

Abstract
Obesity is a pandemic and major risk factor for cancers. The reduction of obesity would have been an effective strategy for cancer prevention, but the reality is that worldwide obesity has kept increasing for decades, remaining a major avoidable cancer risk secondary only to smoke. The present studies suggest that vitamin D may be an effective agent to reduce obesity-associated cancer risks in women. Molecular analyses showed that leptin increased human telomerase reverse transcriptase (hTERT) mRNA expression and cell growth through estrogen receptor alpha (ERα) activation in ovarian cancer (OCa) cells, which was suppressed by 1alpha,25-dihydroxyvitamin D3 [1,25(OH)2D3]. The suppression was compromised when miR-498 induction by the hormone was depleted with miRNA sponges. In mice, high-fat diet (HFD) stimulation of ovarian tumor growth was remarkably suppressed by 1,25(OH)2D3 analogue EB1089, which was also compromised by miR-498 sponges. EB1089 did not alter HFD-induced increase in serum leptin levels but increased miR-498 and decreased the diet-induced hTERT expression in tumors. Quantitative RT-PCR (qRT-PCR) analyses revealed an inverse correlation between hTERT mRNA and miR-498 in response to 1,25(OH)2D3 in estrogen-sensitive ovarian, endometrial and breast cancers. The studies suggest that miR-498-mediated hTERT down regulation is a key event mediating the anti-leptin activity of 1,25(OH)2D3 in estrogen-sensitive tumors in women.
Keywords: Cancer, Estrogen, hTERT, Leptin, MicroRNA, Obesity, Telomerase, Vitamin D, VDR
Introduction
Obesity is a major health problem in the United States and around the world, exceeding 30% in America in most gender and age groups. Its prevalence remained steady in the past decade and, in 2009–2010, the rate was estimated to be 35.5% and 35.8% among adult men and women, respectively. Besides well-known metabolic and cardiovascular complications, obesity has been associated with increased incidence and mortality rates of various cancers and difficulties in treatments. About 20% of all cancers are estimated to be caused by excess body weight with obesity accounting for about 20% of cancer death in women and 14% in men, which is second only to smoking for the number of avoidable cancers (1). Agents capable of mitigating obesity-associated cancer risk will have a great potential to improve the outcomes of cancer intervention.
Among the complex biological mechanisms linking obesity to cancer susceptibility, multiple lines of evidences support a connection between increased production of leptin and obesity-associated cancers, particularly estrogen-sensitive ones in postmenopausal women. Circulating levels of leptin, an adipocyte-derived adipokine that classically plays a crucial role in regulating appetite and energy balance, are strongly correlated with body fat content and markedly elevated in obese individuals (2, 3) as well as in breast, ovarian, and endometrial cancer patients (4, 5). The women’s health initiative studies have identified obesity as a major breast cancer risk factor for postmenopausal women who had never taken hormone replacement therapy (6). Million Women Study has estimated that approximately half of endometrial cancers in postmenopausal women can be attributed to obesity (7). Similarly, multiple studies have revealed a statistically significant association between obesity and the risk of ovarian cancer (OCa) [16–18]. In laboratory studies, leptin is capable of promoting an aggressive cancer phenotype by stimulating growth, migration, invasion and angiogenesis (8, 9). Particularly relevant to estrogen-sensitive cancers, leptin has been shown to amplify estrogen signaling by either increasing aromatase expression in stromal and cancer cells (10, 11) or by activating ERα through ERK and STAT3 signaling pathways (12–14), establishing a functional crosstalk between leptin and estrogen signaling pathways. The leptin-estrogen connection is further supported by studies showing that obesity is associated with an increase in systemic estrogen levels (15, 16) and that obese women exhibit higher levels of ERα expression than lean women in several types of cancers (17).
The active metabolite of vitamin D, 1,25(OH)2D3 or calcitriol, has anti-proliferative and pro-apoptotic properties and is widely recognized as agent with great potential for cancer intervention (18, 19). Previously, we have shown that 1,25(OH)2D3 suppresses the growth of multiple human OCa cells and ovarian tumors in mice (20–25). It induces OCa cell death via telomerase suppression mediated through miR-498 (25, 26), a primate-specific microRNA (miRNA) of which the expression is normally restricted to placenta. In the present study, we present experimental evidences showing that leptin and HFD stimulated the growth of estrogen-sensitive tumors through ERα activation and telomerase induction, which was suppressed by 1,25(OH)2D3 and its analog EB1089 through miR-498. The studies suggest that 1,25(OH)2D3 or its precursors may be used to reduce obesity-associated risks for estrogen-sensitive cancers in women and that clinical trials with obese women may reveal the effectiveness of vitamin D in cancer intervention, a controversial issue debated in both the scientific community and public arena.
Materials and Methods
Materials and cell lines
1,25(OH)2D3, human recombinant leptin, 17β-estradiol and ICI 182,780 were purchased from Sigma (St. Louis, MO). Anti-VDR and anti-hTERT antibodies were from Epitomics (Burlingame, CA). Antibodies against ERα and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). pLEN-hERα, EREelbLuc, pCMVβGal and miR-498 sponges have been used in previous studies (26, 27). BG-1 cells (28) were cultured in DMEM/F-12 medium supplemented with 2 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 5% fetal calf serum (FCS). MCF-7 and Ishikawa (29) cells were maintained in DMEM containing 2 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 5% FCS. PE-04 cells (30) were maintained in RPMI medium containing 2 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% FCS. All cells were maintained at 37°C in a humidified incubator with 5% CO2. ERα expression and estrogen sensitivity of BG-1 (28), PE-04 (30) and Ishikawa (29) cells were validated and morphological analyses and genotyping by Genetics DNA Laboratories (Burlington, NC) were used to ensure that the cells are not cross-contaminated with MCF-7 cells. MCF-7 cells were from American Type Culture Collection and authenticated with its guidelines. For all studies involving estrogen treatments, cells were deprived of estrogens by culturing for 2 days in phenol red-free medium containing 2% charcoal stripped FCS.
