

Abstract
Novel clusters of 3,5-bis(benzylidene)-4-oxo-1-piperidinyl dimers 3–5 were evaluated against human Molt4/C8 and CEM T-lymphocytes and human HeLa cervix adenocarcinoma cells as well as murine L1210 leukemia neoplasms. Several of these compounds demonstrated IC50 values in the submicromolar and low micromolar range and compounds possessing 4-fluoro, 4-chloro and 3,4,5-trimethoxy substituents in the series 3 and 4 were identified as potent molecules. A heat map revealed the very high cytotoxic potencies of representative compounds against a number of additional leukemic and lymphoma cell lines and displayed greater toxicity to these cells than nonmalignant MCF10A and Hs-27 neoplasms. These dienones are more refractory to breast and prostate cancers. The evaluation of representative compounds in series 3–5 against a panel of human cancer cell lines revealed them to be potent cytotoxins with average IC50 values ranging from 0.05 to 8.51 μM In particular, the most potent compound 4g demonstrated over 382-fold and 590-fold greater average cytotoxic potencies in this screen than the reference drugs, melphalan and 5-fluorouracil, respectively. A mode of action investigation of two representative compounds 3f and 4f indicated that they induce apoptosis which is due, at least in part, to the activation of caspase-3 and depolarization of the mitochondrial membrane potential.
Keywords: Unsaturated ketones, Cytotoxicity, Apoptosis, Structureeactivity relationships, 4-Piperidone, Mitochondria
1. Introduction
The principal interest in our laboratories is the design, synthesis and antineoplastic evaluation of conjugated unsaturated ketones. The perceived importance of these compounds is their ability to interact preferentially with thiols in contrast to amino and hydroxyl groups [1,2] which are present in nucleic acids. Thus the genotoxic properties displayed by various alkylating agents in cancer chemotherapy [3] may well be absent in conjugated enones. Initially compounds containing one α,β-unsaturated group were prepared which demonstrated cytotoxic and anticancer properties [4]. However a number of studies showed that an initial chemical insult followed by a second interaction with cellular constituents can be more harmful to tumors than nonmalignant cells [5–7]. Hence an additional conjugated enone group was introduced into the design of candidate cytotoxins leading to the mounting of the 1,5-diaryl-3-oxo-1,4-pentadienyl pharmacophore onto a number of heterocyclic and carbocyclic scaffolds such as series 1 as indicated in Fig. 1. In these compounds the capacity for sequential alkylation of cellular thiols now exists.
Fig. 1.
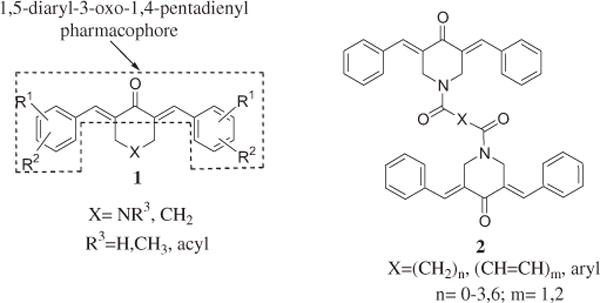
The structures of the compounds in series 1 and 2.
In recent years a novel cluster of cytotoxic agents were developed possessing two 1,5-diaryl-3-oxo-1,4-pentadienyl groups which have the general structure 2 as indicated in Fig.1. In this way multiple sequential alkylations of cellular thiols can take place. Most of the compounds displayed micromolar or submicromolar IC50 and CC50 values against various neoplastic cell lines [8,9]. In general, the greatest potencies were found in those compounds with the shortest linker group X between the two piperidinyl nitrogen atoms. From these studies, two compounds possessing oxalyl and malonyl linkers were identified as the most promising cytotoxic agents and are referred to as 3a and 4a, respectively. An initial study examined the compounds in series 3–5, against HCT116 and HT29 human colon cancer cells [10]. In general, these compounds are potent cytotoxins having IC50 values less than 1 μM in 78% of the bioassays and are substantially more potent than 5-fluorouracil which is used clinically in treating colon cancers. These encouraging results justify the further bioevaluation of series 3–5.
The next phase of the study was threefold. 1. An examination of the effects of placing different substituents into the aryl rings of 3a and 4a on cytotoxic potencies was proposed. The variations in the electronic, hydrophobic and steric properties of the aryl substituents may enable correlations between these physicochemical properties and cytotoxic potencies to be established. The groups chosen had either positive (+) or negative (−) Hammett σ and Hansch π values namely 4-fluoro, 4-chloro and 3,4-dichloro (+,+), 4-methyl and 4-dimethylamino (−,+) and 3,4-dimethoxy, 3,4,5-trimethoxy and 4-methoxy (−,−). In addition, there are substantial differences in the sizes of the aryl groups. For example, the molar refractivity (MR) values of the 4-fluoro and 3,4,5-trimethoxy groups are 0.92 and 23.61, respectively [11]. 2. The aspiration was made to gain some idea of the sensitivity of these compounds towards human lymphomas and leukemia cancers and if tumor selectivity compared to normal cells was demonstrated. 3. An investigation was planned to find some of the ways in which promising lead compounds exert their antineoplastic effects.
The overall objective is that these investigations will enable one or more promising prototypic cytotoxic molecules to be identified which will be developed from the prospective of their drug-likeness properties [12] (see Fig. 2).
Fig. 2.
