

Abstract
We have previously demonstrated that it is possible to effectively vaccinate against long-term murine gammaherpesvirus 68 (γHV68) latency by using a reactivation-deficient virus as a vaccine (S. A. Tibbetts, J. S. McClellan, S. Gangappa, S. H. Speck, and H. W. Virgin IV, J. Virol. 77:2522-2529, 2003). Immune antibody was capable of recapitulating aspects of this vaccination. This led us to determine whether antibody is required for vaccination against latency. Using mice lacking antigen-specific antibody responses, we demonstrate here that antibody and B cells are not required for vaccination against latency. We also show that surveillance of latent infection in normal animals depends on CD4 and CD8 T cells, suggesting that T cells might be capable of preventing the establishment of latency. In the absence of an antibody response, CD4 T cells but not CD8 T cells are required for effective vaccination against latency in peritoneal cells, while either CD4 or CD8 T cells can prevent the establishment of splenic latency. Therefore, CD4 T cells play a critical role in immune surveillance of gammaherpesvirus latency and can mediate vaccination against latency in the absence of antibody responses.
The human gammaherpesviruses Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus cause significant morbidity and mortality worldwide. Primary gammaherpesvirus infection typically produces a mild or subclinical illness associated with a period of lytic replication that is cleared by the immune system (22, 28). However, these viruses evade complete clearance by the host immune response and establish latent infection in cells of the hematopoietic lineage. This capacity to persist despite active immunity leaves the host susceptible to subsequent virus-induced disease. Gammaherpesvirus-associated disease is particularly common in the setting of immunocompromise, an association that has been described for both mice and humans (22, 28, 46). For example, the development of Kaposi's sarcoma in patients with AIDS is strongly associated with prior latent Kaposi's sarcoma-associated herpesvirus infection (3, 22), and the development of posttransplant lymphoproliferatve disease correlates with the level of latent EBV (28, 29). Additionally, B-cell lymphomas and chronic vasculitis develop in immunocompromised mice infected with murine gammaherpesvirus 68 (γHV68) (34, 46; F. Suarez, S. A. Tibbetts, M. Jacoby, S. H. Speck, and H. W. Virgin, unpublished data).
Several groups have tested the hypothesis that high levels of preexisting immunity might attenuate chronic gammaherpesvirus disease by limiting latent infection (reviewed in reference 39). Vaccination against γHV68 infection with single viral antigens attenuates acute infection and decreases the amount of latent infection at early time points (2 to 3 weeks of infection). For example, vaccination against the major membrane glycoprotein gp150 induces a neutralizing antibody response and reduces the number of latently infected cells at day 14 after infection (32). Similarly, T-cell vaccination using immunodominant CD8 T-cell epitopes derived from lytic cycle antigens decreases both acute titer and latency at day 14 after infection (20), and CD8 T cells specific for a latent viral antigen decrease latency early after infection (41). Despite achieving success in the control of acute and early latent infection, these approaches fail to produce a detectable change in long-term latency (day 28 after infection and beyond).
The failure of vaccination with single viral antigens to decrease long-term latency led us to pursue live-attenuated virus vaccination to test the hypothesis that a sufficiently robust preexisting immune response can inhibit or eliminate latent infection. This approach has met with considerable success in other systems. Replication-defective viruses have been used to vaccinate against herpes simplex virus in mice (6, 23, 24), and a live-attenuated varicella-zoster virus vaccine is useful in humans (37, 49). Vaccination of mice with an attenuated murine cytomegalovirus mutant significantly decreases establishment of latency by murine cytomegalovirus (21).
We found that infection with a reactivation-deficient mutant strain of γHV68, γHV68.v-cyclin.LacZ, successfully protects against the establishment of latent infection after challenge with wild-type γHV68 (39). γHV68.v-cyclin.LacZ, generated by replacing the v-cyclin locus with a LacZ expression cassette, establishes both a normal acute infection and a normal level of latent infection but reactivates from latent infection inefficiently (13, 15, 39, 43, 44). Prior infection with γHV68.v-cyclin.LacZ reduces both the acute replication and latency of wild-type challenge virus to undetectable levels. The effect of vaccination is present as late as 125 days postchallenge.
The mechanism responsible for vaccination-mediated protection against γHV68 latency is not completely understood. Vaccination is effective in CD8α-deficient animals, demonstrating that CD8 T cells are not required to achieve vaccination against latency (39). In addition, passive transfer of serum from vaccinated animals to naive mice prevents the establishment of splenic latency upon challenge, demonstrating that antibody can mediate protection against the establishment of latency (39). However, immune serum transfer does not completely recapitulate the protection against latency observed in vaccinated mice, suggesting that additional immune mechanisms are capable of limiting the establishment of a latent infection.
In this study, we first determined whether specific antibody responses were required for vaccination against gammaherpesvirus latency. Interestingly, we found that a virus-specific antibody response was not required for protection against the establishment of latent infection, suggesting that either CD4 T cells or CD8 T cells limited the establishment of latency. Further support for the importance of T cells came from the demonstration that both CD4 and CD8 T cells were required for surveillance against γHV68 latency. Using mice that possess B cells but cannot make antigen-specific antibody responses, we found that CD4 T cells were required to mediate protection against the establishment of latency.
MATERIALS AND METHODS
Viruses and mice.
γHV68 clone WUMS (ATCC VR1465) and γHV68.v-cyclin.LacZ were passed, and titers were determined by plaque assay, on NIH 3T12 cells (13, 39, 43, 44). γHV68.v-cyclin.LacZ contains a LacZ expression cassette (the β-galactosidase gene driven by the human cytomegalovirus immediate-early promoter-enhancer) in place of the first 475 bp of the v-cyclin gene (43). C57BL/6J (B6) and μMT mice on a B6 background (B-cell−/− mice [19]; Jackson Laboratories strain 002288) were obtained from Jackson Laboratories (Bar Harbor, Maine), and were bred at Washington University School of Medicine in accordance with all federal and university guidelines. B-cell−/− mice were bred together with mice that carry transgenes encoding immunoglobulin M (IgM) and IgD heavy- and light-chain cell surface receptors specific for hen egg lysozyme (HEL) (14). These mice (henceforth referred to as HELMET mice) contain normal levels of B cells, but all of the B cells are specific for HEL and the mice fail to produce virus-specific anti-γHV68 antibody responses (K. A. Brett et al., unpublished data). Eight- to 12-week-old animals were used for all experiments.
Vaccination and challenge virus infections.
Mice were vaccinated intraperitoneally (i.p.) with 106 PFU of γHV68.v-cyclin.LacZ diluted in 0.5 ml of Dulbecco's modified Eagle's medium (DMEM) or were mock vaccinated with an NIH 3T12 cell lysate diluted in 0.5 ml of DMEM (39). Twenty-eight days later mice were challenged i.p. with 100 PFU of γHV68 in 0.5 ml of DMEM. At the end of the challenge period, mice were sacrificed and organs from three to five mice per group were harvested and pooled as described previously (13, 39, 43, 44).
In vivo depletion of lymphocyte subsets.
