

Abstract
Knowing how the protein environment modulates ligand pathways and redox centers in the respiratory heme-copper oxidases is fundamental for understanding the relationship between the structure and function of these enzymes. In this study, we investigated the reactions of O2 and NO with the fully reduced G232V mutant of ba3 cytochrome c oxidase from Thermus thermophilus (Tt ba3) in which a conserved glycine residue in the O2 channel of the enzyme was replaced with a bulkier valine residue. Previous studies of the homologous mutant of Rhodobacter sphaeroides aa3 cytochrome c oxidase suggested that the valine completely blocked the access of O2 to the active site [Salomonsson, L., et al. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 11617–11621]. Using photolabile O2 and NO carriers, we find by using time-resolved optical absorption spectroscopy that the rates of O2 and NO binding are not significantly affected in the Tt ba3 G232V mutant. Classical molecular dynamics simulations of diffusion of O2 to the active site in the wild-type enzyme and G232V mutant show that the insertion of the larger valine residue in place of the glycine appears to open up other O2 and NO exit/entrance pathways that allow these ligands unhindered access to the active site, thus compensating for the larger valine residue.
Heme-copper oxidases play a key role in mitochondrial and bacterial respiration, catalyzing the reduction of dioxygen to water and redox-coupled proton translocation.1−3 The resulting electrochemical gradient is used by ATP synthase to make ATP.4 The heme-copper oxidases are classified into the A, B, and C subfamilies.5,6 Ligand channels have been proposed for oxidases from the different subfamilies, including for the bovine enzyme,7Rhodobacter sphaeroides (Rs) aa3,8Paracoccus denitrificans (Pd) aa3,9 and Escherichia coli bo3(10) from the A family, Thermus thermophilus (Tt) ba3 from the B family,11 and Pseudomonas stutzeri cbb3 from the C family.12 A hydrophobic channel originating at a V-shaped cleft formed by subunit III was identified in the crystal structure of Pd aa3.9 A channel starting in subunit I and converging with the channel starting in subunit III was observed in Rs aa3 by trapping Xe atoms in the crystal structure.8
More recently, additional details of the ligand channel in Tt ba3 have been obtained.13,14 The location of Xe atoms in the Tt ba3 crystal structure shows a bifurcated Y-shaped ligand channel 18–20 Å in length, starting from two points, the first between helices II and III of subunit I and the second between helices IV and V of subunit I on the protein exterior.13,14 On the basis of these observations, cavities in the bovine, Rs, and Pd aa3 oxidases were assessed and putative O2 pathways suggested.13 In the bovine, Rs, and Pd aa3 oxidases, there is a narrowing of the ligand channel ∼9 Å from the catalytic site, caused by conserved tryptophan and phenylalanine residues;8,15,16 this narrowing is not present in Tt ba3 in which smaller residues, tyrosine (Y133) and threonine (T231), occupy the positions of the tryptophan and phenylalanine, respectively.11,13 Recent experiments in our laboratory have shown that binding of O2 and NO to the active site in the Y133W and Y133W/T231F mutants of Tt ba3 is ∼5 times slower than in the wild-type enzyme.17 This suggests that the significantly slower ligand binding in the bovine enzyme (1 × 108 M–1 s–1) compared to that in Tt ba3 (1 × 109 M–1 s–1)18,19 is in part due to the tryptophan constriction residue in the ligand channel of the bovine aa3 enzyme (W126) impeding the access of O2 and NO to the active site.17 A hydrophobic pocket in the ligand channel of the bovine enzyme may further impede the access of O2 and NO to the active site.17
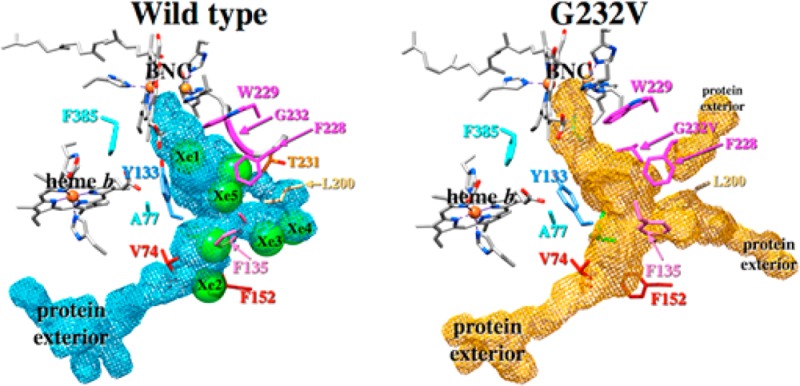
A comparison of amino acid sequences and crystal structures of the heme-copper oxidases has also identified strictly conserved residues in the ligand channels of these enzymes, including that of Tt ba3, which may play important roles in modulating access of ligands to the binuclear active site. One of the highly conserved amino acid residues in the ligand channel is a glycine residue, G232 in Tt ba3.8,10,11,20,21 The crystal structure of Tt ba3 shows that G232 is located between two large residues, tryptophan (W229) and phenylalanine (F228) (Figure 1), which have been suggested to form a hydrophobic and hydrophilic boundary around the binuclear center.11,13 The G232 residue is located behind the Xe1 site, identified in Xe-pressurized crystallographic studies of Tt ba3,13,14 providing a small opening proposed to be the site of entry of ligands into the active site. Mutation of the homologous glycine residue in Rs aa3 oxidase (G283) to a larger valine residue was reported to give rise to significantly faster CO recombination [major phase (50%), 40 μs, compared to 10 ms in the wild-type enzyme], suggesting that the photodissociated CO did not leave the catalytic site cavity of the mutant enzyme.16 The oxidation of the fully reduced G283V mutant upon exposure to dioxygen was found to be significantly slower (minutes) compared to that of the wild-type enzyme (1 ms), which was interpreted as the valine side chain completely blocking access of O2 to the catalytic site in this enzyme.16 Because of the critical involvement of this conserved residue in ligand access in Rs aa3, it is important to know if G232 plays a similar role in Tt ba3, particularly considering that the G232 residue is adjacent to T231 Tt ba3, which in the aa3 oxidases is replaced by the phenylalanine “constriction point” residue.
Figure 1.
Residues lining the ligand channel of the wild-type ba3 cytochrome oxidase (colored yellow, PDB entry 3S8F) and the G232V mutant (blue). The purple spheres are water 618 and the green spheres water 604 as described in the text.