Transfections and reporter assays
Cells were plated in medium containing 5% FCS at 1.5 × 105 cells/well in six-well plates. One day after plating, cells were transfected with Lipofectamine following the protocol from Invitrogen (Life Technologies, Grand island, NY) and were placed in phenol-red free DMEM/F-12 containing 2% charcoal-stripped FCS. 48 h post transfections, cells were treated with vehicle (EtOH), 17β-estradiol or leptin in the presence or absence of 1,25(OH)2D3 for 3 days. For longer treatments, cells were re-fed with fresh culture medium every two days. Luciferase and β-galactosidase (β-gal) activities were determined as previously described (20).
To establish luciferase-marked cells that express miRNA sponges, BG-1 cells were co-transfected with control or miR-498 sponges together with pGL3 control plasmid as previously described (26). Stable clones were isolated after selection with puromycin (2 μg/ml) for about 5 weeks.
Quantitative RT-PCR (qRT-PCR) assays
Total and small RNAs were extracted from cells using the mirVana™ miRNA isolation kit (Ambion, Austin, TX) according to the manufacturer’s instructions. cDNA was reverse-transcribed from 1 μg total cellular RNA with random hexamer primers and thermo-stable reverse transcriptase (Invitrogen). TaqMan assays (Life Technologies) were used to quantify the expression of hTERT mRNA and mature miR-498. GAPDH and U6 were used as controls for hTERT and miR-498 expression studies, respectively. PCR reactions were performed in a 20 μl reaction mixture containing 1.33 μl of RT product, 1.0 μl of corresponding TaqMan probe and 10 μl TaqMan 2× universal PCR master mix. Reactions were run in the ABI Prism 7900 Fast Real-Time PCR system in triplicate with incubation at 95°C for 10 min followed by 40 cycles of denaturation for 15 s at 95°C and annealing at 60°C for 1 min. The target gene levels at each time point were normalized with cognate controls by subtracting the cycle threshold (Ct) value of controls from Ct of target genes to produce a ΔCt. The fold of induction over vehicle controls was calculated based on the formula 2−[ΔCt (treatment) − ΔCt (vehicle)].
Immunoblotting (IB) analyses
Cells were estrogen-starved and treated with vehicle, 17β-estradiol or leptin in the presence or absence of 1,25(OH)2D3 for 3 days and lysed on ice by incubating for 20 min with lysis buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% (v/v) NP-40, 1 mM PMSF, and protease inhibitor cocktail. After centrifugation, protein concentrations of the supernatant were determined using the Bio-Rad assays. Samples of equal amount proteins were separated in SDS-PAGE, transferred to a nitrocellulose membrane and probed with cognate antibodies. Immuno-reacted proteins were detected with ECL plus.
3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assays, apoptosis and cell cycle analyses
To quantify cell growth, MTT assays were performed as described previously (31). Cells were estrogen starved and plated at 5 × 104 in 96-well plates and treated with vehicle, 17β-estradiol or leptin in the presence or absence of 1,25(OH)2D3 for 6 days. MTT reagents were added to each well to give a final concentration of 0.5 mg/ml and incubated with cells for 3 h. After the removal of the media, 200 μl DMSO was added to the cells. The absorption at 595 nm was determined with a microplate reader (DYNEX Technologies, Chantilly, VA).
To determine the apoptotic index, cells were treated with vehicle or leptin in the presence or absence of 1,25(OH)2D3 for 6 days, harvested and washed with phosphate-buffered saline (PBS). Cell suspensions were incubated with FITC and propidium iodide (PI) according to the manufacturer’s protocol (FITC Annexin V kit, BD Biosciences). Flow cytometry was performed in a FACScan (BD Biosciences). For each data point, triplicate samples were analyzed and the experiment was reproduced three times.
To determine cell cycle distribution, cells were harvested, washed with PBS and fixed with 70% ethanol for 2 h. Fixed cells were washed again with PBS, re-suspended in staining solution of 1 ml PBS containing 10 μg/ml DNase-free RNase and propidium iodide and incubated for 3 h at 4°C. Cell populations distributed at different phases of the cell cycle were quantified by FACScan. For each data point, triplicate samples were analyzed and the experiment was repeated three times.
HFD and ovarian tumor studies
Female athymic nu/nu mice of 4–6 week old were obtained from Harlan Sprague Dawley (Indianopolis, IN). Mice were housed at USF animal facility at the appropriate temperature and with a standard 12 h light-dark cycle. All mouse studies were carried out according to procedures approved by the Institutional Animal Care and Use Committee at University of South Florida. For tumor studies, mice were divided into two diet groups (n=20 per group). Group I was fed with vitamin D deficient diet (VDD) containing 22.9%-kcal fat supplemented with 0.47% calcium. Group II received the same calcium-supplemented VDD but containing high fat (HFD) (55%-kcal). The diet was obtained from Harlan Teklad (Madison, WI).
To establish ovarian tumors in mice, BG-1 cells stably transfected with pGL3-control (luciferase) and control or miR-498 sponges were harvested and re-suspended at a concentration of 5 × 106 cells in 100 μl of DMEM/F-12 medium containing 5% FCS and ice-cold Matrigel (1:1) and injected i.p. into mice that have been on VDD or HFD diets for a week. Mice were randomized and treated every other day with either vehicle or EB1089 (Pure Chemistry Scientific Inc., Sugarland, TX) at 0.5 μg/kg body weight diluted in sesame oil in a volume of 50 μl by gavage. All mice were weighed once per week over the course of treatment to monitor changes in body weight. Tumor growths were measured with IVIS-200 (Xenogen, Alameda, CA) live imaging system at 30 and 60 days as described previously (26). All animals were euthanized after treatment for 60 days. Tumor weight and the number of nodules in the abdominal cavity were scored.
Serum leptin measurements
Serum was collected from mice after 60 day treatment. Serum leptin concentrations were measured using a mouse leptin ELISA kit (Crystal Chem Inc., Downers Grove, IL). Serum calcium was measured as previously described (24).
Statistics
All values were presented as means ± standard deviation (SD) of the indicated number of determinations (n). Significant differences were assessed with the student’s t test. All categorical data used numbers, folds and percentages.