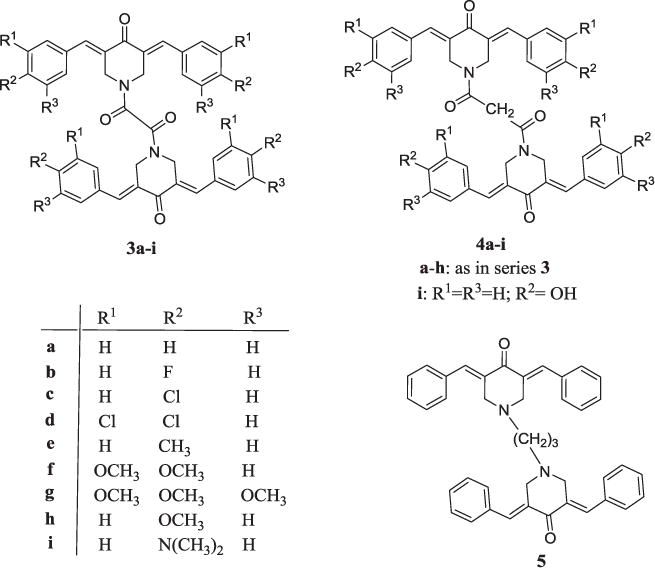
The structures of the analogs 3–5.
2. Results
The syntheses of 3a and 4a has been described previously [8] and a literature method was used for the preparation of 3b–i, 4b–i and 5 [10]. In brief, various aryl aldehydes were condensed with 4-piperidone to produce the corresponding 3,5 bis(benzylidene)-4-piperidones. Acylation of these dienones with oxalyl or malonyl chlorides gave rise to series 3 and 4, respectively. Reaction of 3,5-bis(benzylidene)-4-piperidone with 1,3-dibromopropane led to the synthesis of 5.
The dienones 3a–i, 4a–h and 5 were evaluated against human Molt4/C8 and CEM T-lymphocytes, human cervix carcinoma HeLa cells and murine L1210 leukemic cells. These results are presented in Table 1. The results of the cytotoxic evaluation of 3b,c,e–h, 4b,c,e,f and 5 against a number of lymphomas as well as breast and prostate cancer cell lines are presented as a heat map in Fig. 3 while a summary of these bioevaluations is portrayed in Supplemental Tables S1 and S2. In addition, 3a–d,g, 4a,c,e,g,i and 5 were evaluated against a panel of human cancer cell lines including human colon cancer and leukemic cell lines and these results are summarized in Table 2.
Table 1.
Evaluation of 3a–i, 4a–h and 5 against Molt4/C8, CEM, HeLa and L1210 cells.
| Compound | IC50 (μM)a
|
Average | |||
|---|---|---|---|---|---|
| Molt4/C8 | CEM | HeLa | L1210 | ||
| 3a | 0.462 ± 0.112 | 0.750 ± 0.164 | 1.0 ± 0.1 | 4.46 ± 0.23 | 1.67 |
| 3b | 0.083 ± 0.040 | 0.221 ± 0.146 | 0.388 ± 0.040 | 0.422 ± 0.237 | 0.28 |
| 3c | 0.294 ± 0.006 | 0.793 ± 0.010 | 0.649 ± 0.438 | 0.547 ± 0.279 | 0.57 |
| 3d | 8.46 ± 2.01 | 24.6 ± 4.2 | 27.0 ± 24.3 | 42.0 ± 5.4 | 25.5 |
| 3e | 1.66 ± 0.21 | 7.72 ± 0.91 | 3.79 ± 3.29 | 9.09 ± 1.43 | 5.57 |
| 3f | 0.779 ± 0.097 | 4.36 ± 3.06 | 4.43 ± 0.22 | 6.31 ± 3.46 | 3.97 |
| 3g | 0.047 ± 0.021 | 0.325 ± 0.061 | 0.246 ± 0.163 | 0.182 ± 0.012 | 0.20 |
| 3h | 33.1 ± 2.5 | 95.1 ± 69.1 | 59.9 ± 45.6 | 202 ± 16 | 97.5 |
| 3i | 437 ± 8 | 276 ± 25 | >500 | 297 ± 4 | >378 |
| 4a | 0.073 ± 0.014 | 0.201 ± 0.165 | 0.91 ± 0.07 | 1.23 ± 0.38 | 0.60 |
| 4b | 1.58 ± 0.04 | 2.75 ± 0.67 | 5.61 ± 3.72 | 11.0 ± 2.5 | 5.24 |
| 4c | 1.05 ± 0.62 | 1.06 ± 0.39 | 0.822 ± 0.172 | 0.847 ± 0.358 | 0.95 |
| 4d | 23.5 ±1.0 | 49.9 ± 17.7 | 39.8 ± 27.0 | 30.4 ± 8.8 | 35.9 |
| 4e | 1.89 ± 0.03 | 3.44 ± 1.44 | 2.88 ± 2.72 | 5.38 ± 0.78 | 3.40 |
| 4f | 0.388 ± 0.070 | 0.348 ± 0.064 | 0.370 ± 0.055 | 0.378 ± 0.187 | 0.37 |
| 4g | 0.060 ± 0.023 | 0.284 ± 0.047 | 0.216 ± 0.168 | 0.214 ± 0.082 | 0.19 |
| 4h | 449 ± 4 | 369 ± 45 | 219 ± 17 | >500 | >384 |
| 5 | 1.45 ± 0.06 | 2.52 ± 1.24 | 5.54 ± 3.67 | 2.98 ±1.81 | 3.12 |
| Melphalan | 2.81 ± 0.33 | 1.44 ± 0.20 | 1.70 ± 0.44 | 4.85 ± 0.87 | 2.70 |
| Curcumin | 6.46 ±1.41 | 8.16 ± 1.66 | 21.2 ± 16.1 | 15.1 ± 1.6 | 12.7 |
The IC50 values are the concentrations required to inhibit tumor cell proliferation by 50%.
Fig. 3.
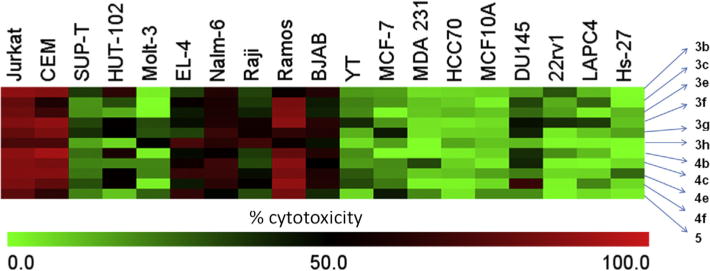
A heat map illustrating the toxicity of 1 μM of 3b,c,e–h, 4b,c,e,f and 5 towards 19 cell lines after 24 h of treatment. The data was generated from three independent determinations.
