

Abstract
Competing mechanisms of long-term potentiation (LTP) and long-term depression (LTD) in principal neurons of the basolateral amygdala (BLA) are thought to underlie the acquisition and consolidation of fear memories, and their subsequent extinction. However, no study to date has examined the locus of action and/or the cellular mechanism(s) by which these processes interact. Here, we report that synaptic plasticity in the cortical pathway onto BLA principal neurons is frequency-dependent and shows a transition from LTD to LTP at stimulation frequencies of ∼10 Hz. At the crossover point from LTD to LTP induction we show that concurrent activation of D1 and group II metabotropic glutamate (mGluR2/3) receptors act to nullify any net change in synaptic strength. Significantly, blockade of either D1 or mGluR2/3 receptors unmasked 10 Hz stimulation-induced LTD and LTP, respectively. Significantly, prior activation of presynaptic D1 receptors caused a time-dependent attenuation of mGluR2/3-induced depotentiation of previously induced LTP. Furthermore, studies with cell type-specific postsynaptic transgene expression of designer receptors activated by designer drugs (DREADDs) suggest that the interaction results via bidirectional modulation of adenylate cyclase activity in presynaptic glutamatergic terminals. The results of our study raise the possibility that the temporal sequence of activation of either presynaptic D1 receptors or mGluR2/3 receptors may critically regulate the direction of synaptic plasticity in afferent pathways onto BLA principal neurons. Hence, the interaction of these two neurotransmitter systems may represent an important mechanism for bidirectional metaplasticity in BLA circuits and thus modulate the acquisition and extinction of fear memory.
Key points
Synaptic plasticity [long-term potentiation (LTP) and long-term depression (LTD) /depotentiation] in principal neurons of the basolateral amygdala (BLA) may underlie the acquisition, consolidation and extinction of fear memories, respectively.
Dopamine-dependent LTP and group II metabotropic glutamate (mGluR2/3) receptor-induced synaptic depotentiation in principal neurons of the BLA are thought to facilitate and attenuate fear memory formation, respectively.
Here, we report that synaptic plasticity in BLA principal neurons is frequency-dependent, with the transition from LTD to LTP occurring at stimulation frequencies at, or around, 10 Hz.
Low frequency paired-pulse stimulation (PP-LFS) of afferents to the BLA elicits mGluR2/3 receptor-dependent LTD, whereas high frequency stimulation elicits D1 receptor-dependent LTP. At intermediate frequencies, synaptic strength is stable due to co-activation of both mGluR2/3 and D1 signalling cascades.
The temporal relationship of mGluR2/3 and D1 signalling is highly relevant for synaptic plasticity, with pre-application of D1 agonists blocking mGluR2/3 receptor-dependent synaptic depression and depotentiation.
Characterization of the functional interactions between these two systems may not only provide important clues to link the hypothesized aberrant dopamine-mediated molecular mechanisms and mGluR2-related treatment to the pathophysiology of these psychiatric disorders, but also represent novel targets for anxiolytic pharmacotherapy in amygdala.
Introduction
Growing evidence indicates that the induction of long-term potentiation (LTP) in basolateral amygdala (BLA) principal neurons underlies the acquisition and consolidation of fear memories (Rogan et al. 1997; Goosens & Maren, 2002), whereas long-term depression (LTD)/depotentiation is thought to facilitate the extinction of learned fear (Dalton et al. 2012). However, mutant mice lacking postsynaptic density-95 (PSD-95) protein that show enhanced hippocampal LTP and reduced LTD have impaired spatial memory (Migaud et al. 1998). A similar spatial memory impairment is seen in mice lacking the insulin receptor substrate p53 (IRSp53) that have enhanced hippocampal LTP but normal LTD (Kim et al. 2009). Conversely, mice expressing a dominant-negative mutant of protein kinase A (PKA) show impaired LTP and enhanced LTD and have deficits in consolidating long term memory but enhanced performance in working memory (Malleret et al. 2010). Together these data suggest that a functional interaction between LTD and LTP is probably required for normal memory formation and extinction.
Although previous studies have reported the induction of LTP and LTD in BLA principal neurons in response to afferent stimulation at 1 and 100 Hz, respectively (Huang & Kandel, 1998; Li et al. 1998, 2011; Lin et al. 2000, 2005), no study to date has systematically examined the response of these neurons to afferent stimulation at intermediate frequencies, or any potential interaction between receptor signalling cascades that might mediate these processes.
Evidence from behavioural and electrophysiological studies indicates that fear memory formation requires dopamine release and the subsequent activation of postsynaptic dopamine D1-like receptors in the BLA (Lamont & Kokkinidis, 1998; Greba & Kokkinidis, 2000; Fadok et al. 2009; Takahashi et al. 2010). Recently, we have shown that a similar sequence of events was necessary for the induction and expression of LTP of cortical inputs onto BLA principal neurons (Li et al. 2011), and that LTP in this pathway was dependent on postsynaptic activation of the adenylyl cyclase (AC)–cAMP–PKA signalling cascade. However, D1 receptors (D1Rs) are also found in presynaptic elements in the BLA, and are often observed on axon terminals making asymmetric contacts with dendritic spines (Muly et al. 2009), suggesting a possible role for these receptors in presynaptic modulation of excitatory transmission. In contrast, activation of group II metabotropic glutamate (mGluR2/3) receptors in the amygdala reduces fear memory formation and elicits an anxiolytic response in both rodents and humans (Helton et al. 1998; Shekhar & Keim, 2000; Walker & Davis, 2002; Walker et al. 2002; Grillon et al. 2003). We have shown that mGluR2/3 receptors are widely distributed in the BLA of rats and non-human primates, where they are primarily located on axon terminals making asymmetric synaptic contacts with dendritic spines of BLA neurons (Muly et al. 2007). Typically mGluR2/3 receptors are coupled to the Gαi/o signal transduction system and function to reduce cAMP levels (Conn & Pin, 1997). Activation of postsynaptic mGluR2/3 receptors directly inhibits BLA principal neurons (Rainnie et al. 1994), whereas activation of presynaptic receptors attenuates excitatory neurotransmission (Rainnie & Shinnick-Gallagher, 1992), induces LTD and de-potentiates previously enhanced synaptic input onto principal neurons (Lin et al. 2000, 2005).
As mGluR2/3 receptors negatively regulate AC activation, synaptic depression and depotentiation in BLA afferents may result from a selective reduction of presynaptic cAMP levels (Prezeau et al. 1992). Conversely, D1Rs are positive modulators of AC activity that could potentially raise presynaptic cAMP levels and facilitate LTP (Clark & White, 1987; Andersen et al. 1990). Together these data suggest that activation of presynaptic mGluR2/3 and D1Rs would have markedly opposing actions on excitatory transmission in the BLA and hence on the formation and consolidation of BLA-dependent fear memory. Significantly, studies in the hippocampus have reported that mGluR2 can be uncoupled from its downstream effectors by cAMP-dependent PKA phosphorylation of a single serine residue on its C terminus, thereby reducing its ability to inhibit excitatory transmission (Kamiya & Yamamoto, 1997; Maccaferri et al. 1998; Schaffhauser et al. 2000). Hence, activation of presynaptic Gαs-coupled D1Rs could significantly attenuate the subsequent response to activation of Gαi/o-coupled receptors, such as mGluR2/3, and vice versa. If a similar process occurs in the BLA, the temporal sequence of either D1R or mGluR2/3 activation could markedly regulate the degree of synaptic plasticity occurring in the input stream onto BLA principal neurons. Consistent with this notion, the Beinenstock, Cooper and Munro theory of experience-dependent synaptic plasticity (BCM theory) has proposed that the activity history of a neuron may influence its subsequent response to synaptic input (Bienenstock et al. 1982), i.e. plasticity of synaptic plasticity, also known as metaplasticity (Abraham & Bear, 1996). A classic form of metaplasticity, observed in multiple brain regions, is the frequency-dependent transition from homosynaptic LTD induction to LTP induction (Bear & Malenka, 1994). In general, stimulation frequencies < 7 Hz induce LTD, whereas stimulation frequencies > 20 Hz favour the induction of LTP both in vitro and in vivo (Blaise & Bronzino, 2003; Froc & Racine, 2005). Moreover, the transition is gradual such that stimulation at ∼10 Hz, paradoxically, results in no net change in synaptic strength (Mayford et al. 1995), suggesting that at this ‘crossover point’ competing cellular processes mediating LTD and LTP may negate one another.
We hypothesized that if a similar frequency-dependent metaplasticity occurs in the BLA, the cellular processes competing at the crossover point may be mediated by activation of presynaptic mGluR2/3 and D1Rs and their respective second messenger signalling cascades. Consistent with this hypothesis, in the prefrontal cortex, ‘priming’ with dopamine prior to low frequency stimulation (LFS), which normally induces LTD, now resulted in the induction of LTP (Matsuda et al. 2006). Conversely, inhibition of LTP by a weak 50 Hz preconditioning stimulus was blocked by co-administration of a non-selective mGluR2/3 antagonist (Gisabella et al. 2003). Hence, the timing of D1R or mGluR2/3 activation may be a critical factor in determining the crossover point of the LTD/LTP frequency–response curve.
The present study was designed to determine whether graded frequency-dependent synaptic plasticity occurs in BLA principal neurons and, if so, determine whether a functional interaction between the mGluR2/3 and D1R systems determines the crossover point for synaptic plasticity in these neurons.
Methods
Animals
A total of 86 male Sprague–Dawley rats (6–8 weeks old, Charles River, Raleigh, NC, USA) were used in this study. Every effort was made to minimize both animal suffering and the number of animals necessary to complete the study. All experimental protocols conform strictly to National Institutes of Health guidelines for the Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee of Emory University.
Slice preparation
Slices of 350 μm thickness containing the BLA were obtained as described previously (Li et al. 2011). Briefly, the brains of isoflurane (Fisher Scientific, Hanoverpark, IL, USA) anaesthetized animals were rapidly dissected out and immersed in a cold (4°C) 95–5% oxygen/carbon dioxide oxygenated ‘cutting solution’ with the following composition (in mm): NaCl (130), NaHCO3 (30), KCl (3.50), KH2PO4 (1.10), MgCl2 (6.0), CaCl2 (1.0) and glucose (10), supplemented with kynurenic acid (2.0). The brain was then blocked and slices containing the BLA were cut using a Leica VTS-1000 vibratome (Leica Microsystems Inc., Bannockburn, IL, USA). After cutting, slices were maintained at 37°C in oxygenated ‘cutting solution’ for at least 50 min before transferring to regular artificial cerebrospinal fluid (ACSF) containing (in mm): NaCl (130), NaHCO3 (30), KCl (3.50), KH2PO4 (1.10), MgCl2 (1.30), CaCl2 (2.50) and glucose (10). Slices were kept in the regular ACSF for at least 30 min before recording.
Patch clamp recording
Individual slices were transferred to a submersion-type recording chamber mounted on the fixed stage of a Leica DM6000 FS microscope (Leica Microsystems), and continuously perfused by gravity-fed oxygenated 32°C ACSF at a flow rate of 1–2 ml min−1. Slices were viewed using differential interference contrast optics and IR illumination with an IR-sensitive CCD camera (Orca-flash2.8, Hamamatsu, Tokyo, Japan). Thin-walled borosilicate glass patch electrodes (WPI, Sarasota, FL, USA), which had a resistance of 4–6 MΩ, were filled with (in mm): 130 potassium gluconate, 2 KCl, 10 Hepes, 3 MgCl2, 2 K-ATP, 0.2 NaGTP and 5 phosphocreatine, adjusted to pH 7.3 with KOH, and having an osmolarity of 280–290 mOsm. Individual BLA projection neurons were visualized in situ using differential interference contrast microscopy in combination with a 40× water immersion objective and displayed in real time on a computer monitor. Projection neurons were identified according to their characteristic size and shape (Rainnie et al. 1993), and were normally located between 50 and 120 μm beneath the surface of the slice. Data acquisition and analysis were performed using a MultiClamp700B amplifier in conjunction with pClamp10.0 software and a DigiData 1320A AD/DA interface (Molecular Devices, Sunnyvale, CA, USA). Whole cell patch clamp recordings were obtained and recorded voltages were low-pass filtered at 5 kHz and digitized at 10–20 kHz. At the start of each experiment, a series of standardized current clamp protocols were performed to validate the identity of BLA projection neurons (Rainnie et al. 1993).