Results
1,25(OH)2D3 dominates over estrogens in controlling miR-498 and telomerase expression as well as the growth of estrogen-sensitive OCa cells
Estrogens have been shown to stimulate hTERT gene expression and telomerase activity in breast (32), ovarian (33, 34) and endometrial (35) cancer cells, which involve estrogen response elements (EREs) present in the hTERT promoter. Our recent studies identified miR-498 as a primary target gene for 1,25(OH)2D3 that binds to hTERT 3′-untranslated region and decreases telomerase activity in OCa cells (26), making it possible for 1,25(OH)2D3 to inhibit estrogen-induced telomerase activity and cell growth through miR-498. To test this idea, we examined whether 1,25(OH)2D3 induced miR-498 in estrogen-sensitive OCa cells and whether the induction occurred in the presence of 17β-estradiol (E2). As shown in Fig. 1A, miR-498 was significantly induced by 1,25(OH)2D3 in BG-1 cells and that the induction occurred similarly in the absence or presence of E2. Consistent with the miR-498 data, 1,25(OH)2D3 suppressed E2-induced hTERT expression at mRNA (Fig. 1B) and protein (Fig. 1C) levels. These analyses show that 1,25(OH)2D3 antagonizes and dominates over estrogen actions in regulating miR-498 and hTERT expression in OCa cells. The 1,25(OH)2D3 dominance was further supported by the findings that 1,25(OH)2D3 suppressed E2 stimulation of BG-1 cell growth (Fig. 1D) and that 1,25(OH)2D3– induced increase in VDR protein expression was not affected by E2 (Fig. 1C). In these analyses, 1,25(OH)2D3 did not alter the ability of E2 to down regulate ERα (Fig. 1C), a process required for efficient ERα transactivation (36), indicating that the dominant effect of 1,25(OH)2D3 over E2 is likely to be exerted at steps downstream of ERα transactivation by its ligands.
Figure 1. 1,25(OH)2D3 suppresses estrogen-induced hTERT expression and cell growth.

BG-1 cells were treated with either ethanol as a vehicle control (Control), 10−7 M 1,25(OH)2D3 (VD), 10−8 M 17β-estradiol (E2) or VD plus E2 for 3 or 6 (panel D only) days. A and B, Small and total RNAs were isolated and the expression of miR-498 (A) and hTERT (B) was determined by qRT-PCR and normalized to U6 and GAPDH, respectively. The values are expressed as fold of the vehicle control. C, Cellular extracts were subjected to IB analyses with indicated antibodies. Protein signals were quantified by ChemiDoc (BioRad) and after being normalized to cognate β-actin signals, are presented as fold of control. D, Cell growth was measured in MTT assays. Data represent three independent experiments. Error bars are SD. Statistical analyses were performed with Student’s t test (n=3 for panels A and B, n=10 for panel D).
Leptin increases hTERT expression and OCa cell growth through ERα
Leptin has been shown to stimulate ERα transactivation (12–14) and as mentioned earlier, estrogens stimulate hTERT expression through EREs present in the hTERT promoter. Thus, it is possible for leptin to stimulate hTERT expression and cell growth through ERα in OCa cells. As shown in Fig. 2A, leptin significantly increased the transcriptional activity of ERα in BG-1 cells in reporter assays, which was blocked by pure ERα antagonist ICI 182,780. Similarly, qRT-PCR analyses and MTT assays respectively revealed that leptin significantly stimulated hTERT expression (Fig. 2B) and cell growth (Fig 2C), which both were blocked by ICI 182,780. These analyses demonstrate that ERα mediates the leptin effect on telomerase expression and cell growth in OCa cells.
Figure 2. Leptin increases hTERT expression and cell growth through ERα activation.
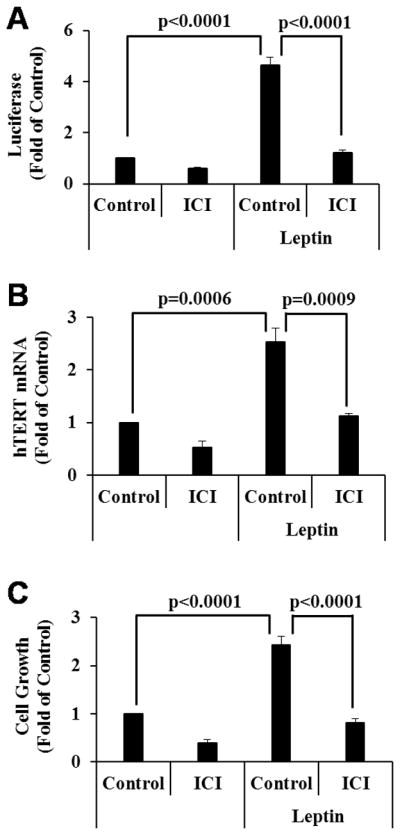
A, BG-1 cells in 6-well plates were transfected with 0.5 μg pLEN-hERα, 0.5 μg EREe1bLuc and 0.2 μg pCMVβGal and 24 h later, treated for 72 h with vehicle (Control), leptin (100 ng/ml), 10−8 M ICI 182,780 (ICI) or leptin plus ICI as indicated. Luciferase activities were determined, normalized with β-gal activity and presented as fold of the vehicle control. B, BG-1 cells were treated as in panel A. Total RNAs were extracted and subjected to qRT-PCR analyses. The levels of hTERT were normalized with corresponding GAPDH and expressed as fold of the vehicle control. C, BG-1 cells were treated as in panel A but for 6 days. Cell growth was measured in MTT assays. Data represent three independent experiments. Error bars are SD. Statistical analyses were performed with Student’s t test (n=3 for panels A and B, n=10 for panel C).
1,25(OH)2D3 suppresses leptin stimulation of hTERT expression and OCa cell growth through miR-498
The ability of 1,25(OH)2D3 to induce miR-498 and its dominance over estrogen actions in BG-1 cells make it likely for the hormone to suppress leptin induction of hTERT expression and OCa cell growth through the miRNA. To test this concept, we first assessed the ability of 1,25(OH)2D3 to induce miR-498 in the presence or absence of leptin in BG-1 cells. As shown in Fig. 3A, 1,25(OH)2D3 stimulated miR-498 expression to comparable levels in the presence or absence of leptin, showing a dominance of 1,25(OH)2D3 over leptin in miR-498 induction. Consistent with the dominance in controlling miR-498 expression, 1,25(OH)2D3 attenuated leptin-induced hTERT mRNA (Fig. 3B) and protein (Fig. 3C) expression as well as BG-1 cell growth (Fig. 3D). In these analyses, leptin decreased the ERα protein expression, likely due to ERα activation by the adipokine, and the decrease also occurred in cells co-treated with 1,25(OH)2D3 (Fig. 3E). On the other hand, 1,25(OH)2D3 increased VDR protein expression, which also occurred in the presence of leptin (Fig. 3E). These analyses show that 1,25(OH)2D3 is dominant over leptin in controlling OCa cell growth and that this dominant action occurs at steps downstream of ERα activation by leptin, which likely involves miR-498-mediated hTERT down regulation.
