

Abstract
We have developed a nickel-catalyzed cross coupling of benzylic ammonium triflates with aryl boronic acids to afford diarylmethanes and diarylethanes. This reaction proceeds under mild reaction conditions and with exceptional functional group tolerance. Further, it transforms branched benzylic ammonium salts to diarylethanes with excellent chirality transfer, offering a new strategy for the synthesis of highly enantioenriched diarylethanes from readily available chiral benzylic amines.
Diarylmethanes and diarylethanes are important molecules in organic synthesis and pharmaceutical development.1 These valuable targets can be prepared via cross couplings of classic electrophiles, such as benzylic halides, with Grignard, organozinc and organoboron reagents.2 In fact, Carretero has shown that enantioenriched diarylethanes can be formed via stereospecific cross couplings of benzylic bromides with aryl Grignard reagents (Scheme 1A-1).2c However, these classic electrophiles are highly reactive, can decompose upon prolonged storage, typically mandate that the cross coupling occur at the beginning of a synthetic sequence, and are difficult to prepare in high enantiopurity. Cross couplings of benzylic carbonates, acetates and phosphates have also been developed to enable use of more stable starting materials.3 Recent reports have also demonstrated that the use of benzylic ethers and alcohols as coupling partners overcomes some of these challenges, offering greater substrate stability and orthogonality.4 Further, the Jarvo group has shown that nickel-catalyzed cross couplings of benzylic ethers with Grignard reagents occurs in a stereospecific fashion (Scheme 1A-2).5,6 In contrast, Tian has reported that copper-catalyzed couplings of benzylic sulfonimides proceed with only low levels of chirality transfer (Scheme 1A-3).7 Notably, almost all cross couplings of benzyl electrophiles to deliver enantioenriched diarylethanes require Grignard or organozinc coupling partners.2b-d,5 No examples of enantioselective couplings of benzylic electrophiles with boronic acids are yet known, and only a single stereospecific coupling of a benzylic α-cyanohydrin mesylate with a boronic acid partner has been reported to our knowledge.8
Scheme 1. Stereospecific Cross Couplings to Form Diarylethanes.

In an alternative bond-disconnection approach to the synthesis of enantioenriched diarylethanes, Crudden has shown that enantioenriched benzylic boronic esters undergo stereospecific cross couplings (Scheme 1A-4).9 Stereospecific protodeborylation of tertiary boronic esters also delivers enantioenriched diarylethanes.6e
Despite these impressive advances in stereospecific cross couplings of benzylic reagents, several challenges remain, including identification of a class of benzylic reagents generally available in exceptional enantiopurity and the ability to employ commercially available and functional group tolerant coupling partners. In considering these challenges, we have been drawn to the use of benzylic ammonium salts as substrates.10 Benzylic ammonium salts are readily prepared from amine precursors.11 Further, these substrates offer a functional group handle orthogonal to both halides and ethers, and both amines and ammonium salts are stable to long-term storage. Importantly, highly enantioenriched benzylic amines are readily available via classical resolution or a variety of catalytic asymmetric methods.12 Csaky et al. have elegantly demonstrated the rhodium-catalyzed cross couplings of boronic acids with ammonium iodides derived from gramines (3-aminomethylindoles) to form achiral 3-benzyl and 3-allyl indoles.10d However, to our knowledge, a general method for the cross coupling of benzylic ammonium salts to yield diarylmethanes and enantioenriched diarylethanes has not yet been realized.
We anticipated that nickel catalysts would enable the cross coupling of a wide variety of benzylic ammonium salts with mild and highly functional group compatible coupling partners, such as commercially available boronic acids. Because ammonium salts can be readily prepared with a wide variety of counter-ions, we envisioned facile tuning of the reactivity of these electrophiles. In particular, we predicted that weakly coordinating counter-ions, such as triflate, would be highly effective. In contrast to halide or alkoxide counter-ions formed in cross couplings with benzylic halides and ethers, the by-products of oxidative addition into an ammonium triflate are neutral trimethylamine and the triflate counter-ion. We envisioned that the resulting oxidative addition intermediate may thus more readily undergo transmetallation with a boronic acid. Such an effect has been observed in cross couplings of aryl ammonium triflates.13
Herein we report the first example of a general method for the cross coupling of benzylic ammonium salts with boronic acids (Scheme 1B). To our knowledge, this is the first cross coupling of an aryl boronic acid with a benzylic amine derivative other than gramine and the first stereospecific coupling of an acyclic benzylic amine derivative with excellent chirality transfer.7,14,15 This method enables the preparation of diarylmethanes with broad substrate scope and diarylethanes with excellent enantioenrichment.
