliferation, cellularity and vascularization after recovery from injury. Am J Physiol Renal Physiol. 2005;288:F568–F577. doi: 10.1152/ajprenal.00330.2004. [DOI] [PubMed] [Google Scholar]
- 128.Basile DP, Fredrich K, Weihrauch DW, Hattan N, Chilian WM. Angiostatin and matrix metalloprotease expression following ischemic acute renal failure. Am J Physiol Renal Physiol. 2004;286:F893–F902. doi: 10.1152/ajprenal.00328.2003. [DOI] [PubMed] [Google Scholar]
- 129.Basile DP, Fredrich K, Chelladurai B, Leonard EC, Parrish AR. Renal ischemia reperfusion inhibits VEGF expression and induces ADAMTS-1, a novel VEGF inhibitor. Am J Physiol Renal Physiol. 2008;294 doi: 10.1152/ajprenal.00596.2007. [DOI] [PubMed] [Google Scholar]
- 130.Kanellis J, Paizis K, Cox AJ, Stacker SA, Gilbert RE, Cooper ME, Power DA. Renal ischemia-reperfusion increases endothelial VEGFR-2 without increasing VEGF or VEGFR-1 expression. Kidney International. 2002;61(5):1696–706. doi: 10.1046/j.1523-1755.2002.00329.x. [DOI] [PubMed] [Google Scholar]
- 131.Johnson RJ, Schreiner GF. Hypothesis: the role of acquired tubulointerstitial disease in the pathogenesis of salt-dependent hypertension. Kidney International. 1997;52(5):1169–79. doi: 10.1038/ki.1997.442. [DOI] [PubMed] [Google Scholar]
- 132.Suga S, Kim YG, Joly A, Puchacz E, Kang DH, Jefferson JA, Abraham JA, Hughes J, Johnson RJ, Schreiner GF. Vascular endothelial growth factor (VEGF121) protects rats from renal infarction in thrombotic microangiopathy. Kidney International. 2001;60(4):1297–308. doi: 10.1046/j.1523-1755.2001.00935.x. [DOI] [PubMed] [Google Scholar]
- 133.Kang DH, Hughes J, Mazzali M, Schreiner GF, Johnson RJ. Impaired angiogenesis in the remnant kidney model: II. Vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function. Journal of the American Society of Nephrology. 2001;12(7):1448–57. doi: 10.1681/ASN.V1271448. [DOI] [PubMed] [Google Scholar]
- 134.Jung YJ, Kim DH, Lee AS, Lee S, Kang KP, Lee SY, Jang KY, Sung MJ, Park SK, Kim W. Peritubular capillary preservation with COMP-angiopoietin-1 decreases ischemia-reperfusion-induced acute kidney injury. American Journal of Physiology - Renal Physiology. 2009;297(4):F952–F960. doi: 10.1152/ajprenal.00064.2009. [DOI] [PubMed] [Google Scholar]
- 135.Zhang B, Chen N, Shi W, Wang W, Shi H, Yu H. Peritubular Capillary Loss Is Ameliorated by Ramipril or Valsartan Treatment. Microcirculation. 2008;15(4):337–348. doi: 10.1080/10739680701743540. [DOI] [PubMed] [Google Scholar]
- 136.Long D, Mu W, Price K, Roncal C, Schreiner GF, Woolf AS, Johnson R. Vascular endothelial growth factor administration does not improve microvascular disease in the salt-dependent phase of post angiotensin II hypertension. Am J Physiol. 2006;291:F1248–54. doi: 10.1152/ajprenal.00096.2006. [DOI] [PubMed] [Google Scholar]
- 137.Chade AR, Kelsen S. Reversal of renal dysfunction by targeted administration of VEGF into the stenotic kidney: a novel potential therapeutic approach. American Journal of Physiology - Renal Physiology. 2012;302(10):F1342–F1350. doi: 10.1152/ajprenal.00674.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Freeburg PB, Abrahamson DR. Hypoxia-Inducible Factors and Kidney Vascular Development. Journal of the American Society of Nephrology. 2003;14(11):2723–2730. doi: 10.1097/01.asn.0000092794.37534.01. [DOI] [PubMed] [Google Scholar]
- 139.Nyengaard J, Rasch R. The impact of experimental diabetes mellitus in rats on glomerular capillary number and sizes. Diabetologia. 1993;36(3):189–194. doi: 10.1007/BF00399948. [DOI] [PubMed] [Google Scholar]
- 140.Ostendorf T, Kunter U, Eitner F, Loos A, Regele H, Kerjaschki D, Henninger DD, Janjic N, Floege Jr. VEGF165 mediates glomerular endothelial repair. The Journal of Clinical Investigation. 1999;104(7):913–923. doi: 10.1172/JCI6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nangaku M, Alpers CE, Pippin J, Shankland SJ, Adler S, Kurokawa K, Couser WG, Johnson RJ. A new model of renal microvascular endothelial injury. Kidney International. 1997;52(1):182–94. doi: 10.1038/ki.1997.318. [DOI] [PubMed] [Google Scholar]