Monoclonal antibodies (MAbs) specific to CD4 (YTS191.1 [5]) and CD8 (H35 [31]) were used to deplete mice of lymphocyte subsets. Hybridomas producing these MAbs were grown in protein-free medium (ADCF-MAb; HyClone, Logan, Utah) in Celline CL1000 flasks (Integra Biosciences, Ijamsville, Md.). Supernatants from these flasks were sterile filtered, and the amount of IgG was quantified by enzyme-linked immunosorbent assay for total rat IgG. Beginning 1 day prior to challenge, 500 μg of lymphocyte-depleting antibody or an isotype-matched control antibody (SFR3-DR5 [IgG2b]; ATCC HB-151 [27]) was administered to each mouse by i.p. injection. MAb treatment was then repeated every fourth day. The efficacy of depletion was monitored by using flow cytometric analysis of splenocytes with anti-mouse CD4-phycoerythrin (BD PharMingen, San Jose, Calif.) and anti-mouse CD8α-Tri-Color (Caltag, Burlingame, Calif.). Efficacy of depletion was measured on the day of harvest (4 days after the final administration of antibody). In experiments involving T-cell depletion, the efficacy of depletion was 98% or greater in each case.
Ex vivo limiting-dilution reactivation analysis.
The frequency of cells reactivating from latency was assayed as described previously (13, 39, 43, 44). Briefly, serial twofold dilutions of harvested cells (24 wells per dilution starting at 1 × 105 cells per well for splenocytes and 4 × 104 cells per well for peritoneal cells) were plated onto permissive mouse embryonic fibroblast monolayers for 21 days and then scored for cytopathic effect due to reactivating virus. To determine whether samples contained preformed infectious virus as a result of persistent replication, replicate cell aliquots were mechanically disrupted in 1/3× DMEM in the presence of 0.5-mm-diameter silica beads prior to limiting dilution and plating. This procedure kills >99% of cells but has at most a twofold effect on viral titer, thus allowing experimental distinction between reactivation from latency (which requires live cells) and persistent replication (45). Unless otherwise noted, samples contained no persistently replicating virus.
Single-well β-galactosidase staining.
In samples from mice that were both vaccinated and challenged, the identity of reactivating virus was determined by staining for the presence of β-galactosidase (γHV68.v-cyclin.LacZ but not γHV68 encodes β-galactosidase). The contents of individual wells containing single reactivation events (cell dilutions that yielded ≤63.2% of wells positive for cytopathic effect) were replated onto fresh mouse embryonic fibroblast monolayers, fixed with 2% paraformaldehyde, and stained overnight at 37°C with X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) [2.2 mM X-Gal, 2 mM MgCl2, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6 · H2O] (2). White or blue monolayer color was evaluated microscopically.
Limiting-dilution PCR analysis.
To determine the frequency of cells carrying the γHV68 (challenge virus) genome, single-copy sensitivity nested PCR for the γHV68 v-cyclin gene (gene 72), was performed on serial dilutions of cells as described previously (39). Primers for v-cyclin were 5′GAGATCTGTACTCAGGCACCTGT3′ and 5′GGATTTCTTGACAGCTCCCTGT3′ for round 1 and 5′TGTCAGCTGTTGTTGCTCCT3′ and 5′CTCCGTCAGGATAACAACGTCT3′ for round 2. One false positive was detected in a total of 210 reactions across all experiments. Positive control reactions with 10, 1, or 0.1 copy of v-cyclin plasmid DNA were positive in 99, 44, and 2% of all reactions, respectively. The single-copy sensitivity nested-PCR assay to determine the frequency of cells carrying the γHV68.v-cyclin.LacZ (vaccine strain) genome has been described previously (39).
Statistical analysis.
All data points represent the mean ± standard error of the mean for two or three experiments with three to five mice per condition per experiment. To quantify the number of cells from which the virus reactivated or which carry latent viral genome, data were subjected to nonlinear regression (sigmoidal dose curve with nonvariable slope) by using Prism (GraphPad, San Diego, Calif.). Frequencies of reactivation events or genome-positive cells were determined by using the Poisson distribution, assuming that the cell number at which 63.2% of the wells scored positive for reactivation or viral genome represented a single event. The reactivation efficiency of a given cell sample has been defined as the frequency of reactivation within that sample divided by the frequency of cells containing viral genome in that sample (expressed as a percentage) (40). To calculate significance, data were statistically analyzed by use of the paired t test over the range of dilutions.
RESULTS
CD4 and CD8 T cells participate in immune surveillance of γHV68 latency.
We and others have demonstrated a role for B cells and antibody in regulation of γHV68 latency (12, 18, 47), and we found that passively transferred immune antibody can partly prevent the establishment of latency (39). Surprisingly, we found that CD8 T cells were not required for vaccination against γHV68 latency (39). This contrasted with the findings of studies using immunodeficient mice, which suggested that CD4 and CD8 T cells are critical for the control of chronic γHV68 infection (1, 40). We therefore questioned whether either CD4 or CD8 T cells are necessary to maintain latent infection at a given frequency after latency is established in a wild-type animal. At 28 days following infection with γHV68, B6 mice were depleted of CD4 T cells, CD8 T cells, or both CD4 and CD8 T cells for 14 days. By 28 days after infection, acute infection has been cleared (35, 45), so any effect of T-cell depletion observed could be attributed to an ongoing role of the T cells in surveillance of γHV68 latency rather than to effects of the T cells on acute γHV68 infection. The efficacy of depletion was monitored by flow cytometry and was >98% in each case. Control mice were treated with an isotype-matched control antibody or vehicle alone.
Depletion of either CD4 (P = 0.01) or CD8 (P = 0.04) cells alone resulted in an approximately threefold increase in the frequency of peritoneal cells that reactivated ex vivo (Fig. 1A) but did not significantly change the frequency of cells carrying the viral genome (Fig. 1C). In contrast to results obtained when either CD4 or CD8 T cells were individually depleted, depletion of both CD4 and CD8 T cells resulted in an approximately 20-fold increase (P < 0.001) in the frequency of peritoneal cells that reactivated ex vivo. This increase was not due solely to an increase in the efficiency of reactivation, since there was a 10-fold increase in the frequency of peritoneal cells carrying the viral genome (Fig. 1C) (P = 0.04). Thus, depletion of both CD4 and CD8 T cells increased the reactivation efficiency of latently infected peritoneal cells to 15%, compared to 6% in cells harvested from the isotype control-treated group. Therefore, depletion of both T-cell subsets increased but the absolute number of latently infected cells had only minor effects on the efficiency with which these cells reactivated ex vivo. Preformed infectious virus, likely the result of reactivation from latency in vivo, was also detected in peritoneal cells after depletion of both CD4 and CD8 T cells but not when either T-cell subset was individually depleted (Fig. 1B). Thus, significant increases in latency and preformed infectious virus were observed only when both CD4 and CD8 T cells were depleted, suggesting that these subsets have partially overlapping roles in the control of latent infection in the peritoneum. In contrast to the significant effect of depletion of both CD4 and CD8 T cells on latency in peritoneal cells (Fig. 1), this depletion regimen had no effect on latency in the spleen (data not shown). We do not understand the reason for this site-specific difference in the effects of T-cell depletion on latency. However, we conclude that either CD4 or CD8 T cells are required to control the level of latency in peritoneal cells after infection. These data are consistent with previous data showing a role for both CD4 and CD8 T cells in the control of chronic γHV68 infection (1, 40).
FIG. 1.