In this study, the CO photolysis and recombination dynamics and the reactions of O2 and NO with the G232V Tt ba3 mutant were investigated using photolabile NO and O2 carriers. The replacement of glycine with the larger valine does not impede ligand access because the resulting structural change appears to open other O2 cavities in the ligand channel.
Methods
Protein Expression, Purification, and Steady-State Activity Measurements
Recombinant wild-type Tt ba3 and the G232V mutant were expressed in Tt HB8 and isolated and purified as previously described.22 The proteins were concentrated to 20 mg/mL in a buffer containing 20 mM Tris (pH 7.6) and 0.05% dodecyl β-d-maltopyranoside (DM) (Affimetrix Inc.). All the samples were stored at 4 °C for future use. The steady-state activities were measured as previously described.17
Crystallization, X-ray Data Collection, and Structure Determination
Crystallization was performed at room temperature in 24-well hanging drop plates.17 The crystals were harvested and immediately flash-frozen in liquid nitrogen. All diffraction data were collected at the Stanford Synchrotron Radiation Lightsource. iMosflm and SCALA were used to integrate, scale, and merge the data sets.23,24 The G232V structure was determined by molecular replacement with Phaser, using the wild-type Tt ba3 structure (PDB entry 3S8F) as the initial search model. The output model was manually adjusted in Coot and refined with Refmac5.25−27
Sample Preparation
The Tt ba3 recombinant wild-type enzyme and G232 mutant were dissolved in 0.1 M HEPES buffer (pH 7.5) containing 0.1% DM. The potassium pentachloronitrosyl-ruthenate(II) photolabile NO complex was obtained from Alfa Aesar, and the (μ-peroxo)(μ-hydroxo)bis[bis(bipyridyl)cobalt(III)] nitrate photolabile O2 complex was synthesized as previously described.28
The fully reduced Tt ba3 was prepared by first taking the oxidized enzyme solution through several alternating cycles of vacuum and nitrogen. For the O2 experiments, glucose, glucose oxidase, and catalase (final concentrations of 3.5, 0.25, and 0.01 mg/mL, respectively) were added to the deoxygenated enzyme solution to remove any residual O2 in the solution. The enzyme was reduced using ascorbic acid and a ruthenium hexamine mediator (final concentrations of 2 mM and 15–25 μM, respectively). For the NO binding studies, a final concentration of phenazine methosulfate (PMS) of 1 μM was used in place of the ruthenium hexamine to prevent a possible reaction with the photolabile NO complex. The enzyme concentration was determined from the spectrum of the fully reduced enzyme using a Δε560–590 of 26 mM–1 cm–1.22 The fully reduced CO-bound enzyme for the CO flash-photolysis experiments was prepared by exposing the fully reduced enzyme to CO for ∼30 min with occasional agitation.
Time-Resolved Optical Absorption (TROA) Measurements
The O2 reaction and NO binding of the fully reduced Tt ba3 G232V mutant were investigated by mixing the mutant with the O2 or NO carrier and photolyzing the respective complex with a 355 nm laser pulse.19 The concentrations of the photoproduced O2 and NO were determined as previously described.18,19
The TROA difference spectra were recorded using a CCD camera and analyzed using singular-value decomposition (SVD) and global exponential fitting, which provided the apparent lifetimes and the associated b-spectra.29,30 The spectra of the intermediates in the O2 reaction of the Tt ba3 G232V mutant were determined on the basis of a proposed kinetic scheme. The plausibility of the scheme was tested by comparing the experimental and model spectra of the proposed intermediates.
Classical Molecular Dynamics Simulations
Classical molecular dynamics simulations of diffusion of O2 to the active site of the fully reduced wild-type ba3 and the G232V mutant were performed using the CHARMM27 force field31 as implemented in GROMACS version 4.5.5.32 The system comprises the enzyme’s catalytic subunit I, two leaflets of 64 dimyristoylphosphatidylcholine (DMPC) lipids each, TIP3P water, and chloride ions to achieve charge neutrality. Initial coordinates for the wild-type system were taken from production simulations of Xe diffusion in fully reduced ba3 oxidase as reported in our recent study,17 where the Xe atom was located near the Xe1 crystallographic binding site.13 The Xe atom was replaced with a dioxygen molecule, and the system was energy minimized using a combination of the conjugate gradient and low-memory BFGS algorithms. Harmonic position restraints were subsequently placed on the dioxygen ligand, and the system was heated to 300 K and then equilibrated at 300 K for 700 ps. The position restraints were then removed from the dioxygen ligand, and twenty 1 ns production simulations were performed.17 Coordinates for the fully reduced G232V mutant were obtained from the production simulations of the wild-type enzyme after replacing the G232 side chain with that of valine in the conformation observed in the crystal structure presented here. The mutant system was then energy minimized, heated, and equilibrated as described above, and fourteen 2 ns production simulations were performed. Additional simulation parameters, including the integration time step, neighbor list update, and nonbonded interactions, can be found in the Supporting Information.
Results
Structure and Activity of the G232V Mutant
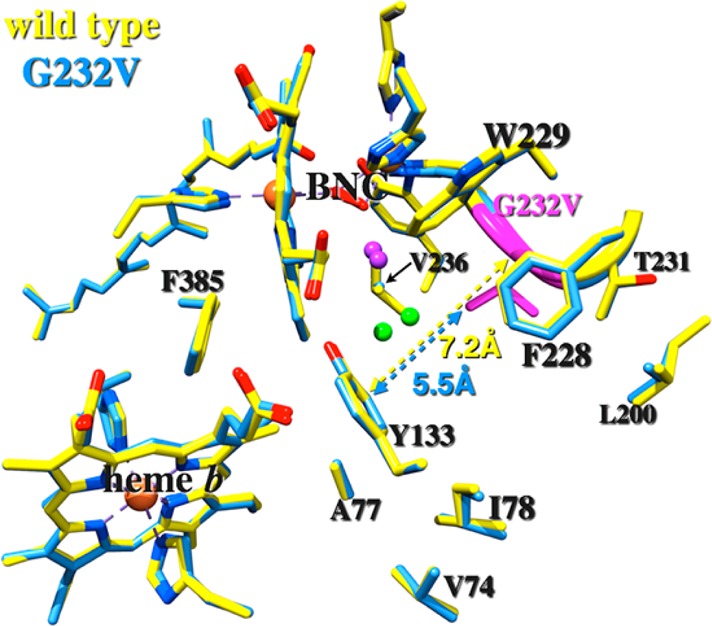
Overall, the crystal structures of wild-type ba3 and the G232V mutant are similar, with an rmsd of ∼0.25 Å (Figure 1). The major difference at residue 232 is that the N–Cα–C plane rotates ∼26° toward residue I235, resulting in a 0.96 Å upward (i.e., toward the P-side of the membrane) displacement of the Cα atom in the G232V mutant compared to that in the wild-type enzyme. The increase in the rise of helix VI as a result of the glycine-to-valine mutation also leads to changes at the CuB site, which will be addressed in more detail in the Discussion. Furthermore, the side chain of V232 in the mutant protrudes ∼2.5 Å toward the oxygen channel such that the CG1 and CG2 atoms enter the channel. This expansion of the side chain into the channel also affects the two water molecules that are bridged to the active site in the native structure (waters 604 and 618 in PDB entry 3S8F), forcing one of them (water 604 in PDB entry 3S8F) to move 1.25 Å away (Figure 1, green spheres). The crystallographic statistics for the refined structure of the G232V mutant are listed in Table S1 of the Supporting Information. The steady-state activity of G232V was the same as that of the wild-type enzyme.