Figure 3. 1,25(OH)2D3 suppresses leptin-induced hTERT expression and cell growth.

A and B, BG-1 cells were treated with EtOH (Control), 10−7 M VD, leptin (100 ng/ml) or VD plus leptin for 3 days. Total and small RNAs were extracted and the expression of miR-498 (A) and hTERT (B) was determined by qRT-PCR and normalized to U6 and GAPDH levels, respectively. The values are presented as fold of the vehicle control. C, BG-1 cells were treated for 3 days with EtOH (Control), 10−7 M VD or 10−8 M ICI in the absence or presence of 100 ng/ml leptin. The levels of hTERT protein expression were determined and quantified as in Figure 1C. D, BG-1 cells were treated as in panel A but for 6 days. Cell growth was measured in MTT assays. E, BG-1 cells were treated as in panel C. ERα and VDR protein levels were determined and quantified as in Figure 1C. Data represent three independent experiments and are presented as mean ± SD. Statistical analyses were performed with Student’s t test (n=3 in panels A and B, n=10 in panel D).
To assess the role of miR-498 in the dominant action of 1,25(OH)2D3 over leptin, control and miR-498 sponges (26) were stably transfected into BG-1 cells and the response of the stable transfectants to leptin and 1,25(OH)2D3 was analyzed. As shown in Fig. 4, 1,25(OH)2D3 decreased leptin-induced hTERT expression (Fig. 4A) and cell growth (Fig 4B) in cells transfected with control sponges but had little effect in cells expressing miR-498 sponges, showing that the suppression of leptin actions was mediated through miR-498. Consistent with its known proliferative activity, leptin increased the percentage of BG-1 cells in S phase, which was also suppressed by 1,25(OH)2D3 in cells transfected with control but not miR-498 sponges (Fig 4C). The studies demonstrate that leptin stimulates OCa cell growth through telomerase induction and that 1,25(OH)2D3 suppresses the leptin effect through miR-498. In the analyses, leptin had little effect on the ability of 1,25(OH)2D3 to induce cell death whereas miR-498 sponges, as expected, significantly relieved the apoptotic induction by the hormone (Fig. 4D), showing that the role of miR-498 in the induction of OCa cell death initially established in OVCAR3 cells is applicable to estrogen-sensitive cells. In BG-1 cells, miR-498 sponges partially relieved the suppressive effect of 1,25(OH)2D3 on leptin stimulation of S phase accumulation, suggesting that the role of miR-498 in 1,25(OH)2D3 actions in estrogen-sensitive OCa cells goes beyond cell death induction.
Figure 4. 1,25(OH)2D3 works through miR-498 to suppress leptin-induced hTERT expression and cell proliferation.

A, Control (C-bSP) or miR-498 (M-bSP) sponges were stably transfected into BG1-cells. The stable clones were treated with EtOH (Control), 10−7 M VD, leptin (100 ng/ml) or 10−7 M VD plus leptin (100 ng/ml) for 3 days. Total RNAs were extracted and the expression of hTERT was determined by qRT-PCR and normalized to GAPDH. The values are expressed as fold of the control. B, Cells were treated as in panel A but for 6 days. Cell growth was measured in MTT assays. C, Cells were treated as in panel B and stained with propidium iodide. Cell cycle distribution was assessed by flow cytometry. D, Cells were treated as in panel B. Apoptosis was measured by FITC Annexin-V analyses. Data represent three independent experiments and are presented as the mean ± SD. Statistical analyses were performed with Student’s t test (n=3 in panels A, C and D, n=10 in panel B).
1,25(OH)2D3 analogue EB1089 suppresses HFD-induced ovarian tumor growth in mice through miR-498
HFD is known to increase serum leptin levels, cause leptin resistance and promote the development of obesity and cancer (37). The dominance of 1,25(OH)2D3 over leptin through miR-498 makes it likely for 1,25(OH)2D3 to suppress HFD-induced ovarian tumor growth. To test this idea, BG-1 cells stably transfected with firefly luciferase together with either control or miR-498 sponges were injected into peritoneal cavity of nude mice. The mice were then treated for up to 60 days with a vehicle or synthetic 1,25(OH)2D3 analogue EB1089 that is known to be less calcemic and more potent in tumor suppression. Tumor growth was monitored by live luciferase imaging (Figs. 5A and 5B), counting numbers and size of tumor nodules (Supplement Fig. s1A) and measuring tumor weight (Supplement Fig. s1B). As shown in Figs. 5A and 5B, the growth of tumors expressing control sponges was significantly suppressed by EB1089 in mice fed with VDD diet. The suppression was largely diminished in tumors expressing miR-498 sponges, showing the importance of miR-498. More importantly, HFD stimulated BG-1 tumor growth by up to 6 folds, which was remarkably suppressed by EB1089 in groups expressing control sponges but not in those expressing miR-498 sponges. In parallel studies, tumors expressing miR-498 sponges grew faster than those expressing control sponges in vehicle-treated groups, revealing a role of basal miR-498 expression in suppressing basal and HFD induced ovarian tumor growth. Ki67 staining and TUNEL assays of tumor tissue sections revealed that that the tumor suppression by EB1089 is associated with decreased proliferation (Supplement Fig. s2) and increased apoptosis (Supplement Fig. s3). Overall, the data show that EB1089 suppresses the HFD-induced ovarian tumor growth and that miR-498 is an important mediator of this antitumor effect.
Figure 5. EB1089 suppresses HFD-induced ovarian tumor growth in vivo through miR-498.