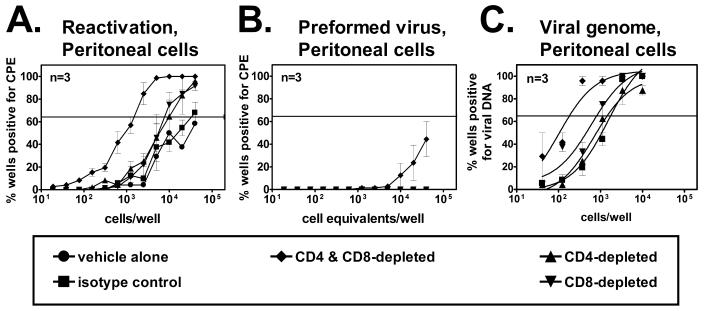
CD4 and CD8 T cells are responsible for surveillance of latent infection in wild-type mice. (A) Reactivation from latency in peritoneal cells from B6 mice treated with an irrelevant control antibody or depleted of CD4 T cells, CD8 T cells, or both CD4 and CD8 T cells beginning 28 days after infection. The horizontal line indicates the 63.2% Poisson distribution line used to calculate the frequency of cells reactivating virus or containing viral DNA. (B) Preformed virus in mechanically disrupted peritoneal cell samples. (C) Frequency of peritoneal cells containing viral DNA.
CD4 and CD8 T cells are not required at time of challenge to mediate protection against latent infection in wild-type mice.
Since our findings indicated that both CD4 and CD8 T cells were involved in the maintenance of stable levels of latency, we questioned whether immune CD4 and/or CD8 T cells would be required for vaccine-mediated protection against the establishment of latency in wild-type animals. B6 mice were vaccinated or mock vaccinated with γHV68.v-cyclin.LacZ, and, beginning 28 days later, vaccinated groups were depleted of CD4, CD8, or both CD4 and CD8 cells. A vaccinated group and the mock-vaccinated control mice were treated with an isotype-matched control antibody. All groups were then challenged with wild-type γHV68 on the subsequent day. The efficacy of depletion was >98% in each case. At day 16 postchallenge, the frequencies of peritoneal cells and splenocytes from mock-vaccinated and challenged mice that reactivated ex vivo were 1 in 635 and 1 in 2100, respectively (Fig. 2). Little to no reactivation of wild-type challenge virus was noted in cells derived from mice that were vaccinated and challenged. Vaccination reduced the frequencies of reactivation in the peritoneal cells and splenocytes by greater than 600-fold (P = 0.0004) and 2,000-fold (P = 0.0007), respectively. Relative to control antibody-treated groups, vaccination followed by depletion of CD8 T cells prior to challenge had no effect on the frequency of reactivation in peritoneal cells or splenocytes (Fig. 2). Depletion of either CD4 T cells or both CD4 and CD8 T cells resulted in increased frequencies of reactivation in the peritoneal cells (Fig. 2A). However, this increase was entirely due to an increase in reactivation of the vaccine strain rather than the challenge strain, since 100% of the reactivating wells (81 of 81 wells, total for all vaccinated and challenged groups) stained positive for β-galactosidase activity (only the vaccine strain contains a LacZ expression cassette). The finding that the vaccine virus reactivated ex vivo following T-cell depletion is consistent with previous studies demonstrating detectable reactivation of γHV68.v-cyclin.LacZ in cells obtained from immunocompromised mice (13, 39). Depletion of T cells from mice that were vaccinated and challenged did not have a statistically significant effect on the frequency of reactivating splenocytes (Fig. 2B). Thus, as judged by ex vivo reactivation of challenge virus, vaccination was protective against latent-infection virus following T-cell depletion.
FIG. 2.
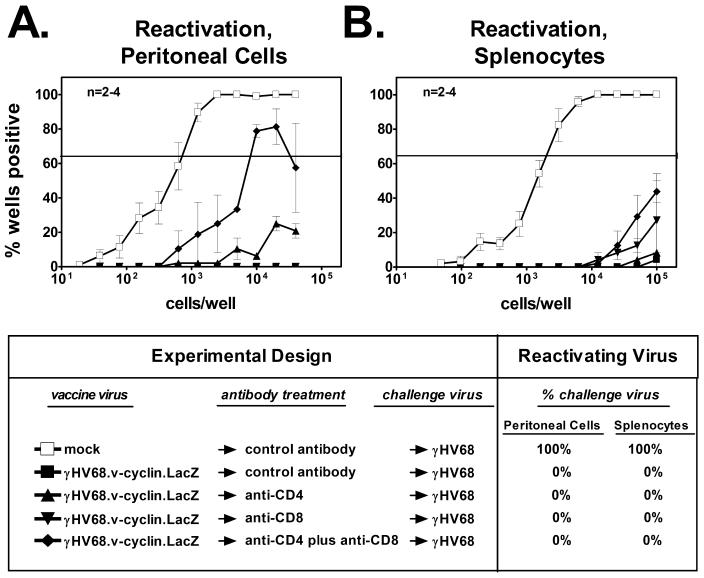
CD4 and CD8 T cells are not required to mediate vaccination against latency in wild-type mice. (A) Reactivation from latency in peritoneal cells from B6 mice that were either mock vaccinated or vaccinated and then treated with control or T-cell-depleting antibodies prior to challenge. Percent challenge virus refers to the percentage of wells containing single reactivation events (wells at cell dilutions which yielded ≤63.2% cytopathic effect for that dilution) that were positive for challenge virus (indicated by the absence of β-galactosidase activity). (B) Reactivation from latency in splenocytes from B6 mice treated as described for panel A.
To test for the presence of the viral genome following vaccination, we used a limiting-dilution nested-PCR assay that detects the challenge virus (wild-type γHV68) but not the vaccine strain and is sensitive to one copy of plasmid DNA (39). In peritoneal cells and splenocytes from mice that were mock vaccinated and challenged, the challenge virus genome was readily detected by PCR (data not shown). In contrast, the challenge virus genome was not detected in either peritoneal cells or splenocytes from any of the vaccinated and challenged groups (data not shown). This confirmed that vaccination against latency was effective even in mice depleted of T cells prior to challenge. While it is possible that we did not fully deplete T cells in these experiments, these data were most consistent with a role for immune antibody in the vaccination effect (12, 18, 39).
B cells are not required to protect against the establishment of latent infection.
Based on the results described above, we sought to determine whether B cells are required for vaccine-mediated protection against the establishment of challenge virus latency. We thus vaccinated and challenged B-cell-deficient mice (19). Efficient reactivation was observed in 1 in 150 peritoneal cells and 1 in 59,000 splenocytes from B-cell−/− animals that were mock vaccinated and then challenged (Fig. 3A and C). Compared to mock-vaccinated groups, vaccination prior to challenge significantly reduced the both the frequency of reactivating peritoneal cells (1 in 1,544; P < 0.001) and the frequency of reactivating splenocytes (less than 1 in 100,000; P < 0.005). Additionally, 30 out of 30 sampled wells containing single reactivation events from the vaccinated and challenged group stained positive for β-galactosidase activity, indicating that all of these reactivation events were attributable to the vaccine strain rather than the challenge strain. This indicated that vaccination against latency was effective even in the absence of B cells.
FIG. 3.