CO Flash-Photolysis and Recombination of the Fully Reduced G232V Tt ba3 Mutant
Time-resolved optical absorption difference spectra (postphotolysis minus prephotolysis) were recorded upon photolysis of CO from heme a3 of the fully reduced CO-bound G232V mutant with a 532 nm laser pulse (Figure S1A of the Supporting Information). The spectra were analyzed by SVD and global exponential fitting, which resolved four apparent lifetimes, 94 μs, 630 μs, 13 ms, and 274 ms. The corresponding b-spectra, which represent the spectral change associated with the individual apparent lifetimes, are presented in the Supporting Information (Figure S1B). The apparent lifetime of 274 ms associated with the major process (∼90% of the overall spectral change) is attributed to CO rebinding to heme a32+ based on the respective b-spectrum (Figure S1B of the Supporting Information, cyan). This lifetime is comparable to the CO rebinding rate of ∼260 ms in the wild-type enzyme.
Reaction of Fully Reduced G232V Tt ba3 with NO and O2
Previous studies in our laboratory have shown that the rate of binding of NO to the a32+ reaction center in wild-type Tt ba3 in the absence of CO is comparable to that of binding of O2 (∼1 × 109 M–1 s–1), supporting the general assumption that the NO ligand entry pathway from the protein exterior to the binuclear center is the same as that of O2.18,19 To determine whether the mutation of the small glycine to a larger valine residue affects the access of ligands to the binuclear active site, we investigated binding of NO to fully reduced G232V ba3 using a photolabile NO complex. TROA difference spectra (postphotolysis minus prephotolysis) (Figure 2A) were subjected to SVD and global exponential fitting, and a single apparent lifetime of 9.5 μs (100 μM NO) was observed for the G232V mutant. The corresponding b-spectrum (Figure 2B, blue) confirms that this process is caused by the binding of NO to heme a32+. The 9.5 μs lifetime yields a second-order rate constant of 1.1 × 109 M–1 s–1 (100 μM NO), which is the same as that observed for wild-type recombinant ba3 (∼1 × 109 M–1 s–1).18 Thus, the glycine-to-valine mutation does not impede the access of NO to the active site.
Figure 2.
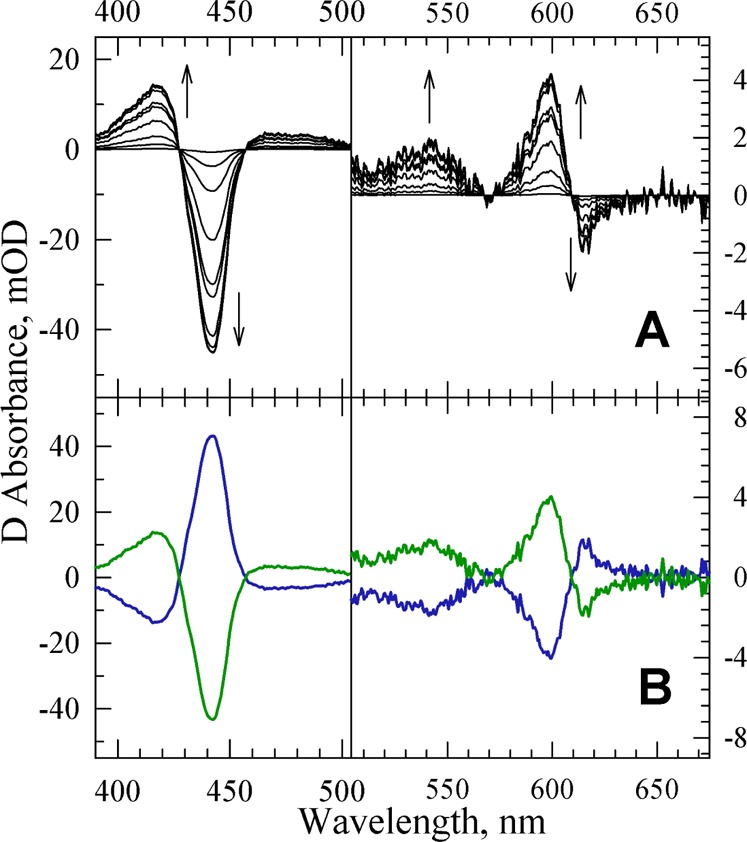
(A) SVD-filtered TROA difference spectra (postphotolysis minus prephotolysis) recorded during the reaction of the fully reduced G232V Tt ba3 mutant with the photoproduced NO. The spectra were recorded at 10 delay times, equally spaced on a logarithmic time scale between 500 ns and 500 μs. The spectra are those obtained following the subtraction of the spectral contribution from the photolabile NO complex, determined in a separate experiment. The arrows represent the direction of the absorption change with time. Conditions: 0.1 M HEPES (pH 7.5) with 0.1% DM; optical path length of 0.5 cm. The effective enzyme concentration was 1.0 μM. The NO concentration was 100 μM. (B) b-spectra resulting from a single-exponential fit of the TROA difference spectra recorded during the reaction of the photoproduced NO with the fully reduced G232V Tt ba3 mutant. The b1 spectrum (blue, apparent lifetime of 9.5 μs) and the non-zero time-dependent b0 spectrum (green), which represents the difference spectrum extrapolated to infinite time.
To investigate whether O2 binding and subsequent electron transfer steps are affected by the G232V mutation, we monitored the reaction of the fully reduced G232V Tt ba3 mutant with photoproduced O2. The TROA difference spectra are shown in Figure 3. SVD-based global exponential fitting resolved four apparent lifetimes of 8 μs, 12 μs, 114 μs, and 4 ms with the corresponding b-spectra (Figure S2 of the Supporting Information).