Luciferase-marked BG-1 cells (5×106) expressing control (C-bSP) or miR-498 (M-bSP) sponges were mixed with Matrigel and i.p injected into female athymic nu/nu mice fed with VDD or HFD. Mice bearing BG-1 xenograft tumors were treated by gavage every other day with EB1089 (0.5 μg/kg) or a vehicle (EtOH) in sesame oil. A, After 30 and 60 days of treatments, tumor growth was monitored by bioluminescent in vivo imaging. B, The bioluminescence intensity, a measurement of viable tumor volume, was quantified and presented as photon counts (photons/sec/cm2/steradian). Statistical analyses were performed with Student’s t test (n=5). #p<0.01; *p=0.002; **p=0.001; ***p<0.0001.
As expected, mice fed with HFD were heavier than mice on VDD diet and the difference became statistically significant (p<0.0001) since 4 weeks of the experiment (Fig. 6A). Serum leptin levels at the end of the experiment were also significantly (p<0.0001) increased in mice on HFD as compared those on VDD diet (Fig. 6B). Neither EB1089 nor miR-498 sponges altered the effect of HFD on body weight and serum leptin levels. EB1089 increase serum calcium levels but did not induce hypercalcemia (Supplement Fig. s4). QRT-PCR (Fig. 6C) and Western blot (Fig. 6D) analyses showed that levels of hTERT mRNA and protein expression in tumors were significantly suppressed by EB1089 in both VDD and HFD groups and that miR-498 sponges diminished the suppressive effect of EB1089. The data suggest that HFD increased leptin to stimulate ovarian tumor growth in vivo and the stimulation of tumor growth was suppressed by EB1089 through miR-498 mediated hTERT down regulation.
Figure 6. EB1089 decreases tumor hTERT levels without an effect on mouse body weight and serum leptin levels.

A, The average body weight of mice was measured on weekly basis and presented. B, Serum leptin levels were measured and expressed as ng/mL. C, Total RNAs were extracted from tumor tissues. hTERT mRNA levels were determined by qRT-PCR, normalized to GAPDH and expressed as fold of the control (C-bSP + EtOH). D, Tumor hTERT protein levels were determined by Western blot and quantified as in Figure 1C. Data are presented as mean ± SD. Statistical analyses were performed with Student’s t test (n=5 for panels A and B, n=3 for panel C). * p<0.0001.
Inverse regulation of miR-498 and hTERT expression by 1,25(OH)2D3 in multiple ERα-positive cancer cell lines
The studies described above have established the concept that 1,25(OH)2D3 and EB1089 suppress BG-1 ovarian tumor growth induced by leptin and HFD through miR-498-mediated telomerase down regulation. To assess whether the concept can be extended to other estrogen-sensitive cancers, a panel of estrogen-sensitive cancer cell lines, including ovarian (PE-04), breast (MCF-7) and endometrial (Ishikawa) cells, were treated with EtOH or 1,25(OH)2D3 for 0, 3 and 6 days and the expression of miR-498 and hTERT mRNA determined by qRT-PCR. As shown in Fig 7, miR-498 (Fig. 7A) and hTERT mRNA (Fig. 7B) expressions was inversely regulated by 1,25(OH)2D3 in a time-dependent manner. As in BG-1 cells, 1,25(OH)2D3 induced apoptosis in PE-04, MCF-7 and Ishikawa cells (Fig 7C), which was not affected by leptin, and suppressed leptin-induced growth (Fig. 7D) of these cells, showing that the concept established in BG-1 tumors (Fig. 7E) can be extended to other estrogen-sensitive OCa as well as breast and uterine cancers. Dosage analyses revealed that 1,25(OH)2D3 induced apoptosis at concentrations as low as 1 nM and that the induction occurred in a dose dependent manner in both BG-1 and MCF-7 cells (Supplement Fig. s5), showing that the hormone killed estrogen-sensitive tumor cells at physiological concentrations. Interestingly, miR-498 induction was not observed in vitamin D-sensitive HCT116 (colon) and LNCaP (prostate) cancer cells (data not shown), showing a specificity to women’s cancers.
Figure 7. 1,25(OH)2D3 induces miR-498 and decreases hTERT mRNA expression in multiple estrogen-sensitive human cancer cells.

A and B, BG-1 (ovarian), PE-04 (ovarian), MCF-7 (breast) and Ishikawa (endometrial) cancer cells were treated with vehicle (EtOH) or 10−7 M 1,25(OH)2D3 (VD) for 0, 3 and 6 days. Total and small RNAs were extracted and the expression of miR-498 (A) and hTERT (B) were determined by qRT-PCR and normalized to U6 and GAPDH, respectively. The expressions are presented as fold of the vehicle control (0 day treatment). C and D, Cells were treated with EtOH (Control), 10−7 M VD, leptin (100 ng/ml) or 10−7 M VD plus leptin (100 ng/ml) for 6 days. Cell death (C) and growth (D) were analyzed by Annexin-V analyses and MTT assays, respectively, as in Fig. 4. Data represent three independent experiments and are presented as mean ± SD. Statistical analysis was performed with Student’s t test (n=3), *p<0.0001. E, Our working hypothesis that explains how HFD and leptin work through ERα activation to stimulate hTERT expression and the growth of estrogen-sensitive cancers in women and how 1,25(OH)2D3 works through miR-498 mediated hTERT destruction to suppress the stimulatory effect of HFD and leptin (See text for details).
Discussion
Telomerase is a hallmark of cancer that cells must possess to overcome two proliferative barriers, senescence and crisis-associated apoptosis, to become cancerous (38). Due to the critically short telomere length, telomerase activity remains rate-limiting in cancer cells, explaining why tumor-promoting factors need to increase telomerase activity in order to increase tumor growth. In the present studies, ICI 182,780, a pure ERα antagonist, diminished the ability of leptin to induce hTERT expression, cell growth and ERα transcriptional activity, defining ERα-mediated hTERT induction as the main mechanism underlying leptin stimulation of estrogen-sensitive OCa growth. 1,25(OH)2D3 suppressed such a leptin action through miR-498-mediated hTERT destruction, which was translated into the suppression of HFD-induced hTERT expression and ovarian tumor growth in vivo in mice (Fig. 7E). In addition to BG-1 cells, miR-498 induction and hTERT down regulation by 1,25(OH)2D3 were also detected in ERα-positive PE-04, MCF-7 and Ishikawa cells, showing that the concept that 1,25(OH)2D3 may prevent obesity-associated cancers can be generalized to estrogen-sensitive ovarian as well as breast and uterine cancers.