To examine the frequency–response curve of synaptic plasticity in BLA projection neurons, pharmacologically isolated excitatory postsynaptic potentials (eEPSPs) were evoked as previously described (Braga et al. 2004). In brief, a concentric bipolar stimulation electrode (FHC, Bowdoinham, ME, USA) was placed approximately 500 μm from the recorded neuron, close to the fibre tract of the external capsule immediately adjacent to the BLA. In all experiments, 50 μm picrotoxin was added to the patch solution to block GABAA currents exclusively in the recorded neuron. In addition, slices were continuously perfused with oxygenated ACSF (32°C) containing the GABAB receptor antagonist CGP36742 (2 μm). This recording configuration allowed stable recording of isolated EPSPs without contamination from epileptiform, recurrent EPSPs.
EPSPs were evoked at 0.05 Hz and adjusted to 30% of the maximal response, and those eEPSPs obtained 10 min immediately before drug treatment were considered as baseline. All EPSPs evoked during and after treatments were normalized to the mean baseline amplitude and expressed as a percentage of baseline amplitude. For LTD and LTP experiments recordings continued for at least 40 min after the induction protocol terminated. The protocols used to induce LTD were: (1) LFS using 900 stimuli at 1 Hz; and (2) paired-pulse LFS (PP-LFS) using 900 paired stimuli with a 50 ms inter-stimulus-interval and delivered at 1 Hz. As previously reported LTP was induced using a 5× 100 Hz protocol, which consisted of five trains of stimulation at 100 Hz for 1 s duration, applied at 20 s intervals. A 2× 100 Hz protocol was used as a subthreshold protocol to examine the effects of drug treatment on LTP threshold (Li et al. 2011). Intervening stimulation frequencies consisted of 900 stimulation pulses at 5, 10 and 30 Hz. A DC holding current was injected to maintain the membrane potential at −70 mV, except during high frequency stimulation (HFS) when the potential was adjusted to −60 mV.
To determine if synaptic plasticity was mediated at pre- or postsynaptic loci, we calculated the paired-pulse ratio (PPR) of eEPSPs during the first and last 30 s of the PP-LFS LTD protocol; PPR was calculated as eEPSP2/eEPSP1, where eEPSP1 and eEPSP2 represent the amplitude of the first and the second eEPSP, respectively. In addition, eEPSPs recorded during a 5 min period directly before or 20 min after a particular treatment were used to calculate the coefficient of variation (CV). When synaptic efficacy changes at a presynaptic locus CV will either increase or decrease (Faber & Korn, 1991). CV was calculated as σ/μ where σ and μ are the standard deviation and mean amplitude of 15 successive eEPSPs, respectively. SD was calculated from the variance taken as the total amplitude variance minus the variance due to background noise. Background noise variance was calculated using the same method as used to measure eEPSP amplitude by using 2 ms traces before stimulus. During each experiment the input resistance of BLA projection neurons was monitored throughout by injecting a transient hyperpolarizing current (0.2 nA, 200 ms) before each successive stimulation. Hence, any effect of drug treatment on resting membrane input resistance could be determined immediately prior to each eEPSP.
Adenoassociated viral vector (AAV) infusion procedure
For these studies 6-week-old rats (n = 16) were anaesthetized with an i.p. injection of a mixture of dexdomitor (0.16 mg kg−1) (Pfizer Animal Health, New York, USA) and ketamine hydrochloride (48 mg kg−1) (Butler-Schein Animal Health, Dublin, OH, USA) and mounted in a stereotaxic frame, the was skull exposed, and a burr hole was drilled above the injection site. A 26-gauge Hamilton microsyringe (Reno, NV, USA) was then lowered towards the BLA using the following coordinates: anterior/posterior, −2.1 mm; medial/lateral, ± 5.0 mm; dorsal/ventral, −8.0 mm, and 1 μl of virus (either AAV5-CaMKIIα-HA-hM4D(Gi)-IRES-mCitrine or AAV5-CaMKIIα-HA-rM3D(Gs)-IRES-mCitrine; University of North Carolina Viral Vector Core, approximate titre 1012) was injected at a rate of 0.1 μl min−1. Incisions in the scalp were closed and the animals were allowed to recover in accordance with IACUC guidelines. Animals were then killed for patch clamp recording 2 weeks after surgery.
Drug application
The following drugs were obtained from Sigma-Aldrich (St Louis, MO, USA): picrotoxin, 3-[[(3, 4-dichlorophenyl) methyl] amino] propyl]diethoxymethyl) phosphinic acid (CGP52432), 7β-acetoxy-8,13-epoxy-1α,6β,9α-trihydroxylabd-14-en-11-one (forskolin), clozapine- N-oxide (CNO), cis-N-(2-phenylcyclopentyl)-azacyclotridec-1-en-2-amine hydrochloride (MDL-12,330A hydrochloride); 3-hydroxytyramine hydrochloride (dopamine hydrochloride); and from Tocris R&D (Ellisville, MO, USA): (E)-2-cyano-3-(3,4-dihydrophenyl)-N-(phenylmethyl)-2-propenamide (AG490), 3-((R)-2-Carboxypiperazin-4-yl)-propyl-1-phosphonic acid ((R)-CPP); (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid (LY341495), (1R,4R,5S,6R)-4-amino-2-oxabicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY379268), 2,2,2-trifluoro-N-[4-(2-methoxypheno-xy)phenyl]-N-(3-pyridinylmethyl) ethanesulfonamide hydrochloride (LY487379 hydrochloride); (R)-(+)-7-Chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH 23390 hydrochloride); (±)-1-phenyl-2,3,4,5-tetrahydro-(1H)-3-benzazepine-7,8-diol hydrobromide (SKF38393 hydrobromide); (R)-adenosine, cyclic 3′,5′-(hydrogenphosphorothioate) triethylammonium (cAMPs-RP), 8-bromo-adenosine-3′,5′-cyclic monophosphate, sodium salt (8Br-cAMP) and 1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]-hexyl]-1H-pyrrole-2,5-dione (U 73122). All drugs were made as concentrated stock solutions in distilled water, except picrotoxin, forskolin, MDL-12230-A and U73122, which were prepared as stocks in 100% DMSO. The final concentration of DMSO was no more than 0.1% on final application. Drugs were applied in the ACSF using a continuous gravity-fed bath application unless specifically stated. An overview of the mechanism of drug action is given in Table 1.
Table 1.
Drug index
| Type | Drug name | Function |
|---|---|---|
| mGluR2/3 | LY341495 | Antagonist |
| LY379268 | Agonist | |
| LY487379 | mGluR2-specific positive allosteric modulator | |
| D1Rs | SCH23390 | Antagonist |
| SKF38393 | Agonist | |
| Dopamine | ||
| PKA | 8-Br-cAMP | Activator |
| Rp-cAMP | Inhibitor | |
| AC | Forskolin | Activator |
| MDL-12230A | Inhibitor | |
| PLC | U73211 | Inhibitor |
| Neomycin | Inhibitor | |
| Others | (R)-CPP | NMDAR antagonist |
| CNO | Agonist of DREADD | |
| AG490 | JAK2 inhibitor |
Statistics
All data are expressed as the mean ± SEM. The amplitudes of the EPSPs were normalized to the average baseline EPSP amplitude. All statistical tests were conducted using Excel 2007 or GraphPad Prism 4.0. Unless specified, Student's t test was used to detect significant differences between the baseline eEPSP amplitude and the eEPSP amplitude 30 min after drug treatment or the induction stimulation. A two-way repeated-measures ANOVA was performed to test the interaction of drug and time on the amplitude of the EPSPs. P < 0.05 was considered statistically significant for all measures.
Results
Frequency-dependent changes in synaptic plasticity in BLA principal neurons
We have shown that 5× HFS (100 Hz) electrical stimulation of cortical afferents in the external capsule (EC) that synapse onto BLA principal neurons produces a robust LTP of evoked monosynaptic eEPSPs (Li et al. 2011). Yet other groups have reported that LFS (1 Hz) of the same pathway induces a depression (LTD) of eEPSPs (Lin et al. 2000). Here, we extend these observations to examine the relative expression of LTD and/or LTP in principal neurons using intermediate afferent stimulation frequencies (5, 10 and 30 Hz). For these studies, monosynaptic eEPSPs were examined in isolation following blockade of GABA receptor-mediated synaptic transmission (see Methods). First we confirmed previous reports that low frequency 1 Hz EC stimulation (LFS; 900 pulses) induced a significant LTD of the eEPSP in principal neurons (eEPSP amplitude 30 min after LFS = 58 ± 9% of baseline values, n = 6, P < 0.01, Fig. 1A, B). Increasing the stimulation frequency to 5 Hz (900 pulses) induced a small but noticeable LTD in all neurons tested (84 ± 7% of baseline values, n = 5, P < 0.05, Fig. 1A). Intriguingly, stimulation at 10 Hz (900 pulses) had no significant net effect on the amplitude of the eEPSP (106 ± 8%, n = 5), whereas LTP was induced by stimulation at 30 Hz (900 pulses, 130 ± 8% of baseline values, n = 5, P < 0.01), as well as by the standard 5× 100 Hz stimulation protocol (152 ± 14%, n = 5). Together these data suggest that plasticity of the cortical input is strongly frequency-dependent, and that the transition from LTD to LTP in BLA principal neurons occurs at stimulation frequencies at, or around, a crossover point of ∼10 Hz.
Figure 1. Frequency-dependent LTP and LTD at intermediate stimulation frequencies is determined by an interaction between mGluR2/3 and D1R activation.
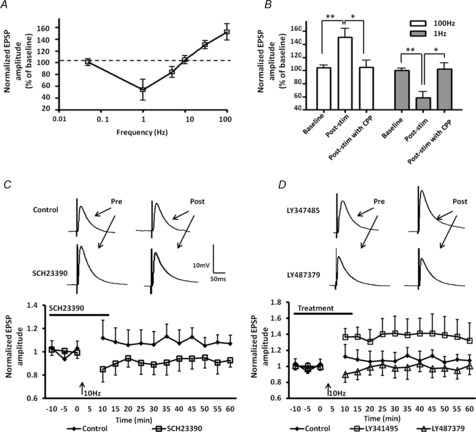
A, frequency versus synaptic strength summarizing the ability of different stimulation frequencies (1, 5, 10, 30 and 100 Hz) to induce robust changes in synaptic strength in the cortical afferent into BLA principal neurons; 0.05 Hz stimulation, which is used to monitor synaptic transmission throughout the experiments, had no effect on synaptic strength. The dashed line at 100% of baseline indicates no net change in synaptic strength. B, bar chart showing that the NMDAR antagonist, (R)-CPP, could block both HFS-induced LTP (n = 6) and LFS-induced LTD (n = 6). C, bath application of the D1R antagonist SCH23390 (10 μm, n = 6) for 10 min prior to and during 10 Hz stimulation unmasked a stimulation-induced LTD. D, application of the mGluR2/3 antagonist LY341495 (100 nm) for 10 min prior to and during 10 Hz stimulation unmasked a stimulation-induced LTP (n = 6). Application of the positive allosteric modulator of mGluR2, LY487379 (30 μm, n = 6), caused a transient depression of evoked EPSPs during the first 10 min after 10 Hz stimulation compared to the control group. **P < 0.01, *P < 0.05, error bars indicate SEM. Upper traces in C and D are representative EPSPs before and after 10 Hz stimulation for the treatment groups indicated.