Figure 3.

SVD-filtered TROA difference spectra (postphotolysis minus prephotolysis) recorded during the reaction of the fully reduced G232V Tt ba3 mutant with photoproduced O2. The spectra were recorded at 17 delay times, equally spaced on a logarithmic time scale between 200 ns and 50 ms. The spectra are those obtained following the subtraction of the spectral contribution of the photolabile O2 complex, determined in a separate experiment. The arrows represent the direction of the absorption change with time. Conditions: 0.1 M HEPES (pH 7.5) with 0.1% DM; optical path length of 0.5 cm. The effective enzyme concentration was 2.6 μM. The O2 concentration was 74 μM.
The spectra of the intermediates generated in the reaction of O2 with the fully reduced G232V mutant were determined on the basis of the slow–fast sequential mechanism that we recently applied to analyze analogous data for the wild-type Tt ba3 enzyme.19 Our previous TROA studies of the reaction of O2 with fully reduced wild-type Tt ba3 showed that the traditional fast–slow mechanism, in which decreasing values of the experimental apparent rate constants were assigned to microscopic rate constants of consecutive steps, did not provide a good correspondence between the experimental spectrum of compound A (AR) and the model spectrum of compound A of the bovine enzyme.19 However, a good correspondence was obtained between the experimental and model spectra using a slow–fast mechanism in which O2 binding was followed by a faster step generating PI.19 Similarly, a good agreement was observed between the experimental intermediate spectra of the G232V mutant (Figure 4A) and those of the recombinant wild-type enzyme (Figure 4B) based on the slow–fast mechanism; minor changes are observed in the spectra between the G232V mutant and the wild-type enzyme. In this mechanism (Scheme 1), the binding of O2 to heme a3 in G232V (12 μs) is followed by a faster cleavage of the O–O bond (8 μs), which generates the PI intermediate, with the concomitant oxidation of heme b.
Figure 4.
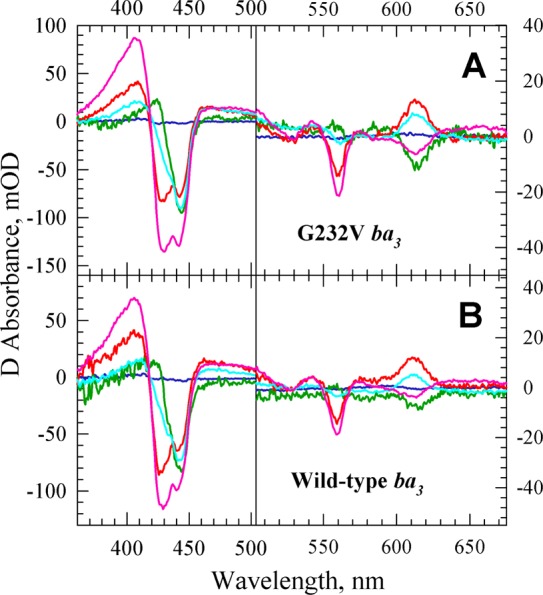
Experimental intermediate spectra for the reaction of the photoproduced O2 with the fully reduced G232V mutant (A) and wild-type ba3 (B). The intermediate spectra are referenced against the reduced enzyme and were extracted on the basis of the slow–fast mechanism in Scheme 1. The intermediate spectra determined are represented with the following colors: blue for R, green for A, red for PI, cyan for PII, and magenta for O.
Scheme 1. Slow–Fast Sequential Scheme for O2 Reduction in the Fully Reduced G232V Tt ba3 Mutant.
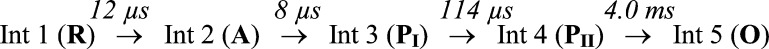
The 12 μs (74 μM O2) lifetime assigned to the O2 binding step in the G232V mutant corresponds to a second-order rate constant of ∼1 × 109 M–1 s–1. This rate constant is the same as that observed for the reaction of reduced recombinant wild-type ba3 with O2 (17 μs at 48 μM O2; 1.2 × 109 M–1 s–1) (this study) and that previously observed for the nonrecombinant wild-type enzyme.19 It is also consistent with the observed rate of NO binding reported above. Furthermore, the rates of the other steps in the reaction mechanism are similar between the ba3 G232V mutant and the recombinant wild-type enzyme (5 μs, 71 μs, and 2.2 ms) or the nonrecombinant wild-type enzyme.19
Classical Simulations of Diffusion of O2 to the Active Site in Wild-Type Tt ba3 and the G232V Mutant
Our TROA measurements show that the rate of O2 or NO binding to the a3 reaction center in Tt ba3 is unaffected upon replacement of the conserved glycine (G232) in ba3 with valine. In contrast, a previous study concluded that the mutation of the conserved glycine to valine in Rs aa3 significantly decreased the level of access of O2 to the catalytic site; this conclusion was based on the much slower oxidation (from ∼1 ms to minutes) of the reduced mutant upon its exposure to air.12 To explore in more detail why NO and O2 ligand access in Tt ba3 is not affected upon replacement of glycine with the bulkier valine residue, we performed a total of 20 and 28 ns classical molecular dynamics simulations of O2 diffusing through subunit I of the wild-type enzyme and the G232V mutant, respectively.
The diffusion of O2 through the ligand channel was monitored on the basis of the distances of O2 from CuB, Y133, and I78 as previously described.17 Figure 5 shows a comparison of the wild-type and G232V mutant time series for these three distances when O2 was initially placed at the Xe1 (A and B), Xe5 (C and D), and Xe2 (E and F) sites identified in the X-ray xenon-pressurized crystallographic study.13 The time series demonstrate that the ligand channel of the G232V mutant is “less sticky” than that of the wild-type enzyme as evidenced by the noisier traces of the mutant enzyme, and the increased frequency of transitions between different regions of the G232V ligand channel when compared to that of the wild-type enzyme (Figure 5). Furthermore, there is a well-defined O2 docking site within the wild-type ligand channel that is characterized by O2–CuB distances of 13—15 Å (e.g., Figure 5E, 200—400 ps); this site is not observed in the time series of the G232V mutant. Moreover, the O2 ligand comes within van der Waals contact of the CuB atom (<5 Å) for tens of picoseconds in the mutant (Figure 5F, 340–360 and 400–445 ps) but not in the wild-type enzyme, suggesting conformational differences in the environment close to the CuB site between the wild-type enzyme and the G232V mutant. This will be discussed in more detail below.
Figure 5.