Previous studies in estrogen-insensitive OCa cells have defined miR-498 as a primary 1,25(OH)2D3 target gene that directly binds to the 3′ UTR of hTERT mRNA to induce cell death (26). The present studies are the first to document an important role for miR-498 in mediating the suppressive effect of 1,25(OH)2D3 on estrogen stimulation of cancer growth. The studies are also the first to document a crosstalk between leptin and 1,25(OH)2D3 and place telomerase and the miRNA pathway at the center stage. It is important to point out that our studies do not project miR-498-mediated hTERT degradation as the sole mechanism underlying the 1,25(OH)2D3 suppression of estrogen and leptin actions in cancer cells. 1,25(OH)2D3 has been reported to decrease ERα expression and activity in MCF-7 cells (39, 40). In BG-1 cells, 1,25(OH)2D3 caused a modest reduction in ERα protein expression (Fig 1C) and decreased its transcriptional activity in reporter assays (data not shown), suggesting that the effect of 1,25(OH)2D3 on ERα expression and/or activity may also contribute to its suppression of leptin actions in OCa cells. Nevertheless, the fact that miR-498 sponges relieved majority of the suppressive effect of 1,25(OH)2D3 on leptin and HFD-induced OCa growth shows that the miR-498 pathway is the main mediator of the 1,25(OH)2D3 effect on leptin.
Although present studies have focused on how 1,25(OH)2D3 suppresses ERα-mediated leptin effect on telomerase, there are no reasons to believe that the impact of 1,25(OH)2D3 action through miR-498 on telomerase will be limited to leptin action or ERα-positive tumors. Recently, hypercholesterolemia, another obesity comorbidity and an independent cancer risk factor in postmenopausal women has been shown to active ERα and liver X receptor to control breast cancer growth and invasion after converting into 27-hydroxycholesterol (41). Although remaining to be determined, 27-hydroxycholesterol may increase telomerase activity through ERα and 1,25(OH)2D3 may prevent hypercholesterolemia-associated cancer risk through miR-498. Likewise, leptin has been shown to increase telomerase activity in breast and hepatocellular carcinoma cells through STAT3 (42, 43) and c-Myc (43), which may not depend on ERα. 1,25(OH)2D3 should also be able to suppress the ERα-independent leptin effect on telomerase through the same miR-498 pathway.
Although controversial, literature information in general supports an association between obesity and the risk of OCa (44–46). Similarly, the estrogen sensitivity of human OCa has been implicated by the facts that hormone replacement therapy increased OCa incidence (47) and that its decreased usage after the Women’s Health Initiative studies was associated with a reduction (48). Despite the estrogen sensitivity of their cancers, OCa patients do not benefit significantly from treatments with anti-estrogens or aromatase inhibitors. 1,25(OH)2D3 may offer an effective approach to block ERα actions, which may be employed to selectively reduce OCa risk in obese women. Consistent with this idea, the well-publicized Cohort Consortium Vitamin D Pooling Project of Rare Cancers revealed an inverse correlation between serum 25-hydroxyvitamin D3 and OCa risk that reached statistical significance only in obese women (49).
The concept that vitamin D may prevent cancers has been supported by epidemiological and molecular studies from many laboratories. However, population-based studies aiming to advance vitamin D into clinics for cancer intervention are complicated by many issues such as timing, dosages and duration, etc. With personalized medicine concept in mind, one may argue that the key to success will be the identification of a sub-population of human subjects who are highly sensitive to vitamin D. The present studies argue that two groups of women may benefit greatly from vitamin D intervention. One group would be women who are overweight or obese. The idea is consistent with literature information that obesity is usually correlated with low serum levels of 25-hydroxyvitamin D3, which was projected to be responsible for 20% of the cancer risk linked to increased body mass index (50). Another group would be postmenopausal women who receive hormone replacement therapy, which are known to increase their cancer risks. Well-controlled clinical trials should be carried out to determine whether vitamin D will offer a nontoxic and economic way of cancer intervention in overweight and obese women and whether its addition to the current hormone replacement therapy formula will cut down the increase in cancer risks.
Supplementary Material
Acknowledgments
We thank Drs. Kenneth S. Korach and Yin Li for the generous gift of BG-1 cells, Dr. J. L Becker for Ishikawa cells, Dr. Toshiyasu Taniguchi for PE-04 cells. The authors also thank the members of small animal imaging lab and tissue core facility at H. Lee Moffitt Cancer Center for help with the in vivo tumor analyses and Dr. Karoly Szekeres of USF flow cytometry core facility for the help with FACS analyses.
Grant Support
The studies were supported by Public Health Service grants CA111334 (to Bai) and CA164147 (to Zhang) from the National Institutes of Health and a program development grant from Ovarian Cancer Research Fund.
Footnotes
Conflicts of Interest: The authors disclose no potential conflicts of interest.
Disclosure of Potential Conflicts of Interest
The authors have no conflicts of interest to report.