Table 2.
Evaluation of 3a–d, g, 4a, c, e, g, i and 5 against some human tumor cell lines.a
| Compound | All cell lines, IC50 (μM)c | Colon cancer cells, IC50 (μM)b
|
Leukemic cells, IC50 (μM)b
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| COLO 205 | HCC-2998 | HCT-15 | KM12 | SW-620 | Ave | HL-60 (TB) | K-562 | RPMI-8226 | SR | Ave | ||
| 3aa | 0.31 | 0.36 | 0.19 | 0.30 | 0.04 | 0.07 | 0.19 | 0.20 | 0.20 | 0.03 | 0.02 | 0.11 |
| 3b | 0.58 | 0.63 | 0.62 | 0.16 | 0.30 | 0.32 | 0.41 | 1.12 | 0.28 | 0.12 | 0.16 | 0.42 |
| 3c | 0.22 | 0.16 | 0.31 | 0.14 | 0.21 | 0.18 | 0.20 | 0.19 | 0.13 | 0.05 | 0.02 | 0.10 |
| 3d | 8.51 | 12.3 | 16.6 | 5.89 | 11.2 | 3.80 | 9.96 | 13.2 | 5.89 | 1.17 | 1.78 | 5.51 |
| 3g | 0.06 | 0.07 | 0.10 | 0.07 | 0.06 | 0.03 | 0.07 | 0.07 | 0.03 | 0.02 | 0.02 | 0.04 |
| 4aa | 0.24 | 0.16 | 0.17 | 0.17 | 0.13 | 0.05 | 0.14 | 0.29 | 0.05 | 0.13 | 0.03 | 0.13 |
| 4c | 0.09 | 0.08 | 0.13 | 0.06 | 0.16 | 0.03 | 0.09 | 0.11 | 0.03 | 0.02 | 0.01 | 0.04 |
| 4e | 0.19 | 0.17 | 0.30 | 0.08 | 0.17 | 0.15 | 0.17 | 0.23 | 0.10 | 0.03 | 0.05 | 0.10 |
| 4g | 0.05 | 0.06 | 0.10 | 0.03 | 0.04 | 0.05 | 0.06 | 0.04 | 0.03 | 0.05 | 0.01 | 0.03 |
| 4i | 1.66 | 1.66 | 1.74 | 1.17 | 0.69 | 0.76 | 1.20 | 1.74 | 0.53 | 0.35 | 0.46 | 0.77 |
| 5 | 0.19 | 0.19 | 0.18 | 0.20 | 0.16 | 0.06 | 0.16 | 0.25 | 0.04 | 0.04 | 0.04 | 0.09 |
| 5-Fluorouracil | 29.5 | 3.39 | 5.75 | 6.61 | 7.94 | 18.6 | 8.46 | 85.1 | 126 | 3.55 | 60.3 | 68.7 |
| Melphalan | 19.1 | 32.4 | 52.5 | 36.3 | 57.5 | 26.9 | 41.1 | 0.38 | 195 | 28.2 | 3.24 | 56.7 |
The data for 3a and 4a have been published previously [9].
The IC50 values are the concentrations of the compounds which inhibit tumor cell growth by 50%.
As indicated in the experimental section, the IC50 values of 3d and 5-fluorouracil towards a few cell lines were not generated due to their being somewhat refractory to the compounds.
The modes of action of the dienones 3f and 4f in CEM cells were also investigated. Both compounds induce cell death via apoptosis, activate caspase-3 and depolarize the mitochondrial membrane potential. These results are presented in Figs. 4–6, respectively.
Fig. 4.
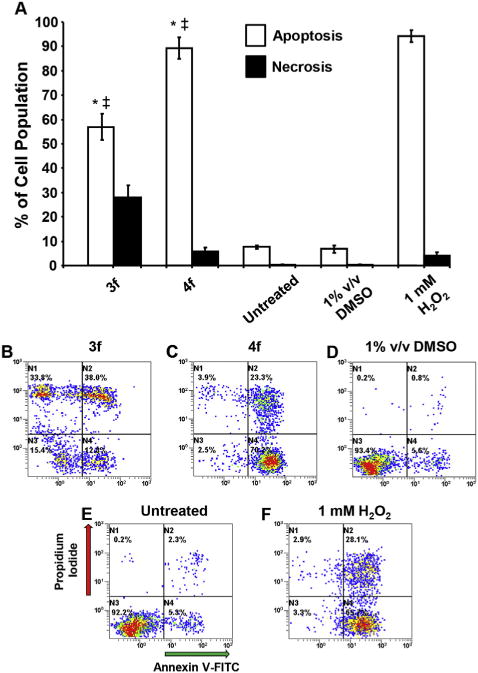
3f and 4f induce apoptotic cell death in a human CEM T-lymphocyte cell line as detected by phosphatidylserine externalization. The CEM cell line was treated with 1 mM concentration of 3f and 4f and controls (media alone, 1% v/v DMSO, or 1 mM H2O2) for 24 h and the level of apoptosis induction was measured by flow cytometry via Annexin V-FITC binding and Propidium iodide (PI) staining. (A) Apoptotic cell distribution is expressed as the sum of the percentages of both early and late phases of apoptosis (Annexin V-FITC positive; white bars), whereas cells that were stained by PI but without FITC signal were considered necrotic cells (black bars). Analysis using the two-tailed Student’s paired t-test of experimental compounds against untreated (*) and DMSO (+/+) controls was consistently at the level of p < 0.001, respectively. Each bar represents the standard deviation of the mean of three independent experiments. Panels B to F are representative flow cytometric analyses utilized to estimate the distribution of apoptosis/necrosis cells, where the FL1 or FL2 detectors settings were accommodated on the x- and y-axes, respectively. Data analysis from quadrant regions in the dot plots are interpreted as follows: the N1 quadrant represents necrotic cells (PI positive): N2 represents cells undergoing late apoptosis (PI and Annexin V-FITC positive); N3 represents unstained viable cells; and N4 represents cells in early apoptosis (Annexin V-FITC positive). Approximately, 3000 events were acquired and analyzed using CXP software (Beckman Coulter).