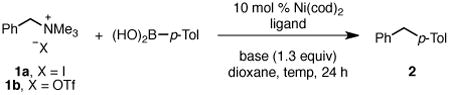
We began by investigating the cross coupling of p-tolyl boronic acid and benzyl ammonium salts 1, which are easily prepared in quantitative yield via methylation of dimethyl benzyl amine.11 Using conditions similar to those reported for Suzuki reactions of aryl ammonium salts,13a low yields of diarylmethane 2 were observed when iodide 1a was employed (Table 1, entry1). In contrast, use of monodentate phosphine ligands, particularly mixed aryl/alkyl phosphines, provided increased yields (entries 2–4). As we had anticipated, the use of the less coordinating triflate counter-ion resulted in greater reactivity. Quantitative yield was observed when triflate 1b was employed as the substrate (entry 5). Lowering the reaction temperature to 40 °C, PPh2Cy proved to be the best ligand (entries 6–8). K3PO4 was as effective as CsF, resulting in nearly quantitative yield (entry 10). Notably, no product is observed in the absence of Ni catalyst (entries 12, 13).16 Re-examination of the N-heterocyclic carbene IMes·HCl under the optimized conditions again showed phosphines to be superior ligands.
Table 1. Optimization of Reaction Conditionsa.
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | X | ligand (mol %) | base | temp (°C) | yield (%)b |
|
| |||||
| 1 | I | IMes·HCl (11) | CsF | 60 | 21 |
| 2 | I | PCy3 (22) | CsF | 60 | 30 |
| 3 | I | PPhCy2 (22) | CsF | 60 | 59 |
| 4 | I | PPh2Cy (22) | CsF | 60 | 54 |
| 5 | OTf | PPh2Cy (22) | CsF | 60 | 100 |
| 6 | OTf | PPh2Cy (22) | CsF | 40 | 96 |
| 7 | OTf | PPhCy2 (22) | CsF | 40 | 16 |
| 8 | OTf | PPh3 (22) | CsF | 40 | 82 |
| 9 | OTf | PPh2Cy (22) | CsF | r.t. | 68 |
| 10 | OTf | PPh2Cy (22) | K3PO4 | 40 | 99 |
| 11 | OTf | PPh2Cy (22) | K3PO4 | r.t. | 96 |
| 12c | OTf | None | CsF | 40 | 0 |
| 13c | OTf | None | K3PO4 | 40 | 0 |
| 14 | OTf | IMes·HCl (11) | CsF | 40 | 28 |
Conditions: ammonium triflate 1 (0.10 mmol, 1.0 equiv), boronic acid (1.2 equiv), Ni(cod)2 (10 mol %), ligand, CsF or K3PO4 (1.3 equiv), dioxane (0.33 M), 24 h, unless noted otherwise.
Determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as internal standard.
No Ni(cod)2 used.
Using the optimized conditions (Table 1, entries 6 and 10), we observed broad substrate scope for both the boronic acid and ammonium triflate in the cross couplings of unbranched benzylic ammonium triflates. We generally observed similar results with CsF and K3PO4. However, for products with base-sensitive functional groups, such as 6 (Scheme 2), CsF proved advantageous. For the boronic acid, substitution was well tolerated at the ortho, meta, and para positions of the aromatic ring. High yields were observed for arylboronic acids with both electron-donating (4) and electron-withdrawing (5–9) substituents. Further, these mild reaction conditions tolerated a wide variety of functional groups, including ethers (4, 10, 14, 17, 18, 21), fluorides (5, 12, 13, 15), esters (6), secondary amides (7), sulfones (8), trifluoromethyls (9), free alcohols (11), and acetals (19). Naphthyl, pyridyl and quinolinyl boronic acids were also effective partners (20–23).17
Scheme 2. Scope in Boronic Acida.

a Conditions: ammonium triflate 1b (0.30 mmol, 1.0 equiv), boronic acid (1.2 equiv), Ni(cod)2 (10 mol %), PPh2Cy (22 mol %), CsF or K3PO4 (1.3 equiv), dioxane (0.33 M), 40 °C, 24 h. Average isolated yield of duplicate experiments reported (±0–12%), unless otherwise noted. b Result of a single experiment.