References
- 1.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–38. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 2.Vendrell J, Broch M, Vilarrasa N, Molina A, Gomez JM, Gutierrez C, et al. Resistin, adiponectin, ghrelin, leptin, and proinflammatory cytokines: relationships in obesity. Obes Res. 2004;12:962–71. doi: 10.1038/oby.2004.118. [DOI] [PubMed] [Google Scholar]
- 3.Owecki M, Nikisch E, Miczke A, Pupek-Musialik D, Sowinski J. Leptin, soluble leptin receptors, free leptin index, and their relationship with insulin resistance and BMI: high normal BMI is the threshold for serum leptin increase in humans. Horm Metab Res. 2010;42:585–9. doi: 10.1055/s-0030-1253422. [DOI] [PubMed] [Google Scholar]
- 4.Tessitore L, Vizio B, Pesola D, Cecchini F, Mussa A, Argiles JM, et al. Adipocyte expression and circulating levels of leptin increase in both gynaecological and breast cancer patients. Int J Oncol. 2004;24:1529–35. [PubMed] [Google Scholar]
- 5.Uddin S, Bu R, Ahmed M, Abubaker J, Al-Dayel F, Bavi P, et al. Overexpression of leptin receptor predicts an unfavorable outcome in Middle Eastern ovarian cancer. Mol Cancer. 2009;8:74. doi: 10.1186/1476-4598-8-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaga T, Inui A, Okita M, Asakawa A, Ueno N, Kasuga M, et al. Modest overexpression of neuropeptide Y in the brain leads to obesity after high-sucrose feeding. Diabetes. 2001;50:1206–10. doi: 10.2337/diabetes.50.5.1206. [DOI] [PubMed] [Google Scholar]
- 7.Reeves GK, Pirie K, Beral V, Green J, Spencer E, Bull D, et al. Cancer incidence and mortality in relation to body mass index in the Million Women Study: cohort study. BMJ. 2007;335:1134. doi: 10.1136/bmj.39367.495995.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garofalo C, Surmacz E. Leptin and cancer. J Cell Physiol. 2006;207:12–22. doi: 10.1002/jcp.20472. [DOI] [PubMed] [Google Scholar]
- 9.Lang K, Ratke J. Leptin and Adiponectin: new players in the field of tumor cell and leukocyte migration. Cell Commun Signal. 2009;7:27. doi: 10.1186/1478-811X-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Catalano S, Marsico S, Giordano C, Mauro L, Rizza P, Panno ML, et al. Leptin enhances, via AP-1, expression of aromatase in the MCF-7 cell line. J Biol Chem. 2003;278:28668–76. doi: 10.1074/jbc.M301695200. [DOI] [PubMed] [Google Scholar]
- 11.Pino AM, Rodriguez JM, Rios S, Astudillo P, Leiva L, Seitz G, et al. Aromatase activity of human mesenchymal stem cells is stimulated by early differentiation, vitamin D and leptin. J Endocrinol. 2006;191:715–25. doi: 10.1677/joe.1.07026. [DOI] [PubMed] [Google Scholar]
- 12.Binai NA, Damert A, Carra G, Steckelbroeck S, Lower J, Lower R, et al. Expression of estrogen receptor alpha increases leptin-induced STAT3 activity in breast cancer cells. Int J Cancer. 2010;127:55–66. doi: 10.1002/ijc.25010. [DOI] [PubMed] [Google Scholar]
- 13.Catalano S, Mauro L, Marsico S, Giordano C, Rizza P, Rago V, et al. Leptin induces, via ERK1/ERK2 signal, functional activation of estrogen receptor alpha in MCF-7 cells. J Biol Chem. 2004;279:19908–15. doi: 10.1074/jbc.M313191200. [DOI] [PubMed] [Google Scholar]
- 14.Choi JH, Lee KT, Leung PC. Estrogen receptor alpha pathway is involved in leptin-induced ovarian cancer cell growth. Carcinogenesis. 2011;32:589–96. doi: 10.1093/carcin/bgq276. [DOI] [PubMed] [Google Scholar]
- 15.Bertone ER, Rosner BA, Hunter DJ, Stampfer MJ, Speizer FE, Colditz GA, et al. Dietary fat intake and ovarian cancer in a cohort of US women. Am J Epidemiol. 2002;156:22–31. doi: 10.1093/aje/kwf008. [DOI] [PubMed] [Google Scholar]
- 16.Verkasalo PK, Thomas HV, Appleby PN, Davey GK, Key TJ. Circulating levels of sex hormones and their relation to risk factors for breast cancer: a cross-sectional study in 1092 pre- and postmenopausal women (United Kingdom) Cancer Causes Control. 2001;12:47–59. doi: 10.1023/a:1008929714862. [DOI] [PubMed] [Google Scholar]
- 17.Moran C, Garcia-Hernandez E, Cortes MA, Calzada L, Salazar L, Bermudez JA. Estradiol and progesterone endometrial receptors and body fat distribution in obese women. Gynecol Obstet Invest. 1996;42:117–9. doi: 10.1159/000291916. [DOI] [PubMed] [Google Scholar]
- 18.Garland CF, Garland FC, Gorham ED, Lipkin M, Newmark H, Mohr SB, et al. The role of vitamin D in cancer prevention. Am J Public Health. 2006;96:252–61. doi: 10.2105/AJPH.2004.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–81. doi: 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- 20.Jiang F, Li P, Fornace AJ, Jr, Nicosia SV, Bai W. G2/M arrest by 1,25-dihydroxyvitamin D3 in ovarian cancer cells mediated through the induction of GADD45 via an exonic enhancer. J Biol Chem. 2003;278:48030–40. doi: 10.1074/jbc.M308430200. [DOI] [PubMed] [Google Scholar]
- 21.Shen Z, Zhang X, Tang J, Kasiappan R, Jinwal U, Li P, et al. The coupling of epidermal growth factor receptor down regulation by 1alpha,25-dihydroxyvitamin D3 to the hormone-induced cell cycle arrest at the G1-S checkpoint in ovarian cancer cells. Mol Cell Endocrinol. 2011;338:58–67. doi: 10.1016/j.mce.2011.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Nicosia SV, Bai W. Vitamin D receptor is a novel drug target for ovarian cancer treatment. Curr Cancer Drug Targets. 2006;6:229–44. doi: 10.2174/156800906776842939. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, Li P, Bao J, Nicosia SV, Wang H, Enkemann SA, et al. Suppression of death receptor-mediated apoptosis by 1,25-dihydroxyvitamin D3 revealed by microarray analysis. J Biol Chem. 2005;280:35458–68. doi: 10.1074/jbc.M506648200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Jiang F, Li P, Li C, Ma Q, Nicosia SV, et al. Growth suppression of ovarian cancer xenografts in nude mice by vitamin D analogue EB1089. Clin Cancer Res. 2005;11:323–8. [PubMed] [Google Scholar]
- 25.Jiang F, Bao J, Li P, Nicosia SV, Bai W. Induction of ovarian cancer cell apoptosis by 1,25-dihydroxyvitamin D3 through the down-regulation of telomerase. J Biol Chem. 2004;279:53213–21. doi: 10.1074/jbc.M410395200. [DOI] [PubMed] [Google Scholar]