Fig. 6.
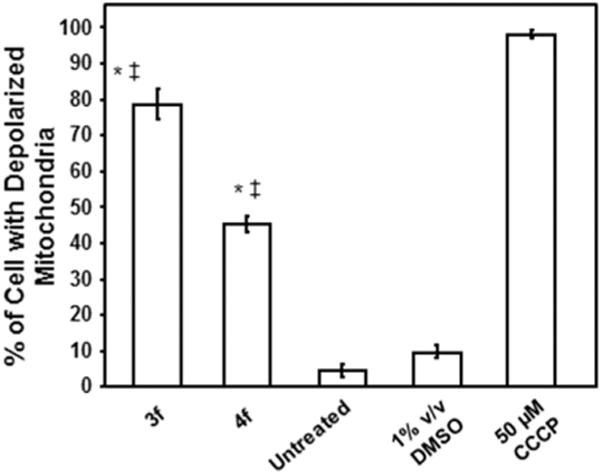
3f and 4f mediated cytotoxicity appears to be initiated via mitochondrial ΔΨm disruption in CEM T-cells. Cells were treated for 8 h with the compounds (1 μM) and changes in the mitochondrial ΔΨm were determined by staining with the aggregate-forming lipophilic cationic fluorophore JC-1 and monitored via flow cytometry. After dissipation of mitochondrial ΔΨm, the JC-1 reagent emitted a green fluorescence signal, whereas cells with polarized mitochondrial membrane emitted a red signal. Cells emitting a green fluorescence signal (y-axis) versus treatment type (x-axis) are depicted. Analysis using the two-tailed Student’s paired t-test of the experimental compounds against untreated (*) and DMSO (+/+) controls was consistently at the level of p < 0.001, respectively. Each bar represents the standard deviation of the mean of three independent assays. Cells exposed to the mitochondrial stressor CCCP (50 μM) were used as positive controls. DMSO solvent and untreated controls were also analyzed in parallel. Approximately 10,000 events were captured and analyzed per sample using CXP software (Beckman Coulter).
3. Discussion
The dienones 3a–i, 4a–h and 5 were evaluated against non-adherent Molt4/C8 and CEM T-lymphocytes as well as adherent HeLa adenocarcinoma cells with a view to determine if the compounds displayed efficacy towards human transformed and neoplastic cells. In addition, compounds which are cytotoxic to non-adherent cells may have the potential to inhibit the growth of tumors with metastatic potential. The murine L1210 assay was employed as a number of anticancer drugs are toxic to this cell line [13] and hence this bioassay may point to compounds with clinical utility.
The biodata presented in Table 1 reveals that the majority of the compounds are potent cytotoxins. No less than 39% of the IC50 values are submicromolar and in particular the IC50 figures of 3b,c,g and 4f,g are submicromolar in all four bioassays. In the case of 3b,g and 4a,g, the IC50 values in the Molt4/C8 screen are in the double digit nanomolar range (10−8 M) and are clearly lead molecules. Furthermore, the relative cytotoxic potencies of the compounds in series 3 and 4 were verified against the four neoplastic cell lines using Kendall’s coefficient of concordance [14]. This non-parametric test is based on ranks and ignores the magnitude of the differences in the potencies of groups of compounds. The derivation of the equation used in these determinations is given in the Supplemental Section. The relative potencies of 3a–i are the same in the Molt4/C8, CEM, HeLa and L1210 bioassays since Kendall’s coefficient of concordance is 0.9812 (p = 0.0001). A similar situation prevails among the analogs 4a–h; in this case, Kendall’s coefficient is 0.9315 (p = 0.0005). Furthermore, when all of the dienones in series 3 and 4 are considered, the relative potencies are the same as revealed by Kendall’s coefficient of 0.9504 (p < 0.0001). Thus it is likely that these alkylating agents which are designed to target thiol macromolecules e.g., glutathione, cysteine, thioredoxin reductases and glutathione S-transferase isozymes appear to act with the same molecular targets in all four cancer cell lines. The variations in potencies include differences in the alignment of the compounds at their binding sites.
In order to make some specific observations regarding the effects of placing different substituents in the aryl rings on cytotoxic potencies, the average IC50 values in the four bioassays presented in Table 1 were computed. Both 3c and 4c possess a para chloro group and display excellent potencies; however the addition of a meta chloro atom leading to 3d and 4d brings about 45- and 38-fold decreases, respectively, in the average IC50 values. Very interestingly, the replacement of a para chloro group in 3c and 4c by a methoxy group led to 3h and 4h, respectively, that were virtually inactive. On the other hand, the addition of a meta methoxy group to 3h and 4h leading to 3f and 4f increased potencies by 25- and >1038-fold, respectively. Still greater increases in potencies were noted with the 3,4,5-trimethoxy analogs 3g and 4g whose average potencies are the highest among the compounds in series 3–5. The replacement of a chloro group in 3c by a fluoro group (3b) in series 3 lead to an increase in the average cytotoxic potency by over 2fold. The relative cytotoxic potencies of the compounds in series 3 and 4 which have different para substituents are F > Cl > H> CH3 > OCH3 and H >Cl > CH3>F > OCH3, respectively. This observation reveals that in addition to the electronic, hydrophobic, and steric properties of the aryl substituents, the nature of the linker groups between the piperidinyl nitrogen atoms influences the magnitude of the IC50 values. This point is further illustrated in comparing the biodata between 4a and 5. Compound 5 which possesses no carboxamide group is 5-fold less potent than 4a, suggesting that the two carbonyl groups in carboxamide group present in 4a contribute to the cytotoxic potencies observed. Linear and semilogarithmic plots between the σ, π and MR constants of the aryl substituents in series 3 and 4 and the IC50 values generated in each of the four bioassays were undertaken. However no correlations (p < 0.05) or trends toward a correlation (p < 0.1) were noted. The mean of the average potencies of 3a–h and 4a–h are 16.9 and >53.8 μM, respectively, revealing that compounds in series 3 are more potent than the series 4. If the data for the two outliers 3h and 4h are discounted, the relevant figures are 5.39 and 6.66 μM, respectively. Hence the oxalyl linker in series 3 is marginally preferred in terms of potency.