Representative time series of the O2–CuB distance (red trace), O2–Y133 distance (blue), and O2–I78 distance (green trace) from classical molecular dynamics simulations of O2 diffusion in the wild type (A, C, and E) and the ba3 G232V mutant (B, D, and F). Simulations were initiated with O2 placed at the Xe1 crystallographic site (A and B), the Xe5 binding site (C and D), or the Xe2 crystallographic site (E and F).13
Discussion
A conserved glycine (G232 in Tt ba3) is highly conserved in the O2 channel of the heme-copper oxidases and has been suggested to play a significant role in facilitating access of ligands to the binuclear active site.16 The glycine-to-valine mutation in Rs aa3 (G283V) was previously reported to have O2 access to the catalytic site significantly slower than in the wild-type enzyme.16 Reduced O2 access was concluded from the slow oxidation rate of the reduced enzyme after its exposure to air; however, the O2 binding and reduction reaction was not investigated directly by fast detection techniques, such as the flow-flash method. In the “flow-flash” study presented here, NO binding and O2 binding and reduction in the G232V Tt ba3 mutant appear to be unaffected by the replacement of the small glycine residue with the significantly larger valine residue. One possible explanation is that the valine side chain exhibits conformational freedom, with the side chain rotating out of the ligand pathway, thus not interfering with ligand access. However, this appears to be unlikely because the G232 residue is “sandwiched” between two large amino acids, W229 and F228, and therefore may not have room to rotate. The lack of conformational freedom of the G232V side chain is confirmed by the classical molecular dynamics simulations, in which the G232V side chain adopts a conformation different from that of the crystal structure for only 13 ps of 27.5 ns of total simulation time. Furthermore, the root-mean-square fluctuations (rmsfs) in atomic positions of the valine side chain CG1 and CG2 atoms (0.72 and 0.73 Å, respectively) are smaller than the average fluctuations for the whole protein (0.97 Å).
In agreement with previous investigations into the protein flexibility of subunit I in Rs aa3,33 our classical simulations find that helices I–VI, X, and XI form a stable, relatively rigid structure in wild-type Tt ba3, as indicated by Cα rmsfs that are ∼1 standard deviation smaller than the average. Helices VII–IX and XII in the wild-type enzyme are slightly more mobile (less rigid) as indicated by rmsfs in atomic positions that are 0–0.5 standard deviation smaller than the average Cα fluctuations. The G232V mutation increases the flexibility of helices VIII–X in the mutant. The rmsfs are presented in Figure 6 where they have been converted to b-factors for the G232V mutant (left) and wild-type ba3 (right). The increase in warm colors (orange and red) in G232V reflects the increased flexibility of this mutant, particularly of helix VIII (circled by an ellipsoid), compared to that of the less flexible wild type, which is depicted in predominately cool colors (green and blue). Furthermore, the classical molecular dynamics simulations of the G232V mutant show that the glycine-to-valine mutation distorts helix VI, causing the cross-linked H233–Y237 CuB ligand to move relative to the atom positions of the wild-type enzyme. This movement severs the H-bond between the phenolic OH of Y237 and the hydroxyethyl moiety of heme a3 and slightly increases the separation between CuB and the heme a3 in the G232V mutant (by 0.2 Å) compared to the separation in the wild type (Figure 7). These changes may explain the small differences in the observed electron transfer rates between the wild-type enzyme and the mutant during O2 reduction. Furthermore, they allow the O2 ligand to approach the binuclear center significantly more closely (∼4 Å from CuB) in the G232V mutant than in the wild-type enzyme (∼5.5 Å from CuB). This could be one reason why the larger residue has no effect on ligand binding in the G232V mutant compared to that in the wild-type enzyme. The simulations also demonstrate that O2 moves much more frequently between different sites within the ligand channel in the G232V mutant than in the wild-type enzyme (Figure 5).
Figure 6.

Main chain flexibility of the G232V mutant (left) and wild-type ba3 (right) presented as b-factors obtained by converting rmsfs in atomic positions from the classical molecular dynamics simulations. Helix VIII is circled by the ellipsoid. CuB is shown as the black sphere, heme a3 is cyan, and heme a is pink. The cyan sphere is Fea3.
Figure 7.
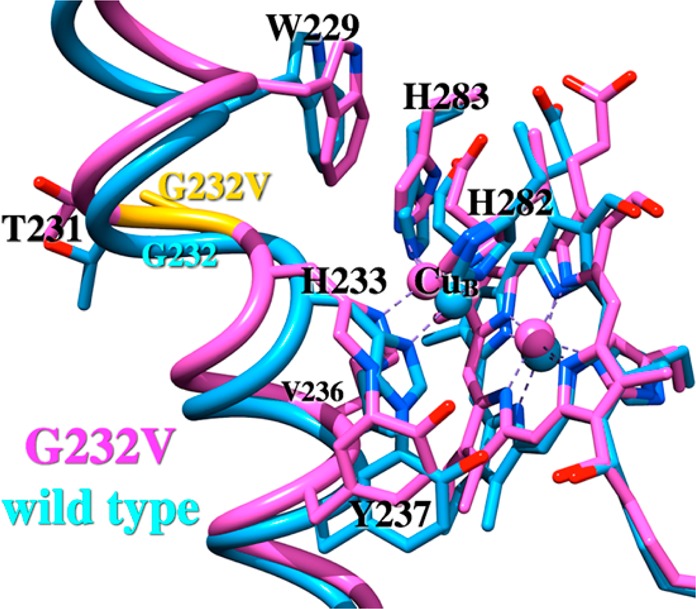
Structural changes to helix VI and CuB due to the G232V mutation. The wild-type enzyme is colored cyan (PDB entry 3S8F), and the G232V mutant enzyme is colored magenta. Mutation G232V is colored yellow.