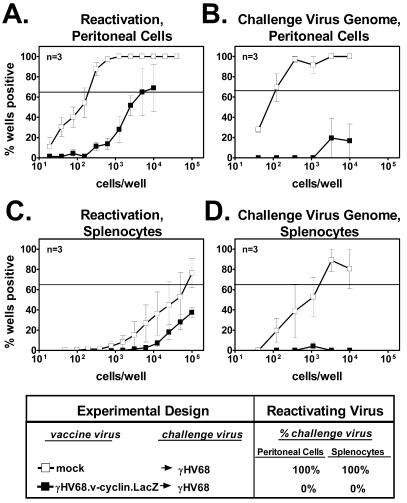
B cells are not required for vaccination against latency. (A) Reactivation from latency in peritoneal cells from B-cell−/− mice that were either vaccinated and challenged or mock vaccinated and challenged. Data are presented as described in the Fig. 2 legend. (B) Frequency of peritoneal cells from B-cell−/− mice containing challenge virus genome. (C) Reactivation from latency in splenocytes from B-cell−/− mice that were either vaccinated and challenged or mock vaccinated and challenged. (D) Frequency of splenocytes from B-cell−/− mice containing challenge virus genome.
Results from a limiting-dilution PCR to detect challenge virus confirmed that vaccination against latency was effective in B-cell−/− animals. The frequencies of γHV68 genome-bearing peritoneal cells and splenocytes derived from mock-vaccinated B-cell−/− mice were 1 in 90 and 1 in 1,200, respectively. In contrast, the frequency of cells containing γHV68 genome was very low or undetectable in mice that were vaccinated prior to challenge (Fig. 3B and D). Vaccination prior to challenge reduced the frequencies of peritoneal cells and splenocytes containing challenge virus genome by at least 110-fold (P < 0.001) and 8-fold (P = 0.02), respectively. This confirmed that protection against latency did not require B cells.
A specific antibody response is not required to mediate vaccination against latency.
B-cell−/− mice provide a useful model to study the importance of B cells in vaccination. However, since B cells are an important reservoir of latent γHV68 infection (10, 11, 36, 48, 50), B-cell−/− animals do not provide a setting in which to examine the effects of vaccination on B-cell latency. To perform an experiment determining whether the presence of B cells might alter the requirement for antibody in prevention of latency, we generated mice (HELMET mice) that contain normal numbers of B cells but are incapable of mounting a virus-specific antibody response (Brett et al., unpublished data).
HELMET mice were either mock vaccinated or vaccinated and then challenged with γHV68. While efficient reactivation was noted in peritoneal cells and splenocytes from mice that were mock vaccinated (frequencies of 1 in 180 and 1 in 32,000, respectively), little reactivation was noted in peritoneal cells and splenocytes derived from mice that were vaccinated prior to challenge (Fig. 4A and C, respectively). The low level of reactivating virus detected in these cells was due exclusively to reactivation of γHV68.v-cyclin.LacZ, as determined by staining for β-galactosidase activity. Limiting-dilution PCR to detect challenge virus confirmed the absence of the challenge virus genome in HELMET mice that were vaccinated and challenged (Fig. 4B and D). Vaccination prior to challenge reduced the frequency of peritoneal cells and splenocytes containing the challenge virus genome by at least 150-fold (P < 0.001) and 5-fold (P = 0.03), respectively. Therefore, as observed in mice completely devoid of B cells (Fig. 3), vaccination of HELMET mice prior to challenge significantly reduced the establishment of latency by the challenge strain in both the peritoneal cells and splenocytes.
FIG. 4.
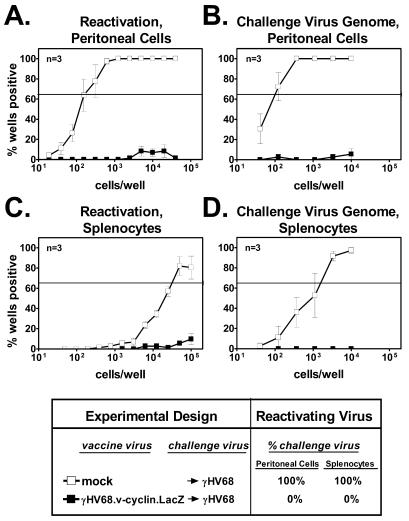
An antiviral antibody response is not required for vaccination against latency. (A) Reactivation from latency in peritoneal cells from HELMET mice that were either vaccinated and challenged or mock vaccinated and challenged. Data are presented as described in the Fig. 2 legend. (B) Frequency of peritoneal cells from HELMET mice containing challenge virus genome. (C) Reactivation from latency in splenocytes from HELMET mice that were either vaccinated and challenged or mock vaccinated and challenged. (D) Frequency of splenocytes from HELMET mice containing challenge virus genome.
CD4, but not CD8, T cells are required for vaccination against the establishment of latency in peritoneal cells in the absence of antibody.
Given that CD4 T cells, CD8 T cells, B cells, and antibody are each individually nonessential for protection against latency (Fig. 2, 3, and 4), we questioned whether T cells and antibody may play overlapping roles during vaccination. We therefore sought to determine whether CD4 or CD8 T cells are necessary for vaccination in the absence of a specific antibody response. To address this question, HELMET mice were vaccinated and depleted of CD4, CD8, or both CD4 and CD8 T cells beginning 1 day prior to challenge. The efficacy of depletion was >98% in each case. As noted above, the frequency of reactivating peritoneal cells from mock-vaccinated and challenged HELMET mice was 1 in 180 (Fig. 4A and 5A). In contrast, little to no reactivation was noted in peritoneal cells from mice that were vaccinated and depleted of CD8 T cells prior to challenge with wild-type γHV68 (Fig. 5A). Thus, even following CD8 T-cell depletion of HELMET mice, vaccination reduced the frequency of peritoneal cells that reactivated by at least 220-fold (P < 0.001). Within the vaccinated groups that were given either control antibody or CD8 T-cell-depleting antibody, all wells from peritoneal cell dilutions that contained reactivating virus also stained positive for β-galactosidase activity, indicating that the reactivation observed was solely due to vaccinating virus rather than to the wild-type challenge virus. Furthermore, the wild-type γHV68 challenge virus genome was not detected in peritoneal cells from groups that were treated with either isotype control or CD8-depleting antibody (Fig. 5B). Thus, confirming earlier studies using CD8−/− mice (39), CD8 T cells are not required to protect against latent infection in the peritoneal cell population even in the absence of a specific antibody response.
FIG. 5.
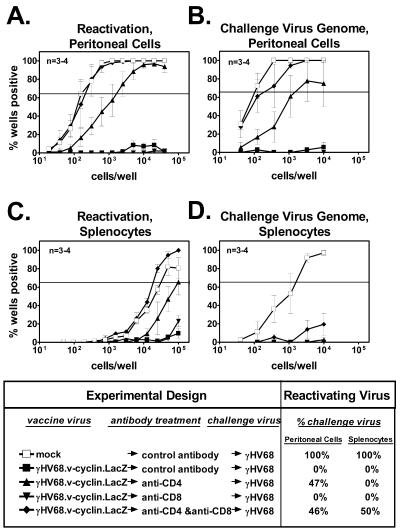
CD4 T cells are required to mediate vaccination against latency in the absence of an antiviral antibody response. (A) Reactivation from latency in peritoneal cells from HELMET mice that were either vaccinated and challenged or mock vaccinated and challenged and then treated with control antibodies or T-cell-depleting antibodies. Data are presented as described in the Fig. 2 legend. (B) Frequency of peritoneal cells from HELMET mice, treated as described for panel A, containing challenge virus genome. (C) Reactivation from latency in splenocytes from HELMET mice treated as described for panel A. (D) Frequency of splenocytes that contain challenge virus genome from HELMET mice treated as described for panel A. Data from Fig. 4 are repeated for clarity.