For the ammonium triflate partner, a variety of functional groups were tolerated at the ortho, meta, and para positions of the phenyl group (Scheme 3). Products containing ether (24, 26–28), trifluoromethyl (25), ketone (29) and fluoride (30) substituents were formed in high yields. Notably, these reaction conditions selectively activate the benzylic C–N bond in the presence of either aryl or benzylic C–O bonds (24, 26–28), highlighting the complementarity of the benzylic ammonium salts to ethers. This method also provides orthogonal reactivity to aryl halides. Although chloride-substituted benzyl ammonium triflates undergo cross coupling at both the C–Cl and C–NMe3 bonds, aryl bromides can be effectively cross coupled in the presence of benzylic dimethylamino groups.18 Finally, naphthyl and indolyl10d substrates are also effective in this reaction (31, 32).
Scheme 3. Scope in Ammonium Salta.

a Conditions: ammonium triflate (0.20 mmol, 1.0 equiv), boronic acid (1.2 equiv), Ni(cod)2 (10 mol %), PPh2Cy (22 mol %), CsF or K3PO4 (1.3 equiv), dioxane (0.33 M), 40 °C, 24 h. Average isolated yield of duplicate experiments reported (±0–9%), unless otherwise noted. b Result of a single experiment.
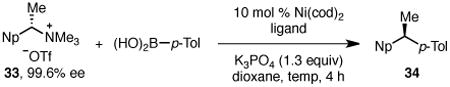
Encouraged by this broad substrate scope, we turned to the cross coupling of branched benzylic ammonium triflate 33 (Np = 2-naphthyl), which was readily prepared from (1S)-1-(2-naphthyl)ethanamine, purchased in 99.6% ee. Although application of the previously optimized conditions gave no desired product at 40 °C, high yields of diarylethane 34 were realized at a higher reaction temperature (Table 2, entry 1). However, only moderate chirality transfer was observed. A screen of ligands revealed that ligand affects the chirality transfer with 9,9-dimethyl-4,5-bis(di-tert-butylphosphino)xanthene (t-Bu-XantPhos) and tri(o-tolyl)phosphine proving best (entries 4 and 6). Due to ligand cost, we selected P(o-Tol)3 for further optimization and found that the reaction proceeds in high yield and enantiospecificity at 70 °C in either dioxane (entry 7) or PhMe (entry 8). Under these conditions, the reaction proceeds with excellent chirality transfer and yield, providing a powerful route to enantioenriched diarylethanes. Notably, no product is observed in the absence of nickel catalyst at these elevated temperatures (entries 9 and 10).16
Table 2. Optimization of Branched Ammonium Salta.
| ||||
|---|---|---|---|---|
|
| ||||
| entry | ligand (mol %) | temp (° C) | yield (%)b | ee (%)c |
|
| ||||
| 1 | PPh2Cy (22) | 100 | 84 | 81 |
| 2 | PPhCy2 (22) | 100 | 97 | 79 |
| 3 | PPh3 (22) | 100 | 83 | 81 |
| 4 | t-Bu-XantPhos (12) | 100 | (91) | 98 |
| 5 | XantPhos (12) | 100 | 15 | 40 |
| 6 | P(o-Tol)3 (22) | 100 | 71 | 98 |
| 7 | P(o-Tol)3 (22) | 70 | 94 | 98 |
| 8d | P(o-Tol)3 (22) | 70 | 95 | 98 |
| 9e | None | 70 | 0 | n.d.f |
| 10e | None | 100 | 0 | n.d.f |
Conditions: ammonium triflate 33 (0.10 mmol, 1.0 equiv), boronic acid (1.2 equiv), Ni(cod)2 (10 mol %), ligand, K3PO4 (1.3 equiv), dioxane (0.4 M), 4 h, unless noted otherwise.
Determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as internal standard.
Determined by chiral HPLC.
PhMe replaced dioxane as solvent.
No Ni(cod)2 used.
n.d. = not determined.