The ligand channels within subunit I of the wild-type ba3 and the G232V mutant were obtained from the simulation data using O2 occupation counts on a grid of spatial points surrounding the ligand using UCSF Chimera.34 The ligand channels so obtained are presented in Figure 8A for wild-type ba3 and in Figure 8B for the G232V mutant. For comparison, the seven crystallographic Xe binding sites of ref (13) are shown in Figure 8 after rmsd fitting the Cα atoms of the Xe-pressurized wild-type crystal structure to the simulated wild-type structure. The ligand channels derived from the simulations have good overlap with all seven crystallographic Xe sites, which validates the use of occupation data to locate ligand cavities within proteins. While the overall size and shape of the ligand channels in the wild type and G232V mutant are similar (the cavity volumes are 1061 and 1005 Å3 for the wild type and mutant, respectively), the mutation does alter the ligand channel in important ways. Figure 8B clearly shows that in the G232V mutant, the valine side chain protrudes into the ligand channel at the location of the Xe6 binding site (behind the magenta F228), narrowing the mutant ligand channel slightly at this location to 4.2 Å from 6.6 Å in the wild-type enzyme; these distances are obtained by measuring the diameter of the cavity generated from the O2 occupation data. Furthermore, in the G232V mutant, the O2 ligand channel shifts “up” (i.e., toward the P-side of the membrane) near the point mutation compared to the cavity position in the wild-type enzyme. Additional structural changes observed in the ligand channel of the G232V mutant are the ligand exit pathways between helices IV and V (Figure 8B, bottom right “protein exterior”) and helices V and VI (top right “protein exterior”). These ligand exit (and presumably entrance) sites are not used by the O2 ligand in simulations of the wild-type enzyme. Hence, the theoretical calculations show that the insertion of the larger valine residue in place of the glycine opens other “cavities” that allow O2 and NO unhindered access to the active site, thus compensating for the larger valine residue.
Figure 8.
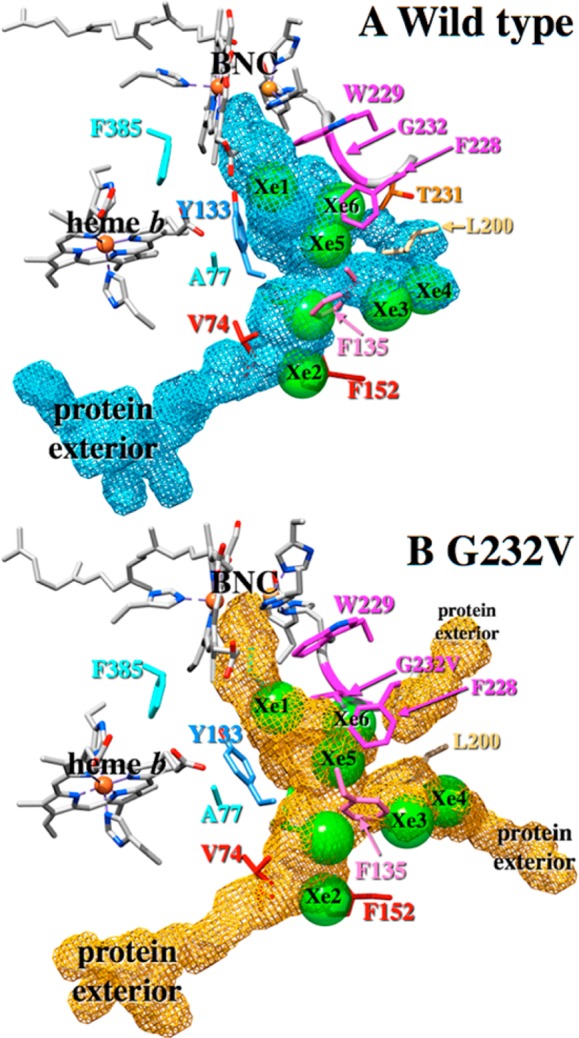
Ligand cavities in subunit I of wild-type ba3 (A) and the G232V mutant (B). In panels A and B, the seven crystallographic Xe binding sites are shown as green spheres (PDB entry 3BVD).13 The cavities were obtained from the classical molecular dynamics simulations using O2 occupation counts on a grid of points surrounding the ligand using UCSF Chimera.33
A model of the putative ligand channel in the G283V mutant of Rs aa3 cytochrome oxidase is presented in Figure 9. This model was constructed by replacing the side chain of the conserved glycine (G283) in the fully reduced wild-type Rs crystal structure (PDB entry 3FYE)35 with that of valine in the conformation found in the crystal structure of the G232V ba3 enzyme presented here. The fully reduced Rs aa3 structure was used to be consistent with the simulations, which are based on the fully reduced ba3 structure. The wild-type fully reduced Rs aa3 enzyme has some structural similarities with the ba3 G232V mutant, including the lack of a direct H-bond between the phenolic OH of the cross-linked histidine-tyrosine and the hydroxyethyl moiety of heme a3 as discussed above. However, in the crystal structure of fully reduced Rs aa3, there is a bridging water molecule that forms H-bonds with both the phenolic OH and the hydroxyethyl moiety of the high-spin heme; this bridging water is not present in the G232V mutant. Furthermore, the CuB–Fea3 separation is larger in the fully reduced Rs aa3 structure than in the fully reduced G232V ba3 mutant. The distance between the CG1 carbon atom of the valine side chain and the CH2 carbon atom of the tryptophan 172 side chain in the Rs model G283V mutant is 3.38 Å. This is considerably shorter than the distance of 5.5 Å between the CG1 atom of the valine 232 side chain and the CE2 carbon atom of the homologous Y133 side chain in the G232V ba3 enzyme (cf. Figure 1).
Figure 9.
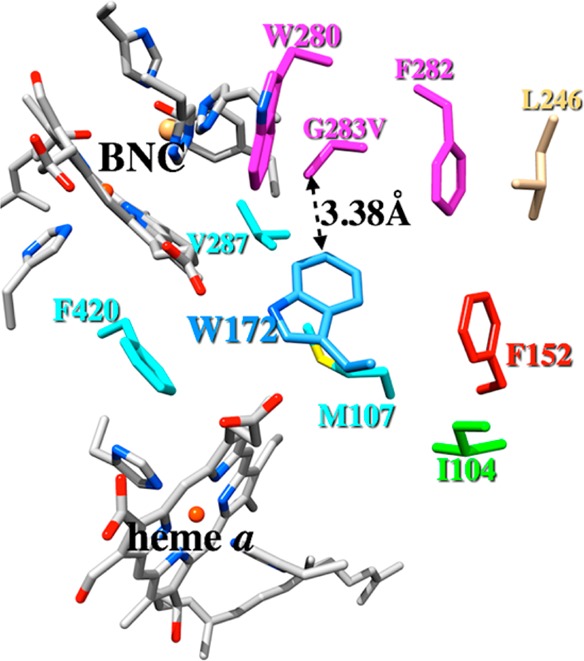
Model of the R. sphaeroides cytochrome oxidase G283V mutant showing residues that line the postulated ligand channel. The model was constructed by replacing the native G283 residue in the fully reduced wild-type Rs aa3 (PDB entry 3FYE) with valine in the conformation observed in the G232V ba3 crystal structure presented here.