Compared to the isotype control-treated and CD8 T-cell-depleted groups, depletion of CD4 T cells from HELMET mice prior to challenge resulted in at least a 30-fold increase (P < 0.001) in the frequency of peritoneal cells that reactivated from latency (Fig. 5A). Seventeen of 36 wells (47%) containing single reactivation events were negative for β-galactosidase activity, indicating that a portion of peritoneal cells from these animals were latently infected with the challenge wild-type γHV68. Simultaneous depletion of both CD4 and CD8 T cells resulted in an even greater increase (>200-fold; P < 0.0001) in the frequency of peritoneal cells that reactivated. Eighteen of 39 wells (46%) containing single reactivation events after simultaneous CD4 and CD8 T-cell depletion were attributable to the challenge virus as judged by β-galactosidase staining. Additionally, using a limiting-dilution PCR assay, we detected the wild-type γHV68 genome in peritoneal cells from groups that were vaccinated and treated either with anti-CD4 or both anti-CD4 and anti-CD8 prior to challenge (1 in 1,900 and 1 in 300 cells, respectively) (Fig. 5B). In contrast, very little challenge virus genome was detected in mice that were vaccinated and treated with control antibody prior to challenge. Relative to the isotype-control treated group, vaccination combined with depletion of CD4 T cells or both CD4 T cells and CD8 T cells prior to challenge resulted in a significant increase in the frequency of cells scoring positive for the challenge virus genome (P = 0.02 and P = 0.001, respectively). Additionally, only peritoneal cells from these two groups contained a detectable amount of preformed infectious virus (data not shown). This confirmed that CD4 T cells were required to protect against the establishment of latency in peritoneal cells in the absence of a specific antibody response.
Compared to depletion of CD4 T cells alone, depletion of CD4 and CD8 T cells simultaneously prior to challenge resulted in a statistically significant increase in wild-type γHV68 latency (Fig. 5A and B). This was true for latency measured by ex vivo reactivation and for the frequency of viral genome-bearing cells (P < 0.01 and P < 0.05, respectively). Thus, while CD4 T cells are required for efficient protection against latent infection in HELMET mice, vaccine-primed CD8 T cells also play a role in limiting viral latency in peritoneal cells.
Either CD4 or CD8 T cells can mediate protection against latency in the spleen in the absence of antibody.
Efficient reactivation was observed in splenocytes derived from HELMET mice that were mock vaccinated and then challenged (1 in 32,000 cells reactivating ex vivo) (Fig. 4C and 5C). In contrast, little reactivation was noted in splenocytes from vaccinated and challenged mice that were treated with anti-CD8 or isotype control antibody and then challenged. Only depletion of both CD4 and CD8 T cells from vaccinated and then challenged mice resulted in a significantly increased frequency of reactivation relative to the isotype control-treated group (P = 0.02) (Fig. 5C). In splenocytes derived from the vaccinated and double-depleted group, 51% (19 of 37 wells) of the single reactivation events scored negative for β-galactosidase activity. This indicated that roughly half of these events were due to reactivation of the challenge virus. Furthermore, the wild-type γHV68 genome was detected only in splenocytes derived from mice which were mock vaccinated (1 in 1,850) or vaccinated and depleted of both CD4 and CD8 T cells prior to challenge (Fig. 5D). The frequency of genome-bearing cells in the latter case did not reach 63.2% and thus could not be calculated. Thus, simultaneous depletion of both CD4 and CD8 cells was most effective at counteracting the effects of vaccination on the establishment of latency in the spleens of HELMET animals.
In addition to B cells and macrophages, dendritic cells have been identified as sites of latent γHV68 infection (10, 48). A subset of dendritic cells express CD8α (30) and are theoretically susceptible to depletion by anti-CD8 antibody. Thus, it was possible that an otherwise positive effect of T-cell depletion was masked by the simultaneous depletion of latently infected cells. To rule out the possibility that this was confounding our results, we utilized a LacZ-specific PCR assay (described previously [39]) to measure the frequency of cells latently infected with the vaccine strain (γHV68.v-cyclin.LacZ). We reasoned that if anti-CD8 treatment depleted a significant portion of the pool of latently infected cells, we would observe a decrease in the frequency of cells bearing the vaccine virus genome following anti-CD8 treatment. However, we did not observe a decrease in vaccine virus latency in either peritoneal cells or splenocytes following treatment with either anti-CD8 or anti-CD4 antibody (data not shown). Indeed, anti-CD8 treatment resulted in an eightfold (P = 0.04) increase in the frequency of splenocytes bearing the viral genome (data not shown), despite the fact that vaccination against challenge virus latency was still effective in this setting (Fig. 5D).
DISCUSSION
The successful protection against the establishment of long-term γHV68 latency obtained after infection with a reactivation-deficient γHV68 mutant (39) provided us the opportunity to identify the components of the immune system that are required to protect against latent infection. Based on two observations, we were encouraged to pursue the possibility that CD4 or CD8 T cells might be able to prevent establishment of latency in the absence of antibody. First, CD8 T cells specific for the M2 latency protein of γHV68 significantly limited the establishment of latency early after infection (41, 42). Second, we found that simultaneous depletion of both CD4 and CD8 T cells from latently infected wild-type mice can increase the level of viral latency (Fig. 1), a finding consistent with studies by Christensen et al. that employed T-cell depletion in the setting of latently infected B-cell−/− mice (4). We show here that CD4 T cells, even in the absence of antibody responses, can have significant and lasting effects on the establishment of gammaherpesvirus latency. This is confirmed by studies reported in the accompanying paper (31a). Overall, immune CD4 T cells and the antiviral antibody response provided redundant protection against latent infection. These findings suggest that vaccines that prime either CD4 T cells or antibody-secreting B cells may be sufficient to protect against latent gammaherpesvirus infection. However, an optimal vaccine strategy should target both the CD4 T-cell response and the B-cell response.
Both CD4 and CD8 T cells participate in the surveillance of established latent infections.
A central goal of this study was to identify the immune mechanisms responsible for vaccination against latency. We hypothesized that these mechanisms might overlap significantly with those responsible for surveillance of latent infection once established. We thus sought to further define immune components active in surveillance of latency. Studies using knockout mouse models have provided evidence that both CD4 and CD8 T cells are critical components of the immune response to chronic γHV68 infection (1, 40). However, since knockout mice are immunodeficient throughout infection (including during the acute phases of infection), this approach does not assess the importance of a given immune component to the control of latent infection specifically. By administering T-cell-depleting antibodies to B6 mice after the resolution of the acute phase of infection, we were able to assess the contribution of CD4 and CD8 T cells to the surveillance of latent infection directly. Interestingly, only depletion of both subsets simultaneously resulted in a significant increase in latency in the peritoneal cells, suggesting functional redundancy between these T-cell subsets.