We next examined what cellular processes mediate the frequency dependence of LTD and LTP. In the hippocampus, LFS-LTD and HFS-LTP are both reported to be sensitive to blockade by NMDA receptor antagonists (Malenka, 1994). To test if LTD and LTP in BLA principal neurons showed the same sensitivity, slices were pretreated with the NMDAR antagonist (R)-CPP (10 μm), for 10 min prior to, and during either LFS or HFS. Bath application of (R)-CPP completely blocked LFS-induced LTD (103 ± 8% of baseline values, n = 6, Fig. 1B, grey bars), as well as HFS-induced LTP (102 ± 11% of baseline values, n = 6, Fig. 1B, white bars), suggesting that NMDAR activation is necessary for synaptic plasticity at the extreme poles of the stimulus–response curve. Notably in the hippocampus NMDA receptor-dependent LTD is sensitive to blockade of the Janus kinase (JAK2)/signal transducer and activator of transcription (STAT3) pathway (Nicolas et al. 2012), whereas LTP is insensitive to blockade of this pathway. To test if NMDAR-LTD in BLA principal neurons was also dependent on activation of the JAK2/STAT3 pathway, we examined the effect of a selective JAK2 inhibitor, AG490 (10 μm), on LFS-induced LTD. Significantly, bath application of AG490 10 min prior to and during LFS markedly attenuated LFS-induced LTD (84 ± 7% of baseline values, n = 4, P < 0.05; data not shown) compared to control LTD (58 ± 9% of baseline values). Hence, activation of the JAK2 pathway is at least partially involved in LFS-induced LTD in BLA principal neurons. We next examined what processes may modulate synaptic plasticity at intermediate stimulation frequencies.
Activation of mGluR2/3 and D1Rs underlies competing synaptic plasticity at intermediate stimulation frequencies
Concurrent activation of NMDAR and D1Rs in the BLA is necessary for the induction of LTP (Li et al. 2011), whereas Lin et al. (2000) have reported a pharmacologically induced form of mGluR2/3-dependent LTD in BLA principal neurons that was insensitive to NMDAR antagonists (Lin et al. 2000). Hence, we reasoned that at intermediate stimulation frequencies competition between D1Rs and mGluRs may significantly affect the direction and magnitude of synaptic plasticity. To test this hypothesis, we first examined the effects of prior application of the D1R antagonist SCH23390 (10 μm) on the direction and magnitude of synaptic plasticity induced in BLA principal neurons following 10 Hz stimulation. Bath application of SCH23390 for 10 min prior to and during 10 Hz stimulation favoured the induction of LTD compared to the control 10 Hz stimulation group (89 ± 8% of baseline values, n = 6, Fig. 1C, squares). Two-way ANOVA showed a significant time and treatment effect compared to the control 10 Hz stimulation group (F1,117 = 21.4, P < 0.001). Hence, 10 Hz stimulation resulted in a significant activation of D1Rs that may occlude processes mediating LTD.
Conversely, bath application of an mGluR2/3 antagonist, LY341495 (100 nm), prior to and during 10 Hz stimulation shifted the frequency–response curve in favour of LTP induction (141 ± 19% of baseline values, n = 6, Fig. 1D, squares). A two-way ANOVA showed a significant time and treatment effect compared to the control 10 Hz stimulation group (F2,195 = 19.85, P < 0.001), suggesting that 10 Hz stimulation is also capable of significantly activating mGluR2/3. Because LY341495 does not distinguish between the relative contributions of mGluR2 and/or mGluR3 activation in the response to 10 Hz stimulation we next examined the effect of application of an mGluR2-specific positive allosteric modulator (PAM), LY487379. Compared to the control group, application of LY487379 (30 μm) caused a transient depression of the eEPSP amplitude during the first 10 min after 10 Hz stimulation (93 ± 10% of baseline values, n = 6, P < 0. 05, Fig. 1D, triangles), which then quickly returned to baseline. Together, these results suggest that at the 10 Hz crossover point, competition between D1R and mGluR2/3 signalling cascades nullify one another to prevent any net change in synaptic strength in BLA principal neurons. Hence, factors affecting the efficacy of either of these two signalling cascades could have a significant impact on frequency-dependent synaptic plasticity in BLA principal neurons particularly at input frequencies at or around the crossover point.
PP-LFS stimulation induces an mGluR2/3-dependent LTD in BLA principal neurons
We next determined whether a single stimulation paradigm could elicit an mGluR2/3-dependent form of LTD in BLA principal neurons. Elsewhere in the brain, an NMDAR-independent form of LTD has been identified that was mGluR-dependent, and which was evoked using a PP-LFS protocol (Domenici et al. 1998; Kemp et al. 2000; Martin et al. 2006; Kasanetz et al. 2010). Here, we examined whether the same PP-LFS protocol (50 ms PP interval, 900 paired stimuli at 1 Hz) could induce LTD in BLA principal neurons. As illustrated in Fig. 2A, PP-LFS of the EC induced an LTD of the eEPSP (69 ± 6% of baseline values, n = 6, P < 0.01, diamonds) that was insensitive to prior application of (R)-CPP (67 ± 9% of baseline values, n = 6, P < 0.01, squares). Similarly, application of the JAK2 inhibitor AG490 prior to and during PP-LFS failed to block PP-LFS LTD (74 ± 12% of baseline values, n = 5, P < 0.01; data not shown). As the mGluR2 PAM caused a transient facilitation of LTD following 10 Hz stimulation, we tested the sensitivity of PP-LFS LTD to prior application of the selective mGluR2/3 antagonist, LY347495. Application of LY347495 (100 nm) for 10 min prior to PP-LFS significantly attenuated the magnitude of LTD (89 ± 9% of baseline values, n = 6, P < 0.001, Fig. 2B, squares). Application of LY341495 alone caused no enduring change in the membrane input resistance of principal neurons at any time point measured (F10,70 = 0.72, P = 0.8466, two-way ANOVA, data not shown). In the hippocampus, PP-LTP was reported to be mediated by activation of mGluR1/5. Hence, we next examined whether the residual PP-LFS LTD observed in the presence of the mGluR2/3 antagonist could be blocked following application of MCCG (100 μm), a non-selective antagonist at both mGluR1/5 and mGluR2/3. MCCG fully blocked PP-LFS-induced LTD in BLA principal neurons (102 ± 7% of baseline values, n = 6, P < 0.001, Fig. 2C). Application of MCCG alone caused no enduring change in the membrane input resistance at any time point measured (F10,88 = 0.68, P = 0.6528, two-way ANOVA). Together, these results suggest that, unlike the hippocampus, PP-LFS-induced LTD in BLA principal neurons is primarily dependent on mGluR2/3 activation; however, there may also be a residual mGluR1/5 receptor-dependent component to the response.
Figure 2. PP-LFS elicits an mGluR2 receptor-dependent form of LTD in BLA principal neurons that was attenuated by an mGluR2/3 selective antagonist and mimicked by an mGluR2/3 agonist.
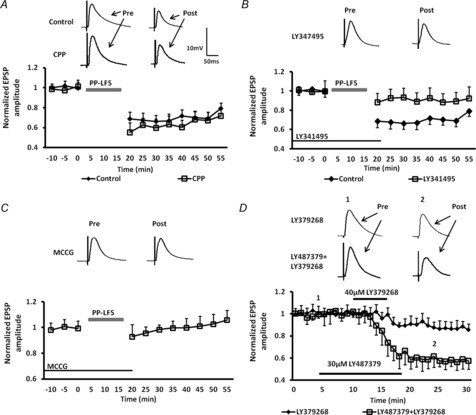
A, A PP-LFS at 1 Hz induced LTD (diamonds, n = 6) that was insensitive to the NMDAR antagonist, (R)-CPP (squares, n = 6). B, PP-LFS-induced LTD was markedly attenuated by the specific mGluR2/3 antagonist, LY341495 (100 nm, n = 6). C, PP-LFS-induced LTD was blocked by the non-selective mGluR1/5 and mGluR2/3 antagonist, MCCG (100 μm, n = 6). D, co-incubation with the positive allosteric modulator of mGluR2, LY487379 (30 μm), caused a 4-fold increase in the potency of the mGluR2/3 agonist LY379268 (40 μm, n = 5). Upper traces in A–D are representative EPSPs before and after PP-LFS for the treatment groups indicated.
Previous studies using exogenous agonist application to induce mGluR2/3-dependent synaptic depression had reported that the site of action was at presynaptic terminals. Consequently, we examined the PPR of eEPSPs during the first and last 30 s of the PP-LFS LTD protocol. Alterations in the PPR suggest a presynaptic site of action. Consistent with a presynaptic site of action, PPR changed from 1.32 ± 0.07 at the onset of the protocol to 1.56 ± 0.12 at the termination of the protocol (n = 7, data not shown). To further confirm a presynaptic locus we next examined the effect of application of the non-selective mGluR2/3 agonist LY379268 on the CV of eEPSP amplitude. Here, LY379268 (40–100 μm) caused a significant and dose-dependent depression in the amplitude of the eEPSP (LY-SD). Thus, 40 μm LY379268 reduced the eEPSP amplitude by 12 ± 3% (Fig. 2D, diamonds, n = 5), whereas 100 μm LY379268 reduced the eEPSP amplitude by 53 ± 8% (n = 6, P < 0.001, Fig. 3A). Analysis of the eEPSP amplitude revealed that 100 μm LY379268 increased the CV to 342 ± 47% of baseline in all cells tested (n = 6, P < 0.001, data not shown), further suggesting that LY-LFS LTD was presynaptic in origin. Moreover, as illustrated in Fig. 2D (squares), co-administration of the mGluR2 PAM, LY487379 (30 μm), caused an almost 4-fold increase in the potency of 40 μm LY379268, which now reduced the eEPSP amplitude to 58 ± 7% of baseline (Fig. 2D, n = 6, P < 0.001). Application of LY487379 alone caused a significant change in the CV for eEPSPs in any neuron tested (132 ± 27% of baseline; n = 6, data not shown). Hence, mGluR2/3-induced LTD in the BLA appears to be modulated, at least in part, by activation of presynaptic mGluR2.
Figure 3. The group II mGluR agonist, LY379268, causes a dose-dependent synaptic depression in BLA principal neurons.
A, application of 40 μm LY379268 elicits a modest synaptic depression of eEPSPs (n = 6), whereas application of 100 μm LY379268 elicits robust synaptic depression (n = 6). B, bar chart showing the facilitatory effect of co-application of the positive allosteric modulator of mGluR2, LY487379, on synaptic depression induced by 40 μm LY379268. **P < 0.01. Error bars indicate SEM. Upper traces in A are representative EPSPs evoked at time points 1 and 2.