Comparisons were made between the cytotoxic properties of the compounds in series 3–5 and two reference compounds, melphalan and curcumin. Melphalan is an alkylating agent used in cancer treatment, while there is considerable interest at the present time in curcumin as an antineoplastic agent [15]. The IC50 values of 3–5 are lower than the figures for melphalan in 53% of the comparisons made. Curcumin contains an arylidene keto and aryl vinyl groups and therefore bears some structural similarity to the compounds in series 3–5. In 72% of the comparisons made, the IC50 values of 3–5 were lower than the figures for curcumin. In particular, the average IC50 values of 3g and 4g are 13.5 and 14.2 times lower than the figure for melphalan and are 63.8 and 66.8 times more potent than curcumin.
An evaluation of the biodata was undertaken with a view to determine if the compounds varied in their potency towards each of the four cell lines. In other words, is tumor-selectivity observed? The average IC50 values of 3a–g, 4a–g and 5 (the outliers 3h,i and 4h are omitted) towards Molt4/C8, CEM, HeLa and L1210 cells are 2.79, 6.62, 6.24 and 7.70 μM, respectively. Thus clearly Molt4/C8 cells are more sensitive to 3a–g, 4a–g and 5 than CEM, HeLa and L1210 cells while the murine L1210 neoplasms are the most refractory. In general, tumor-selectivity was demonstrated. The importance of this observation is that compounds which vary in their potencies to different cells may have the capacity to display greater toxicity to tumors than nonmalignant cells.
The biodata in Table 1 are encouraging and thus the extent to which representative compounds are effective against other neoplasms was addressed. Many of the analogs in series 3–5 are effective against Molt4/C8 and CEM T-lymphocytes and L1210 leukemic cells. Hence the cytotoxic potencies of representative compounds against T-cell derived lymphoma and leukemic cell lines (Jurkat, CEM, SUP-T, HUT-102, Molt-3 and EL-4) were compared with B-cell derived lymphoma and leukemic cells (Nalm-6, Raji, Ramos and BJAB). In addition, the YT neoplasm, which is a natural killer-like leukemic cell line, was included in the array of cell lines. In view of the high mortalities caused by breast and prostate cancers, the efficacy of various analogs against several of these neoplastic cell lines was considered viz breast (MCF-7, MDA-231 and HCC70) and prostate cancers (DU145, 22rv1 and LAPC4). Two non-transformed cell lines were also utilized in order to observe the relative toxicity towards nonmalignant tissues namely breast epithelial cells (MCF10A) and foreskin fibroblasts (Hs-27). The cells are of human origin except for murine EL-4 cells and are non-adherent except for the breast, prostate and Hs-27 cell lines which are adherent in nature.
The results obtained for representative compounds in series 3–5 are presented as a heat map in Fig. 3 while specific figures are available in Supplemental Tables 1 and 2. The following figures in parentheses are the average of the percentage of dead cells caused by 3b,c,e–h,4b,c,e,f and 5 towards one or more cell lines. The following generalizations of the data obtained were made. First, Fig. 3 reveals clearly the greater lethality of the compounds towards lymphoma and leukemic cell lines (49.8) than either breast (11.9) or prostate (19.7) cell lines. The most sensitive cells are Jurkat (81.9), CEM (78.3) and Ramos (76.9). In addition, the compounds are more effective against B-cells (59.0) than T-cells (48.6). Secondly, the cytotoxic potencies of the lymphatic and leukemic cells (49.8) and to a lesser extent the DU145, 22rv1 and LAPC4 prostate cancers (19.7) compare favorably with the non-toxic effects of the compounds on the non-transformed Hs-27 control cell line (7.33). On the other hand, the toxicity of the compounds towards MCF-7, MDA 231 and HCC70 breast cancer cells (11.9) is similar to the effect on the MCF10A cell lines (8.50). Thirdly, somewhat surprising is the extent of cell death of the lymphoma and leukemic cells being influenced more by the cell line than the structures of the compounds as may be observed in Fig. 3. Thus the average percentage of dead lymphoma and leukemic cells was within the relatively narrow range of 43.0–58.3. The most potent compounds towards leukemia and lymphoma cells are 3f (58.3), 4e (56.4), 4f (53.4) and 3g (52.5) suggesting the importance of the 3,4-dimethoxy aryl substitution pattern present in 3f and 4f to cytotoxicity. One may also note that the two most potent compounds towards prostate cancers are 3f (39.8) and 4f (30.0). In summary, the compounds are toxic against a range of leukemic and lymphoma cells which are non-adherent and therefore these compounds may have the capacity to combat metastasis. In addition, the relatively low toxicity towards nonmalignant MCF10A and Hs-27 cells is encouraging.
The data generated in this study for the compounds in series 3–5 is that a number of these dienones display marked cytotoxic potencies for various leukemia and lymphoma cell lines. A previous study indicated that many of these compounds are very toxic towards human HCT116 and HT29 colon cancer cells [12] Thus the next phase of the current investigation consisted of examining additional colon cancers and leukemic cells in order to confirm their potency to these tumors. Representative compounds in series 3–5 were evaluated against approximately 59 human tumor cell lines from nine different neoplastic conditions, viz leukemia, melanoma and non-small cell lung, central nervous system, colon, ovarian, renal, prostate and breast cancers [16]. Some of these data are presented in Table 2.