- 142.Iruela-Arispe L, Gordon K, Hugo C, Duijvestijn A, Claffey K, Reilly M, Couser W, Alpers C, Johnson R. Participation of glomerular endothelial cells in the capillary repair of glomerulonephritis. Am J Pathol. 1995;147(6):1715–1727. [PMC free article] [PubMed] [Google Scholar]
- 143.Floege J, Burns MW, Alpers CE, Yoshimura A, Pritzl P, Gordon K, Seifert RA, Bowen-Pope DF, Couser WG, Johnson RJ. Glomerular cell proliferation and PDGF expression precede glomerulosclerosis in the remnant kidney model. Kidney Int. 1992;41(2):297–309. doi: 10.1038/ki.1992.42. [DOI] [PubMed] [Google Scholar]
- 144.Hohenstein B, Kuo MC, Addabbo F, Yasuda K, Ratliff B, Schwarzenberger C, Eckardt KU, Hugo CPM, Goligorsky MS. Enhanced progenitor cell recruitment and endothelial repair after selective endothelial injury of the mouse kidney. American Journal of Physiology - Renal Physiology. 2010;298(6):F1504–F1514. doi: 10.1152/ajprenal.00025.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Basile DP, Friedrich JL, Spahic J, Knipe NL, Mang HE, Leonard EC, Ashtiyani SC, Bacallao RL, Molitoris BA, Sutton TA. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. American Journal of Physiology - Renal Physiology. 2011 doi: 10.1152/ajprenal.00546.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lin S, Chang F, Schrimpf C, Chen Y, Wu C, Wu V, Chiang W, Kuhnert F, Kuo C, Chen Y, Wu K, Tsai T, duffield J. Targetting endothelium pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol. 2011;178:911–23. doi: 10.1016/j.ajpath.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schrimpf C, Xin C, Campanholle G, Gill S, Stallcup W, Lin S, David G, Gharib S, Humphreys BD, Duffield JS. Pericyte TIMP3 and ADAMTS1 modulate vascular stability after kidney injury. Journal of the American Society of Nephrology. 2012 doi: 10.1681/ASN.2011080851. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of Putative Progenitor Endothelial Cells for Angiogenesis. Science. 1997;275(5302):964–966. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 149.Kogler G, Critser P, Trapp T, Yoder M. Future of cord blood for non-oncology uses. Bone Marrow Transplant. 2009;44(10):683–697. doi: 10.1038/bmt.2009.287. [DOI] [PubMed] [Google Scholar]
- 150.Patschan D, Patschan S, Muller GA. Endothelial Progenitor Cells in Acute Ischemic Kidney Injury: Strategies for Increasing the Cells' Renoprotective Competence. International Journal of Nephrology. 2011;2011 doi: 10.4061/2011/828369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, Krasich R, Temm CJ, Prchal JT, Ingram DA. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109(5):1801–1809. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Asahara T, Kawamoto A, Masuda H. Concise Review: Circulating Endothelial Progenitor Cells for Vascular Medicine. Stem Cells. 2011;29(11):1650–1655. doi: 10.1002/stem.745. [DOI] [PubMed] [Google Scholar]
- 153.Rookmaaker MB, Verhaar MC, van Zonneveld AJ, Rabelink TJ. Progenitor cells in the kidney: biology and therapeutic perspectives. Kidney International. 2004;66(2):518–22. doi: 10.1111/j.1523-1755.2004.761_10.x. [DOI] [PubMed] [Google Scholar]
- 154.Li J, Deane JA, Campanale NV, Bertram JF, Ricardo SD. Blockade of p38 Mitogen-Activated Protein Kinase and TGF-beta/Smad Signaling Pathways Rescues Bone Marrow–Derived Peritubular Capillary Endothelial Cells in Adriamycin-Induced Nephrosis. Journal of the American Society of Nephrology. 2006;17(10):2799–2811. doi: 10.1681/ASN.2006020130. [DOI] [PubMed] [Google Scholar]