Prior activation of D1Rs attenuates mGluR2/3-induced synaptic depression via a presynaptic AC-PKA pathway
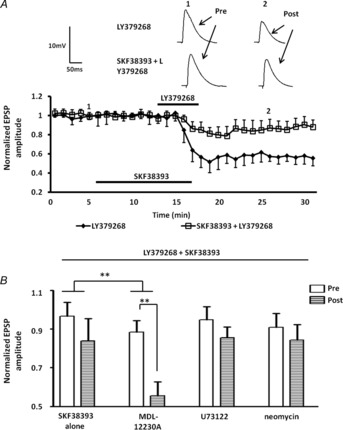
To directly examine the temporal dynamics of the interaction between D1R activation and mGluR2/3 activation, we next examined the effects of prior application of the D1R agonist, SKF38393, on LY-SD. As illustrated in Fig. 4, prior application of SKF38393 (50 μm for 10 min) significantly attenuated LY-SD (Fig. 4A, B, squares, 84 ± 11% of baseline values, n = 5). A two-way ANOVA showed a significant interaction of drug and time compared to the response to LY379268 application alone (F12,58 = 2.88, P < 0.01). Moreover, application of SKF38393 alone caused no enduring changes in either the amplitude of the eEPSP (107 ± 12% of baseline values, n = 10) or the membrane input resistance (P = 0.6828). As our results above confirmed previous observations (Lin et al. 2000) that LY-SD was mediated by presynaptic receptors, we next analysed the CV of the eEPSP amplitude following SKF38393 and LY379268 application to determine if the D1R agonist-mediated attenuation of LY-SD also had a possible presynaptic locus. Prior application of SKF38393 significantly attenuated the CV increase induced by LY379268 in all cells tested (165 ± 6% of baseline; n = 5, P < 0.001, data not shown). Together these data suggest that the attenuation of LY-SD by D1R activation was at least in part due to an interaction at a presynaptic locus.
Figure 4. Prior activation of D1Rs attenuated mGluR2/3-induced synaptic depression through activation of a presynaptic AC pathway.
A, prior application of 50 μm SKF38393 for 10 min significantly suppressed the LY-SD (n = 5). B, bar chart summarizing the effect of AC inhibitors and PLC inhibitors on SKF38393-induced suppression of LY-SD. Pre-incubation with the membrane-permeable AC inhibitor MDL-12230A (30 μm, n = 5) blocked the attenuation of LY-SD induced by SKF38393. Bath application of the membrane-permeable PLC inhibitor U73122 (10 μm, n = 4), or intracellular application of the impermeable PLC inhibitor neomycin (0.5 mm, n = 4) failed to block D1R agonist effect. ***P < 0.001, **P < 0.01. Error bars indicate SEM. Upper traces in A are representative EPSPs evoked at time points 1 and 2 for each treatment group.
To assess whether the D1R-mediated attenuation of LY-SD was dependent on AC activation, and to further determine its site of action we next examined the effect of intracellular application of the membrane-impermeable AC inhibitor 2′,5′-dideoxy-3′-ATP, or bath application of the membrane-permeable AC inhibitor MDL-12230A, on the D1R-mediated response. Intracellular application of 2′,5′-dideoxy-3′-ATP (1 μm) failed to block the D1R-mediated attenuation of the LY-SD response (79 ± 11% of baseline values, n = 5). Similarly, bath application of MDL-12230A (30 μm) 10 min prior to LY-SD also failed to block the effect of SKF38393 (86 ± 8% of baseline values, n = 5, data not shown). However, pre-incubation of slices with MDL-12230A for over 30 min prior to recording significantly attenuated the effect of SKF38393 (Fig. 4B, 55 ± 7% of baseline values, P < 0.01, n = 5). A two-way ANOVA showed a significant interaction of drug and time compared to the response to SKF38393 application alone (F12,64 = 2.94, P < 0.01). However, D1Rs can couple not only to Gαs and AC activation, but also to Gαq and phospholipase C (PLC) activation (Wang et al. 1995). To ensure that the response to D1R activation was not due to activation of the PLC pathway we tested the effects of prior application of two PLC inhibitors on D1R-mediated attenuation of LY-SD. Bath application of the membrane-permeable PLC inhibitor, U73122 (10 μm), or intracellular application of the membrane-impermeable PLC inhibitor, neomycin (0.5 mm), had no effect on the D1R-induced attenuation of LY-SD (Fig. 4B, U73122: 86 ± 6% of baseline values, n = 4; neomycin: 84 ± 8% of baseline values, n = 4). Application of U73122 (P = 0.7361) or neomycin (P = 0.5382) alone caused no enduring change in the membrane input resistance. Together these data suggest that D1Rs may activate an AC signalling pathway in glutamatergic terminals to attenuate LY-SD.
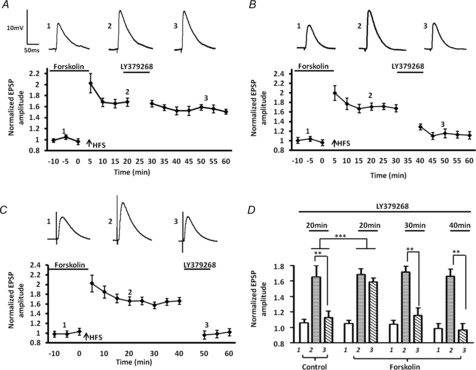
To further confirm that activation of presynaptic AC signalling pathway may attenuate mGluR2/3-induced synaptic depression, we next determined if forskolin, a non-specific activator of AC, could mimic the D1 agonist response. Pretreatment with forskolin (20 μm) for 10 min prior to application of LY379268 significantly reduced the magnitude of the LY-SD from 47 ± 9% of baseline in control ACSF (Fig. 5A, B, diamonds, n = 5) to 97 ± 5% of baseline (Fig. 5A, B, squares, n = 6). A two-way ANOVA showed a significant interaction of drug and time compared to the response of LY379268 application alone (F12,62 = 2.43, P < 0.001). Conversely, intracellular application of the PKA activator, 8-Br-cAMP (100 μm), via the patch pipette had no effect on LY-SD (Fig. 5B, 57 ± 8% of baseline values, P < 0.001, n = 6). Similarly, intracellular application of the selective PKA inhibitor, Rp-cAMP (100 μm), failed to block the effect of forskolin on LY-SD (Fig. 5C, 88 ± 6% of baseline values, n = 6). Application of Rp-cAMP (P = 0. 2436) and 8-Br-cAMP (P = 0.4875) alone had no effect on the membrane input resistance of BLA principal neurons. In contrast, application of the membrane-permeable AC inhibitor, MDL-12230A (>30 min), significantly reduced the effects of forskolin on LY-SD (Fig. 5C, 65 ± 6% of baseline, P < 0.001, n = 6). A two-way ANOVA showed a significant interaction of drug and time compared to the response to forskolin application alone (F12,60 = 3.41, P < 0.001). Together, these data further support the hypothesis that D1R-mediated attenuation of LY-SD is probably mediated by activation of a presynaptic AC-PKA signalling cascade.
Figure 5. Blockade of LY379268-induced synaptic depression by forskolin is independent of postsynaptic AC-PKA pathway activation.
A, bath application of foskolin (20 μm) fully blocked LY379268-induced synaptic depression (n = 6). B, bar chart comparing the effects of forskolin and intracellular 8-Br-cAMP (100 μm, n = 6) application on LY379268-induced synaptic depression. Inclusion of the AC activator, 8-Br-cAMP, in the postsynaptic compartment failed to mimic the effects of forskolin. C, bar chart comparing the effects of intracellular application of a membrane-impermeable PKA inhibitor, Rp-cAMPS (100 μm, n = 6), or bath application (>30 min) of a membrane-permeable AC inhibitor, MDL-12230A (30 μm, n = 6), on LY379268-induced synaptic depression. Pre-incubation with MDL-12230A markedly attenuated the forskolin effect (n = 6), whereas intracellular Rp-cAMPS failed to block the forskolin effect. ***P < 0.001, **P < 0.01. Error bars indicate SEM. Upper traces in A are representative EPSPs evoked before and after co-application of forskolin and LY379268.
The buffering capacity of presynaptic D1R activation on mGluR2/3-induced depotentiation is time-dependent
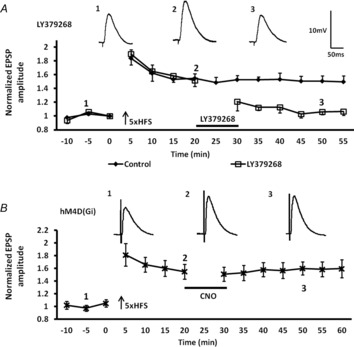
Previous studies have shown that HFS-induced LTP in the BLA can be de-potentiated by subsequent activation of mGluR2/3 (Lin et al. 2005). However, studies in the hippocampus have shown that depotentiation, and its timing, is critically dependent on the activity of a family of protein phosphatases (Jouvenceau et al. 2006; Genoux et al. 2011). Hence, we reasoned that if prior activation of D1Rs could attenuate LY-SD by activating an AC signalling pathway in glutamatergic terminals, then the kinetics of inactivation of the AC-PKA signalling cascade may also play a pivotal role in regulating the efficacy of mGluR2/3 to induce depotentiation of previously enhanced synaptic plasticity. First, we confirmed results from earlier studies and showed that the mGluR2/3 agonist, LY379268, could induce robust depotentiation of LTP in BLA principal neurons (Lin et al. 2005). As illustrated in Fig. 6A (open squares), a 10 min application of LY379268 (100 μm) starting 20 min after HFS resulted in significant depotentiation of LTP. A two-way ANOVA showed a significant interaction of drug and time compared to the response of the control group (F13,126 = 3.04, P < 0.001). When measured at 50 min after 5× HFS, the amplitude of the eEPSP was 106 ± 4% of baseline levels after LY379268 treatment, compared to 151 ± 8% of baseline values in the control 5× HFS group (P < 0.001, n = 5).
Figure 6. The mGluR2/3-dependent depotentiation of 5× HFS-induced LTP was not mimicked by DREADD-induced postsynaptic Gαi activation.
A, application of the mGluR2/3 agonist, LY379268 (100 μm), 20 min after 5× 100 Hz stimulation depotentiated (n = 5, squares) LTP in BLA afferents (n = 6, diamonds). B, activation of postsynaptic DREADD hM4D (Gi) receptors in BLA principal neurons following bath application of the selective ligand, CNO (10 μm, n = 5), failed to mimic the LY379268-induced depotentiation of LTP. Upper traces in A and B are representative EPSPs evoked at time points 1, 2 and 3.
To control for a postsynaptic site of the action of the mGluR2/3-induced depotentiation, we examined the effect of selective expression of a Gαi-coupled DREADD (designer receptors exclusively activated by designer drugs) in BLA principal neurons. Here, we used an AAV expressing a CaMKIIα promoter sequence to drive expression of a mutated muscarinic receptor, hM4D(Gi), in BLA principal neurons. We could then selectively examine the effect of postsynaptic inhibition of AC by exogenous application of the small molecule agonist of hM4D(Gi), CNO (Armbruster et al. 2007). First, we tested whether activating hM4D(Gi) by applying CNO (10 μm) 20 min after HFS could mimic the mGluR2/3 agonist-induced depotentiation. As shown in Fig. 6B, 10 min application of CNO (10 μm) starting 20 min after 5× HFS failed to induce depotentiation in BLA principal neurons from the AAV-CaMKIIα-hM4D(Gi) injected animals (Fig. 6B, LTP pre CNO = 154 ± 9%; after CNO = 156 ± 11% of baseline, n = 5). Hence, activation of presynaptic Gαi-coupled mGluR2 may play a key role in mediating mGluR2/3-induced depotentiation in the BLA. As a positive control for hM4D(Gi) activation, we examined the effect of CNO application before and during LTP induction using a standard 5× HFS protocol. As shown in Fig. 7A, CNO significantly attenuated LTP in BLA principal neurons from hM4D(Gi) injected animals (Fig. 7A, stars, 123 ± 10% of baseline, n = 5, P < 0.05), whereas normal LTP was induced in control animals in the presence of CNO (Fig. 7A, diamonds, 151 ± 7% of baseline, n = 6). Application of CNO (P = 0.6531) alone caused no enduring change in the membrane input resistance.