As noted previously, the major structural difference between the O2 pathway in Tt ba3 and the bovine, Rs, and Pd aa3 oxidases is the narrowing of the ligand channel in the aa3 oxidases caused by conserved tryptophan and phenylalanine residues;8,15,16 in Tt ba3, these residues are replaced with tyrosine (Y133) and threonine (T231), respectively.11,13 Specifically, the distances between the Cα atom of the glycine and the CH2 carbon atom of the tryptophan 172 side chain in Rs aa3 is 5.27 Å, while the distance between the Cα atom of the glycine and the CE2 carbon atom of the homologous Y133 side chain in the ba3 enzyme is 8.31 Å. Therefore, the ligand channel is also narrower in the wild-type Rs aa3 enzyme than in wild-type ba3 at the location of the glycine. This is also true of the bovine enzyme. The very narrow ligand channel of G283V Rs aa3 is consistent with previous experiments that showed that the rate of O2 reduction was slowed from ∼1 ms in the wild-type enzyme to minutes in the G283V mutant,16 while the wider, more open ligand channel of the G232V Tt ba3 mutant agrees with our results indicating that this mutation has no effect on ligand access to the binuclear center. Under microaerobic conditions, such as those present in hypersaline microbial mats or at oxic–anoxic interfaces inhabited by the T. thermophilus bacterium,36 O2 binding may indeed be rate-limiting. Therefore, a more open O2 channel in Tt ba3 may reflect an evolutionary adaptation in Tt ba3 that increases the rate of O2 diffusion to the active site at low O2 concentrations to maintain physiologically relevant reaction rates.17 This is consistent with phylogenetic analysis that has shown that of the three heme-copper oxidase superfamilies, the A family is the most ancient, with the B and C families evolving later.36
Conclusions
The G232 residue in Tt ba3 has been postulated to create a small passageway for ligands to pass through en route to the active site, with the glycine residue being sandwiched between two larger amino acid residues (W229 and F228 in Tt ba3). Considering the high degree of conservation of the glycine in the ligand channels of the heme-copper oxidases, this residue would be predicted to play a crucial role in ligand access to the binuclear active site and/or in O2 reduction. However, the replacement of G232 in Tt ba3 with valine does not appear to affect the rate of O2 or NO binding, although the crystal structure shows that the two arms of the valine side chain protrude ∼2.5 Å into the ligand channel. Importantly, there is a distortion in helix VI, resulting in an increased separation between CuB+ and heme a3. Furthermore, the histidine–tyrosine cross-link moves significantly, resulting in an increase of 0.67 Å in the distance between heme a3 and the OH group of the cross-linked tyrosine, which has been proposed to act as an electron and/or proton donor during the reaction of the mixed-valence enzyme with O2.37−40 The increased separation between the OH of Y237 and the hydroxyethyl moiety of heme a3 results in a loss of the hydrogen bonding interaction in the G232V mutant, which may affect proton-coupled electron transfer involving the cross-linked tyrosine in the mixed valence mutant and/or disrupt the K proton channel. Moreover, the O2 ligand is able to diffuse much closer to the active site in the G232V mutant than in the wild-type enzyme, and the glycine-to-valine mutation appears to open other O2 exit and entrance pathways to the active site, allowing O2 and NO unhindered access to the active site.
Acknowledgments
Portions of this research were conducted at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including Grant P41GM103393), and the National Center for Research Resources (Grant P41RR001209).
Glossary
Abbreviations
- SVD
singular-value decomposition
- b-spectrum
spectral changes associated with an apparent rate (lifetime)
- Tt ba3
T. thermophilus ba3 cytochrome oxidase
- Rs
Rhodobacter sphaeroides
- Pd
Paracoccus denitrificans
- PDB
Protein Data Bank.
Supporting Information Available
Additional details regarding the simulation protocol and system preparation and equilibration for the classical molecular dynamics simulations of wild-type Tt ba3 and the G232V mutant, a summary of data collection parameter and refinement statistics for the G232V mutant (Table S1), TROA difference spectra and b-spectra resulting from the exponential fit of the CO recombination flash-photolysis data of the fully reduced CO-bound G232V mutant (Figure S1), and the b-spectra resulting from the multiexponential fit to the TROA difference spectra recorded during the reaction of the photoproduced O2 with the fully reduced G232V mutant (Figure S2). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Y.L.: Program in Cellular and Molecular Medicine, Boston Children’s Hospital, Harvard Medical School, 3 Blackfan Circle, Boston, MA 02115.
Author Contributions
W.M. and C.F. contributed equally to this work.
This work was supported by National Science Foundation Grant CHE-1158548 to Ó.E. and National Institutes of Health Grant GM035342 to C.D.S.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Brzezinski P.; Larsson G. (2003) Redox-driven proton pumping by heme-copper oxidases. Biochim. Biophys. Acta 1605, 1–13. [DOI] [PubMed] [Google Scholar]
- Einarsdóttir Ó. (1995) Fast reactions of cytochrome oxidase. Biochim. Biophys. Acta 1229, 129–147. [DOI] [PubMed] [Google Scholar]
- Ferguson-Miller S.; Babcock G. T. (1996) Heme/copper terminal oxidases. Chem. Rev. 96, 2889–2907. [DOI] [PubMed] [Google Scholar]
- Wikström M. K. F. (1977) Proton pump coupled to cytochrome c oxidase in mitochondria. Nature 266, 271–273. [DOI] [PubMed] [Google Scholar]
- Hemp J.; Gennis R. B. (2008) Diversity of the heme-copper superfamily in Archaea: Insights from genomics and structural modeling. Results Probl. Cell Differ. 45, 1–31. [DOI] [PubMed] [Google Scholar]
- Pereira M. M.; Santana M.; Teixeira M. (2001) A novel scenario for the evolution of haem-copper oxygen reductases. Biochim. Biophys. Acta 1505, 185–208. [DOI] [PubMed] [Google Scholar]
- Tsukihara T.; Aoyama H.; Yamashita E.; Tomizaki T.; Yamaguchi H.; Shinzawa-Itoh K.; Nakashima R.; Yaono R.; Yoshikawa S. (1996) The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science 272, 1136–1144. [DOI] [PubMed] [Google Scholar]