- 155.Duffield JS, Park KM, Hsiao LL, Kelley VR, Scadden DT, Ichimura T, Bonventre JV. Restoration of tubular epithelial cells during repair of the postischemic kidney occurs independently of bone marrow-derived stem cells. Journal of Clinical Investigation. 2005;115(7):1743–55. doi: 10.1172/JCI22593. see comment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yoder MC. Defining human endothelial progenitor cells. Journal of Thrombosis and Haemostasis. 2009;7(s1):49–52. doi: 10.1111/j.1538-7836.2009.03407.x. [DOI] [PubMed] [Google Scholar]
- 157.Assmus B, Honold J, Schächinger V, Britten MB, Fischer-Rasokat U, Lehmann R, Teupe C, Pistorius K, Martin H, Abolmaali ND, Tonn T, Dimmeler S, Zeiher AM. Transcoronary Transplantation of Progenitor Cells after Myocardial Infarction. New England Journal of Medicine. 2006;355(12):1222–1232. doi: 10.1056/NEJMoa051779. [DOI] [PubMed] [Google Scholar]
- 158.Napoli C, Hayashi T, Cacciatore F, Casamassimi A, Casini C, Al-Omran M, Ignarro LJ. Endothelial progenitor cells as therapeutic agents in the microcirculation: An update. Atherosclerosis. 2010;215(1):9–22. doi: 10.1016/j.atherosclerosis.2010.10.039. [DOI] [PubMed] [Google Scholar]
- 159.Rehman J, Li J, Orschell CM, March KL. Peripheral Blood Endothelial Progenitor Cells Are Derived From Monocyte/Macrophages and Secrete Angiogenic Growth Factors. Circulation. 2003;107(8):1164–1169. doi: 10.1161/01.cir.0000058702.69484.a0. [DOI] [PubMed] [Google Scholar]
- 160.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner JM. Bone Marrow Origin of Endothelial Progenitor Cells Responsible for Postnatal Vasculogenesis in Physiological and Pathological Neovascularization. Circulation Research. 1999;85(3):221–228. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 161.Yasuda K, Park HC, Ratliff B, Addabbo F, Hatzopoulos AK, Chander P, Goligorsky MS. Adriamycin Nephropathy: A Failure of Endothelial Progenitor Cell-Induced Repair. Am J Pathol. 2010;176(4):1685–1695. doi: 10.2353/ajpath.2010.091071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Patschan D, Krupincza K, Patschan S, Zhang Z, Hamby C, Goligorsky MS. Dynamics of mobilization and homing of endothelial progenitor cells after acute renal ischemia: modulation by ischemic preconditioning. American Journal of Physiology - Renal Physiology. 2006;291(1):F176–F185. doi: 10.1152/ajprenal.00454.2005. [DOI] [PubMed] [Google Scholar]
- 163.Schwarz TM, Leicht SF, Radic T, Rodriguez-Araboalaza I, Hermann PC, Berger F, Saif J, Becker W, Ellwart JW, Aicher A, Heeschen C. Vascular Incorporation of Endothelial Colony-Forming Cells Is Essential for Functional Recovery of Murine Ischemic Tissue Following Cell Therapy. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;32(2):e13–e21. doi: 10.1161/ATVBAHA.111.239822. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 164.Ingram DA, Caplice NM, Yoder MC. Unresolved questions, changing definitions, and novel paradigms for defining endothelial progenitor cells. Blood. 2005;106(5):1525–31. doi: 10.1182/blood-2005-04-1509. [DOI] [PubMed] [Google Scholar]
- 165.Yoon CH, Hur J, Park KW, Kim JH, Lee CS, Oh IY, Kim TY, Cho HJ, Kang HJ, Chae IH, Yang HK, Oh BH, Park YB, Kim HS. Synergistic Neovascularization by Mixed Transplantation of Early Endothelial Progenitor Cells and Late Outgrowth Endothelial Cells. Circulation. 2005;112(11):1618–1627. doi: 10.1161/CIRCULATIONAHA.104.503433. [DOI] [PubMed] [Google Scholar]
- 166.Yoder MC. Is Endothelium the Origin of Endothelial Progenitor Cells? Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(6):1094–1103. doi: 10.1161/ATVBAHA.109.191635. [DOI] [PubMed] [Google Scholar]
- 167.Basile DP, Zeng P, Friedrich JL, Leonard E, Yoder MC. Low proliferative potential and impaired angiogenesis of cultured rat kidney endothelial cells. Microcirculation. 2012;19:598–609. doi: 10.1111/j.1549-8719.2012.00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]