Figure 7. Modulation of postsynaptic AC levels following DREADD receptor activation in BLA principal neurons bidirectionally regulates LTP induction.
A, bath application of CNO (10 μM) significantly attenuated the magnitude of LTP induced by 5× HFS in BLA principal neurons transfected with hM4D Gi-coupled DREADD receptors (n = 5, stars). B, conversely, bath application of CNO facilitated the induction of LTP by a sub-threshold stimulation protocol (2× HFS) in BLA principal neurons transfected with rM3D Gs-coupled DREADD receptors (n = 5, stars). Upper traces in A and B are representative EPSPs evoked before and after CNO application.
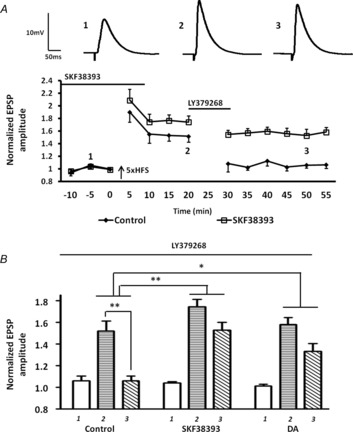
We then artificially elevated AC-PKA activity by applying the D1 agonist, SKF38393 (50 μm) before and during 5× HFS, and investigated the efficacy of mGluR2/3-induced depotentiation by applying LY379268 20 min after 5× HFS. Pretreatment with SKF38393 significantly attenuated the LY379268-induced depotentiation of LTP in BLA principal neurons (Fig. 8A, B, squares, LTP pre LY379268 = 174 ± 9%; post LY379268 = 152 ± 10%, n = 5). A two-way ANOVA comparing LY379268 treatment alone with SKF38393 pretreatment revealed a significant interaction of drug and time (F7,64 = 12.9, P < 0.01). As expected, the SKF38393 response was mimicked by bath application of 50 μm dopamine, which although not as potent as SKF38393 still buffered against the LY379268-induced depotentiation (Fig. 8B, LTP pre LY379268 = 157 ± 6%; post LY379268 = 133 ± 7%, n = 5). A two-way ANOVA revealed a significant interaction of drug and time comparing dopamine pretreatment group with the LY379268 treatment alone group (F7,64 = 18.3, P < 0.05).
Figure 8. Prior activation of D1Rs markedly attenuated the mGluR2/3-induced depotentiation of LTP following 5× HFS.
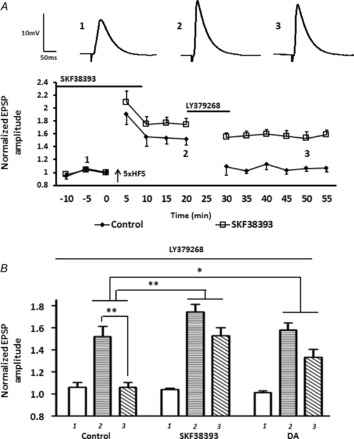
A, prior activation of D1Rs by SKF38393 (50 μm; n = 5) suppressed the LY379268-induced depotentiation of LTP. B, bar chart of group data showing the normalized EPSP amplitude before and after LTP induction (labelled 1, 2 and 3). Prior application of dopamine (n = 4) and SKF38393 (n = 5) significantly attenuated LY379268-induced depotentiation. **P < 0.01, *P < 0.05. Error bars show ± SEM. Upper traces in A are representative EPSPs evoked at time points 1, 2 and 3.
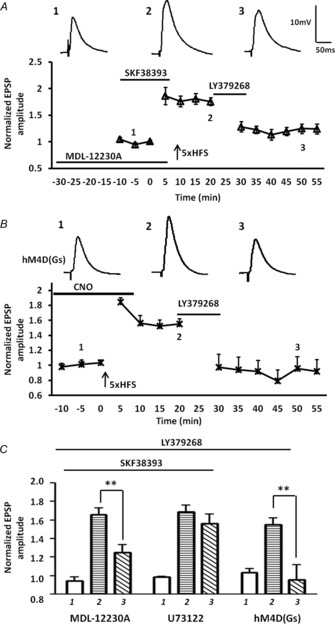
Significantly, pre-incubation with the membrane-permeable AC inhibitor, MDL-12230A (30 μm; 30 min) alone, followed by SKF38393 application during baseline and 5× HFS completely blocked the ability of SKF38393 to buffer against the LY379268-induced depotentiation when measured 50 min after 5× HFS (Fig. 9A, C, LTP pre LY379268 = 175 ± 7%; post LY379268 = 125 ± 9%, P < 0.01, n = 5). A two-way ANOVA showed a significant interaction of drug and time compared to the response to the SKF38393 pretreatment alone group (F12,103 = 2.87, P < 0.01). In contrast prior incubation with the PLC inhibitor, U73122 (10 μm), for 30 min failed to block the SKF38393-induced attenuation of depotentiation (Fig. 9C, U73122: LTP pre LY379268 = 178 ± 7%; post LY379268 = 167 ± 10%, n = 5).
Figure 9. Presynaptic inhibition of AC activity blocked the inhibitory effects of prior D1R activation on the LY379268-induced depotentiation of LTP following 5× HFS.
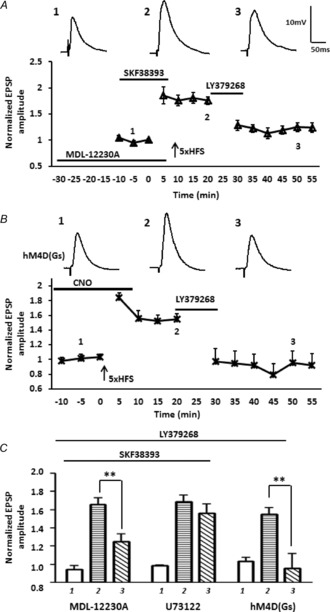
A, bath application of the membrane-permeable AC inhibitor, MDL-12230A (30 μm, n = 5), significantly reduced the ability of prior activation of D1Rs to attenuate LY379268-induced depotentiation of the eEPSP amplitude. B, enhancing postsynaptic AC levels by activating DREADD rM3D Gαs-coupled receptors in BLA principal neurons with exogenous CNO application during baseline and 5× HFS failed to attenuate LY379268-induced depotentiation of LTP (n = 5). C, bar chart illustrating the group effect of three different drug treatments on the magnitude of LTP before (1) and after (2, and 3) LTP induction. MDL-12230A significantly attenuated the effect of prior SKF38393 application on LY379268-induced depotentiation. Application of the PLC inhibitor U73122 (10 μm, n = 5) failed to block the SKF38393-mediated attenuation of LY379268-induced depotentiation. Selectively enhancing postsynaptic AC levels with Gs-coupled DREADDs failed to attenuate the LY379268-induced depotentiation of LTP. **P < 0.01. Error bars show ± SEM. Upper traces in A and B are representative EPSPs evoked at time points 1, 2 and 3.
To confirm that the site of action of the D1R-mediated attenuation of mGluR2/3-induced depotentiation was presynaptic, we next examined the effect of expressing a Gαs-coupled DREADD, AAV-CaMKIIα-rM3D(Gs), in BLA principal neurons. In this case postsynaptic AC activity is enhanced when CNO binds to rM3D(Gs). Application of CNO (10 μm) before and during 5× HFS in rM3D(Gs) injected animals failed to attenuate the LY379268-induced depotentiation of LTP in BLA principal neurons (Fig. 9B, C, LTP pre LY379268 = 156 ± 11%; post LY379268 = 95 ± 16%, n = 5, P < 0.01). Together, these data further support the hypothesis that D1R-mediated attenuation of LY379268-induced depotentiation is probably occurring through activation of a presynaptic AC-PKA signalling cascade. To validate that activated rM3D(Gs) elevated postsynaptic AC-PKA activity in BLA principal neurons we examined the effects of CNO application on the threshold of LTP induction using a sub-threshold 2× HFS protocol (Li et al. 2011). Application of CNO before and during 2× HFS, induced a robust LTP in rM3D(Gs) injected animals (Fig. 7B, 140 ± 13% of baseline, P < 0.01), whereas LTP was never induced in tissue from control animals in the presence of CNO (Fig. 7B, 117 ± 11% of baseline).
Finally, we examined the effects of artificially elevating AC-PKA activity by applying forskolin before and during HFS, and then investigated the time dependency of the efficacy of depotentiation by applying LY379268 for 10 min at 20, 30 or 40 min after HFS. As illustrated in Fig. 10, the depotentiation of LTP induced by LY379268 20 min after HFS was almost completely abolished by prior forskolin treatment (Fig. 10A, LTP pre LY379268 = 168 ± 8%; post LY379268 = 159 ± 5%, n = 5). Moreover, a two-way ANOVA showed a significant interaction of drug and time compared to the response to LY379268 treatment alone group (F10,88 = 2.53, P < 0.01). Significantly, prior forskolin application failed to affect depotentiation when LY379268 was applied 30 min after HFS (Fig. 10B, LTP pre LY379268 = 172 ± 8%; post LY379268 = 116 ± 9%, n = 4, P < 0.01), suggesting that the protective effects of forskolin against depotentiation are transient and last no longer than 30 min. Note the first measured time points after LY379268 application were always significantly higher than the baselines values, suggesting that a residual effect of forskolin was still present at that time (baseline levels: 104 ± 5%; post LY379268: 129 ± 6%, P < 0.05). However, the amplitude of the eEPSP at subsequent points was not significantly different from baseline values. As expected, prior application of forskolin had no effect on the depotentiation induced by LY379268 when it was applied 40 min after 5× HFS (Fig. 10C, LTP pre LY379268 = 166 ± 9%; post LY379268 = 96 ± 9%, P < 0.01, n = 4). For comparison purposes, group data are illustrated in Fig. 10D.
Figure 10. Forskolin causes a time-dependent suppression of LY379268-induced depotentiation following 5× HFS.
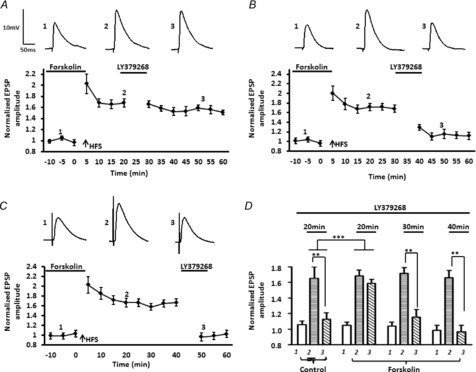
A, the depotentiation induced by LY379268 was almost completely abolished if LY379268 was applied 20 min after 5× HFS in the presence of forskolin (n = 5). B and C, prior application of forskolin failed to affect LY379268-induced depotentiation when LY379268 was applied 30 min (B, n = 4) or 40 min (C, n = 4) after 5xHFS. D, bar chart summarizing the group effect of foskolin on the response to LY379268 at three different time points after 5× HFS. Similar to controls, application of LY379268 30 40 min after 5× HFS in the presence of forskolin significantly depotentiated LTP. ***P < 0.001, **P < 0.01. Error bars show ± SEM. Upper traces in A–C are representative EPSPs evoked at time points 1, 2 and 3.