In contrast to the effect on peritoneal latency, depletion of both CD4 and CD8 T cells did not result in an increase in latency in the spleen. This finding may appear to conflict with published reports demonstrating defective control of splenic latency in CD8α−/− mice (40). This discrepancy may be due in part to the fact that in CD8α−/− mice, loss of CD8 T-cell function is not limited to the latent phases of infection. Also, once the immune system has been activated by infection, the effect of T-cell depletion on latency may be blunted by compensatory protection by other immune components, such as antiviral antibody. This phenomenon has been observed in analogous T-cell depletion experiments with wild-type mice latently infected with murine cytomegalovirus (26). Overall, our findings indicate that both CD4 and CD8 T cells contribute to the ongoing surveillance of latent infection and are thus in agreement with studies using knockout mouse models.
Role of B cells and antibody in the protection against latent infection.
Previous studies have shown that immune control of γHV68 infection in naive animals occurs via multiple mechanisms, including T cells and antibody (7, 9, 12, 18, 20, 40). In this study, we have demonstrated that effective vaccination against latency virus occurs through several redundant immune effector mechanisms. Surprisingly, mice that possessed an intact humoral immune response did not require CD4 or CD8 T cells during the challenge phase to mediate protection against the establishment of latent infection (Fig. 2). This led to the hypothesis that antiviral antibody is a mediator of vaccination against latent infection. This is consistent with several of our previous findings: (i) transfer of serum from vaccinated mice to naive mice can recapitulate the effect of vaccination in the spleen, and (ii) CD8-deficient mice can be effectively vaccinated (39). This is also consistent with the observation that induction of a neutralizing antibody response can attenuate latent infection at early time points (32).
While the antibody response clearly contributes to vaccination, antibody is not required to mediate vaccination, since vaccination was protective against the establishment of latent infection in B-cell−/− mice. The use of B-cell−/− mice in this model is problematic, however, since the B-cell compartment is an important site of latent infection (10, 11, 36, 48, 50). We thus took advantage of the availability of HELMET mice, which possess B cells but do not produce antiviral antibody. Importantly, like in wild-type and B-cell−/− mice, latent γHV68 infection is established in HELMET mice (Fig. 4) (Brett et al., unpublished data). As observed in wild-type and B-cell−/− mice, the protective effect of vaccination against latency was quite robust in HELMET animals. We cannot rule out the possibility than HELMET mice and B-cell−/− possess immune defects other than the inability to mount an antiviral antibody response, but such putative defects do no prevent a protective vaccination response against the establishment of latency.
That CD4 T cells were required in the absence of antibody (Fig. 5) suggests that antibody and T cells play redundant roles in protection against the establishment of latency. However, it should be acknowledged that, in a background in which components of the immune response are absent (such as in HELMET mice), it is conceivable that compensation from other immune components could be more robust than would be observed in wild-type mice.
Role of CD4 T cells and CD8 T cells in vaccination against latency.
Although humoral immunity was clearly involved in limiting latent infection following vaccination, antibody was not required for protection against gammaherpesvirus latency (Fig. 3 and 4). In the absence of a specific antibody response, CD4 but not CD8 T cells were required to prevent the establishment of latent infection in the peritoneal cells. This contrasted with the finding by Liu et al. that vaccination with immunodominant major histocompatibility complex (MHC) class I epitopes but not a class II epitope could attenuate peak splenic latency (20). These conflicting findings may be reconciled by the fact that our vaccination protocol is most likely priming a response to many class II epitopes rather than a single one. Indeed, the requirement of CD4 T cells and MHC class II molecules for normal control of chronic infection in class II−/− mice is well documented (1).
Depletion of CD8 T cells from vaccinated HELMET animals did not result in a loss of the protective effect of vaccination, suggesting that CD8 T cells were not required for protection against the establishment of latency. This does not indicate, however, that immune CD8 T cells have no role in vaccination against latency. Indeed, in HELMET mice that were vaccinated and challenged, simultaneous depletion of both CD4 and CD8 T cells resulted in significantly more establishment of challenge virus latency than did depletion of CD4 T cells alone. Thus, induction of a CD8 T-cell response contributed to limiting the establishment of peritoneal latency. This finding is consistent with studies demonstrating that vaccination with MHC class I epitopes from either of two lytic cycle antigens or the latency-associated antigen M2 induces a specific CD8 T-cell response that correlates with a reduction in peak latency (16, 41, 42) and a decrease in latency early after infection. Thus, it is possible that the loss of protection observed when CD4 T cells were depleted may in part reflect a loss of CD4 T-cell help to CD8 T cells.
Differences observed in the roles of CD4 and CD8 T cells in the peritoneum compared to the spleen.
The protective effect of vaccination against latency was particularly robust in the spleen. In contrast to the case for the peritoneum, vaccination was able to prevent the establishment of latent infection even following the depletion of CD4 T cells from antibody-deficient mice. This is consistent with previous findings that the immune effector mechanisms that control latency in the spleen are distinct from those controlling latency in the peritoneum. Specifically, the cytokine gamma interferon has been shown to be critical to the control of latency in the peritoneum, while control of splenic latency is perforin dependent (40). We do not know which cells are the primary sources of these molecules in either the spleen or peritoneum, but site-specific differences in either the production of these molecules or their target might partially underlie the differences in vaccination against latency reported here.
The studies reported here do not fully explain the mechanism of protection in the spleen. Even following depletion of both CD4 and CD8 T cells from antibody-deficient mice, vaccination resulted in significant protection against the establishment of challenge virus latency in the spleen (Fig. 5C and D). One explanation for this would be the presence of residual T cells following depletion that were sufficient to limit latent infection. This is unlikely, since depletion was >98% effective. Vaccination may have also primed other components of the adaptive immune response, such as γδ T cells, that are protective against the establishment of splenic latency.
Prior infection with γHV68.v-cyclin.LacZ may also have rendered the spleen partially resistant to challenge virus latency via mechanisms that are not antigen specific. For example, vaccination may have induced a nonspecific inflammatory or antiviral state that prevented the spread of the challenge virus to the spleen. Prior i.p. infection with vaccinia virus does not result in an alteration in γHV68 latency upon subsequent challenge (39), but we cannot rule out the possibility that a nonspecific herpesvirus-induced inflammatory state contributed to protection against challenge virus latency in the spleen. It is also possible that the vaccine virus may have “saturated” a limited set of splenocytes that are permissive to subsequent latent infection, rendering the spleen resistant to the challenge virus. The finding that the magnitude of latent infection is independent of the infective dose and route may indicate that there is indeed a latency “niche” that can be saturated (38). However, while non-antigen-specific effects of vaccination may have a role, protection against latency in both the spleen and peritoneal cells was at least partially immunologic in nature, since depletion of CD4 T cells (Fig. 5A and B) or of both CD4 and CD8 T cells (Fig. 5) diminished the protective effect of vaccination.