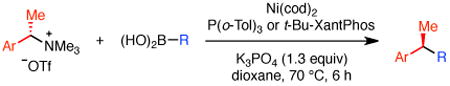
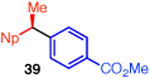
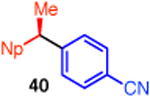
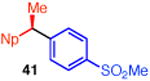
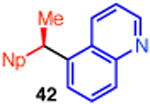
With these conditions in hand, we examined the scope of this transformation. Upon increasing the reaction scale, we found that the catalyst loading could be lowered to 3 mol % without detrimental effect on yield or enantioselectivity of diarylethane 34 (Table 3, entry 1). In some cases, lower catalyst loading resulted in higher enantiospecificities, suggesting a possible Ni-mediated epimerization pathway (entries 1 vs 2, 12 vs 13). Electron-rich and electron-neutral aryl boronic acids perform well in this reaction (entries 1–11). Further, a wide variety of functional groups are tolerated, including ethers, olefins, fluorides, esters, nitriles, and sulfones. In terms of electron-poor boronic acids, we were surprised that the cross coupling of 4-fluorophenyl boronic acid and ammonium triflate 33 resulted in low yield when K3PO4 was used as base (entry 6). However, exceptional yield and chirality transfer were observed when CsF was employed (entry 7). Notably, the successful formation of ester 39 (entry 8), nitrile 40 (entry 9), and sulfone 41 (entry 10) particularly highlights the advantage of using a boronic acid as the coupling partner. For some of these base-sensitive functional groups, we replaced K3PO4 with CsF (entries 8 and 9). Finally, we were pleased to observe that the reaction of vinyl boronic acids also proceeds in high yield and enantiospecificity (entry 13). Such enantioenriched allyl arenes are known precursors to α-arylpropanoic acid analgestics, such as naproxen.19
Table 3. Formation of Enantioenriched Diarylethanesa.
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | product | mol % Ni | ligand (mol %) | yield (%)b | Prod ee(%)c |
|
| |||||
| 1 |
|
3 | P(o-Tol)3 (7) | 60 | 99 |
| 2d,e | 10 | P(o-Tol)3 (22) | 72 | 97 | |
| 3 |
|
3 | P(o-Tol)3 (7) | 82 | 99 |
| 4 |
|
3 | P(o-Tol)3 (7) | 51 | 95 |
| 5 |
|
3 | P(o-Tol)3 (7) | 68 | 98 |
| 6 |
|
10 | P(o-Tol)3 (22) | (15) | n.d.f |
| 7d,g | 10 | P(o-Tol)3 (22) | 94 | 98 | |
| 8g |
|
10 | P(o-Tol)3 (22) | 71 | 98 |
| 9d,h |
|
1 | P(o-Tol)3 (3) | 76 | 95 |
| 10 |
|
3 | P(o-Tol)3 (7) | 94 | 97 |
| 11d,i |
|
10 | P(o-Tol)3 (22) | 53 | 52 |
| 12j |
|
2 | P(o-Tol)3 (5) | 96 | 99 |
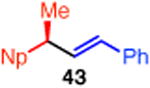
| 13d,i | 10 | P(o-Tol)3 (22) | (91) | 77 | |
| 14d,e,k |
|
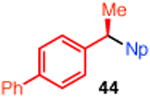
10 | t-Bu-XantPhos (12) | 46 | 98 |
| 15e |
|
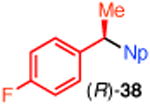
10 | t-Bu-XantPhos (12) | 37 | 95 |
| 16e |
|
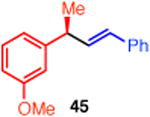
15 | P(o-Tol)3 (32) | 56 | 98 |
| 17e |
|
15 | P(o-Tol)3 (32) | 54 | 91 |
Conditions: ammonium triflate (0.26 mmol, 1.0 equiv), boronic acid (1.2 equiv), Ni(cod)2, P(o-Tol)3, K3PO4 (1.3 equiv), dioxane (0.4 M), 70 °C, 6 h unless otherwise noted. Starting materials were ≥99% ee, unless otherwise noted.
Average isolated yield of duplicate experiments (±5%). Yields in parentheses determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as internal standard.
Average ee from duplicate experiments as determined by chiral HPLC analysis (±1%).
Result of a single experiment.
Performed on 0.2 mmol scale.
n.d. = not determined.
CsF used instead of K3PO4.
Performed on 0.5 mmol scale.
Performed on 0.1 mmol scale.
Performed on 1.36 mmol scale.
Starting material was 98% ee.
With respect to the scope of the ammonium salt, electron-poor aryl groups were well tolerated (entries 14–17).20 In every case, the benzylic ammonium triflates are available in greater than 98% ee, highlighting one of the advantages of using benzylic amine derivatives in these stereospecific cross couplings. However, with these substrates, higher catalyst loadings are required. In some cases, we also observed higher yields and greater chirality transfer when t-Bu-XantPhos was employed as ligand (entries 14 and 15). Although we do not yet fully understand the basis for the effect of ligand on the chirality transfer, the benefit of higher catalyst loadings for these substituted benzene derivatives suggests that a nickel-catalyzed epimerization pathway is not significant for these substrates.