Discussion
To the best of our knowledge, this study is the first of its kind to show the frequency–response characteristics for changes in the valence of synaptic plasticity induced by stimulation of afferent inputs onto BLA principal neurons. Consistent with observations elsewhere in the brain, stimulation frequencies below 10 Hz induced LTD, whereas stimulation frequencies above 10 Hz induced LTP. Intriguingly, both the LFS-induced LTD and the HFS-induced LTP were seen to be NMDAR-dependent, whereas LTD induced by a PP-LFS protocol was dependent on mGluR2/3 activation and insensitive to NMDAR antagonists. Significantly, at the crossover point for LTD and LTP induction (10 Hz) we show that concurrent activation of presynaptic D1Rs and mGluR2/3 act to nullify any net change in synaptic strength. However, blockade of either D1Rs or mGluR2/3 can unmask 10 Hz stimulation-induced LTD and LTP, respectively. Moreover, consistent with previous studies we show that activation of mGluR2/3 could induce synaptic depression of evoked EPSPs, and depotentiate LTP induced by HFS. We extend these studies to show that prior activation of presynaptic D1Rs could buffer against the mGluR2/3 effect by activating a presynaptic AC signalling cascade. However, the buffering effect of presynaptic D1R activation on the mGluR2/3-induced depotentiation was transient, lasting no longer than 30 min. The results of our study raise the possibility that the temporal sequence of activation of either presynaptic D1Rs or mGluR2/3 may critically regulate the direction of synaptic plasticity in afferent pathways onto BLA principal neurons. Hence, the interaction of these two neurotransmitter systems may represent an important mechanism for bidirectional metaplasticity in BLA circuits and thus modulate the acquisition and extinction of fear memory.
Wang and Gean (1999) reported that LTD induced by LFS of afferent pathways from the lateral nucleus to the BLA required activation of both NMDA and mGluRs (Wang & Gean, 1999). In contrast, Li et al. (1998) reported that LFS of the EC induced a persistent enhancement of evoked synaptic potentials in the BLA, and LTD was only induced when LFS was applied after a brief period of HFS (Li et al. 1998). In the present study, we have shown that two pharmacologically independent forms of LTD can occur in BLA projection neurons depending on the stimulation protocol used, which may reconcile these two apparently conflicting observations: an LTD induced by LFS of the EC at 1 Hz that is NMDAR-dependent, and one that is induced by an LFS 1 Hz PP paradigm that was mostly dependent on activation of mGluR2/3. The difference between the results of the Li et al. (1998) study and our own with regard to the valence of the response to LFS stimulation of the EC may be attributed to several variables, including the composition of the patch recording solution. They used a high chloride patch solution, whereas we used a gluconate-based patch solution. However, the most parsimonious explanation may be the age of the animals used in the two studies. Li and colleagues used 3- to 5-week-old adolescent animals, whereas we used 6- to 8-week-old animals. Recently we have shown that early in development major changes occur in the physiological properties of BLA principal neurons (Ehrlich et al. 2012) which are accompanied by concomitant changes in synaptic transmission (Ehrlich et al. 2013). It is possible that the expression of LTD in response to LFS in the BLA does not appear until rats approach adulthood.
In agreement with studies eleswhere in the brain (Bear & Malenka, 1994; Blaise & Bronzino, 2003; Froc & Racine, 2005), we have shown that the response of BLA principal neurons to EC stimulation is highly frequency-dependent. In particular, at stimulation frequencies around 10 Hz, there is no net change in synaptic strength. The competing cellular processes of LTP and LTD may be simultaneously activated in this frequency range. Consequently, any factor that could modulate one or other of these processes would be predicted to have a significant effect on the direction of synaptic plasticity at this intermediate stimulation frequency. Consistent with this hypothesis, we have shown that blockade of either D1Rs or mGluR2/3 facilitate the induction of LTD or LTP, respectively, in response to 10 Hz stimulation. Hence, D1R and mGluR2/3 activation appear to act in opposition following afferent input in the high theta (∼10 – 12 Hz) range. These results then raised the question of how the activation of either D1Rs or mGluR2/3 could regulate the direction of synaptic plasticity in afferent pathways onto BLA principal neurons in a temporally dynamic manner.
Although not addressed in this study, a question arises as to how 10 Hz stimulation of the external capsule may trigger dopamine release. Two processes may contribute to this scenario. (1) Glutamatergic afferents in the BLA are in close proximity to dopaminergic terminals, and axo-axonic contacts may trigger DA release. A similar process has been reported for BLA regulation of DA release in the nucleus accumbens. (2) It is possible that charge accumulation induced by 10 Hz stimulation of the external capsule may be sufficient to directly trigger dopamine efflux from local DA terminals.
Consistent with the studies of Lin et al. (2000, 2005), we have shown that bath application of the mGluR2/3 agonist, LY379268, induced a long lasting form of synaptic depression (LY-SD) and a depotentiation of LTP induced by HFS of cortical afferents onto BLA principal neurons. Intriguingly, Lin et al. (2000) reported that the induction of mGluR2/3 synaptic depression was activity-dependent and required concurrent synaptic stimulation (Lin et al. 2000). In our hands transient application of LY379268 could induce synaptic depression even in the absence of concurrent pathway stimulation. It is not immediately apparent why our results differ from those of Lin et al.; however, our results are consistent with those of Robbe et al. (2002) who have also shown an mGluR2/3-dependent form of LTD in the nucleus accumbens that did not require concurrent synaptic activation (Robbe et al. 2002). Similarly, mGluR2/3-dependent suppression of synaptic transmission has been reported in multiple brain regions including the nucleus tractus solitarius (Chen et al. 2002), striatum (Lovinger & McCool, 1995) and hippocampus (Kamiya et al. 1996), and in the spinal cord (Gerber et al. 2000).
To circumvent problems associated with global mGluR2/3 activation, recently selective PAMs of mGluRs have been proposed as a promising new approach for the treatment of schizophrenia, anxiety disorders and some forms of depression (Krystal et al. 2010). We have shown that the positive allosteric modulator of mGluR2 receptors, LY487379, significantly enhanced the efficacy of the non-selective mGluR2/3 agonist, LY379268, to induce synaptic depression in BLA principal neurons. Hence, mGluR2 may play a key role in mediating synaptic depression in this region. As PAMs would act to modulate the activity of endogenous glutamate release, and many of the disorders mentioned above are associated with abnormally high amygdala activation in response to sensory stimuli (Stein et al. 2007; Rauch et al. 2010; Zhong et al. 2011), the data presented here would suggest that PAMs of mGluR2 may represent a useful approach for treating these affective disorders.
Several studies have reported that activation of the AC/cAMP/PKA-dependent signalling pathway is both necessary and sufficient for the induction of pre- and postsynaptic LTP at cortical input to lateral amygdala synapses (Huang & Kandel, 1998; Fourcaudot et al. 2008). Conversely, Lin et al. (2000) reported that mGluR2/3 agonist-induced LTD is mediated by presynaptic inhibition of AC, resulting in decreased cAMP formation and PKA activation. Consistent with these studies, we have shown that activation of AC by the D1R agonist, SKF 39393, or the AC activator, forskolin, markedly attenuated the mGluR2/3 agonist-induced synaptic depression, an effect that was independent of the postsynaptic AC/cAMP/PKA-dependent signalling pathway, suggesting a presynaptic site of action. Together these results suggest that D1R modulation of the mGluR2/3 response occurs through activation of presynaptic AC/PKA cascades. A similar AC/PKA-dependent modulation of mGluR2/3 LTD has been reported in the nucleus accumbens (Robbe et al. 2002).
Many neuronal models of memory storage include bidirectional modifications of synaptic strength, namely synaptic potentiation and depression (Sejnowski, 1977), both of which are thought to be necessary for error correcting (Willshaw & Dayan, 1990; Hancock et al., 2011). Consistent with this hypothesis, genetic distuption of sub-synaptic proteins, which attenuate hippocampal LTD and facilitate LTP, has been shown to impair spatial learning and memory (Migaud et al. 1998; Kim et al. 2009), suggesting that a functional interaction between LTD and LTP in hippocampal circuits is essential for normal spatial learning and memory. Bidirectional synaptic plasticity also occurs in the BLA, and the competitive interraction between synaptic depression/depotentiation and LTP may underlie a form of competition at synapses to provide specificity in the formation of fear memories. Growing evidence indicates that the induction of LTP in BLA principal neurons underlies the acquisition and consolidation of fear memories (Rogan et al. 1997; Goosens & Maren, 2002), whereas LTD/depotentiation is thought to mediate the extinction of learned fear (Dalton et al. 2012). We would contend that an interaction between LTD and LTP in BLA circuits is most likely required for normal fear memory formation and extinction. For example, the pathway from the medial prefontal cortex to the BLA is reportedly resistant to LTP induction, whereas LFS of these inputs can induce robust LTD (Maroun, 2006). However, prior exposure to inescapable stress inhibits the expression of LTD in this pathway and facilitates the expression of LTP. Stress is known to increase release of dopamine in the BLA (Inglis & Moghaddam, 1999; Kienast et al. 2008), and our results would suggest that the switch from LTD to LTP following exposure to stress may result from a D1R-mediated attenuation of mGluR2/3-dependent LTD.
Under basal conditions the in vivo firing rate of many cortical neurons (Ji & Neugebauer, 2012; Mahon & Charpier, 2012; Hengen et al. 2013; Tsubo et al. 2013) and BLA neurons is very low, ∼1–2 Hz (Rosenkranz & Grace, 1999; Pare & Collins, 2000; Likhtik et al. 2005). If this firing rate, which is the same rate as the LFS used to induce LTD, also reflects the resting state frequency of afferent input to the BLA there may be little or no dopamine release and the default state for excitatory synaptic transmission in BLA circuits may be synaptic depression. Moreover, in response to sensory stimulation pyramidal neurons of the visual cortex shift from regular firing to doublet firing (Rumberger et al. 1998). Cells firing in doublets and/or transient bursts of action potentials may be more likely to induce a PP-LFS form of LTD. Cells firing in doublets are not unique to the cortex. A recent publication by Senn et al. (2014) has shown that different populations of BLA principal neurons shift from regular firing to doublet or ‘burst’ firing during fear conditioning and extinction. Hence, LTD of extrinsic and intrinsic afferents onto BLA neurons may be differentially modulated depending on the activation state of the upstream neuron. Similarly, during states of hyperarousal the rate of afferent input to the BLA would be expected to increase dramatically in those pathways carrying salient sensory information and could effectively reach 50–100 Hz; at this firing frequency endogenous dopamine would be released and facilitate the induction of LTP. If activity-dependent release of dopamine occurs simultaneously with increased glutamate release, as has been suggested, then activation of presynaptic D1Rs may buffer this pathway against subsequent depotentiation. Conversely, pathways that do not see concurrent release of glutamate and dopamine may be suppressed due to activation of presynaptic mGluR2/3. Hence, presynaptic D1R activation that attenuates mGluR2-dependent excitatory synaptic depression and depotentiation may serve as a heterosynaptic mechanism to conserve input fidelity in pathways carrying salient sensory information, while simultaneously reducing the signal-to-noise ratio in pathways carrying irrelevant information. Moreover, the buffering effect of D1R activation terminated approximately 30 min after receptor activation, suggesting that there is an optimal time window during which incoming salient sensory information would be buffered against subsequent degradation.
Excessive dopamine release has been implicated in the psychopathology of several psychiatric disorders including post-traumatic stress disorder (PTSD), depression (Jovanovic & Ressler) and schizophrenia (Carlsson & Lindqvist, 1963), as well as drug misuse (Volkow et al. 2004). Each of these disorders has also been associated with abnormally high levels of amygdala activation in response to emotional stimuli (Schneider et al. 1998; Etkin & Wager, 2007; Yang et al. 2010). Hence, aberrant activation of D1R may contribute to enhanced synaptic transmission in these pathological conditions. Our data suggest that selective activation of mGluR2 could play a key role in treatment strategies aimed at alleviating these disorders. Consistent with this hypothesis, Patil et al. (2007) have shown that treatment with a novel class of mGluR2/3 agonists lead to a significant improvement in all primary outcome measures of patients with chronic schizophrenia. Our findings provide for the first time strong evidence that presynaptic modulation of mGluR2 activity by dopamine release may provide important clues to link the hypothesized aberrant dopamine-mediated molecular mechanisms and mGluR2-related treatment to the pathophysiology of these psychiatric disorders.