- 26.Kasiappan R, Shen Z, Tse AK, Jinwal U, Tang J, Lungchukiet P, et al. 1,25-Dihydroxyvitamin D3 Suppresses Telomerase Expression and Human Cancer Growth through MicroRNA-498. J Biol Chem. 2012;287:41297–309. doi: 10.1074/jbc.M112.407189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bao J, Cao C, Zhang X, Jiang F, Nicosia SV, Bai W. Suppression of beta-amyloid precursor protein signaling into the nucleus by estrogens mediated through complex formation between the estrogen receptor and Fe65. Mol Cell Biol. 2007;27:1321–33. doi: 10.1128/MCB.01280-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baldwin WS, Curtis SW, Cauthen CA, Risinger JI, Korach KS, Barrett JC. BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro. In Vitro Cell Dev Biol Anim. 1998;34:649–54. doi: 10.1007/s11626-996-0015-9. [DOI] [PubMed] [Google Scholar]
- 29.Pinelli DM, Drake J, Williams MC, Cavanagh D, Becker JL. Hormonal modulation of Ishikawa cells during three-dimensional growth in vitro. J Soc Gynecol Investig. 1998;5:217–23. doi: 10.1016/s1071-5576(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 30.Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009;69:6381–6. doi: 10.1158/0008-5472.CAN-09-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 32.Kyo S, Takakura M, Kanaya T, Zhuo W, Fujimoto K, Nishio Y, et al. Estrogen activates telomerase. Cancer Res. 1999;59:5917–21. [PubMed] [Google Scholar]
- 33.Kimura A, Ohmichi M, Kawagoe J, Kyo S, Mabuchi S, Takahashi T, et al. Induction of hTERT expression and phosphorylation by estrogen via Akt cascade in human ovarian cancer cell lines. Oncogene. 2004;23:4505–15. doi: 10.1038/sj.onc.1207582. [DOI] [PubMed] [Google Scholar]
- 34.Misiti S, Nanni S, Fontemaggi G, Cong YS, Wen J, Hirte HW, et al. Induction of hTERT expression and telomerase activity by estrogens in human ovary epithelium cells. Mol Cell Biol. 2000;20:3764–71. doi: 10.1128/mcb.20.11.3764-3771.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou C, Steplowski TA, Dickens HK, Malloy KM, Gehrig PA, Boggess JF, et al. Estrogen induction of telomerase activity through regulation of the mitogen-activated protein kinase (MAPK) dependent pathway in human endometrial cancer cells. PLoS One. 2013;8:e55730. doi: 10.1371/journal.pone.0055730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lonard DM, Nawaz Z, Smith CL, O’Malley BW. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol Cell. 2000;5:939–48. doi: 10.1016/s1097-2765(00)80259-2. [DOI] [PubMed] [Google Scholar]
- 37.Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–4. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- 38.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 39.Swami S, Krishnan AV, Feldman D. 1alpha,25-Dihydroxyvitamin D3 down-regulates estrogen receptor abundance and suppresses estrogen actions in MCF-7 human breast cancer cells. Clin Cancer Res. 2000;6:3371–9. [PubMed] [Google Scholar]
- 40.Demirpence E, Balaguer P, Trousse F, Nicolas JC, Pons M, Gagne D. Antiestrogenic effects of all-trans-retinoic acid and 1,25-dihydroxyvitamin D3 in breast cancer cells occur at the estrogen response element level but through different molecular mechanisms. Cancer Res. 1994;54:1458–64. [PubMed] [Google Scholar]
- 41.Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342:1094–8. doi: 10.1126/science.1241908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ren H, Zhao T, Wang X, Gao C, Wang J, Yu M, et al. Leptin upregulates telomerase activity and transcription of human telomerase reverse transcriptase in MCF-7 breast cancer cells. Biochem Biophys Res Commun. 2010;394:59–63. doi: 10.1016/j.bbrc.2010.02.093. [DOI] [PubMed] [Google Scholar]
- 43.Stefanou N, Papanikolaou V, Furukawa Y, Nakamura Y, Tsezou A. Leptin as a critical regulator of hepatocellular carcinoma development through modulation of human telomerase reverse transcriptase. BMC Cancer. 2010;10:442. doi: 10.1186/1471-2407-10-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leitzmann MF, Koebnick C, Danforth KN, Brinton LA, Moore SC, Hollenbeck AR, et al. Body mass index and risk of ovarian cancer. Cancer. 2009;115:812–22. doi: 10.1002/cncr.24086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schouten LJ, Rivera C, Hunter DJ, Spiegelman D, Adami HO, Arslan A, et al. Height, body mass index, and ovarian cancer: a pooled analysis of 12 cohort studies. Cancer Epidemiol Biomarkers Prev. 2008;17:902–12. doi: 10.1158/1055-9965.EPI-07-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olsen CM, Green AC, Whiteman DC, Sadeghi S, Kolahdooz F, Webb PM. Obesity and the risk of epithelial ovarian cancer: a systematic review and meta-analysis. Eur J Cancer. 2007;43:690–709. doi: 10.1016/j.ejca.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 47.Anderson GL, Judd HL, Kaunitz AM, Barad DH, Beresford SA, Pettinger M, et al. Effects of estrogen plus progestin on gynecologic cancers and associated diagnostic procedures: the Women’s Health Initiative randomized trial. JAMA. 2003;290:1739–48. doi: 10.1001/jama.290.13.1739. [DOI] [PubMed] [Google Scholar]
- 48.Yang HP, Anderson WF, Rosenberg PS, Trabert B, Gierach GL, Wentzensen N, et al. Ovarian cancer incidence trends in relation to changing patterns of menopausal hormone therapy use in the United States. J Clin Oncol. 2013;31:2146–51. doi: 10.1200/JCO.2012.45.5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng W, Danforth KN, Tworoger SS, Goodman MT, Arslan AA, Patel AV, et al. Circulating 25-hydroxyvitamin D and risk of epithelial ovarian cancer: Cohort Consortium Vitamin D Pooling Project of Rarer Cancers. Am J Epidemiol. 2010;172:70–80. doi: 10.1093/aje/kwq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lagunova Z, Porojnicu AC, Grant WB, Bruland O, Moan JE. Obesity and increased risk of cancer: does decrease of serum 25-hydroxyvitamin D level with increasing body mass index explain some of the association? Mol Nutr Food Res. 2010;54:1127–33. doi: 10.1002/mnfr.200900512. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.