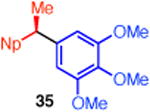
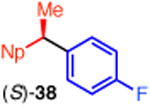
We established the absolute configuration of diarylethane 35, whose racemate displays anti-cancer activity,1d, e by comparison of its optical rotation to the reported value.5b The absolute configuration of (R)-38 was determined by X-ray crystallography (Figure 1).11, 21 Collectively, these assignments demonstrate that these reactions proceed with overall inversion of configuration.
Figure 1. Crystal Structure of (R)-38a.

a Molecular diagram of (R)-38 with ellipsoids at 30% probability. Tertiary H-atom depicted with arbitrary radius. All other H-atoms and a second symmetry-unique compound molecule are omitted for clarity.
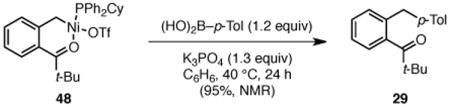
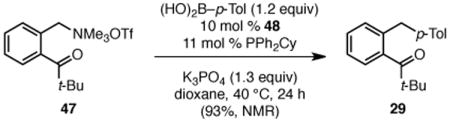
To begin to understand the mechanism of this transformation, we studied the stoichiometric reaction of ammonium triflate 47 with Ni(cod)2 and PPh2Cy. This reaction produced alkylnickel(II) triflate 48 in 51% isolated yield (Scheme 4). The structure of this complex was confirmed by X-ray crystallography, as well as 1H, 13C, 19F, and 31P NMR analysis.11, 21 It is consistent with oxidative addition of nickel into the benzylic C–N bond. The chelating carbonyl likely stabilizes this complex in the η1 form. Upon treatment with p-tolylboronic acid, complex 48 is converted to diarylmethane 29 in 95% yield (1H NMR, eq 1). Further, complex 48 is catalytically competent; product 29 was formed in quantitative yield (1H NMR) when 48 was used as catalyst (eq 2). These results suggest that complex 48 is a viable intermediate in the catalytic reaction. We thus propose that these reactions proceed via oxidative addition of the electron-rich Ni(0) complex into the C–N bond to generate either an η1- or η3-bound benzyl nickel(II) intermediate.22 Transmetallation with the activated boronic acid and subsequent reductive elimination then delivers the diaryl product.
Scheme 4. Synthesis and Crystal Structure of Oxidative Addition Complex 48a.
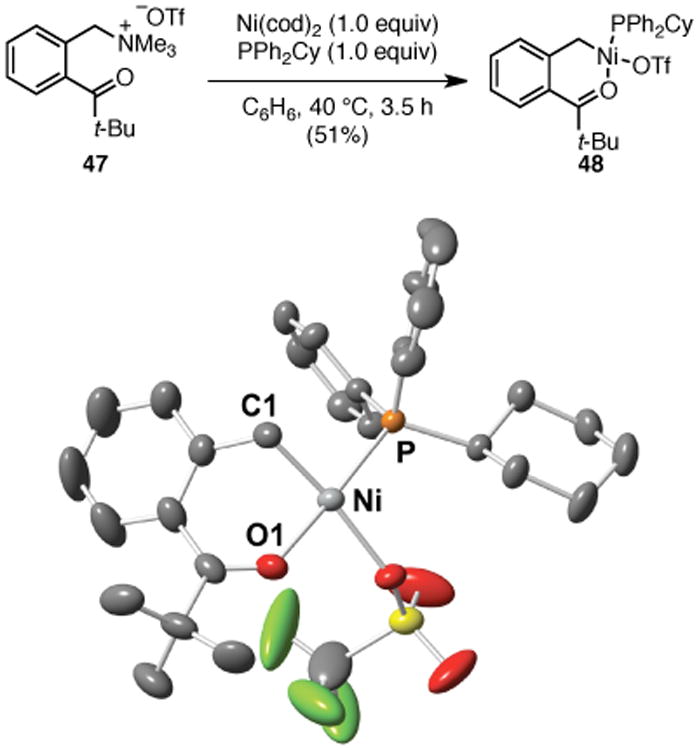
a Molecular diagram of 48 with ellipsoids at 30% probability. H-atoms omitted for clarity.