The results obtained confirm the marked cytotoxic potencies of most of the compounds towards colon cancer cells. With regard to the IC50 values towards the five colon cancer cell lines, no less than 86% are submicromolar and 29% are in the double digit nanomolar range. The most potent compounds are 3g and 4g displaying average IC50 values of 70 and 60 nM, respectively, which are 121 and 141 times lower, respectively, than 5-fluorouracil which is used clinically in treating colon cancers. The same compounds have marked antileukemic properties as the data in Table 2 reveals. In this case, the IC50 values are submicromolar and in the double digit nanomolar range in 86% and 50% of the bioassays, respectively. The most potent compounds are also 3g and 4g with average IC50 figures of 40 and 30 nM, respectively, which are 1418 and 1890 times lower than the average IC50 value of melphalan which is used in treating various leukemias. In addition, 4c is a useful antileukemic lead compound displaying an average IC50 value of 40 nM indicating that 4c has more than double the potency of 3c.
The final segment of this investigation was directed to gaining some ideas of the way in which representative compounds caused cytotoxicity. The dienones 3f and 4f were chosen which have the same aryl substituents but differ in the nature of the X-linker group. Many antineoplastic agents exert their cytotoxicity by causing apoptosis [17]. Hence in order to explore this possibility, CEM cells were treated with 3f and 4f for 24 h and the results were determined by flow cytometry using annexin-FITC and Propidium stains. The results, which are portrayed in Fig. 4, revealed that both 3f and 4f induce apoptosis and to a lesser extent necrosis. One may also note that the extent of apoptosis and necrosis caused by 3f and 4f differs which may contribute to some of the variations in the cytotoxic potencies of these two compounds.
There are two principal apoptotic signaling pathways, namely the intrinsic mitochondrial and extrinsic death receptor routes. In the case of the mitochondrial pathway, apoptosis can be induced by different mechanisms some of which involve activation of caspases while others do not [18]. After incubation of CEM cells with either 3f or 4f, a cell permeable caspase-3 fluorogenic dye was employed. The intensity of the stain reveals the extent of caspase-3 activity in cells with intact cell membranes. The result is presented in Fig. 5 which reveals that both compounds activate caspase-3 which is an important way by which apoptosis takes place.
Fig. 5.
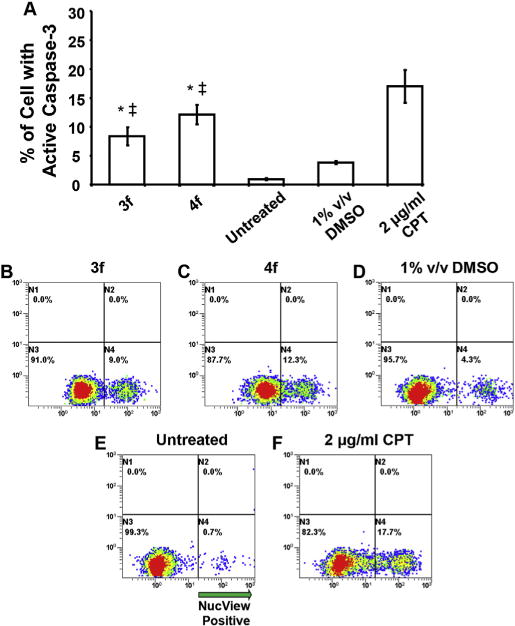
3f and 4f induce apoptotic cell death via caspase-3 activation in the CEM T-lymphocyte cell line. Cells were incubated with compounds (1 μM each) for 8 h and then labeled with NucView 488 caspase-3 substrate and examined via flow cytometry. In panel A, the total numbers of cells with active caspase-3 are graphed along the y-axis, whereas different treatments are plotted along the x-axis. Each bar represents the standard deviation of the mean of three independent experiments. Analysis using the two-tailed Student’s paired t-test of the experimental compounds against untreated (*) and DMSO (+/+) controls was consistently p < 0.01, respectively. Panels B to F are representative flow cytometric analyses dot plots used to determine the distribution of cells with active caspase-3, where the FL1 and FL2 detectors settings were set on the x-and y-axes, respectively. Cells exposed to 2 μg/ml of camptothecin (CPT) were used as positive controls. Untreated and DMSO solvent treated cells were analyzed concurrently. Approximately, 10,000 events were acquired and analyzed using CXP software (Beckman Coulter).
Apoptosis can also be caused by compounds affecting the mitochondrial membrane potential (MMP). In the absence of any cytotoxic effects, the MMP is polarized. If 3f and 4f act via the mitochondrial pathway, disruption of the MMP can take place leading to depolarization which may be detected using the JC-1 dye. Fig. 6 reveals that both 3f and 4f cause significant depolarization of CEM cells which indicates that these compounds cause apoptosis, at least in part, via the mitochondrial pathway.
In summary, while the ways in which 3f and 4f exert their bioactivity are likely multifactorial, this study has revealed that the compounds cause apoptosis by processes which include phosphatidylserine translocation, caspase-3 activation and disruption of the MMP which is one the biochemical hallmarks of apoptosis.
4. Conclusions
This study has led to the discovery of a novel group of potent antineoplastic agents which demonstrate toxicity to a diverse array of human tumors. The aryl substitution pattern has a profound effect on cytotoxic potencies and in general the 4-fluoro, 4-chloro and 3,4,5-trimethoxy analogs have the lowest IC50 values. The bioevaluations against a wide range of tumors revealed that leu-kemias (Tables 1 and 2, Fig. 3), lymphomas (Table 1, Fig. 3) and colon cancers (Table 2) are the most sensitive to these compounds while breast and prostate cancers are more refractory (Fig. 3). Encouragingly, some of the representative compounds in series 3–5 displayed only marginal toxicity to nonmalignant MCF10A and Hs-27 cells. The cytotoxicity of two representative compounds 3f and 4f towards CEM T-lymphocytes is caused, inter alia, by interfering with the mitochondrial intrinsic pathway. Future studies include in vivo evaluations of the promising lead molecules against different human leukemias, lymphomas and colon cancer xeno-grafts. New analogs and prodrugs based on series 3–5 possessing suitable pharmacokinetic properties [19,20] will be developed. The objective is to obtain one or more preclinical candidates to treat conditions for which there are considerable unmet medical needs.