One additional concern regarding T-cell depletion studies is that, in addition to expression on CD8 T cells, CD8α is expressed on a subset of dendritic cells. Thus, it is possible that the use of anti-CD8 antibody treatment might mask an otherwise positive effect of CD8 T-cell depletion by simultaneously destroying an important site of latent infection. However, Flaño et al. reported that in the spleen, dendritc cells represent roughly 40% of the total population of latently infected splenocytes recovered following cell sorting (10), and of the total splenic dendritic cell population, only 23% are CD8α+ (30). Thus, even if anti-CD8 treatment destroyed 100% of the CD8α+ dendritc cell population, the maximum effect on the frequency of latently infected cells would be approximately 10%. Furthermore, we did not observe a significant decrease in the level of latent infection by the vaccine virus following administration of either anti-CD8 or anti-CD4 antibody (data not shown). Thus, it is very unlikely that depletion of dendritic cells by anti-CD8 antibody confounds any of our results.
Mechanisms of T-cell vaccination against latency.
With regard to protection against latency, CD4 T cells were effective in the absence of both CD8 T cells and an antiviral antibody response. This suggests that CD4 T cells directly limit infection rather than provide help to B cells or CD8 T cells. However, the mechanisms by which CD4 T cells directly mediate protection against latency are not known. EBV-specific CD4 T-cell effectors have been shown to be cytotoxic to EBV-transformed cell lines, possibly via the perforin, Fas/Fas ligand, or TRAIL pathways (17, 25, 33). Studies using the γHV68 system have demonstrated the importance of gamma interferon (4, 8, 40) and perforin (40) in the control of latent infection. Further studies will be necessary to determine whether these or other CD4 T-cell effector mechanisms are responsible for protection against the establishment of latent gammaherpesvirus infection. These studies will be facilitated by the development of the transgenic CD4 T-cell system reported by Sparks-Thissen et al. (31a).
Acknowledgments
This work was supported by grant RO1 CA96511 to H.W.V. J.S.M. was supported by a predoctoral training grant in tumor immunology from the Cancer Research Institute. S.A.T. is a Leukemia and Lymphoma Society Special Fellow (grant 3460-04).
We thank Darren Kreamalmeyer for his assistance in mouse breeding and the members of the Virgin lab and Sam Speck's lab for helpful discussions.
REFERENCES
- 1.Cardin, R. D., J. W. Brooks, S. R. Sarawar, and P. C. Doherty. 1996. Progressive loss of CD8+ T cell-mediated control of a gamma-herpesvirus in the absence of CD4+ T cells. J. Exp. Med. 184:863-871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang, J. Y., E. M. Johnson, Jr., and P. D. Olivo. 1991. A gene delivery/recall system for neurons which utilizes ribonucleotide reductase-negative herpes simplex viruses. Virology 185:437-440. [DOI] [PubMed] [Google Scholar]
- 3.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 4.Christensen, J. P., R. D. Cardin, K. C. Branum, and P. C. Doherty. 1999. CD4(+) T cell-mediated control of a gamma-herpesvirus in B cell-deficient mice is mediated by IFN-gamma. Proc. Natl. Acad. Sci. USA 96:5135-5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cobbold, S. P., A. Jayasuriya, A. Nash, T. D. Prospero, and H. Waldmann. 1984. Therapy with monoclonal antibodies by elimination of T-cell subsets in vivo. Nature 312:548-551. [DOI] [PubMed] [Google Scholar]
- 6.Da Costa, X. J., C. A. Jones, and D. M. Knipe. 1999. Immunization against genital herpes with a vaccine virus that has defects in productive and latent infection. Proc. Natl. Acad. Sci. USA 96:6994-6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doherty, P. C., J. P. Christensen, G. T. Belz, P. G. Stevenson, and M. Y. Sangster. 2001. Dissecting the host response to a gamma-herpesvirus. Philos. Trans. R. Soc. London B 356:581-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dutia, B. M., C. J. Clarke, D. J. Allen, and A. A. Nash. 1997. Pathological changes in the spleens of gamma interferon receptor-deficient mice infected with murine gammaherpesvirus: a role for CD8 T cells. J. Virol. 71:4278-4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ehtisham, S., N. P. Sunil-Chandra, and A. A. Nash. 1993. Pathogenesis of murine gammaherpesvirus infection in mice deficient in CD4 and CD8 T cells. J. Virol. 67:5247-5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flaño, E., S. M. Husain, J. T. Sample, D. L. Woodland, and M. A. Blackman. 2000. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J. Immunol. 165:1074-1081. [DOI] [PubMed] [Google Scholar]
- 11.Flaño, E., I. J. Kim, D. L. Woodland, and M. A. Blackman. 2002. γ-Herpesvirus latency is preferentially maintained in splenic germinal center and memory B cells. J. Exp. Med. 196:1363-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gangappa, S., S. B. Kapadia, S. H. Speck, and H. W. Virgin IV. 2002. Antibody to a lytic cycle viral protein decreases gammaherpesvirus latency in B-cell-deficient mice. J. Virol. 76:11460-11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gangappa, S., L. F. Van Dyk, T. J. Jewett, S. H. Speck, and H. W. Virgin IV. 2002. Identification of the in vivo role of a viral bcl-2. J. Exp. Med. 195:931-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodnow, C. C., J. Crosbie, S. Adelstein, T. B. Lavoie, S. J. Smith-Gill, R. A. Brink, H. Pritchard-Briscoe, J. S. Wotherspoon, R. H. Loblay, and K. Raphael. 1988. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334:676-682. [DOI] [PubMed] [Google Scholar]
- 15.Hoge, A. T., S. B. Hendrickson, and W. H. Burns. 2000. Murine gammaherpesvirus 68 cyclin D homologue is required for efficient reactivation from latency. J. Virol. 74:7016-7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Husain, S. M., E. J. Usherwood, H. Dyson, C. Coleclough, M. A. Coppola, D. L. Woodland, M. A. Blackman, J. P. Stewart, and J. T. Sample. 1999. Murine gammaherpesvirus M2 gene is latency-associated and its protein a target for CD8(+) T lymphocytes. Proc. Natl. Acad. Sci. USA 96:7508-7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khanolkar, A., H. Yagita, and M. J. Cannon. 2001. Preferential utilization of the perforin/granzyme pathway for lysis of Epstein-Barr virus-transformed lymphoblastoid cells by virus-specific CD4+ T cells. Virology 287:79-88. [DOI] [PubMed] [Google Scholar]
- 18.Kim, I. J., E. Flaño, D. L. Woodland, and M. A. Blackman. 2002. Antibody-mediated control of persistent γ-herpesvirus infection. J. Immunol. 168:3958-3964. [DOI] [PubMed] [Google Scholar]
- 19.Kitamura, D., J. Roes, R. Kuhn, and K. Rajewsky. 1991. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 350:423-426. [DOI] [PubMed] [Google Scholar]