 |
(1) |
 |
(2) |
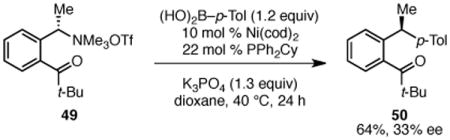
We have attempted to use a similar substrate to determine whether the oxidative addition of nickel into the C–N bond occurs with retention or inversion of configuration. However, the cross coupling of branched ammonium triflate 49 occurs with poor chirality transfer, delivering diarylethane in only 33% ee (eq 3). This result suggests that the carbonyl group promotes an epimerization pathway. Unfortunately, attempts to isolate the oxidative addition adducts of substrates without such chelating groups have been unsuccessful to date. Nonetheless, retention of configuration during transmetallation and reductive elimination is well precedented for alkyl metal species.23 Thus, we propose that the oxidative addition of the nickel catalyst into the benzylic C–N bond likely occurs via an SN2 mechanism, resulting in inversion of configuration of the benzylic stereocenter (Scheme 5). This mechanistic proposal is consistent with the overall inversion of configuration observed in this cross coupling reaction.
Scheme 5. Proposed Reaction Mechanism.

 |
(3) |
In summary, we have developed mild conditions for a high-yielding cross coupling of benzyl ammonium triflates with aryl boronic acids to give diarylmethanes. This nickel-catalyzed reaction displays exceptional functional group tolerance and substrate scope in both the ammonium salt and boronic acid. Further, the nickel-catalyzed cross coupling of chiral ammonium salts was developed to yield diarylethanes in high enantiospecifities. To our knowledge, this is the first Suzuki coupling of a benzylic electrophile that provides highly enantioenriched products. Given the ready availability of highly enantioenriched chiral amines, this method offers a powerful approach to the synthesis of enantioenriched diarylethanes. Studies to determine further mechanistic details of this cross coupling as well as to expand the scope of this transformation are underway in our laboratories.
Supplementary Material
Acknowledgments
Acknowledgement is made to NIH COBRE (P20 RRO17716) and the Donors of the American Chemical Society Petroleum Research Fund for generous support of this research. We also thank AstraZeneca for donated boronic acids. NMR and other data were acquired at UD on instruments obtained with the assistance of NSF and NIH funding (NSF MIR 0421224, NSF CRIF MU CHE0840401, NIH P20 RR017716).
Funding Sources: No competing financial interests have been declared.
Footnotes
Supporting Information. Experimental procedures, characterization data and spectra of new compounds, X-ray crystal structures of (R)-38 and 48. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Zhang J, Bellomo A, Creamer AD, Dreher SD, Walsh PJ. J Am Chem Soc. 2012;134:13765. doi: 10.1021/ja3047816. [DOI] [PubMed] [Google Scholar]; (b) Cheltsov AV, Aoyagi M, Aleshin A, Yu ECW, Gilliland T, Zhai D, Bobkov AA, Reed JC, Liddington RC, Abagyan R. J Med Chem. 2010;53:3899. doi: 10.1021/jm901446n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huang Z, Ducharme Y, Macdonald D, Robichaud A. Curr Opin Chem Biol. 2001;5:432. doi: 10.1016/s1367-5931(00)00224-6. [DOI] [PubMed] [Google Scholar]; (d) Messaoudi S, Hamze A, Provot O, Tréguier B, Rodrigo De Losada J, Bignon J, Liu JM, Wdzieczak-Bakala J, Thoret S, Dubois J, Brion JD, Alami M. ChemMedChem. 2011;6:488. doi: 10.1002/cmdc.201000456. [DOI] [PubMed] [Google Scholar]; (e) Alami M, Messauoudi S, Hamze A, Provot O, Brion JD, Liu JM, Bignon J, Bakala J. WO2009/147217. Dihydro-Iso-Ca-4 and Analogues: Potent Cytotoxics, Inhibitors of Tubulin Polymerization. 2009; f Moree WJ, Li BF, Jovic F, Coon T, Yu J, Gross RS, Tucci F, Marinkovic D, Zamani-Kord S, Malany S, Bradbury MJ, Hernandez LM, O'Brien Z, Wen J, Wang H, Hoare SRJ, Petroski RE, Sacaan A, Madan A, Crowe PD, Beaton G. J Med Chem. 2009;52:5307. doi: 10.1021/jm900933k. [DOI] [PubMed] [Google Scholar]; (g) Beaton G, Moree WJ, Jovic F, Coon T, Yu J. US2006/14797. Sleep Inducing Compound and Methods Relating Thereto. 2006
- 2.For examples, see: Molander GA, Elia MD. J Org Chem. 2006;71:9198. doi: 10.1021/jo061699f.Arp FO, Fu GC. J Am Chem Soc. 2005;127:10482. doi: 10.1021/ja053751f.López-Pérez A, Adrio J, Carretero JC. Org Lett. 2009;11:5514. doi: 10.1021/ol902335c.Binder JT, Cordier CJ, Fu GC. J Am Chem Soc. 2012;134:17003. doi: 10.1021/ja308460z.