Glossary
- AAV
adenoassociated viral vector
- AC
adenylyl cyclase
- ACSF
artificial cerebrospinal fluid
- BLA
basolateral amygdala
- CNO
clozapine-N-oxide
- CV
coefficient of variation
- D1R
D1 receptor
- DREADD
designer receptors exclusively activated by designer drugs
- EC
external capsule
- EPSPs
excitatory postsynaptic potentials
- HFS
high frequency stimulation
- LFS
low frequency stimulation
- LTD
long-term depression
- LTP
long-term potentiation
- LY-SD
LY379268-induced synaptic depression
- mGluR2/3
group II metabotropic glutamate receptors
- PAM
positive allosteric modulator
- PKA
protein kinase A
- PLC
phospholipase C
- PP-LFS
paired pulse-low frequency stimulation
- PPR
paired-pulse ratio
Additional information
Competing interests
None declared.
Author contributions
C.L. designed and performed experiments, analysed data and wrote the paper; D.G.R. designed experiments, analysed the data and wrote the paper. Both authors read and approved the final manuscript.
Funding
This work was supported by NIMH grant MH069852 to D.G.R. and a Base Centre grant to the Yerkes National Primate Research Center (#RR-00165), Animal Resource Program at NIH.
Author's present address
C. Li: McGovern Institute for Brain Research and Department of Brain & Cognitive Sciences, MIT, Cambridge, MA 02139, USA.
References
- Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Andersen PH, Gingrich JA, Bates MD, Dearry A, Falardeau P, Senogles SE, Caron MG. Dopamine receptor subtypes: beyond the D1/D2 classification. Trends Pharmacol Sci. 1990;11:231–236. doi: 10.1016/0165-6147(90)90249-8. [DOI] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaise JH, Bronzino JD. Effects of stimulus frequency and age on bidirectional synaptic plasticity in the dentate gyrus of freely moving rats. Exp Neurol. 2003;182:497–506. doi: 10.1016/s0014-4886(03)00136-5. [DOI] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Li H. The physiological role of kainate receptors in the amygdala. Mol Neurobiol. 2004;30:127–141. doi: 10.1385/MN:30:2:127. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M. Effect of chlorpromazine or haloperidol on formation of 3methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol (Copenh) 1963;20:140–144. doi: 10.1111/j.1600-0773.1963.tb01730.x. [DOI] [PubMed] [Google Scholar]
- Chen CY, Ling Eh EH, Horowitz JM, Bonham AC. Synaptic transmission in nucleus tractus solitarius is depressed by Group II and III but not Group I presynaptic metabotropic glutamate receptors in rats. J Physiol. 2002;538:773–786. doi: 10.1113/jphysiol.2001.012948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark D, White FJ. D1 dopamine receptor – the search for a function: a critical evaluation of the D1/D2 dopamine receptor classification and its functional implications. Synapse. 1987;1:347–388. doi: 10.1002/syn.890010408. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Dalton GL, Wu DC, Wang YT, Floresco SB, Phillips AG. NMDA GluN2A and GluN2B receptors play separate roles in the induction of LTP and LTD in the amygdala and in the acquisition and extinction of conditioned fear. Neuropharmacology. 2012;62:797–806. doi: 10.1016/j.neuropharm.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Domenici MR, Berretta N, Cherubini E. Two distinct forms of long-term depression coexist at the mossy fiber-CA3 synapse in the hippocampus during development. Proc Natl Acad Sci U S A. 1998;95:8310–8315. doi: 10.1073/pnas.95.14.8310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich DE, Ryan SJ, Hazra R, Guo JD, Rainnie DG. Postnatal maturation of GABAergic transmission in the rat basolateral amygdala. J Neurophysiol. 2013;110:926–941. doi: 10.1152/jn.01105.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich DE, Ryan SJ, Rainnie DG. Postnatal development of electrophysiological properties of principal neurons in the rat basolateral amygdala. J Physiol. 2012;590:4819–4838. doi: 10.1113/jphysiol.2012.237453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin A, Wager TD. Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry. 2007;164:1476–1488. doi: 10.1176/appi.ajp.2007.07030504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber DS, Korn H. Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys. J. 1991;60:1288–1294. doi: 10.1016/S0006-3495(91)82162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok JP, Dickerson TM, Palmiter RD. Dopamine is necessary for cue-dependent fear conditioning. J Neurosci. 2009;29:11089–11097. doi: 10.1523/JNEUROSCI.1616-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourcaudot E, Gambino F, Humeau Y, Casassus G, Shaban H, Poulain B, Lüthi A. cAMP/PKA signaling and RIM1α mediate presynaptic LTP in the lateral amygdala. Proc Natl Acad Sci U S A. 2008;105:15130–15135. doi: 10.1073/pnas.0806938105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froc DJ, Racine RJ. Interactions between LTP- and LTD-inducing stimulation in the sensorimotor cortex of the awake freely moving rat. J Neurophysiol. 2005;93:548–556. doi: 10.1152/jn.00253.2004. [DOI] [PubMed] [Google Scholar]
- Genoux D, Bezerra P, Montgomery JM. Intra-spaced stimulation and protein phosphatase 1 dictate the direction of synaptic plasticity. Eur J Neurosci. 2011;33:1761–1770. doi: 10.1111/j.1460-9568.2011.07669.x. [DOI] [PubMed] [Google Scholar]
- Gerber G, Zhong J, Youn D, Randic M. Group II and group III metabotropic glutamate receptor agonists depress synaptic transmission in the rat spinal cord dorsal horn. Neuroscience. 2000;100:393–406. doi: 10.1016/s0306-4522(00)00269-4. [DOI] [PubMed] [Google Scholar]
- Gisabella B, Rowan MJ, Anwyl R. Mechanisms underlying the inhibition of long-term potentiation by preconditioning stimulation in the hippocampus in vitro. Neuroscience. 2003;121:297–305. doi: 10.1016/s0306-4522(03)00440-8. [DOI] [PubMed] [Google Scholar]
- Goosens KA, Maren S. Long-term potentiation as a substrate for memory: evidence from studies of amygdaloid plasticity and Pavlovian fear conditioning. Hippocampus. 2002;12:592–599. doi: 10.1002/hipo.10099. [DOI] [PubMed] [Google Scholar]
- Greba Q, Kokkinidis L. Peripheral and intraamygdalar administration of the dopamine D1 receptor antagonist SCH 23390 blocks fear-potentiated startle but not shock reactivity or the shock sensitization of acoustic startle. Behav Neurosci. 2000;114:262–272. doi: 10.1037//0735-7044.114.2.262. [DOI] [PubMed] [Google Scholar]
- Grillon C, Cordova J, Levine LR, Morgan CA., 3rd Anxiolytic effects of a novel group II metabotropic glutamate receptor agonist (LY354740) in the fear-potentiated startle paradigm in humans. Psychopharmacology (Berl) 2003;168:446–454. doi: 10.1007/s00213-003-1444-8. [DOI] [PubMed] [Google Scholar]
- Hancock PJS, Leslie S, Phillips, William A. A biologically supported error-correcting learning rule. Neural Computation. 1991;3:201–212. doi: 10.1162/neco.1991.3.2.201. [DOI] [PubMed] [Google Scholar]
- Helton DR, Tizzano JP, Monn JA, Schoepp DD, Kallman MJ. Anxiolytic and side-effect profile of LY354740: a potent, highly selective, orally active agonist for group II metabotropic glutamate receptors. J Pharmacol Exp Ther. 1998;284:651–660. [PubMed] [Google Scholar]
- Hengen KB, Lambo ME, Van Hooser SD, Katz DB, Turrigiano GG. Firing rate homeostasis in visual cortex of freely behaving rodents. Neuron. 2013;80:335–342. doi: 10.1016/j.neuron.2013.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron. 1998;21:169–178. doi: 10.1016/s0896-6273(00)80524-3. [DOI] [PubMed] [Google Scholar]
- Inglis FM, Moghaddam B. Dopaminergic innervation of the amygdala is highly responsive to stress. J Neurochem. 1999;72:1088–1094. doi: 10.1046/j.1471-4159.1999.0721088.x. [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Modulation of medial prefrontal cortical activity using in vivo recordings and optogenetics. Mol Brain. 2012;5:36. doi: 10.1186/1756-6606-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenceau A, Hedou G, Potier B, Kollen M, Dutar P, Mansuy IM. Partial inhibition of PP1 alters bidirectional synaptic plasticity in the hippocampus. Eur J Neurosci. 2006;24:564–572. doi: 10.1111/j.1460-9568.2006.04938.x. [DOI] [PubMed] [Google Scholar]
- Jovanovic T, Ressler KJ. How the neurocircuitry and genetics of fear inhibition may inform our understanding of PTSD. Am J Psychiatry. 167:648–662. doi: 10.1176/appi.ajp.2009.09071074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Shinozaki H, Yamamoto C. Activation of metabotropic glutamate receptor type 2/3 suppresses transmission at rat hippocampal mossy fibre synapses. J Physiol. 1996;493:447–455. doi: 10.1113/jphysiol.1996.sp021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Yamamoto C. Phorbol ester and forskolin suppress the presynaptic inhibitory action of group-II metabotropic glutamate receptor at rat hippocampal mossy fibre synapse. Neuroscience. 1997;80:89–94. doi: 10.1016/s0306-4522(97)00098-5. [DOI] [PubMed] [Google Scholar]
- Kasanetz F, Deroche-Gamonet V, Berson N, Balado E, Lafourcade M, Manzoni O, Piazza PV. Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science. 2010;328:1709–1712. doi: 10.1126/science.1187801. [DOI] [PubMed] [Google Scholar]
- Kemp N, McQueen J, Faulkes S, Bashir ZI. Different forms of LTD in the CA1 region of the hippocampus: role of age and stimulus protocol. Eur J Neurosci. 2000;12:360–366. doi: 10.1046/j.1460-9568.2000.00903.x. [DOI] [PubMed] [Google Scholar]
- Kienast T, Hariri AR, Schlagenhauf F, Wrase J, Sterzer P, Buchholz HG, Smolka MN, Gründer G, Cumming P, Kumakura Y, Bartenstein P, Dolan RJ, Heinz A. Dopamine in amygdala gates limbic processing of aversive stimuli in humans. Nat Neurosci. 2008;11:1381–1382. doi: 10.1038/nn.2222. [DOI] [PubMed] [Google Scholar]
- Kim MH, Choi J, Yang J, Chung W, Kim JH, Paik SK, Kim K, Han S, Won H, Bae YS, Cho SH, Seo J, Bae YC, Choi SY, Kim E. Enhanced NMDA receptor-mediated synaptic transmission, enhanced long-term potentiation, and impaired learning and memory in mice lacking IRSp53. J Neurosci. 2009;29:1586–1595. doi: 10.1523/JNEUROSCI.4306-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Mathew SJ, D'Souza DC, Garakani A, Gunduz-Bruce H, Charney DS. Potential psychiatric applications of metabotropic glutamate receptor agonists and antagonists. CNS Drugs. 2010;24:669–693. doi: 10.2165/11533230-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Lamont EW, Kokkinidis L. Infusion of the dopamine D1 receptor antagonist SCH 23390 into the amygdala blocks fear expression in a potentiated startle paradigm. Brain Res. 1998;795:128–136. doi: 10.1016/s0006-8993(98)00281-9. [DOI] [PubMed] [Google Scholar]