5. Experimental
5.1. Syntheses of series 3–5
The preparation of the compounds in series 3–5 has been described previously [8,10]. The detailed characterization data for 3a–i, 4a–i and 5 and the 1H and 13C NMR spectra of three representative compounds (one from each series) namely 3e, 4e and 5 are provided in the Supplementary Section.
5.2. Statistical analyses
Linear and semilogarithmic plots were made between the IC50 values of 3a–i in the Molt4/C8, CEM, HeLa and L1210 bioassays which are presented in Table 1 and the σ, π and MR values of the aryl substituents using a commercial software package [21]. The same evaluation was undertaken with the analogs 4a–h.
A description of the derivation of Kendall’s coefficient of concordance is presented in the Supplemental Section.
5.3. Cytotoxic assays
5.3.1. Evaluation of 3–5 against Molt4/C8, CEM, HeLa and L1210 cells
A literature procedure was utilized to evaluate the compounds in series 3–5, melphalan and curcumin against Molt4/C8, CEM, HeLa and L1210 cells [22]. In brief, different concentrations of the compounds were incubated with the appropriate cell line in RPMI 1640 medium at 37C for 72 h (Molt4/C8, CEM T-lymphocytes and HeLa cells) or 48 h (L1210 cells). Cell survival was determined using a Coulter counter. The IC50 values are expressed as the mean ± SD from the concentrationeresponse curves of at least three independent experiments.
5.3.2. Evaluation of 3b,c,e-h,4b,c,e, f and 5 against some lymphoma, leukemic, breast, prostate and nonmalignant cells
A number of compounds were evaluated against some lymphoma, leukemic, breast, prostate cancers and nonmalignant cells using a published procedure [23]. In brief, solutions of the dienones in dimethylsulfoxide were added to different cell lines in RPMI media except in the case of Hs-27 fibroblasts, DMEM was used. After 24 h incubation at 37°C, cytotoxicity was noted by the disruption of the cell membrane using Propidium iodide. The average cytotoxicity of three independent experiments was expressed as the percentage of dead cells. These results are presented in Supplemental Tables 1 and 2.
5.3.3. Evaluation of 3a–d, g 4a, c, e, g i and 5 against a panel of human tumor cells
The data in Table 2 was obtained by a literature procedure [16]. The GI50 figures are IC50 values except for 3d (the IC50 values using four cell lines are greater than 32.4 μ,M) and 5-fluorouracil (for three of the cell lines the IC50 values are in excess of 2.512 mM). All of the data for the colon cancers and leukemic cells are IC50 values. The compounds were evaluated against 59 cell lines except for 3a (60 cell lines) as well as 5 and 5-flurouracil (57 cell lines).
5.4. The induction of phosphatidylserine externalization, caspase activation and depolarization of the mitochondrial membrane potential in CEM cells by 3f and 4f
CEM cells (ATCC, Manassas, VA) were grown in RPMI-1640 medium (HyClone, Logan, UT) supplemented with 10% heat-inactivated fetal bovine serum (HyClone), 100 U/mL penicillin and 100 (μg/mL streptomycin (Lonza, Walkersville, MD). Cells grown exponentially were counted and seeded into 24-well plate formats at a density of 50,000 cells in 500 μL culture media per well. The dienones 3f and 4f were dissolved in dimethylsulfoxide and ali-quots were added to the plates containing the CEM cells in the culture media. All incubation conditions were conducted in a humidified 5% carbon dioxide atmosphere at 37°C. To guarantee high viability, the cells were prepared and cultured as described previously [24]. All treatment and controls were undertaken in triplicate.
5.4.1. Phosphatidylserine externalization assay
The evaluation of the ability of 3f and 4f to induce apoptosis in CEM cells was undertaken as follows. CEM T-lymphocytes were incubated with 1 μM of 3f or 4f for 24 h and then the cells from each well were collected in a pre-chilled cytometric tube, washed and processed essentially as described previously [25]. In brief, cells were stained with a solution of Annexin V-FITC and Propidium iodide (PI) in 100 μLof binding buffer (Beckman Coulter, Miami, FL). After 15 min of incubation on ice in the dark, 400 μL of ice-cold binding buffer was added to cell suspensions and immediately analyzed by flow cytometry (Cytomics FC 500, Beckman Coulter, Miami, FL). These results are presented in Fig. 4.
5.4.2. Caspase-3 assay
The activation of caspase-3 by 3f and 4f was demonstrated using the following methodology. The CEM cells were seeded in 24-well plates vide supra and treated with 1 μM of 3f or 4f for 8 h. Caspase-3 activation was determined using a fluorogenic NucView 488 caspase-3/7 substrate for living cells (Biotium, Hayward, CA) following the Vendor’s instructions. This substrate is permeable to cells with intact plasma membranes and the emission of a green fluorescent signal indicates caspase-3 activation in real-time via flow cytometry (Cytomics FC 500). The results obtained are portrayed in Fig. 5.
5.4.3. Mitochondrial membrane polarization assay
The depolarization of the mitochondrial membrane potential in CEM cells by 3f and 4f was demonstrated by the following methodology. Using a concentration of 1 μM, the dienones were added to CEM cells and after incubation for 8 h, the cells were stained with 2 μM of the JC-1 fluorophore (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetrae-thylbenzimidazoly-carbocyanine iodide, MitoProbe, Life Technologies, Grand Island, NY) following the manufacturer’s protocol. This result is presented in Fig. 6.