- 3.(a) Kuwano R. J Synth Org Chem, Jpn. 2011;69:1263. [Google Scholar]; (b) Kuwano R, Yokogi M. Chem Commun VL. 2005:5899. doi: 10.1039/b513372f. [DOI] [PubMed] [Google Scholar]; (c) Kuwano R, Yokogi M. Org Lett. 2005;7:945. doi: 10.1021/ol050078q. [DOI] [PubMed] [Google Scholar]; (d) Kuwano R, Yu JY. Heterocycles. 2007;74:233. [Google Scholar]; (e) Ohsumi M, Kuwano R. Chemistry Letters. 2008;37:796. [Google Scholar]; (f) Yu JYJ, Kuwano RR. Org Lett. 2008;10:973. doi: 10.1021/ol800078j. [DOI] [PubMed] [Google Scholar]; (g) Kofink CC, Knochel P. Org Lett. 2006;8:4121. doi: 10.1021/ol0616790. [DOI] [PubMed] [Google Scholar]; (h) McLaughlin M. Org Lett. 2005;7:4875. doi: 10.1021/ol0517271. [DOI] [PubMed] [Google Scholar]
- 4.(a) Guan BT, Xiang SK, Wang BQ, Sun ZP, Wang Y, Zhao KQ, Shi ZJ. J Am Chem Soc. 2008;130:3268. doi: 10.1021/ja710944j. [DOI] [PubMed] [Google Scholar]; (b) Yu DG, Wang X, Zhu RY, Luo S, Zhang XB, Wang BQ, Wang L, Shi ZJ. J Am Chem Soc. 2012;134:14639. doi: 10.1021/ja307045r. [DOI] [PubMed] [Google Scholar]
- 5.(a) Taylor BLH, Harris MR, Jarvo ER. Angew Chem, Int Ed. 2012;51:7790. doi: 10.1002/anie.201202527. [DOI] [PubMed] [Google Scholar]; (b) Taylor BLH, Swift EC, Waetzig JD, Jarvo ER. J Am Chem Soc. 2011;133:389. doi: 10.1021/ja108547u. [DOI] [PubMed] [Google Scholar]; (c) Greene MAM, Yonova IMI, Williams FJF, Jarvo ERE. Org Lett. 2012;14:4293. doi: 10.1021/ol300891k. [DOI] [PubMed] [Google Scholar]
- 6.For other methods to form enantioenriched diarylethane derivatives, see: Bolshan Y, Chen Cy, Chilenski JR, Gosselin F, Mathre DJ, O'shea PD, Roy A, Tillyer RD. Org Lett. 2004;6:111. doi: 10.1021/ol0361655.Fessard TC, Andrews SP, Motoyoshi H, Carreira EM. Angew Chem, Int Ed. 2007;46:9331. doi: 10.1002/anie.200702995.Lewis CA, Chiu A, Kubryk M, Balsells J, Pollard D, Esser CK, Murry J, Reamer RA, Hansen KB, Miller SJ. J Am Chem Soc. 2006;128:16454. doi: 10.1021/ja067840j.Pathak TP, Gligorich KM, Welm BE, Sigman MS. J Am Chem Soc. 2010;132:7870. doi: 10.1021/ja103472a.Nave S, Sonawane RP, Elford TG, Aggarwal VK. J Am Chem Soc. 2010;132:17096. doi: 10.1021/ja1084207.Roesner S, Casatejada JM, Elford TG, Sonawane RP, Aggarwal VK. Org Lett. 2011;13:5740. doi: 10.1021/ol202251p.
- 7.Li MB, Tang XL, Tian SK. Adv Synth Catal. 2011;353:1980. [Google Scholar]
- 8.He A, Falck JR. J Am Chem Soc. 2010;132:2524. doi: 10.1021/ja910582n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Imao D, Glasspoole BW, Laberge VS, Crudden CM. J Am Chem Soc. 2009;131:5024. doi: 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]; (b) Dreher SD, Dormer PG, Sandrock DL, Molander GA. J Am Chem Soc. 2008;130:9257. doi: 10.1021/ja8031423. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sandrock DL, Jean-Gerard L, Chen Cy, Dreher SD, Molander GA. J Am Chem Soc. 2010;132:17108. doi: 10.1021/ja108949w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For examples of allylic and benzylic ammonium salts in other reactions, see: Langlois Y, Van Bac N, Fall Y. Tetrahedron Lett. 1985;26:1009.Dressaire G, Langlois Y. Tetrahedron Lett. 1980;21:67.Gupton JT, Layman WJ. J Org Chem. 1987;52:3683.de la Herrán G, Segura A, Csákÿ AG. Org Lett. 2007;9:961. doi: 10.1021/ol063042m.Hosomi A, Hoashi K, Tominaga Y, Otaka K, Sakurai H. J Org Chem. 1987;52:2947.