- 20.Liu, L., E. J. Usherwood, M. A. Blackman, and D. L. Woodland. 1999. T-cell vaccination alters the course of murine herpesvirus 68 infection and the establishment of viral latency in mice. J. Virol. 73:9849-9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacDonald, M. R., X. Y. Li, R. M. Stenberg, A. E. Campbell, and H. W. Virgin IV. 1998. Mucosal and parenteral vaccination against acute and latent murine cytomegalovirus (MCMV) infection by using an attenuated MCMV mutant. J. Virol. 72:442-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore, P. S., and Y. Chang. 2001. Kaposi's sarcoma-associated herpesvirus, p. 2803-2833. In D. M. Knipe and P. Howley (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 23.Morrison, L. A., and D. M. Knipe. 1994. Immunization with replication-defective mutants of herpes simplex virus type 1: sites of immune intervention in pathogenesis of challenge virus infection. J. Virol. 68:689-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrison, L. A., and D. M. Knipe. 1997. Contributions of antibody and T cell subsets to protection elicited by immunization with a replication-defective mutant of herpes simplex virus type 1. Virology 239:315-326. [DOI] [PubMed] [Google Scholar]
- 25.Munz, C., K. L. Bickham, M. Subklewe, M. L. Tsang, A. Chahroudi, M. G. Kurilla, D. Zhang, M. O'Donnell, and R. M. Steinman. 2000. Human CD4(+) T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA1. J. Exp. Med. 191:1649-1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polic, B., H. Hengel, A. Krmpotic, J. Trgovcich, I. Pavic, P. Lucin, S. Jonjic, and U. H. Koszinowski. 1998. Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J. Exp. Med. 188:1047-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radka, S. F., C. Machamer, P. Cresswell, D. D. Kostyu, F. E. Ward, and D. B. Amos. 1983. SFR3-DR5, a monoclonal antibody with HLA-DR5 specificity. J. Immunol. 130:1863-1866. [PubMed] [Google Scholar]
- 28.Rickinson, A. B., and E. Kieff. 2001. Epstein-Barr virus, p. 2575-2627. In D. M. Knipe and P. Howley (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 29.Savoie, A., C. Perpete, L. Carpentier, J. Joncas, and C. Alfieri. 1994. Direct correlation between the load of Epstein-Barr virus-infected lymphocytes in the peripheral blood of pediatric transplant patients and risk of lymphoproliferative disease. Blood 83:2715-2722. [PubMed] [Google Scholar]
- 30.Shortman, K., and Y. J. Liu. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151-161. [DOI] [PubMed] [Google Scholar]
- 31.Smith, S. C., and P. M. Allen. 1991. Myosin-induced acute myocarditis is a T cell-mediated disease. J. Immunol. 147:2141-2147. [PubMed] [Google Scholar]
- 31a.Sparks-Thissen, R. L., D. C. Braaten, S. Kreher, S. H. Speck, and H. W. Virgin IV. 2004. An optimized CD4 T-cell response can control productive and latent gammherpesvirus infection. J. Virol. 78:6827-6835. [DOI] [PMC free article] [PubMed]
- 32.Stewart, J. P., N. Micali, E. J. Usherwood, L. Bonina, and A. A. Nash. 1999. Murine gamma-herpesvirus 68 glycoprotein 150 protects against virus-induced mononucleosis: a model system for gamma-herpesvirus vaccination. Vaccine 17:152-157. [DOI] [PubMed] [Google Scholar]
- 33.Sun, Q., R. L. Burton, and K. G. Lucas. 2002. Cytokine production and cytolytic mechanism of CD4(+) cytotoxic T lymphocytes in ex vivo expanded therapeutic Epstein-Barr virus-specific T-cell cultures. Blood 99:3302-3309. [DOI] [PubMed] [Google Scholar]
- 34.Sunil-Chandra, N. P., J. Arno, J. Fazakerley, and A. A. Nash. 1994. Lymphoproliferative disease in mice infected with murine gammaherpesvirus 68. Am. J. Pathol. 145:818-826. [PMC free article] [PubMed] [Google Scholar]
- 35.Sunil-Chandra, N. P., S. Efstathiou, J. Arno, and A. A. Nash. 1992. Virological and pathological features of mice infected with murine gammaherpesvirus 68. J. Gen. Virol. 73:2347-2356. [DOI] [PubMed] [Google Scholar]
- 36.Sunil-Chandra, N. P., S. Efstathiou, and A. A. Nash. 1992. Murine gammaherpesvirus 68 establishes a latent infection in mouse B lymphocytes in vivo. J. Gen. Virol. 73:3275-3279. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi, M., T. Otsuka, Y. Okuno, Y. Asano, and T. Yazaki. 1974. Live vaccine used to prevent the spread of varicella in children in hospital. Lancet ii:1288-1290. [DOI] [PubMed]
- 38.Tibbetts, S. A., J. Loh, V. van Berkel, J. S. McClellan, M. A. Jacoby, S. B. Kapadia, S. H. Speck, and H. W. Virgin IV. 2003. Establishment and maintenance of gammaherpesvirus latency are independent of infective dose and route of infection. J. Virol. 77:7696-7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tibbetts, S. A., J. S. McClellan, S. Gangappa, S. H. Speck, and H. W. Virgin IV. 2003. Effective vaccination against long-term gammaherpesvirus latency. J. Virol. 77:2522-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tibbetts, S. A., L. Van Dyk, S. H. Speck, and H. W. Virgin IV. 2002. Immune control of the number and reactivation phenotype of cells latently infected with a gamma-herpesvirus. J. Virol. 76:7125-7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Usherwood, E. J., D. J. Roy, K. Ward, S. L. Surman, B. M. Dutia, M. A. Blackman, J. P. Stewart, and D. L. Woodland. 2000. Control of gammaherpesvirus latency by latent antigen-specific CD8(+) T cells. J. Exp. Med. 192:943-952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Usherwood, E. J., K. A. Ward, M. A. Blackman, J. P. Stewart, and D. L. Woodland. 2001. Latent antigen vaccination in a model gammaherpesvirus infection. J. Virol. 75:8283-8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Dyk, L. F., H. W. Virgin IV, and S. H. Speck. 2000. The murine gammaherpesvirus 68 v-cyclin is a critical regulator of reactivation from latency. J. Virol. 74:7451-7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Dyk, L. F., H. W. Virgin IV, and S. H. Speck. 2003. Maintenance of gammaherpesvirus latency requires viral cyclin in the absence of B lymphocytes. J. Virol. 77:5118-5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weck, K. E., M. L. Barkon, L. I. Yoo, S. H. Speck, and H. W. Virgin IV. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 70:6775-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weck, K. E., A. J. Dal Canto, J. D. Gould, A. K. O'Guin, K. A. Roth, J. E. Saffitz, S. H. Speck, and H. W. Virgin. 1997. Murine gammaherpesvirus 68 causes severe large vessel arteritis in mice lacking interferon-gamma responsiveness: a new model for virus induced vascular disease. Nature Medicine. 3:1346-1353. [DOI] [PubMed] [Google Scholar]
- 47.Weck, K. E., S. S. Kim, H. W. Virgin IV, and S. H. Speck. 1999. B cells regulate murine gammaherpesvirus 68 latency. J. Virol. 73:4651-4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weck, K. E., S. S. Kim, H. W. Virgin IV, and S. H. Speck. 1999. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J. Virol. 73:3273-3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weibel, R. E., B. J. Neff, B. J. Kuter, H. A. Guess, C. A. Rothenberger, A. J. Fitzgerald, K. A. Connor, A. A. McLean, M. R. Hilleman, E. B. Buynak, et al. 1984. Live attenuated varicella virus vaccine. Efficacy trial in healthy children. N. Engl. J. Med. 310:1409-1415. [DOI] [PubMed] [Google Scholar]
- 50.Willer, D. O., and S. H. Speck. 2003. Long-term latent murine gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J. Virol. 77:8310-8321. [DOI] [PMC free article] [PubMed] [Google Scholar]