Supplementary Material
Acknowledgments
Funding for this study was provided from a number of sources which are acknowledged with gratitude. A CIHR-RPP Saskatchewan grant was awarded to J. R. Dimmock and U. Das. The Belgian Concerted Research Actions (GoA 10/014) provided funds to J. Balzarini which enabled Lizette van Berckelaer to undertake the bioevaluations which are presented in Table 1. The National Cancer Institute, USA, kindly undertook the antineoplastic evaluations of representative compounds and these data are portrayed in Table 2. Funding to R. Aguilera was provided by a NIGMS grant ISC3GM10371. Robles-Escajeda, Santiago-Vazquez and Ortega were supported by NIGMS RISE training grant 5 R25 GM069621-10. Appreciation is also extended to the Border Biomedical Research Center at the University of Texas at El Paso (8G12MD007592) funded by the National Institute on Minority Health and Health Disparities and Research Centers for Minority Institutions which generated the results presented in Figs. 3–6.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2014.03.009.
References
- 1.Das U, Pati H, Sakagami H, Hashimoto K, Kawase M, Balzarini J, De Clercq E, Dimmock JR. Journal of Medicinal Chemistry. 2011;54:3445–3449. doi: 10.1021/jm101595p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dimmock JR, Kumar P, Chen M, Quail JW, Yang J, Allen TM, Kao GY. Pharmazie. 1995;50:449–453. [PubMed] [Google Scholar]
- 3.Okey AB, Harper PA. In: Principles of Medical Pharmacology. seventh. Galant H, Grant DM, Mitchell J, editors. Elsevier; Toronto: 2007. p. 902. [Google Scholar]
- 4.Dimmock JR, Taylor WG. Journal of Pharmaceutical Sciences. 1975;64:241–249. doi: 10.1002/jps.2600640210. [DOI] [PubMed] [Google Scholar]
- 5.Chen G, Waxman DJ. Biochemical Pharmacology. 1994;47:1079–1087. doi: 10.1016/0006-2952(94)90420-0. [DOI] [PubMed] [Google Scholar]
- 6.Tsutsui K, Komuro C, Ono K, Nishidia T, Shibamoto Y, Takahashi M, Abe M. International Journal of Radiation Oncology Biology Physics. 1996;12:1183–1186. doi: 10.1016/0360-3016(86)90254-3. [DOI] [PubMed] [Google Scholar]
- 7.Howard EW, Lee DT, Chiu YT, Chua CW, Wang X, Wong UC. International Journal of Cancer. 2008;122:1941–1948. doi: 10.1002/ijc.23355. [DOI] [PubMed] [Google Scholar]
- 8.Das S, Das U, Varela-Ramírez A, Lema C, Aguilera RJ, Balzarini J, De Clercq E, Dimmock SG, Gorecki DKJ, Dimmock JR. ChemMedChem. 2011;6:1892–1899. doi: 10.1002/cmdc.201100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das S, Das U, Sakagami H, Umemura N, Iwamoto S, Matsuta T, Kawase M, Molnár J, Serly J, Gorecki DKJ, Dimmock JR. European Journal of Medicinal Chemistry. 2012;51:193–199. doi: 10.1016/j.ejmech.2012.02.042. [DOI] [PubMed] [Google Scholar]
- 10.Das S, Das U, Michel D, Gorecki DKJ, Dimmock JR. European Journal of Medicinal Chemistry. 2013;64:321–328. doi: 10.1016/j.ejmech.2013.03.055. [DOI] [PubMed] [Google Scholar]
- 11.Hansch C, Leo AJ. Substituent Constants for Correlation Analysis in Chemistry and Biology. John Wiley and Sons; New York: 1979. p. 49. [Google Scholar]
- 12.Rydzewski RM. Real World Drug Discovery. Elsevier; Oxford: 2008. pp. 289–293. [Google Scholar]
- 13.Suffness M, Douros T. In: Methods of Cancer Research, Part A, vol XVI. De Vita VT Jr, Busch H, editors. Academic Press; New York: 1979. p. 84. [Google Scholar]
- 14.Sheskin D. Handbook of Parametric and Nonparametric Statistical Procedures. Chapman and Hall; London: 2004. pp. 1093–1107. [Google Scholar]
- 15.Goel A, Kunnumakkara AB, Aggarwal BB. Biochemical Pharmacology. 2008;75:787–809. doi: 10.1016/j.bcp.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 16.Boyd MR, Paull KD. Drug Development Research. 1995;34:91–109. [Google Scholar]
- 17.Los M. European Journal of Pharmacology. 2009;625:1–5. doi: 10.1016/j.ejphar.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 18.Rahman MA. International Journal of Pharmaceutical Sciences and Research. 2013;3:1946–1954. [Google Scholar]
- 19.Kerns EH, Di L. Drug-like Properties: Concepts, Structure Design and Methods: from ADME to Toxicity Optimization. Elsevier; Burlington, MA: 2008. p. 45. [Google Scholar]
- 20.Artursson P, Palm K, Luthman K. Advanced Drug Delivery Reviews. 2001;46:27–43. doi: 10.1016/s0169-409x(00)00128-9. [DOI] [PubMed] [Google Scholar]
- 21.Statistical Package for Social Sciences SPSS for Windows, Release 14.0.0. SPSS Inc; Chicago: 2005. [Google Scholar]
- 22.Baraldi PG, del M, Nunez C, Tabrizi MA, De Clercq E, Balzarini J, Bermejo J, Estévez F, Romagnoli K. Journal of Medicinal Chemistry. 2004;47:2877–2886. doi: 10.1021/jm031104y. [DOI] [PubMed] [Google Scholar]
- 23.Elie BT, Levine C, Ubarretxena-Be′landia I, Varela-Ramírez A, Aguilera RJ, Ovalle R, Contel M. European Journal of Inorganic Chemistry. 2009:3421–3430. doi: 10.1002/ejic.200900279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lema C, Varela-Ramírez A, Aguilera RJ. Current Cellular Biochemistry. 2011;1:1–14. [PMC free article] [PubMed] [Google Scholar]
- 25.Varela-Ramírez A, Costanzo M, Carrasco YP, Pannell KH, Aguilera RJ. Cell Biology and Toxicology. 2011;27:159–168. doi: 10.1007/s10565-010-9178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.