- 11.See Supporting Information for details.
- 12.(a) Nugent TC. Chiral Amine Synthesis. Wiley-VCH Verlag GmbH & Co; KGaA: Weinheim: 2010. [Google Scholar]; (b) Mereyala HB, Pola P. Tetrahedron: Asymmetry. 2003;14:2683. [Google Scholar]; (c) Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J, Devine PN, Huisman GW, Hughes G. J Science. 2010;329:305. doi: 10.1126/science.1188934. [DOI] [PubMed] [Google Scholar]; (d) Vries T, Wynberg H, van Echten E, Koek J, ten Hoeve W, Kellogg RM, Broxterman QB, Minnaard A, Kaptein B, van der Sluis S, Hulshof L, Kooistra J. Angew Chem, Int Ed. 1998;37:2349. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2349::AID-ANIE2349>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 13.(a) Blakey S, MacMillan D. J Am Chem Soc. 2003;125:6046. doi: 10.1021/ja034908b. [DOI] [PubMed] [Google Scholar]; (b) Wenkert E, Han AL, Jenny C. J Chem Commun. 1988:975. [Google Scholar]; (c) Reeves JT, Fandrick DR, Tan Z, Song JJ, Lee H, Yee NK, Senanayake CH. Org Lett. 2010;12:4388. doi: 10.1021/ol1018739. [DOI] [PubMed] [Google Scholar]; (d) Buszek KR, Brown N. Org Lett. 2007;9:707. doi: 10.1021/ol063027h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Xie LG, Wang ZX. Angew Chem, Int Ed. 2011;50:4901. doi: 10.1002/anie.201100683. [DOI] [PubMed] [Google Scholar]; (f) Zhang XQ, Wang ZX. J Org Chem. 2012;77:3658. doi: 10.1021/jo300209d. [DOI] [PubMed] [Google Scholar]; (g) Blessley G, Holden P, Walker M, Brown JM, Gouverneur V. Org Lett. 2012;14:2754. doi: 10.1021/ol300977f. [DOI] [PubMed] [Google Scholar]
- 14.For an example of an enantioselective reaction of a benzylic amine derivative, see: Weng ZT, Li Y, Tian SK. J Org Chem. 2011;76:8095. doi: 10.1021/jo2014142.
- 15.High stereospecificity has been observed in Negishi cross couplings of indene-derived aziridines. See: Huang CY, Doyle AG. J Am Chem Soc. 2012;134:9541. doi: 10.1021/ja3013825.
- 16.These control reactions were performed with new stirbars.
- 17.Lower yields were observed for pyridine boronic acids without substituents ortho to the nitrogen, such as 3-pyridinylboronic acid. Ortho substituents likely hinder pyridine from binding the Ni catalyst.
- 18.For an example, see the preparation of 1-(biphenyl-4-yl)-N,N-dimethylethanamine in the Supporting Information.
- 19.Li CC, Xing JJ, Zhao JJ, Huynh PP, Zhang WW, Jiang PP, Zhang YJY. Org Lett. 2012;14:390. doi: 10.1021/ol203154j. [DOI] [PubMed] [Google Scholar]
- 20.The cross coupling of the 1-naphthyl substituted ammonium salt proceeded in only moderate enantioselectivity under the optimized reaction conditions (67% ee). See Supporting Information for more detail.
- 21.CCDC-899360 ((R)-38) and -899359 (48) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
- 22.Both the η1 and η3 forms of benzyl nickel(II) intermediates have been observed. See: Matsubara R, Gutierrez AC, Jamison TF. J Am Chem Soc. 2011;133:19020. doi: 10.1021/ja209235d.
- 23.(a) Stille JK. Oxidative Addition and Reductive Elimination. In: Hartley FR, Patai S, editors. The Chemistry of the Metal–Carbon Bond. Vol. 2. John Wiley & Sons, Ltd; New York: 1985. p. 625. [Google Scholar]; (b) Netherton MR, Fu GC. Angew Chem, Int Ed. 2002;41:3910. doi: 10.1002/1521-3773(20021018)41:20<3910::AID-ANIE3910>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.