- Svensson-Ek M.; Abramson J.; Larsson G.; Törnroth S.; Brzezinski P.; Iwata S. (2002) The X-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J. Mol. Biol. 321, 329–339. [DOI] [PubMed] [Google Scholar]
- Iwata S.; Ostermeier C.; Ludwig B.; Michel H. (1995) Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature 376, 660–669. [DOI] [PubMed] [Google Scholar]
- Abramson J.; Riistama S.; Larsson G.; Jasaitis A.; Svensson-Ek M.; Laakkonen L.; Puustinen A.; Iwata S.; Wikström M. (2000) The structure of the ubiquinol oxidase from Escherichia coli and its ubiquinone binding site. Nat. Struct. Biol. 7, 910–917. [DOI] [PubMed] [Google Scholar]
- Soulimane T.; Buse G.; Bourenkov G. P.; Bartunik H. D.; Huber R.; Than M. E. (2000) Structure and mechanism of the aberrant ba3-cytochrome c oxidase from Thermus thermophilus. EMBO J. 19, 1766–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschmann S.; Warkentin E.; Xie H.; Langer J. D.; Ermler U.; Michel H. (2010) The structure of cbb3 cytochrome oxidase provides insights into proton pumping. Science 329, 327–330. [DOI] [PubMed] [Google Scholar]
- Luna V. M.; Chen Y.; Fee J. A.; Stout C. D. (2008) Crystallographic studies of Xe and Kr binding within the large internal cavity of cytochrome ba3 from Thermus thermophilus: Structural analysis and role of oxygen transport channels in the heme-Cu oxidases. Biochemistry 47, 4657–4665. [DOI] [PubMed] [Google Scholar]
- Luna W. M.; Fee J. A.; Deniz A. A.; Stout C. D. (2012) Mobility of Xe atoms within the oxygen diffusion channel of cytochrome ba3 oxidase. Biochemistry 51, 4669–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker I.; Schulten K. (1998) Oxygen and proton pathways in cytochrome c oxidase. Proteins: Struct., Funct., Genet. 30, 100–107. [PubMed] [Google Scholar]
- Salomonsson L.; Lee A.; Gennis R. B.; Brzezinski P. (2004) A single-amino-acid lid renders a gas-tight compartment within a membrane-bound transporter. Proc. Natl. Acad. Sci. U.S.A. 101, 11617–11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald W.; Funatogawa C.; Li Y.; Szundi I.; Chen Y.; Fee J. A.; Stout C. D.; Einarsdóttir Ó. (2013) Ligand access to the active site in Thermus thermophilus ba3 and bovine heart aa3 cytochrome oxidases. Biochemistry 52, 640–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einarsdóttir Ó.; Funatogawa C.; Soulimane T.; Szundi I. (2012) Kinetic studies of the reactions of O2 and NO with reduced Thermus thermophilus ba3 and bovine aa3 using photolabile carriers. Biochim. Biophys. Acta 1817, 672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szundi I.; Funatogawa C.; Fee J. A.; Soulimane T.; Einarsdóttir Ó. (2010) CO impedes superfast O2 binding in ba3 cytochrome oxidase from Thermus thermophilus. Proc. Natl. Acad. Sci. U.S.A. 107, 21010–21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepke J.; Olkhova E.; Angerer H.; Muller H.; Peng G.; Michel H. (2009) High resolution crystal structure of Paracoccus denitrificans cytochrome c oxidase: New insights into the active site and the proton transfer pathways. Biochim. Biophys. Acta 1787, 635–645. [DOI] [PubMed] [Google Scholar]
- Muramoto K.; Hirata K.; Shinzawa-Itoh K.; Yoko-o S.; Yamashita E.; Aoyama H.; Tsukihara T.; Yoshikawa S. (2007) A histidine residue acting as a controlling site for dioxygen reduction and proton pumping by cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 104, 7881–7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Hunsicker-Wang L.; Pacoma R. L.; Luna E.; Fee J. A. (2005) A homologous expression system for obtaining engineered cytochrome ba3 from Thermus thermophilus HB8. Protein Expression Purif. 40, 299–318. [DOI] [PubMed] [Google Scholar]
- Battye T. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. (2011) iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D62, 72–82. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubak P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard-Jones A. R.; Adam V.; Cowley A.; Baldwin J. E.; Bourgeois D. (2009) Cryophotolysis of a caged oxygen compound for use in low temperature biological studies. Photochem. Photobiol. Sci. 8, 1150–1156. [DOI] [PubMed] [Google Scholar]
- Georgiadis K. E.; Jhon N.-I.; Einarsdóttir Ó. (1994) Time-resolved optical absorption studies of intramolecular electron transfer in cytochrome c oxidase. Biochemistry 33, 9245–9256. [DOI] [PubMed] [Google Scholar]
- Szundi I.; Van Eps N.; Einarsdóttir Ó. (2003) pH dependence of the reduction of dioxygen to water by cytochrome c oxidase. 2. Branched electron transfer pathways linked by proton transfer. Biochemistry 42, 5074–5090. [DOI] [PubMed] [Google Scholar]
- Feller S. E.; MacKerell A. D. (2000) An improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B 104, 7510–7515. [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Buhrow L.; Ferguson-Miller S.; Kuhn L. A. (2012) From static structure to living protein: Computational analysis of cytochrome c oxidase main-chain flexibility. Biophys. J. 102, 2158–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Qin L.; Liu J.; Mills D. A.; Proshlyakov D. A.; Hiser C.; Ferguson-Miller S. (2009) Redox-dependent conformational changes in cytochrome c oxidase suggest a gating mechanism for proton uptake. Biochemistry 48, 5121–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H.; Hemp J.; Pace L. A.; Ouyang H.; Ganesan K.; Roh J. H.; Daldal F.; Blanke S. R.; Gennis R. B. (2011) Adaptation of aerobic respiration to low O2 environments. Proc. Natl. Acad. Sci. U.S.A. 108, 14109–14114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg M. R. A.; Siegbahn P. E. M.; Babcock G. T.; Wikström M. (2000) O-O bond splitting mechanism in cytochrome oxidase. J. Inorg. Biochem. 80, 261–269. [DOI] [PubMed] [Google Scholar]
- McDonald W. J.; Einarsdóttir Ó. (2010) Solvent effects on the physicochemical properties of the cross-linked histidine-tyrosine ligand of cytochrome c oxidase. J. Phys. Chem. B 114, 6409–6425. [DOI] [PubMed] [Google Scholar]
- Proshlyakov D. A.; Pressler M. A.; Babcock G. T. (1998) Dioxygen activation and bond cleavage by mixed-valence cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 95, 8020–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sucheta A.; Szundi I.; Einarsdóttir Ó. (1998) Intermediates in the reaction of fully reduced cytochrome c oxidase with dioxygen. Biochemistry 37, 17905–17914. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.