- Li C, Dabrowska J, Hazra R, Rainnie DG. Synergistic activation of dopamine D1 and TrkB receptors mediate gain control of synaptic plasticity in the basolateral amygdala. PLoS One. 2011;6:e26065. doi: 10.1371/journal.pone.0026065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Weiss SR, Chuang DM, Post RM, Rogawski MA. Bidirectional synaptic plasticity in the rat basolateral amygdala: characterization of an activity-dependent switch sensitive to the presynaptic metabotropic glutamate receptor antagonist 2S-α-ethylglutamic acid. J Neurosci. 1998;18:1662–1670. doi: 10.1523/JNEUROSCI.18-05-01662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Pelletier JG, Paz R, Pare D. Prefrontal control of the amygdala. J Neurosci. 2005;25:7429–7437. doi: 10.1523/JNEUROSCI.2314-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Lee CC, Huang YC, Wang SJ, Gean PW. Activation of group II metabotropic glutamate receptors induces depotentiation in amygdala slices and reduces fear-potentiated startle in rats. Learn Mem. 2005;12:130–137. doi: 10.1101/lm.85304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Wang SJ, Luo MZ, Gean PW. Activation of group II metabotropic glutamate receptors induces long-term depression of synaptic transmission in the rat amygdala. J Neurosci. 2000;20:9017–9024. doi: 10.1523/JNEUROSCI.20-24-09017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, McCool BA. Metabotropic glutamate receptor-mediated presynaptic depression at corticostriatal synapses involves mGLuR2 or 3. J Neurophysiol. 1995;73:1076–1083. doi: 10.1152/jn.1995.73.3.1076. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, Toth K, McBain CJ. Target-specific expression of presynaptic mossy fibre plasticity. Science. 1998;279:1368–1370. doi: 10.1126/science.279.5355.1368. [DOI] [PubMed] [Google Scholar]
- Mahon S, Charpier S. Bidirectional plasticity of intrinsic excitability controls sensory inputs efficiency in layer 5 barrel cortex neurons in vivo. J Neurosci. 2012;32:11377–11389. doi: 10.1523/JNEUROSCI.0415-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC. Synaptic plasticity in the hippocampus: LTP and LTD. Cell. 1994;78:535–538. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- Malleret G, Alarcon JM, Martel G, Takizawa S, Vronskaya S, et al. Bidirectional regulation of hippocampal long-term synaptic plasticity and its influence on opposing forms of memory. J Neurosci. 2010;30:3813–3825. doi: 10.1523/JNEUROSCI.1330-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroun M. Stress reverses plasticity in the pathway projecting from the ventromedial prefrontal cortex to the basolateral amygdala. Eur J Neurosci. 2006;24:2917–2922. doi: 10.1111/j.1460-9568.2006.05169.x. [DOI] [PubMed] [Google Scholar]
- Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A. Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci. 2006;9:868–869. doi: 10.1038/nn1713. [DOI] [PubMed] [Google Scholar]
- Matsuda Y, Marzo A, Otani S. The presence of background dopamine signal converts long-term synaptic depression to potentiation in rat prefrontal cortex. J Neurosci. 2006;26:4803–4810. doi: 10.1523/JNEUROSCI.5312-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Wang J, Kandel ER, O'Dell TJ. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell. 1995;81:891–904. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Muly EC, Mania I, Guo JD, Rainnie DG. Group II metabotropic glutamate receptors in anxiety circuitry: correspondence of physiological response and subcellular distribution. J Comp Neurol. 2007;505:682–700. doi: 10.1002/cne.21525. [DOI] [PubMed] [Google Scholar]
- Muly EC, Senyuz M, Khan ZU, Guo JD, Hazra R, Rainnie DG. Distribution of D1 and D5 dopamine receptors in the primate and rat basolateral amygdala. Brain Struct Funct. 2009;213:375–393. doi: 10.1007/s00429-009-0214-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas CS, Peineau S, Amici M, Csaba Z, Fafouri A, Javalet C, Collett VJ, Hildebrandt L, Seaton G, Choi SL, Sim SE, Bradley C, Lee K, Zhuo M, Kaang BK, Gressens P, Dournaud P, Fitzjohn SM, Bortolotto ZA, Cho K, Collingridge GL. The Jak/STAT pathway is involved in synaptic plasticity. Neuron. 2012;73:374–390. doi: 10.1016/j.neuron.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare D, Collins DR. Neuronal correlates of fear in the lateral amygdala: multiple extracellular recordings in conscious cats. J Neurosci. 2000;20:2701–2710. doi: 10.1523/JNEUROSCI.20-07-02701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, Avedisova AS, Bardenstein LM, Gurovich IY, Morozova MA, Mosolov SN, Neznanov NG, Reznik AM, Smulevich AB, Tochilov VA, Johnson BG, Monn JA, Schoepp DD. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- Prezeau L, Manzoni O, Homburger V, Sladeczek F, Curry K, Bockaert J. Characterization of a metabotropic glutamate receptor: direct negative coupling to adenylyl cyclase and involvement of a pertussis toxin-sensitive G protein. Proc Natl Acad Sci U S A. 1992;89:8040–8044. doi: 10.1073/pnas.89.17.8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Intracellular recordings from morphologically identified neurons of the basolateral amygdala. J Neurophysiol. 1993;69:1350–1362. doi: 10.1152/jn.1993.69.4.1350. [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Holmes KH, Shinnick-Gallagher P. Activation of postsynaptic metabotropic glutamate receptors by trans-ACPD hyperpolarizes neurons of the basolateral amygdala. J Neurosci. 1994;14:7208–7220. doi: 10.1523/JNEUROSCI.14-11-07208.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG, Shinnick-Gallagher P. Trans-ACPD and L-APB presynaptically inhibit excitatory glutamatergic transmission in the basolateral amygdala (BLA) Neurosci Lett. 1992;139:87–91. doi: 10.1016/0304-3940(92)90864-4. [DOI] [PubMed] [Google Scholar]
- Rauch AV, Reker M, Ohrmann P, Pedersen A, Bauer J, Dannlowski U, Harding L, Koelkebeck K, Konrad C, Kugel H, Arolt V, Heindel W, Suslow T. Increased amygdala activation during automatic processing of facial emotion in schizophrenia. Psychiatry Res. 2010;182:200–206. doi: 10.1016/j.pscychresns.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Chaumont S, Bockaert J, Manzoni OJ. Role of p/q-Ca2+ channels in metabotropic glutamate receptor 2/3-dependent presynaptic long-term depression at nucleus accumbens synapses. J Neurosci. 2002;22:4346–4356. doi: 10.1523/JNEUROSCI.22-11-04346.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA. Modulation of basolateral amygdala neuronal firing and afferent drive by dopamine receptor activation in vivo. J Neurosci. 1999;19:11027–11039. doi: 10.1523/JNEUROSCI.19-24-11027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumberger A, Schmidt M, Lohmann H, Hoffmann KP. Correlation of electrophysiology, morphology, and functions in corticotectal and corticopretectal projection neurons in rat visual cortex. Exp Brain Res. 1998;119:375–390. doi: 10.1007/s002210050353. [DOI] [PubMed] [Google Scholar]
- Schaffhauser H, Cai Z, Hubalek F, Macek TA, Pohl J, Murphy TJ, Conn PJ. cAMP-dependent protein kinase inhibits mGluR2 coupling to G-proteins by direct receptor phosphorylation. J Neurosci. 2000;20:5663–5670. doi: 10.1523/JNEUROSCI.20-15-05663.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider F, Weiss U, Kessler C, Salloum JB, Posse S, Grodd W, Müller-Gärtner HW. Differential amygdala activation in schizophrenia during sadness. Schizophr Res. 1998;34:133–142. doi: 10.1016/s0920-9964(98)00085-1. [DOI] [PubMed] [Google Scholar]
- Sejnowski TJ. Statistical constraints on synaptic plasticity. J Theor Biol. 1977;69:385–389. doi: 10.1016/0022-5193(77)90146-1. [DOI] [PubMed] [Google Scholar]
- Senn V, Wolff SB, Herry C, Grenier F, Ehrlich I, Gründemann J, Fadok JP, Müller C, Letzkus JJ, Lüthi A. Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron. 2014;81:428–437. doi: 10.1016/j.neuron.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Shekhar A, Keim SR. LY354740, a potent group II metabotropic glutamate receptor agonist prevents lactate-induced panic-like response in panic-prone rats. Neuropharmacology. 2000;39:1139–1146. doi: 10.1016/s0028-3908(99)00215-4. [DOI] [PubMed] [Google Scholar]
- Stein MB, Simmons AN, Feinstein JS, Paulus MP. Increased amygdala and insula activation during emotion processing in anxiety-prone subjects. Am J Psychiatry. 2007;164:318–327. doi: 10.1176/ajp.2007.164.2.318. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Takano H, Kodaka F, Arakawa R, Yamada M, Otsuka T, Hirano Y, Kikyo H, Okubo Y, Kato M, Obata T, Ito H, Suhara T. Contribution of dopamine D1 and D2 receptors to amygdala activity in human. J Neurosci. 2010;30:3043–3047. doi: 10.1523/JNEUROSCI.5689-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubo Y, Isomura Y, Fukai T. Neural dynamics and information representation in microcircuits of motor cortex. Front Neural Circuits. 2013;7:85. doi: 10.3389/fncir.2013.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Swanson JM. Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol Psychiatry. 2004;9:557–569. doi: 10.1038/sj.mp.4001507. [DOI] [PubMed] [Google Scholar]
- Walker DL, Davis M. The role of amygdala glutamate receptors in fear learning, fear-potentiated startle, and extinction. Pharmacol Biochem Behav. 2002;71:379–392. doi: 10.1016/s0091-3057(01)00698-0. [DOI] [PubMed] [Google Scholar]
- Walker DL, Rattiner LM, Davis M. Group II metabotropic glutamate receptors within the amygdala regulate fear as assessed with potentiated startle in rats. Behav Neurosci. 2002;116:1075–1083. doi: 10.1037//0735-7044.116.6.1075. [DOI] [PubMed] [Google Scholar]
- Wang HY, Undie AS, Friedman E. Evidence for the coupling of Gq protein to D1-like dopamine sites in rat striatum: possible role in dopamine-mediated inositol phosphate formation. Mol Pharmacol. 1995;48:988–994. [PubMed] [Google Scholar]
- Wang SJ, Gean PW. Long-term depression of excitatory synaptic transmission in the rat amygdala. J Neurosci. 1999;19:10656–10663. doi: 10.1523/JNEUROSCI.19-24-10656.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willshaw D, Dayan P. Optimal plasticity from matrix memories: what goes up must come down. Neural Comput. 1990;2:85–93. [Google Scholar]
- Yang TT, Simmons AN, Matthews SC, Tapert SF, Frank GK, Max JE, Bischoff-Grethe A, Lansing AE, Brown G, Strigo IA, Wu J, Paulus MP. Adolescents with major depression demonstrate increased amygdala activation. J Am Acad Child Adolesc Psychiatry. 2010;49:42–51. doi: 10.1097/00004583-201001000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, Wang X, Xiao J, Yi J, Zhu X, Liao J, Wang W, Yao S. Amygdala hyperactivation and prefrontal hypoactivation in subjects with cognitive vulnerability to depression. Biol Psychol. 2011;88:233–242. doi: 10.1016/j.biopsycho.2011.08.007. [DOI] [PubMed] [Google Scholar]